

Abstract
Mutations in the FBXO7 (PARK15) gene have been implicated in a juvenile form of parkinsonism termed parkinsonian pyramidal syndrome (PPS), characterized by Parkinsonian symptoms and pyramidal tract signs. FBXO7 (F‐box protein only 7) is a subunit of the SCF (SKP1/cullin‐1/F‐box protein) E3 ubiquitin ligase complex, but its relevance and function in neurons remain to be elucidated. Here, we report that the E3 ligase FBXO7‐SCF binds to and ubiquitinates the proteasomal subunit PSMA2. In addition, we show that FBXO7 is a proteasome‐associated protein involved in proteasome assembly. In FBXO7 knockout mice, we find reduced proteasome activity and early‐onset motor deficits together with premature death. In addition, we demonstrate that NEX (neuronal helix–loop–helix protein‐1)‐Cre‐induced deletion of the FBXO7 gene in forebrain neurons or the loss of FBXO7 in tyrosine hydroxylase (TH)‐positive neurons results in motor defects, reminiscent of the phenotype in PARK15 patients. Taken together, our study establishes a vital role for FBXO7 in neurons, which is required for proper motor control and accentuates the importance of FBXO7 in proteasome function.
Keywords: PARK15, FBXO7, ubiquitination, parkinsonism, PSMA2
Subject Categories: Neuroscience; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Parkinson's disease (PD) is a highly debilitating movement disorder of mostly idiopathic origin. Recently, mutations in the FBXO7 gene were associated with early‐onset parkinsonism (PARK15) (Shojaee et al, 2008; Di Fonzo et al, 2009; Paisan‐Ruiz et al, 2010; Gunduz et al, 2014; Yalcin‐Cakmakli et al, 2014). The disease is characterized by motor deficits typically found in Parkinson's disease patients in addition to pyramidal tract signs (Shojaee et al, 2008; Di Fonzo et al, 2009; Paisan‐Ruiz et al, 2010).
Interestingly, examination of skin fibroblasts isolated from affected PARK15 patients revealed the loss of the F‐box protein FBXO7, indicating a decrease in protein stability due to the mutations (Zhao et al, 2011). PARK15 patients respond to Levodopa treatment, suggesting a dopamine deficiency characteristic of PD, but show severe side effects such as dyskinesia and behavioral disturbances (Di Fonzo et al, 2009; Paisan‐Ruiz et al, 2010). While some of the PARK15 patients present with mild cortical atrophy and presynaptic abnormalities in brain scans (Di Fonzo et al, 2009; Lohmann et al, 2009), the neuropathology of patients remains to be determined.
FBXO7 is a subunit of the cullin‐1‐based SCF (SKP1/cullin‐1/F‐box protein) complex (Zheng et al, 2002; Cardozo & Pagano, 2004; Jin et al, 2004). The scaffold proteins cullin‐1 and SKP1, the RING subunit Rbx1 together with the substrate‐recruiting subunit FBXO7 form a functional E3 ubiquitin ligase. Initially introduced as a potential oncogene (Laman et al, 2005), interesting lines of research link FBXO7 to mitochondrial and proteasomal regulation. Together with parkin and PINK1, FBXO7 was implicated in mitochondrial maintenance and mitophagy (Burchell et al, 2013). Due to its interaction with the proteasomal inhibitor 31 (PI31), FBXO7 is speculated to be involved in regulation of proteasomal activity (Kirk et al, 2008). Taken together, while the PARK15 mutations link FBXO7 to parkinsonism, FBXO7's relevance in the brain and its role in neurons remain to be explored.
In this study, we examined the systemic loss of the PARK15 gene in mice on motor behavior and neuropathology. In addition, we generated conditional mouse models to establish the relevance of FBXO7 in neurons. At the molecular level, we characterized a newly identified interactor and substrate of the E3 ligase FBXO7‐SCF in the control of proteasome activity.
Results
Systemic loss of FBXO7 in mice triggers motor defects and premature death
Motor symptoms in patients carrying mutations in the FBXO7 gene suggest a role for FBXO7 in the brain. To investigate the function of FBXO7 in neurons, we studied its expression in the rodent brain. We first confirmed endogenous FBXO7 protein expression in the brain and spinal cord as well as heart, kidney, liver, and spleen in rat (Fig 1A). Moreover, FBXO7 shows a stable expression from embryonic to adult stages in cortex and hippocampus and a declining expression in the cerebellum (Fig EV1A–C).
Figure 1. Characterization of FBXO7 −/− mice.
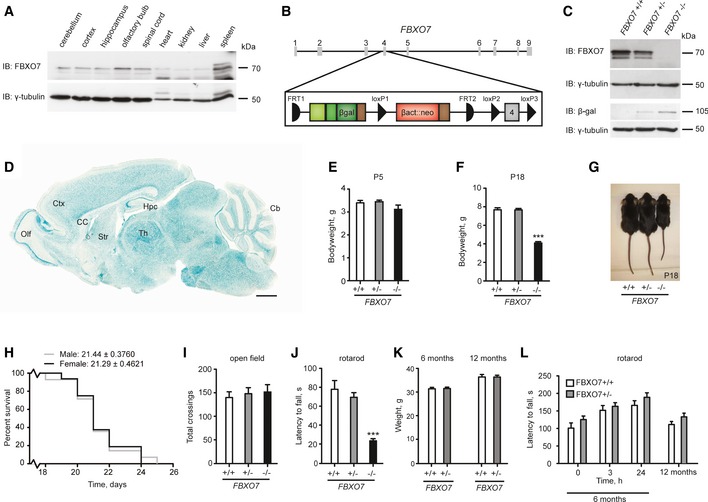
-
ALysates of the indicated tissues isolated from P30 rat were immunoblotted with FBXO7 and γ‐tubulin antibodies. The latter served as a loading control.
-
BSchematic of the genetically modified PARK15 gene locus located on chromosome 22.
-
CBrain lysates from P5 FBXO7 +/+, FBXO7 +/−, or FBXO7 −/− mice were immunoblotted with FBXO7, βgal or γ‐tubulin antibodies. The latter served as a loading control.
-
DLacZ staining of P18 FBXO7 −/− sagittal brain section. Ctx = cortex, Hpc = hippocampus, Cb = cerebellum, Olf = olfactory bulb, CC = corpus callosum, Str = striatum, Th = thalamus. Scale bar = 1 mm.
-
E, FBody weight of P5 or P18 FBXO7 +/+, FBXO7 +/−, or FBXO7 −/− mice. n = 17, 46, 16 (E) and n = 16, 25, 11 (F), respectively (ANOVA, ***P < 0.001, mean ± s.e.m.).
-
GImage of representative P18 FBXO7 +/+, FBXO7 +/− or FBXO7 −/− mice.
-
HKaplan–Meier survival curve of male and female FBXO7 −/− mice. n = 14 and 16, respectively.
-
I, JP18 FBXO7 +/+, FBXO7 +/−, or FBXO7 −/− littermates were tested in the open field (I) and on the rotarod (J). n = 20, 23, 16, respectively (ANOVA, ***P < 0.001, mean ± s.e.m.).
-
KAverage weight of 6‐month‐ and 12‐month‐old FBXO7 +/+ and FBXO7 +/− littermates. n = 15 and 18, respectively (ANOVA, mean ± s.e.m.).
-
L6‐month‐ and 12‐month‐old FBXO7 +/+ and FBXO7 +/− littermates were examined on the rotarod. n = 15 and 18, respectively (ANOVA, mean ± s.e.m.).
Source data are available online for this figure.
Figure EV1. Characterization of FBXO7 −/− mice.
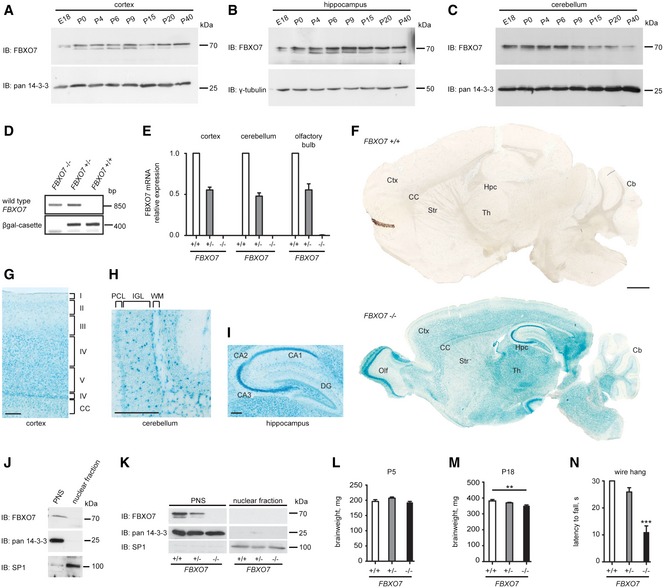
-
A–CLysates of cortices (A), hippocampi (B), and cerebella (C) of the indicated age isolated from rat were immunoblotted with FBXO7, pan 14‐3‐3, or γ‐tubulin antibodies. The latter two served as loading controls.
-
DGenotyping of FBXO7 +/+, FBXO7 +/−, and FBXO7 −/− mice. Detection of wild‐type and mutant FBXO7 alleles.
-
EQuantitative PCR of FBXO7 +/+, FBXO7 +/− and FBXO7 −/− brain tissue. n = 4, 2, and 4, respectively (mean ± s.e.m.).
-
FLacZ staining of P18 FBXO7 +/+ and FBXO7 −/− sagittal brain sections. Ctx = cortex, Hpc = hippocampus, Cb = cerebellum, Olf = olfactory bulb, CC = corpus callosum, Str = striatum, Th = thalamus. Scale bar = 1 mm.
-
G–IHigher magnification of LacZ‐stained P18 FBXO7 −/− sagittal brain: cortex (G), cerebellum (H), and hippocampus (I). CC = corpus callosum, PCL = Purkinje cell layer, IGL = internal granule cell layer, WM = white matter, DG = dentate gyrus, CA1, 2, 3 = cornus ammonis 1, 2, 3. Scale bars = 200 μm.
-
J, KCultured rat cerebellar granule neurons (J) or mouse cortical tissue (K) was subjected to subcellular fractionation analyses. Nuclear fraction and postnuclear supernatant (PNS) were immunoblotted with FBXO7, SP1, and pan 14‐3‐3 antibodies. The latter two served as quality control for the nuclear fraction and PNS, respectively.
-
L, MBrain weight of P5 and P18 FBXO7 +/+, FBXO7 +/−, or FBXO7 −/− mice. n = 8, 20, and 7 (L) and n = 6, 11, and 8 (M), respectively (ANOVA, **P < 0.01, mean ± s.e.m.).
-
NP18 littermates were tested on the hanging wire. n = 20, 23, and 16, respectively (ANOVA, ***P < 0.001, mean ± s.e.m.).
Source data are available online for this figure.
A recent report demonstrated the loss of FBXO7 in patients harboring compound mutations in the FBXO7 gene (Zhao et al, 2011). We thus generated a conventional FBXO7 knockout mouse (C57B6/6N JM8.N4 Fbxo7tm1a(EUCOMM)Hmgu) (Fig 1B), in which the fourth exon of FBXO7 was deleted, resulting in a truncated protein. Genotyping and immunoblotting analyses confirmed disruption of the FBXO7 gene together with expression of the LacZ reporter cassette in brain lysates of postnatal day (P) 5 mice (Figs 1C and EV1D), thus validating the specificity of the FBXO7 antibody. We also confirmed the absence of full‐length FBXO7 mRNA in FBXO7 −/− mouse brains (Fig EV1E). To characterize the spatial distribution of FBXO7 expression in the brain, we took advantage of the FBXO7 promoter‐driven LacZ cassette. Sagittal brain sections of P18 FBXO7 −/− mice displayed a specific and widespread β‐galactosidase staining in different regions including cortex, hippocampus, cerebellum, olfactory bulb, midbrain, and striatum (Figs 1D and EV1F–I). To resolve the subcellular localization of FBXO7 in murine neuronal tissue, we used the validated FBXO7 antibody and found endogenous FBXO7 to be mainly localized to the cytoplasm (Fig EV1J and K).
PARK15 patients show an early onset of severe motor symptoms followed by rapid decline; hence, we characterized the FBXO7 −/− mice from an early age. Pups obtained from matings of FBXO7 +/− mice were born at expected Mendelian ratio (+/+: +/−: −/− = 1.02: 2.12: 0.86), excluding embryonic lethality. At P5, we observed no difference in body or brain weight of FBXO7 −/− and control littermates (Figs 1E and EV1L), while at P18 the FBXO7 −/− mice displayed significantly lower body and brain weights (Figs 1F and G, and EV1M), and obvious motor deficits. FBXO7 −/− mice expired without exception at the beginning of the 4th postnatal week, independently of gender (Fig 1H).
To quantify the motor phenotype of the FBXO7 −/− mice, we tested P18 pups as older mice deteriorated rapidly. With the open field test, we monitored the mice's ambulance, but despite a slight stiffness in gait, there was no difference in distance travelled between genotypes (Fig 1I). FBXO7 −/− mice however were unable to perform the rotarod test (Fig 1J). In accordance with this, FBXO7 −/− mice were also unable to complete the wire hang test (Fig EV1N), suggesting reduced muscle strength. FBXO7 heterozygosity was sufficient to sustain weight (Fig 1K) and motor skills since FBXO7 +/− mice performed well on the rotarod during the testing period of 12 months (Fig 1L). Our results demonstrate that systemic loss of FBXO7 in the mouse is characterized by early‐onset motor deficits and premature death.
Since PARK15 patients are diagnosed with a variant of juvenile parkinsonism and have been reported to respond to L‐dopa treatment (Di Fonzo et al, 2009; Lohmann et al, 2015), we first inquired whether loss of FBXO7 affected the dopamine level in the striatum. Using HPLC analysis, we measured dopamine and its metabolites but found no change in the striatum of P18 FBXO7 −/− mice as compared to their littermates (Fig 2A–D). Consistently, stereological analysis revealed no difference in TH+ neurons in the substantia nigra of FBXO7 −/− mice (Fig 2E). Further neuropathological investigations of the FBXO7 −/− brain did not reveal any α‐synuclein+ protein deposits, the hallmark of PD (Appendix Fig S1A). We also did not find a change in the number of Iba1+ microglia in FBXO7 −/− brains, but we observed astrogliosis as the area occupied by GFAP+ astrocytes increased significantly (Fig 2F and Appendix Fig S1B). While the SN showed no loss of TH+ neurons, we found a slight but significant increase in TUNEL+ apoptotic cells in the cortex (Fig 2G) and a non‐significant increase in apoptotic cells in cerebellum and hippocampus of FBXO7 −/− brains (Fig 2H). We also determined the number of apoptotic, cleaved caspase‐3+ cells, and found an upward trend in the FBXO7 −/− cortices (Appendix Fig S1C). We then determined if FBXO7 contributes to neuronal integrity by examining its potential prosurvival function in a less supportive environment using cultured cortical neurons. Using FBXO7 shRNA, which were validated both in HEK293T cells and in cultured cortical neurons (Appendix Fig S1D and E), we transfected these neurons with control vector, a functional FBXO7 shRNA plasmid, or a non‐functional FBXO7 shRNA plasmid and counted apoptotic neurons 5 days later. We found a 2.5‐fold increase in cell death upon knockdown of FBXO7 (Fig 2I). These experiments indicate that systemic loss of FBXO7 induces astrogliosis and may negatively affect the neurons' health.
Figure 2. Histological analyses of the FBXO7 −/− mice.
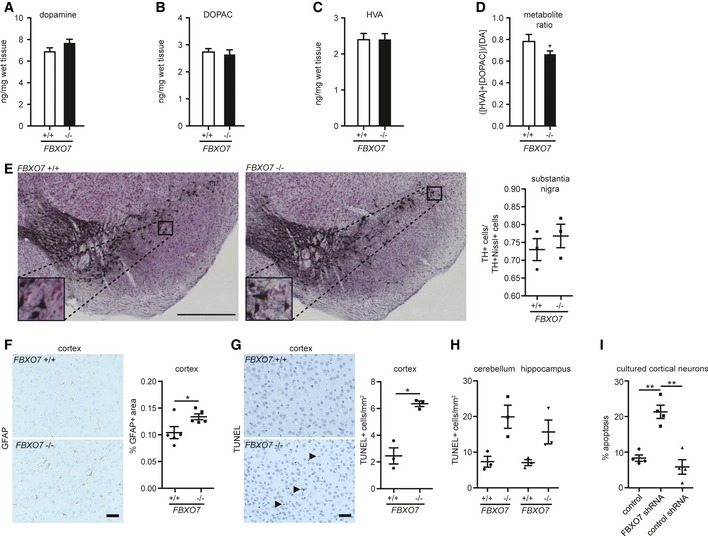
-
A–DStriatal tissue from P18 FBXO7 +/+ and FBXO7 −/− mice was subjected to HPLC analyses to determine dopamine (A), 3,4‐dihydroxyphenylacetic acid (DOPAC) (B), and homovanillic acid (HVA) (C) concentrations along with the metabolite ratio (D). n = 12 and 13, respectively (unpaired t‐test, *P < 0.05, mean ± s.e.m.).
-
ERepresentative images from FBXO7 +/+ and FBXO7 −/− midbrains. Insets display higher magnification of TH+ neurons of substantia nigra. Scale bar = 500 μm. Stereological analysis of the ratio of TH+Nissl+ cells/Nissl+ cells in the substantia nigra. Three independent litter pairs were analyzed (paired t‐test, mean ± s.e.m.)
-
FSagittal paraffin sections of brains from P18 FBXO7+/+ and FBXO7−/− mice were subjected to immunohistochemistry using GFAP antibody. Five independent litter pairs were analyzed (paired t‐test, *P < 0.05, mean ± s.e.m.). Scale bar = 40 μm.
-
GRepresentative images of sagittal paraffin sections of cortices from P18 FBXO7 +/+ and FBXO7 −/− mice that were subjected to TUNEL staining. Three independent litter pairs were analyzed (paired t‐test, *P < 0.05, mean ± s.e.m.). Arrows indicate TUNEL+ cells. Scale bar = 40 μm.
-
HAnalyses of TUNEL staining of cerebellum and hippocampus. Three independent litter pairs were examined (paired t‐test, mean ± s.e.m.).
-
ICortical neurons were transfected at day in vitro (DIV) 3 with control vector, a functional FBXO7 shRNA plasmid, or a non‐functional FBXO7 shRNA plasmid together with a transfection marker. At DIV6, apoptotic neurons were counted. Four independent experiments were included in the analysis (ANOVA, **P < 0.01, mean ± s.e.m.).
FBXO7 interacts directly with the proteasome by binding to its novel ubiquitination target PSMA2
Disruption of the FBXO7 gene in mice shows that a loss of the E3 ubiquitin ligase FBXO7 has detrimental consequences for the organism. While recent studies have implicated FBXO7 in several cellular systems (Nelson et al, 2013), mechanistic insight into its neuronal function is sparse. As a component of an SCF E3 ligase, FBXO7 is anticipated to serve as its substrate‐recruiting subunit (Jin et al, 2004), but this function however remains underexplored. To identify neuronal FBXO7‐SCF ligase substrates, we carried out a yeast two‐hybrid screen using a fetal brain library. The analysis revealed two already known FBXO7 interaction proteins: the SCF subunit SKP1 and the proteasomal regulator PI31 (Kirk et al, 2008). In addition, we identified the proteasomal subunit α2 (PSMA2) as a potential novel interactor. PSMA2 is part of the proteasomal core particle (CP) that consists of two heptameric α‐rings that bind the regulatory particle (RP), and two heptameric β‐rings, harboring proteolytic activity (Forster et al, 2013; Tomko & Hochstrasser, 2013). This finding is particularly interesting given that mass spectrometry studies identified FBXO7 and SCF components as proteasome‐associated proteins (Bousquet‐Dubouch et al, 2009; Fabre et al, 2015).
We first validated the interaction by forward and reverse co‐immunoprecipitation analyses in heterologous cells (Figs 3A and EV2A) and by in vitro interaction analysis of purified FBXO7 and PSMA2 (Fig 3B). We then carried out mapping analyses, for which we generated various FBXO7 deletion mutants (Fig 3C) and uncovered the ubiquitin‐related domain (UbRD) as the PSMA2‐binding region (Fig 3D). The naming of this N‐terminal domain is in contrast to other reports, since our sequence search (including Ensembl, NCBI, Smart, Pfam) for FBXO7 revealed only a UbRD domain and not a ubiquitin‐like (Ubl) domain. As a positive control for the searches, we entered the E3 ubiquitin ligase parkin, which harbors a Ubl. Having identified the UbRD as binding site excludes the binding of the potential FBXO7 isoform 2 to PSMA2 as it lacks the UbRD domain and hence stresses the selectivity of the FBXO7 isoform 1/PSMA2 interaction.
Figure 3. FBXO7 interacts with the proteasomal subunit PSMA2 and binds to the proteasome.
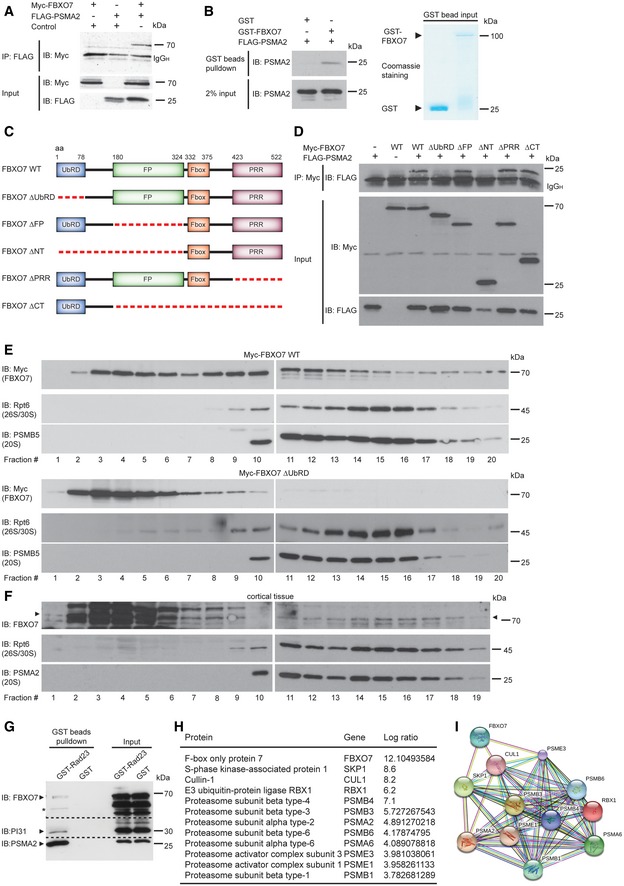
- Lysates of HEK293T cells, transfected with the indicated plasmids, were subjected to immunoprecipitation (IP) with FLAG antibody (PSMA2), followed by immunoblotting (IB) with myc antibody (FBXO7).
- Recombinant PMSA2 together with glutathione bead‐coupled GST‐FBXO7 or GST only was incubated in Co‐IP buffer. Precipitated proteins were subjected to immunoblotting with PSMA2 antibody. 2% input was used as a loading control for PSMA2. GST‐coupled proteins were visualized by Coomassie staining.
- Schematic depicts full‐length FBXO7 and FBXO7 deletion mutants used in mapping analyses. UbRD = ubiquitin‐related domain, FP = FBXO7/PI31 domain, F‐box = F‐box domain, PRR = proline‐rich region.
- The indicated FBXO7 expression plasmids were used to transfect HEK293T cells together with the FLAG‐PSMA2 plasmid. Lysates were then subjected to IP with myc antibody followed by IB with FLAG antibody.
- HEK293T cells were transfected with full‐length myc‐FBXO7 or ΔUbRD and subjected to fractionation using a 10–40% linear glycerol gradient. Fractions were immunoblotted for myc (FBXO7), Rpt6 (26S/30S proteasome), and PSMB5 (20S proteasome).
- Cortical lysates were subjected to fractionation using a 10–40% linear glycerol gradient. Fractions were immunoblotted for FBXO7, Rpt6 (26S/30S proteasome), and PSMA2 (20S proteasome).
- GST or GST‐Rad23 bound to glutathione–Sepharose were used to precipitate proteasomes from brain lysates followed by IB with FBXO7, PI31, or PSMA2 antibody.
- HEK293T control lysates and FLAG‐FBXO7 cells were subjected to immunoprecipitation, followed by digestion. The resulting peptide mixtures were analyzed by MS; ratio FBXO7 versus control; mean of three independent measurements based on the intensity of the relevant protein.
- STRING analysis of the proteins listed in (H) (string‐db.org). PSMA2, PSMA6, PSMB1, PSMB3, PSMB4, PSMB6 are members of the gene ontology term (GO) “proteasome core complex” (GO: 0005839).
Source data are available online for this figure.
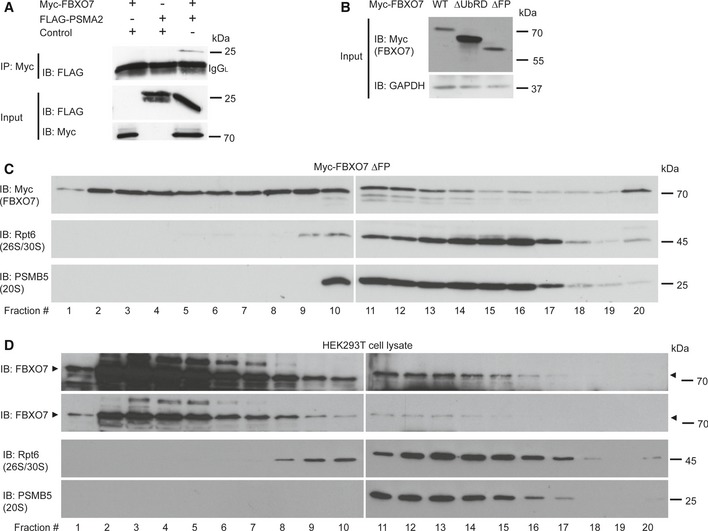
Figure EV2. FBXO7 interacts with the proteasomal subunit PSMA2 and binds to the proteasome.
- Lysates of HEK293T cells, transfected with the indicated plasmids, were subjected to IP with myc antibody for FBXO7, followed by IB with FLAG antibody for PSMA2.
- Input from HEK293T cells, transfected with full‐length myc‐FBXO7, ΔUbRD, or ΔFP (panel C and Fig 3E), was immunoblotted with myc (FBXO7) or GAPDH antibody. The latter served as a loading control.
- HEK293T cells were transfected with myc‐FBXO7 ΔFP and subjected to fractionation using a 10–40% linear glycerol gradient. Fractions were immunoblotted for myc (FBXO7), Rpt6 (26S/30S proteasome), and PSMB5 (20S proteasome).
- HEK293T cells were subjected to fractionation using a 10–40% linear glycerol gradient. Fractions were immunoblotted for FBXO7, Rpt6 (26S/30S proteasome), and PSMB5 (20S proteasome).
Source data are available online for this figure.
To underscore the association of FBXO7 with the proteasome, we carried out fractionation analyses and found that myc‐tagged FBXO7 co‐fractionated with 20S and 26S proteasomes in HEK293T cells (Fig 3E). Deletion of the UbRD domain (FBXO7 ΔUbRD) led to a complete loss of co‐fractionation in the proteasome‐enriched fractions (Fig 3E). We also ruled out PI31‐mediated association of FBXO7 with the proteasome, as the FBXO7 ΔFP mutant, which fails to bind to PI31, co‐fractionated with the proteasome (Fig EV2B and C). To confirm the association of endogenous FBXO7 with the proteasome, we performed further fractionation experiments using HEK293T cells and cortical tissue. Again, we observed that a fraction of FBXO7 co‐fractionated with the proteasome both in heterologous cells and in the brain (Figs 3F and EV2D). To confirm this finding, we carried out pull‐down experiments for which the ubiquitin‐like (Ubl) domain of RAD23B was used to precipitate proteasomes from mouse brain lysates. Subsequent immunoblotting revealed that FBXO7 co‐precipitated with the proteasomal subunit PSMA2 and PI31 (Fig 3G).
To bolster the close contact of FBXO7 with the proteasome, we used Flag‐sepharose to precipitate proteins from control or FLAG‐FBXO7 expressing HEK293T cells and analyzed the precipitated proteins by nano‐LC‐coupled mass spectrometry. We found not only a more than 10‐fold enrichment of SCF subunits, but also of at least eight proteasomal subunits including PSMA2 (Fig 3H). Analysis of this core interactome using STRING (v10, http://string-db.org) demonstrated a tight protein association network (Fig 3I). These results demonstrate that FBXO7 binds to the proteasome via PSMA2 but not PI31 and that FBXO7‐SCF associates with the proteasome holoenzyme.
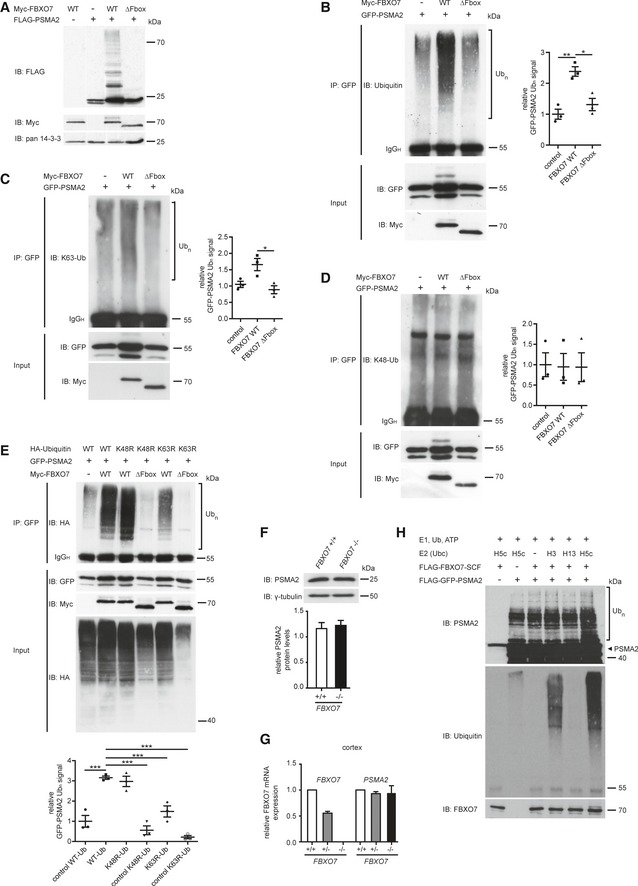
To determine whether PSMA2 is a substrate of the E3 ligase FBXO7‐SCF, we tested whether PSMA2 responded to the active ligase. We expressed PSMA2 together with FBXO7 or FBXO7 ΔF‐box, which fails to form an active SCF E3 ligase complex (Appendix Fig S2A), and examined PSMA2 with immunoblotting. PSMA2 displayed a smear only in the presence of wild‐type FBXO7 (Fig 4A), indicative of ubiquitination. To confirm this, we carried out cell‐based ubiquitination assays for which we transfected HEK293T cells with the PSMA2 expression plasmid together with empty control vector, the FBXO7 or FBXO7 ΔF‐box plasmid. The lysates were subjected to a boiling protocol to prevent non‐specific binding of ubiquitinated proteins to PSMA2. We then analyzed PSMA2 and found that only the presence of active ligase induced a significant polyubiquitination smear, detected by two different ubiquitin antibodies (Fig 4B and Appendix Fig S2B).
Figure 4. FBXO7 ubiquitinates PSMA2.
-
ALysates of HEK293T cells, transfected with empty control vector, myc‐FBXO7, or myc‐FBXO7 ΔF‐box plasmids together with the FLAG‐PSMA2 expression plasmid, were immunoblotted with FLAG, myc, or pan 14‐3‐3 antibody.
-
BLysates of HEK293T cells, transfected with the indicated plasmids, were subjected to boiling, followed by IP with GFP antibody (PSMA2), followed by IB with ubiquitin (DAKO) antibody (upper panel). Inputs were immunoblotted with GFP or myc antibody (lower panels). Three independent blots were quantified and band intensity normalized to control average (ANOVA, *P < 0.05, **P < 0.001, mean ± s.e.m.)
-
C, DLysates of HEK293T cells, transfected as in (B), were first subjected to a denaturing protocol, then to IP with GFP antibody, followed by IB with K63‐specific ubiquitin antibody (C) or K48‐specific antibody (D). Inputs were immunoblotted with GFP or myc antibody (lower panel). Three independent blots were quantified and band intensity normalized to control average (ANOVA, *P < 0.05, mean ± s.e.m.).
-
ELysates of HEK293T cells, transfected with the indicated HA–ubiquitin, GFP‐PSMA2, or myc‐FBXO7 plasmids, were subjected to boiling, followed by IP with GFP antibody (PSMA2), followed by IB with HA antibody (upper panel). Inputs were immunoblotted with GFP, myc or HA antibody (lower panels). Three independent blots were quantified and band intensity normalized to control (lane 2) average (ANOVA, Dunnett's multiple comparison ***P < 0.001, mean ± s.e.m.).
-
FBrain lysates from FBXO7 +/+ and FBXO7 −/− mice immunoblotted with PSMA2 or γ‐tubulin antibody. The latter served as a loading control. Ten brains from each genotype were included in the quantitative analyses (paired t‐test, mean ± s.e.m.).
-
GcDNA generated from cortex isolated from FBXO7 +/+, FBXO7 +/−, and FBXO7 −/− mice, was analyzed with quantitative PCR using FBXO7‐ and PSMA2‐specific primers along with the housekeeping gene β‐actin. n = 4, 2, and 4, respectively (unpaired t‐test, mean ± s.e.m.)
-
HFor the in vitro ubiquitination assay, purified PSMA2, tagged and immunopurified FBXO7, and SCF core components from HEK293T cells, E1 enzyme, ubiquitin, and ATP together with the indicated E2 enzymes were incubated for 1 h at 37°C. The reactions were immunoblotted with PSMA2, ubiquitin, or FBXO7 antibodies.
Source data are available online for this figure.
Ubiquitination serves as an instructive code to modify protein function. Polyubiquitination of proteins by lysine (K) 48‐linked ubiquitin chains triggers their proteasomal degradation while K63‐linked ubiquitin chains predominantly represent non‐proteolytic modifications (Ciechanover et al, 2000; Pickart, 2000; Peng et al, 2003). To further analyze the observed polyubiquitination smear, we performed a cell‐based ubiquitination assay on PSMA2 and found an increase in reactivity for K63 but not for K48 chains (Fig 4C and D). Furthermore, we analyzed PSMA2 polyubiquitination in the presence of ubiquitin mutants K48R or K63R, which are unable to produce K48 or K63 polyubiquitin chains, respectively. Our results revealed a decrease in PSMA2 ubiquitination only with the K63R variant (Fig 4E). These results indicate that FBXO7 facilitates predominantly K63‐linked polyubiquitin chains and thus a non‐proteolytic modification of PSMA2. Consistent with this finding, we found no difference in PSMA2 protein levels in FBXO7 −/− as compared to wild‐type brains (Fig 4F), which was consistent with unchanged transcription of the PSMA2 gene (Fig 4G).
To underscore the FBXO7‐mediated ubiquitination of PSMA2 by usage of K63‐capable E2 enzymes, we carried out in vitro ubiquitination assays. Here, we purified PSMA2 as well as FBXO7 together with exogenous SCF core complex proteins cullin‐1, SKP1, and ROC1 from heterologous cells. These proteins were mixed with E1 enzyme, ubiquitin, ATP in reaction buffer and the E2 enzymes UbcH3, UbcH5c, or UbcH13. The mix was incubated at 37°C and followed by immunoblotting analyses with ubiquitin or PSMA2 antibody. While we observed no response in the control conditions (Fig 4H, lanes 1–3) and no response with the E2 UbcH13 (Fig 4H, lane 5), a ubiquitination response appeared in the presence of the E3 ligase together with the E2 enzyme UbcH5c and less efficiently with UbcH3 (Fig 4H, lanes 6 and 4). UbcH5c has been shown to mediate the transfer of different ubiquitin chains in vitro, including K63 (Jin et al, 2008), supporting our finding that FBXO7‐SCF has the potential to transfer K63 polyubiquitin chains.
FBXO7 affects proteasome function in vivo
To determine the functional relevance of the FBXO7‐PSMA2 interaction, we investigated proteasome activity by measuring the chymotrypsin‐like activity of the proteasome using a fluorogenic peptide substrate. Prior to testing, we confirmed the specific readout of this assay using the proteasome inhibitor lactacystin. We then examined lysates of HEK293T cells, transfected with empty control vector or functional/non‐functional RNAi plasmids for PSMA2 or FBXO7 isoform 1 (Fig EV3A and B). Knockdown of PSMA2 was used as a proof‐of‐principle to demonstrate that reduction in the proteasomal subunit PSMA2 lowered proteasome activity by approximately 20% (Fig 5A). Interestingly, knockdown of FBXO7 also led to a 20% reduction in activity (Fig 5B), indicating the requirement of FBXO7 for fully functional proteasomes. We also observed a significantly reduced proteasome activity in the FBXO7 −/− brains as compared to the wild‐type brains (Figs 5C and EV3C). While we observed the reported destabilization of PI31 in the FBXO7 −/− brains (Fig EV3D), PI31's influence on proteasome activity was negligible (Fig EV3E and F). This is consistent with previous findings showing that neither overexpression nor knockdown of PI31 affects proteasome activity in intact cells (Zaiss et al, 2002; Li et al, 2014).
Figure EV3. FBXO7 ubiquitinates PSMA2 and regulates proteasome assembly.
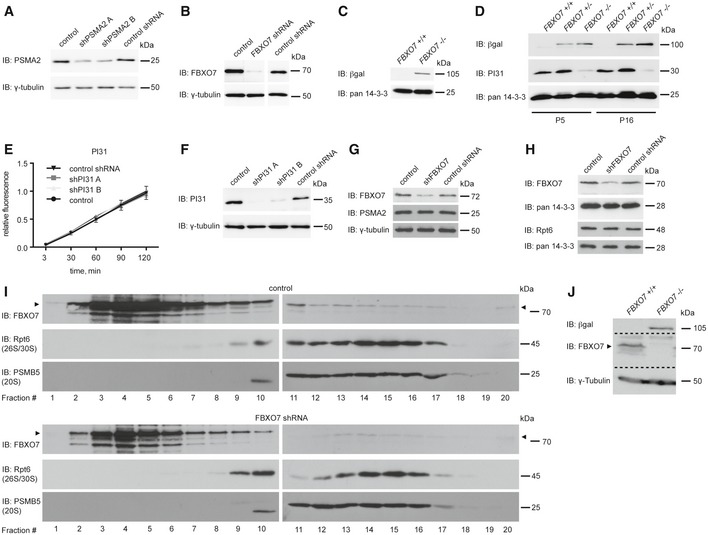
- Control of knockdown for Fig 5A. Lysates of HEK293T cells transfected with empty control vector or PSMA2 shRNA plasmids were immunoblotted with PSMA2 or γ‐tubulin antibody. The latter served as a loading control.
- Control of knockdown for Fig 5B. Lysates of HEK293T cells transfected with empty control vector, FBXO7 shRNA, or non‐functional FBXO7 shRNA plasmids, were immunoblotted with FBXO7 or γ‐tubulin antibody. The latter served as a loading control.
- Control of knockout for Fig 5C. Brain lysates from FBXO7 +/+ or FBXO7 −/− mice were immunoblotted with βgal or pan 14‐3‐3 antibody. The latter served as a loading control.
- Whole brain lysates from FBXO7 +/+ , FBXO7 +/−, and FBXO7 −/− mice at age P5 and P16 were subjected to immunoblotting with βgal and PI31 antibodies. Pan 14‐3‐3 was used as a loading control.
- Lysates from HEK293T cells, transfected with empty control vector, functional PI31 shRNA, or non‐functional PI31 shRNA, were analyzed for chymotrypsin‐like proteasome activity assay (LLVY‐AMC). Three independent experiments were carried out (ANOVA, mean ± s.e.m.).
- Input from (E) was immunoblotted with PI31 or γ‐tubulin antibody. The latter served as a loading control.
- Control of knockdown and loading for Fig 5E. Lysates of HEK293T cells transfected with empty control vector or FBXO7 shRNA plasmids were immunoblotted with FBXO7, PSMA2, or γ‐tubulin antibody. The latter served as a loading control.
- Control of knockdown and loading for Fig 5F. Lysates of HEK293T cells transfected with empty control vector or FBXO7 shRNA plasmids were immunoblotted with FBXO7, Rpt6, or pan14‐3‐3 antibody. The latter served as a loading control.
- HEK293T cells transfected with empty control vector or the FBXO7 RNAi plasmid were subjected to fractionation using a 10–40% linear glycerol gradient. Fractions were subjected to immunoblotting with FBXO7, Rpt6 (26S/30S proteasome), or PSMB5 (20S proteasome) antibody.
- Control of knockout and loading for Fig 5H. Brain lysates from FBXO7 +/+ or FBXO7 −/− mice were immunoblotted with βgal, FBXO7, or γ‐tubulin antibody. The latter served as loading control.
Source data are available online for this figure.
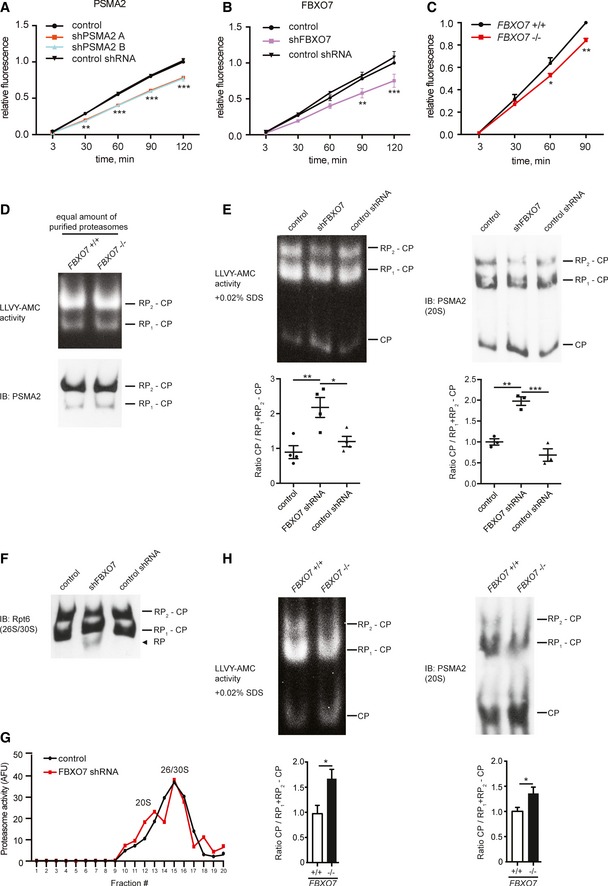
Figure 5. FBXO7 ubiquitinates PSMA2 and regulates proteasome assembly.
-
ALysates from HEK293T cells, transfected with empty control vector, PSMA2 shRNA A, PSMA2 shRNA B, or non‐functional PSMA2 shRNA, were subjected to chymotrypsin‐like proteasome activity assay. Three independent experiments were included in the analysis (ANOVA, **P < 0.01, ***P < 0.001, mean ± s.e.m.).
-
BLysates from HEK293T cells, transfected with control vector, FBXO7 shRNA, or non‐functional FBXO7 shRNA, were subjected to a chymotrypsin‐like proteasome activity assay. Three independent experiments were included in the analysis (ANOVA, **P < 0.01, ***P < 0.001, mean ± s.e.m.).
-
CBrain lysates from wild‐type or homozygous FBXO7 mice were analyzed for chymotrypsin‐like proteasome activity. Three independent experiments were included in the analysis (paired t‐test, *P < 0.05, **P < 0.01, mean ± s.e.m.).
-
DProteasomes purified from FBXO7 +/+ or FBXO7 −/− brains were analyzed by native PAGE, followed by in‐gel chymotrypsin‐like activity assay and immunoblotting with PSMA2 antibody.
-
E, FLysates from HEK293T cells, transfected with empty control vector, FBXO7 shRNA, or non‐functional FBXO7 shRNA, were analyzed by native PAGE followed by in‐gel chymotrypsin‐like proteasome activity assay (LLVY‐AMC), in the presence of SDS, and followed by immunoblotting with PSMA2 (E) or Rpt6 (F) antibody. In (E), ratio of free CPs to RP1 + RP2 − CP was calculated for each lane and normalized to control average. Quantification includes four (activity) and three (IB) independent experiments (ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001, mean ± s.e.m.).
-
GHEK293T cells were transfected with empty control vector or the FBXO7 shRNA plasmid and subjected to fractionation using a 10–40% linear glycerol gradient and fractions were analyzed for chymotrypsin‐like proteasome activity.
-
HBrain lysates from FBXO7 +/+ and FBXO7 −/− mice were analyzed on native PAGE followed by in‐gel chymotrypsin‐like proteasome activity assay and by IB with PSMA2 antibody. Quantification includes six wild‐type and seven FBXO7 −/− brains (activity) and five wild‐type and five FBXO7 −/− brains (IB) (unpaired t‐test, *P < 0.05, mean ± s.e.m.).
Source data are available online for this figure.
PSMA2 together with the other α‐subunits of the 20S core particle regulates the access to the proteolytic chamber of the core particle and directly bind to β‐subunits, some of which harbor protease activity (Finley, 2009). To examine whether the decrease in proteasome activity was due to less efficient gating and thus proteolytic activity, we determined the activity of holoenzymes purified from FBXO7 −/− or wild‐type brains using a native PAGE method followed by an in‐gel proteasome activity assay. Importantly, we found that an equal amount of proteasome holoenzyme displayed equal activity (Fig 5D), indicating that the core particle gating and the proteolytic activity of proteasome holoenzymes are unaffected.
Since PSMA2 and the other α‐subunits are also critical for the binding of the 19S regulatory particle (RP) with the 20S core particle (CP) (Tian et al, 2011; Pathare et al, 2012), we went on to analyze the assembly state of the total proteasome pool in lysates. Proteasome holoenzymes harbor one or two RPs (26/30S), and a minor fraction of proteasomes exist as CPs without RPs. We used HEK293T cells, transfected with empty control vector or FBXO7 RNAi plasmids (Fig EV3G), and subjected the lysates to native PAGE. We found that in‐gel proteasome activity assay as well as subsequent immunoblotting analyses revealed a significant increase in free CPs at the expense of assembled proteasome holoenzymes in the FBXO7 knockdown lysates (Fig 5E). Consistent with an increase in free CPs, we observed an increase in free RPs (Figs 5F and EV3H).
To confirm the change in proteasome assembly state, we carried out fractionation analyses of control and FBXO7 RNAi HEK293T cell lysates (Fig EV3I) and measured the proteasome activity of the fractions. A similar analysis was carried out by Bedford and colleagues who investigated the loss of proteasome holoenzyme function in the brain (Bedford et al, 2008). While 26S/30S fractions typically formed a distinct activity peak, free CPs generated no obvious peak (Fig 5G, control). An increase in free CPs is anticipated to lead to an increase in activity in the 20S fractions and thus to a defined second peak, which we observed upon knockdown of FBXO7 (Fig 5G, FBXO7 shRNA).
To determine whether FBXO7 deficiency in the brain leads to a similar change in the assembly state of the proteasome population, we performed native PAGE analysis using wild‐type and FBXO7 −/− brain lysates. In‐gel proteasome activity as well as subsequent immunoblotting analysis also revealed a significant increase in free CPs in the FBXO7 −/− brain lysates (Figs 5H and EV3J). These data suggest that knockdown or loss of FBXO7 affects the proteasome assembly state, which leads to a redistribution of the proteasome pool and a subsequent reduction in proteasome activity.
Loss of FBXO7 in mouse forebrain causes early‐onset motor deficits reminiscent of pyramidal tract signs
PARK15 patients show a spectrum of symptoms including spasticity, hyperreflexia, and Babinski sign, indicating pyramidal tract lesions. This prompted us to examine whether the conditional loss of FBXO7 in the forebrain of mice triggers motor symptoms. First, we generated FBXO7 mice, which carry the loxP‐flanked exon 4 (→ FBXO7 fl/fl). These mice were indistinguishable from wild‐type mice and had no motor deficits after 12 months (Fig EV4A–F). We then mated the FBXO7 fl/fl mice with the NEX (neuronal helix–loop–helix protein‐1)‐Cre driver line to delete FBXO7 from pyramidal neurons of the cortex and hippocampus (Goebbels et al, 2006) (NEX‐Cre+/− ;FBXO7 fl/fl short: Nex‐Cre;fl/fl). Disruption of the gene was confirmed by genotyping and immunoblotting (Fig 6A and B). We found that Nex‐Cre;fl/fl mice showed normal weight at 2 months of age, but then stagnated (Fig 6C). When we examined the cohorts of 2‐ and 4‐month‐old mice, we observed strong hind limb clasping in Nex‐Cre;fl/fl mice at both ages, but not in NEX‐Cre and FBXO7 fl/fl age‐matched control mice (Fig 6D). In the open field test, the Nex‐Cre;fl/fl mice ran significantly farther than the control mice at 2 and 4 months of age, indicating hyperactivity (Fig 6E), but performed significantly worse than their controls on the rotarod at 2 months of age (Fig 6F). At 4 months of age, Nex‐Cre;fl/fl mice were barely able to perform on the rotarod at all (Fig 6F). The defects in motor coordination of the Nex‐Cre;fl/fl mice seen on the rotarod was confirmed on the balance beam. Nex‐Cre;fl/fl mice slipped significantly more often than the controls (Fig 6G). This resulted in increased time to cross both the 12‐mm beam and the more challenging 6‐mm beam, and worsened with age (Fig 6G). These results indicate that loss of FBXO7 from forebrain neurons triggers progressive motor deficits, which can be translated into pyramidal tract signs found in PARK15 patients.
Figure EV4. Deletion of FBXO7 in the forebrain triggers early‐onset motor impairment.
-
AAverage body weight of 12‐month‐old FBXO7 +/+ or FBXO7 fl/fl mice. n = 13 and 10, respectively (t‐test, mean ± s.e.m.).
-
BHind limb clasping of 12‐month‐old FBXO7 +/+ or FBXO7 fl/fl mice. n = 13 and 10, respectively (Mann–Whitney U‐test, mean ± s.e.m.).
-
C–FOpen field test (C), balance beam test (time to cross and coordination score were measured, D), rotarod (E), and pole test (F) of 12‐month‐old FBXO7 +/+ or FBXO7 fl/fl mice. n = 13 and 10, respectively (unpaired t‐test or Mann–Whitney U‐test (coordination score), mean ± s.e.m.).
-
GSagittal paraffin sections from NEX‐Cre, FBXO7 fl/fl, or NEX‐Cre;fl/fl mice cortices were subjected to TUNEL staining. Three mice per genotype were included in the analysis (ANOVA, mean ± s.e.m.). Scale bar = 40 μm.
Figure 6. Deletion of FBXO7 in the forebrain triggers early‐onset motor impairment.
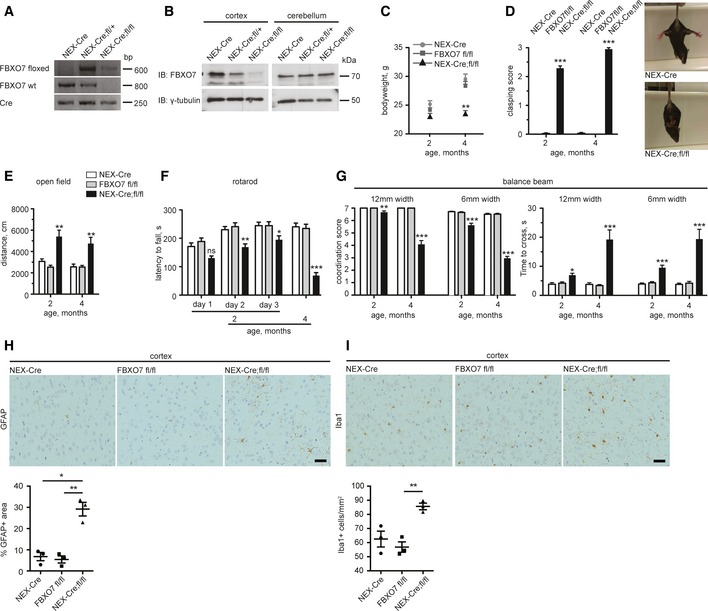
-
AGenotyping PCR of NEX‐Cre, NEX‐Cre;fl/+, or NEX‐Cre;fl/fl mice using primers for floxed allele, wild‐type FBXO7 and Cre.
-
BCortical and cerebellar lysates from NEX‐Cre, NEX‐Cre;fl/+, or NEX‐Cre;fl/fl mice were immunoblotted with FBXO7 or γ‐tubulin antibody. The latter served as a loading control.
-
CBody weight of 2‐ and 4‐month‐old NEX‐Cre, FBXO7 fl/fl, or NEX‐Cre;fl/fl mice. n = 11, 14, and 15, respectively (ANOVA, **P < 0.01, mean ± s.e.m.).
-
DHind limb clasping of NEX‐Cre, FBXO7 fl/fl, or NEX‐Cre;fl/fl mice. 0 = normal, 3 = worst manifestation of clasping. n = 11, 14, and 15, respectively (Kruskal–Wallis test, Dunn's multiple comparison, ***P < 0.001, mean ± s.e.m.). Representative images of Nex‐Cre and Nex‐Cre;fl/fl mice.
-
EOpen field test of NEX‐Cre, FBXO7 fl/fl, or NEX‐Cre;fl/fl mice. n = 11, 14, and 15, respectively (ANOVA, **P < 0.01, mean ± s.e.m.).
-
FRotarod test of NEX‐Cre, FBXO7 fl/fl, or NEX‐Cre;fl/fl mice. n = 11, 14, and 15, respectively (ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001, mean ± s.e.m.).
-
GBalance beam test of NEX‐Cre, FBXO7 fl/fl, or NEX‐Cre;fl/fl mice. Coordination score and time to cross were measured. n = 11, 14, and 15, respectively (ANOVA (time), Kruskal–Wallis test, Dunn's multiple comparison (coordination score), *P < 0.05, **P < 0.01, ***P < 0.001, mean ± s.e.m.).
-
H, ISagittal paraffin sections from NEX‐Cre, FBXO7 fl/fl, or NEX‐Cre;fl/fl mice cortices were subjected to immunohistochemistry with GFAP (H) or Iba1 (I) antibody. Three mice per genotype were included in the analysis (ANOVA, *P < 0.05, **P < 0.01, mean ± s.e.m.). Scale bar = 40 μm.
Source data are available online for this figure.
At the histological level, we found no increase in TUNEL+, apoptotic cells in the cortex, or hippocampus of the Nex‐Cre;fl/fl mice (Fig EV4G), but we observed an increase in GFAP+ cells and thus astrogliosis in the cortex (Fig 6H). We also found an increase in Iba1+ microglia in the cortex (Fig 6I) adding to the notion that loss of FBXO7 in the forebrain triggers an inflammatory response.
Conditional deletion of the FBXO7 gene in tyrosine hydroxylase (TH)‐expressing neurons triggers late‐onset progressive motor symptoms and chronic dopamine deficiency in mice
PARK15 patients present with bradykinesia, rigidity, postural instability, and the initial response to L‐dopa, indicative of nigrostriatal dysfunction. We therefore investigated how loss of FBXO7 in TH+ neurons affects the motor behavior in mice. For this, we mated the FBXO7 fl/fl line with the TH‐Cre driver line (Savitt et al, 2005) and confirmed the recombination with genotyping (Fig 7A). TH‐Cre+;FBXO7 fl/fl mice (short: TH‐Cre;fl/fl) were viable and the majority lived for at least 12 months. Young adult TH‐Cre;fl/fl mice (2 months old) were indistinguishable from control groups in terms of weight (Fig EV5A) and motor performance (Fig EV5B–D). At 6 months however, we observed that the weight of half the TH‐Cre;fl/fl group was significantly higher than the one of the TH‐Cre+ control group (Fig 7B). To correct for these weight differences, we only included mice between 30 and 40 g (Fig 7C). TH‐Cre;fl/fl mice were equally ambulant in the open field test (Fig 7D), crossed the balance beam as readily as the control group (Fig 7E), and passed the pole test, which examines bradykinesia (Fig 7F). The performance of the TH‐Cre;fl/fl mice on the rotarod however was significantly worse (Fig 7G), indicating an emerging motor deficit. To characterize this in a more precise fashion, we used the DigiGait™ system that measures a variety of fine gait parameters including stance, swing, stride, propulsion, and brake (Amende et al, 2005). We found that the 8‐month‐old TH‐Cre;fl/fl mice showed reduced paw area and width of the footprints analyzed, reduced stride duration and length, reduced stance and propulsion, as well as less rapid deceleration (Figs 7H and EV5E). Interestingly, many of these parameters are also changed in the DigiGait analyses of the MPTP mouse modeling PD (Amende et al, 2005).
Figure 7. Loss of FBXO7 in TH + cells results in late‐onset motor deficits.
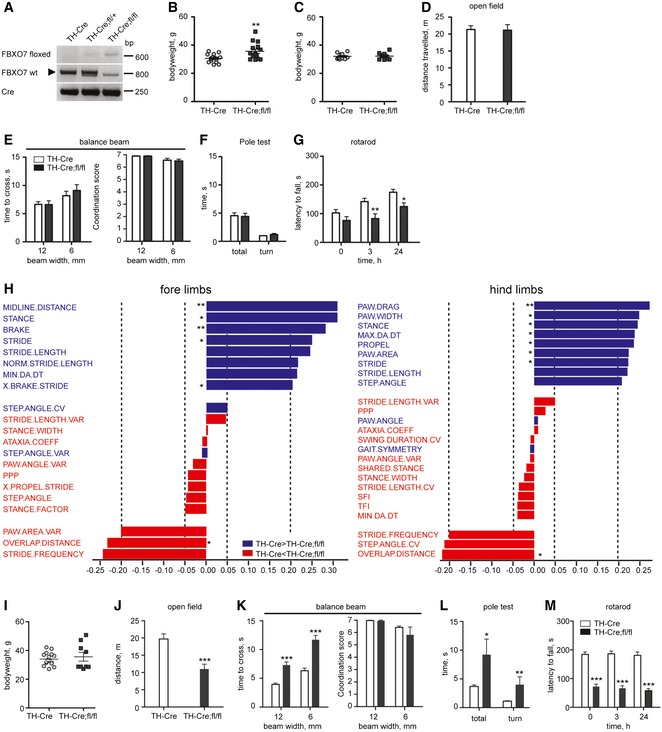
-
AGenotyping PCR of TH‐Cre, TH‐Cre;fl/+, or TH‐Cre;fl/fl mice using primers for floxed allele, wild‐type FBXO7 and Cre.
-
BAverage body weight of 6‐month‐old TH‐Cre and TH‐Cre;fl/fl mice. n = 16 and 15, respectively (unpaired t‐test, **P < 0.01, mean ± s.e.m.).
-
CAverage body weight of the weight‐corrected cohort of 6‐month‐old TH‐Cre and TH‐Cre;fl/fl mice. n = 11 and 10, respectively (unpaired t‐test, mean ± s.e.m.).
-
D–FOpen field (D), balance beam (E) or pole test (F) of 6‐month‐old TH‐Cre and TH‐Cre;fl/fl mice. n = 11 and 10, respectively (unpaired t‐test or Mann–Whitney U‐test (coordination score), mean ± s.e.m.).
-
GRotarod test of 6‐month‐old TH‐Cre and TH‐Cre;fl/fl mice. n = 11 and 10, respectively (unpaired t‐test, *P < 0.05, **P < 0.01, mean ± s.e.m.).
-
HDigiGait analysis of 8‐month‐old TH‐Cre and TH‐Cre;fl/fl mice. n = 7 for both groups (unpaired t‐test, *P < 0.05, **P < 0.01, mean ± s.e.m.). Data were visualized using a partial least squares (PLS) regression with orthogonal signal correction model.
-
IAverage body weight of 12‐month‐old TH‐Cre and TH‐Cre;fl/fl mice. n = 16 and 9, respectively (unpaired t‐test, mean ± s.e.m.).
-
J–MOpen field (J), balance beam (K), pole test (L), rotarod (M) of 12‐month‐old TH‐Cre and TH‐Cre;fl/fl mice. n = 16 and 9, respectively (unpaired t‐test or Mann–Whitney U‐test (coordination score), *P < 0.05, **P < 0.01, ***P < 0.001, mean ± s.e.m.).
Figure EV5. Loss of FBXO7 in TH + cells results in late‐onset motor deficits.
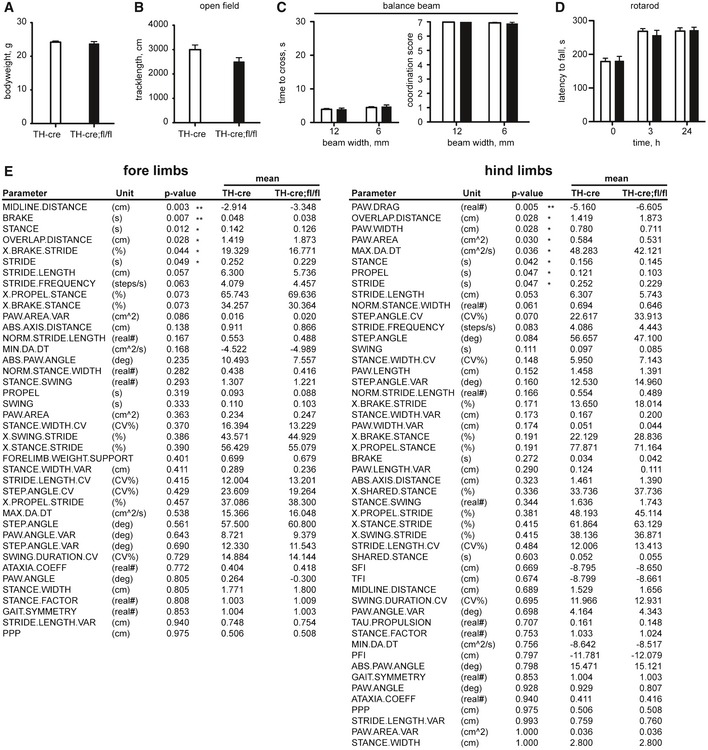
-
AAverage body weight of 2‐month‐old TH‐Cre and TH‐Cre;fl/fl mice. n = 15 and 9, respectively (unpaired t‐test, mean ± s.e.m.).
-
B–DOpen field test (B), balance beam test (time to cross and coordination score were measured, C), and rotarod (D) of 2‐month‐old TH‐Cre and TH‐Cre;fl/fl mice. n = 15 and 9, respectively (unpaired t‐test or Mann–Whitney U‐test (coordination score), mean ± s.e.m.).
-
EParameters tested in DigiGait analysis (unpaired t‐test, *P < 0.05, **P < 0.01, mean ± s.e.m.).
To follow the impending motor impairment, we examined the mice at 12 months of age. The overall health and motor performance of the TH‐Cre;fl/fl mice worsened dramatically. Compared to the age‐matched TH‐Cre+ control mice, we lost six out of the initial 15 mice, which resulted in two weight groups, one with higher and one with lower than average weight (Fig 7I). For all motor tests, we examined the weight groups separately at first, but then combined the results, as the groups performed equally poor. TH‐Cre;fl/fl mice travelled significantly shorter in the open field compared to control (Fig 7J). On the balance beam, it took the TH‐Cre;fl/fl mice significantly longer to cross, but in contrast to the Nex‐Cre;fl/fl mice they retained their coordination ability (Fig 7K). With the pole test, we also observed clear differences between the TH‐Cre;fl/fl and the control mice (Fig 7L), and their rotarod performance had further declined as compared to 6‐month‐old mice (Fig 7M). Slowness of movements combined with reduced mobility and a characteristic change in gait parameters show that deletion of FBXO7 in catecholaminergic neurons causes symptoms reminiscent of the bradykinesia and rigidity seen in PARK15 patients.
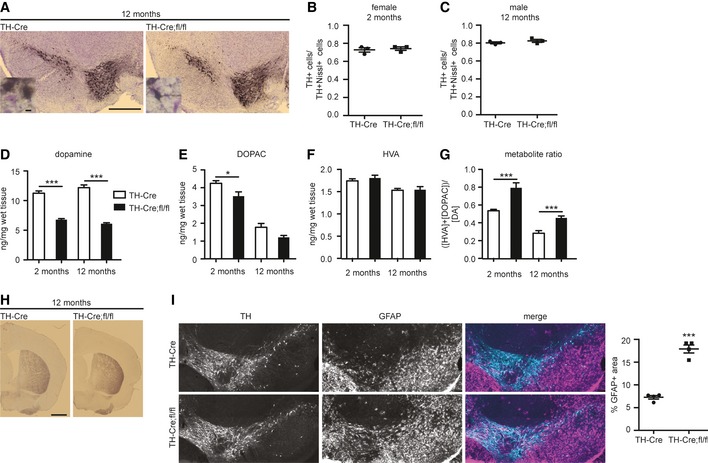
To determine the consequence of loss of FBXO7 in dopaminergic neurons, we performed stereological counting of TH+ neurons in the substantia nigra in relation to total neuron number. We found that loss of FBXO7 does not affect the number of dopaminergic neurons in either 2‐month‐old or 12‐month‐old TH‐Cre;fl/fl mice as compared to their age‐matched control mice (Fig 8A–C). We then subjected striata from the aforementioned mice to HPLC analysis and found that the 12‐month‐old TH‐Cre;fl/fl mice had a 50% reduction in dopamine as compared to controls, and variable reductions in metabolites (Fig 8D–G). Interestingly, we also found a 50% reduction in 2‐month‐old mice, which as described earlier, did not show any motor symptoms. As TH immunostaining in 12‐month‐old TH‐Cre;fl/fl striatum was comparable to control (Fig 8H), a severe loss of axons may have not occurred. The neuropathological examination revealed no increase in Iba1+ microglia in the 12‐month‐old TH‐Cre;fl/fl mice (Appendix Fig S3A), but an increase in astrogliosis in the substantia nigra and adjacent regions (Fig 8I). These results demonstrate that loss of FBXO7 in catecholaminergic neurons in the TH‐Cre;fl/fl mice leads to chronic dopamine deficiency in the striatum that precedes the onset of severe motor deficits.
Figure 8. Loss of FBXO7 in TH + cells leads to reduced striatal dopamine.
-
AImages of representative 12‐month‐old TH‐Cre and TH‐Cre;fl/fl midbrains immunostained with TH antibody (black) and Nissl stain (purple). Scale bar = 500 μm. Scale bar of inset = 10 μm.
-
B, CStereological analysis of ratio of TH+Nissl+ cells/Nissl+ cells in the substantia nigra (A) from 2‐month‐old female (B) and 12‐month‐old male (C) TH‐Cre and TH‐Cre;fl/fl mice. Three animals from each genotype were analyzed (unpaired t‐test, mean ± s.e.m.).
-
D–GStriatal tissue from P18 TH‐Cre and TH‐Cre;fl/fl mice was subjected to HPLC analyses to determine dopamine (D), 3,4‐dihydroxyphenylacetic acid (DOPAC) (E), and homovanillic acid (HVA) (F) concentrations along with the metabolite ratio (G). N = 7 and 9 (2 months) and n = 15 and 12 (12 months) (unpaired t‐test, *P < 0.05, ***P < 0.001, mean ± s.e.m.)
-
HImages of representative TH‐Cre and TH‐Cre;fl/fl striatal sections immunostained with TH antibody. Scale bar = 1 mm.
-
IImmunohistochemistry of midbrain section TH‐Cre and TH‐Cre;fl/fl mice using TH and GFAP antibody. Quantitative measurements of GFAP stainings from four animals per genotype (unpaired t‐test, ***P < 0.001, mean ± s.e.m.).
Discussion
Mutations in the FBXO7 (PARK15) gene are associated with parkinsonian pyramidal syndrome, which is characterized by a spectrum of motor symptoms. We demonstrate that both systemic loss and neuronal loss of the FBXO7 gene in mice cause severe motor deficits reminiscent of the symptoms seen in PARK15 patients. This contrasts with several other PARK gene knockout mice with deletion of, for example, the PARK1/4 (SNCA, α‐synuclein), PARK8 (LRRK2), PARK2 (parkin), PARK6 (PINK1), or PARK7 (DJ‐1) gene, which show little or no effect on mouse behavior and viability (Abeliovich et al, 2000; Goldberg et al, 2003; Kim et al, 2005; Gautier et al, 2008; Andres‐Mateos et al, 2009; Tong et al, 2010).
Systemic loss of FBXO7 is likely to have a negative impact on additional organs other than the brain. We thus used conditional FBXO7 knockout mice to demonstrate the neuronal contribution to the motor phenotype. With the deletion of FBXO7 in the forebrain, we recapitulate spasticity and early‐onset motor defects, critical aspects of the PARK15 syndrome. The FBXO7 midbrain knockout shows late‐onset bradykinesia and hypolocomotion and even presents with gait features that are specifically changed in MPTP‐treated mice (Amende et al, 2005), a widely used PD mouse model. The diverse motor symptoms as a consequence of FBXO7 dysfunction or loss in different brain regions together with the genetic background might therefore contribute to the variable severity of pyramidal tract signs and parkinsonian symptoms among affected PARK15 families.
So far, the neuropathology of PARK15 patients remains elusive. FBXO7 knockout mice show a moderate increase in cell death in the cortex. Interestingly, some PARK15 patients present with cortical atrophy (Paisan‐Ruiz et al, 2010; Gunduz et al, 2014). Another feature observed in all FBXO7 knockout mice is astrogliosis in the affected brain regions. On the one hand, astrogliosis can accompany dead or damaged cells as seen in the FBXO7 knockout as well as in both neurotoxic (MPTP) (Kato et al, 2003) and genetic (ATP13a2) PD mouse models (Kett et al, 2015). On the other hand, astrocytes are also suggested to serve a protective role in PD and precede neuronal loss (Sortwell et al, 2000; Saura et al, 2003), which could partially explain the absence of cell loss in the conditional knockout mice. The absence of protein aggregates in the FBXO7 −/− mouse brain is not too surprising since deposits are only found in a few mouse models to begin with and are typically not observed in brains of juvenile parkinsonism patients (Poulopoulos et al, 2012).
While we do not observe a loss of TH+ neurons in the substantia nigra, we measured an early‐onset deficit in striatal dopamine, suggesting a failure to release or produce dopamine in the TH+ neurons or a decreased number of terminals. By the time the TH‐Cre;fl/fl mice show symptoms, they have been under chronic dopamine deficiency for 4 months. Chronic dopamine deficiency is likely to explain the observed functional impairment. Interestingly, PD patients treated with levodopa for 40 weeks and tested after a washout period of 2 weeks were functionally better than PD patients who remained untreated and thus with a dopamine deficiency for 42 weeks—even though PD patients treated with levodopa showed a trend for a more pronounced decline in the number of dopaminergic neurons as reported by beta‐CIT SPECT (Fahn et al, 2004). Together with our findings, this suggests that chronic dopamine deficiency can lead to rapid functional deterioration even if it is non‐progressive. A similar reduction in striatal dopamine has been found in the SNCA −/− mouse and is hypothesized to be due to dysfunctional synaptic properties (Abeliovich et al, 2000). Given that FBXO7 is involved in several cellular processes, further research will be required to elucidate how loss of FBXO7 may interfere with dopamine production and delivery, and if FBXO7 has any importance in synapse function. The mouse models characterized in this study will be useful tools for future investigations of altered synaptic connections in the brain.
There is a long‐standing relationship between the ubiquitin proteasome system (UPS) and Parkinson's disease. Ubiquitin‐laden protein aggregates in postmortem tissue of PD patients suggested a dysfunction of the UPS (Lennox et al, 1988; Lowe et al, 1988) corroborated by the discovery that mutations in the PARK2 gene, encoding the E3 ubiquitin ligase parkin, are associated with familial parkinsonism (Kitada et al, 1998). In this study, we show that the PARK15 gene product FBXO7 associates with the proteasome by binding to the core subunit PSMA2 via its UbRD domain. In contrast to parkin, which harbors a Ubl domain, sequence analyses revealed a more distant relative of the Ubl domain for FBXO7. While the Ubl domains have been demonstrated to mediate the interaction with proteasomal cap subunits as shown for the binding of parkin with Rpn10 (Sakata et al, 2003), the UbRD of FBXO7 mediates the interaction with PSMA2. Additionally, we observe that loss of FBXO7 affects proteasome assembly, potentially through ubiquitination of PSMA2. This establishes FBXO7 as a novel proteasome‐interacting E3 ubiquitin ligase, which is further supported by our study and recent mass spectrometry analyses by others that identified FBXO7 and its complex subunits as proteasome‐associated components (Bousquet‐Dubouch et al, 2009; Fabre et al, 2015). Ubiquitination sites of the subunit PSMA2 have been identified by various mass spectrometry analyses, in which both the ubiquitinated proteasome and proteome were analyzed (Kim et al, 2011; Moiseeva et al, 2013; Zong et al, 2014) (www.phosphosite.org). Specific E3 ligases that mediate PSMA2 ubiquitination however remained to be uncovered. Our results suggest that the E3 ligase FBXO7‐SCF ubiquitinates PSMA2 via K63‐linked ubiquitin chains resulting in functional modification of PSMA2. SCF complexes are versatile ligases, which in addition to K48 polyubiquitination mediate the transfer of K63 polyubiquitin chains. This has previously been demonstrated for the F‐box proteins β‐TRCP and in particular SKP2 (Kumar et al, 2007; Chan et al, 2012; Jin et al, 2015; Lee et al, 2015). The ubiquitination of PSMA2 is potentially crucial as PSMA2 engages in essential interactions with subunits of the regulatory particle of the proteasome holoenzyme (Satoh et al, 2001; Tian et al, 2011; da Fonseca et al, 2012; Pathare et al, 2012). The modification of PSMA2 could thus be essential in establishing and maintaining proper proteasome function. Consistent with this notion, our findings show that loss or knockdown of FBXO7 and hence a lost FBXO7‐PSMA2 interaction subjects cells to a chronic dysfunction of the proteasome. As a result of the redistribution of the proteasome pool, increased amounts of isolated 20S core particles bring about a reduction in proteasome activity in vivo, as 20S CPs are unable to process ubiquitinated proteins under physiological conditions (Finley, 2009). While our data show that FBXO7 has certainly an important function in proteasome regulation, further in‐depth analyses are required to gain a better understanding of FBXO7's actions.
The recent implications of FBXO7‐mediated recruitment of parkin to mitochondria to initiate mitophagy underscore the essential function of FBXO7 in cells and suggest that loss of FBXO7 might also lead to mitochondrial dysfunction (Burchell et al, 2013). Mitochondrial defects together with the increase in reactive oxygen species has been reported to result in proteasomal disassembly (Livnat‐Levanon et al, 2014). Conversely, proteasomal inhibition has been demonstrated to impair mitochondrial homeostasis and turnover (Sullivan et al, 2004). Our results along with the published data demonstrate a regulatory role of FBXO7 in two integral cellular systems: mitochondria and proteasomes. Since FBXO7 may affect these systems in a cell type‐specific manner, the trigger of dysfunction might be mitochondrial or proteasomal. But owing to the interdependence of the systems, this may cause a chain reaction that ultimately results in high levels of stress in the cell. Collectively, our findings demonstrate that proteasome integrity and activity are FBXO7 dependent and point toward a potential susceptibility to proteasome dysfunction in parkinsonism.
Materials and Methods
Ethics statement
All experiments involving live animals have been conducted according to the animal protocol approved by the “Verbraucherschutz und Lebensmittelsicherheit” of Lower Saxony, Germany (33.11.42502‐04‐11/0632).
Plasmids, antibodies, and molecular weight marker
Full‐length FBXO7, PSMA2, or PI31 was cloned either into the p3xFLAG‐CMV™‐10, pCMV‐Myc, pEGFP‐C1, or the pcDNA3.0 vector as indicated. Untagged full‐length FBXO7 was cloned into pcDNA3.0. Deletion mutants were created by standard PCR amplification of fragments, or fusion PCR of two separate fragments to omit an internal sequence followed by cloning into the pCMV‐Myc vector. Ubl RAD23B was cloned into the pGEX‐4T‐1 vector. UIM S5a was cloned into the pET28a vector. Flag‐PSMA2 and FBXO7 were cloned into the pGEX‐6P‐1 vector. HA‐tagged wild‐type, K63R, and K48R ubiquitin were a kind gift from Dr. Hiroshi Kawabe.
Short hairpin RNA was generated using the pSUPER RNAi System™. Following target sequences against consensus sequences of human, mouse and rat were used:
Fbxo7 shRNA, bp 461–480 human isoform 1: 5′‐GAAGAGACCTTGGCTTCATA‐3′;
Fbxo7 shRNA (non‐functional): 5′‐GAAACTACGCATCTTCCGAC‐3′;
Psma2 shRNA A: 5′‐GAAGTGTACACAAAGTAGAAC‐3′;
Psma2 shRNA B: 5′‐GGATTACTTGGCTGCCATAGC‐3′;
Psma2 shRNA (non‐functional): 5′‐GCTGACTACATTCAGCCC‐3′;
Pi31 shRNA A: 5′‐GGGCTACGATGACATGTACC‐3′;
Pi31 shRNA B: 5′‐GGGAAGTGGTGACAAACGGCT‐3′;
Pi31 shRNA (non‐functional): 5′‐GAACAGCAATAAAGAACTGTA 3′.
Antibodies used for immunoblot analysis were as follows: ms α‐FBXO7 (1:50 dilution; Santa Cruz, sc‐271763), ms α‐γ‐tubulin (1:1,000 dilution; Sigma‐Aldrich, T6557), rb α‐PSMA2 (1:1,000 dilution; Cell Signaling, 2455), rb α‐PSMA2 (in vitro ubiquitination assay, 1:500 dilution; LSBio, L‐C138512), gt α‐PI31 (1:500 dilution; Sigma‐Aldrich, SAB2500788), ms α‐Myc (1:500 dilution; Santa Cruz, sc‐40), ms α‐Flag (1:1,000 dilution; Sigma‐Aldrich, F1804), ms α‐pan 14‐3‐3 (1:5,000 dilution; Santa Cruz, sc‐1657), ms α‐α‐synuclein (1:1,000 dilution; Cell Signaling, 4179), ms α‐β‐galactosidase (1:500 dilution; Santa Cruz, sc‐65670), ms α‐SP1 (1:500 dilution; Santa Cruz, sc‐14027), ms α‐Rpt6 (1:500 dilution; Enzo Lifescience, p45‐110), ms α‐K63‐linked ubiquitin (1:200 dilution; Millipore, 05‐1308), ms α‐K48‐linked ubiquitin (1:200 dilution; Millipore, 05‐1307), rb α‐ubiquitin (1:500 dilution; DAKO, Z0458), ms α‐ubiquitin P4D1 (1:500 dilution; Santa Cruz, sc‐8017), ms α‐GFP (1:500 dilution; Santa Cruz, sc‐9996), ms α‐HA (1:500 dilution; Santa Cruz, sc‐805), ms α‐cullin‐1 (1:500 dilution; Santa Cruz, sc‐17775), ms α‐GAPDH (1:500 dilution; mAbcam 9484).
HRP‐coupled secondary antibodies were goat α‐rabbit (1:1,000; Dianova, 115‐035‐144), goat α‐mouse (1:1,000; Dianova, 11‐035‐146), donkey α‐goat (1:1,000 Santa Cruz, sc‐2020).
Antibodies used for immunohistochemistry were as follows: ms α‐TH (1:1,000 dilution; Sigma, T2928), rb α‐TH (1:1,000 dilution; Zytomed, 620‐0336), ms α‐Iba1 (1:1,000 dilution; WAKO, 019‐19741), ms α‐GFAP (1:200 dilution; Nova Castra, NCL‐GFAP‐GA5), ms α‐α‐synuclein (1:250 dilution; Cell Signaling, 4179), ms α‐Mac3 (1:200 dilution; BD Pharmingen, 553322).
Antibodies used for immunocytochemistry were as follows: rb α‐cleaved caspase‐3 (1:200 dilution; Cell Signaling, 9661), rb α‐GFP (1:1,000 dilution; Invitrogen, A6455).
Cy3/Alexa488‐coupled secondary antibodies were goat α‐rabbit and goat α‐mouse (1:1,000; Dianova, 111‐160‐144/111‐485‐144).
We used the PageRuler 10–180 kD prestained protein ladder (red 70 kD protein band is highlighted in Source Data).
Generation of FBXO7 transgenic mice
FBXO7‐deficient mice were created by homologous recombination in C57BL/6 mouse JM8.N4 ES cells (Fbxo7tm1a(EUCOMM)Hmgu). FBXO7 genomic sequence containing exon 4 was exchanged with a neomycin/lacZ cassette driven by the L1L2_Bact_P promoter. Chimeras were generated by injection of targeted ES cells into C57BL/6N blastocysts. Homozygous transgene mice were then bred to the EIIa‐cre (Holzenberger et al, 2000) or 129S4/SvJaeSor‐Gt(ROSA)26Sortm1(FLP1)Dym/J to create conventional or conditional FBXO7 knockout mouse lines, respectively. Homozygous conditional FBXO7 floxed animals were further bred to either the B6.Cg‐Tg(TH‐cre)1Tmd/J (Savitt et al, 2005) or NEX‐Cre (Goebbels et al, 2006) line.
Mice were genotyped using the following primers:
LacZ cassette; 5′‐attccagctgagcgccggtcgc‐3′ (forward)
5′‐gcgagctcagaccataacttcgtata‐3′ (reverse)
Floxed allele; 5′‐tcagcatgggtttgttaagcatctacta‐3′ (forward)
5′‐ggtctagatatctcgacataacttcgtata‐3′ (reverse)
Wild‐type FBXO7; 5′‐gggctgtatgaaggaagtgctatt‐3′ (forward)
5′‐ ccctgagagtgaagggtgctgttc‐3′ (reverse)
Cre; 5′‐cagggtgttataagcaatccc‐3′ (forward)
5′‐cctggaaaatgcttctgtccg‐3′ (reverse)
Quantitative mass spectrometry
HEK293T cells were transfected with empty vector pCMV‐3XFLAG or the FLAG‐FBXO7 expression plasmid and lysed in NP‐40 buffer (1% NP‐40, 10% glycerol, 150 mM NaCl, 20 mM Tris–HCl pH 7.5, 1 mM EDTA pH 7.5). Immunoprecipitation using anti‐FlagM2 agarose was performed according to the manufacturer's instructions. Precipitated protein complexes were treated for MS analysis as described previously (Turriziani et al, 2014). Briefly, after the immunoprecipitation, anti‐FlagM2 agarose beads were washed thrice with IP buffer followed by three washes with IP buffer without detergent. Dried beads of each IP (three independent experiments in total) were resuspended in 10% formic acid (FA) digested for 1 h at room temperature in 2 M urea, 50 mM Tris–HCl pH 7.5, and 5 μg/ml trypsin, followed by two washes with 2 M urea, 50 mM Tris–HCl pH 7.5, and 1 mM DTT. The pooled supernatants were left to digest overnight at RT. After iodoacetamide modification and acidification of the samples, the peptide mixtures were desalted using homemade C18 tips. The desalted and lyophilized peptides were resuspended in 10% FA and subjected to analysis by reversed‐phase nano‐LC‐MS/MS using a nano‐Ultimate 3000 liquid chromatography system and an Orbitrap Elite mass spectrometer (both Thermo Scientific).
Peptide trapping was performed for 10 min on a C18 precolumn (Acclaim PepMap100, C18, 5 μm, 100 Å, 300 μm i.d. × 5 mm, Thermo Scientific) in Buffer A (0.1% FA) followed by separation on an analytical column (Acclaim PepMap100, C18, 5 μm, 100 Å, 75 μm i.d. × 25 cm, Thermo Scientific) using a 130‐min gradient (0–10 min: 5% buffer B (80% acetonitrile, 0.1% FA), 10–105 min: 10–45% buffer B, 105–107 min: 45–100% buffer B; 107–113 min 100% buffer B; 113–130 min 5% buffer B) at 230 nl/min. Every sample was measured in duplicate. The mass spectrometer was operated in data‐dependent mode with a 20‐s dynamic exclusion range. Full‐scan spectra recording in the Orbitrap was in the range of m/z 350 to m/z 1,500 (resolution: 120,000; AGC: 5e5 ions). The top 10 precursors of each full scan were fragmented using CID in the ion trap utilizing collision energy of 35%. The raw data were analyzed using MaxQuant (version 1.5.2.8) (Cox & Mann, 2008). The spectra were searched against the human SwissProt database version 06/2015 using the built‐in Andromeda search engine (Cox et al, 2011). MaxQuant default settings were used (including mass tolerance), with trypsin as the protease (allowing two missed cleavages). Fixed modification: carbamidomethylation (Cys), variable modifications: oxidation (Met), phosphorylation (Ser, Thr, Tyr), and N‐terminal protein acetylation. The false discovery rate was 0.01 on both peptide and protein level. The minimum peptide length was seven amino acids. The protein intensity of the individual runs was used to compare the composition of the complexes described in Fig 3H.
DigiGait recordings
Coordinated locomotion in mice was investigated using the DigiGait™ imaging system (Mouse Specifics, Inc., Boston) (Hampton et al, 2004; Kale et al, 2004). The animals are placed on a speed‐adjustable motorized treadmill equipped with a transparent treadmill belt. A high‐speed digital video camera positioned below the belt captured images of the underside of the walking mouse. The movies were acquired with the DigiGait™ Imager software (version 12.5).
TH‐Cre and TH‐Cre;fl/fl animals were tested at 8 months and acclimated to the testing room for at least 1 week before the test. Prior to recordings, the animals were allowed to explore the treadmill compartment for 5 min, with the motor speed set to zero. Then, the speed was slowly increased up to 25 cm/s. The recording session consisted in sequential running bouts separated by a few minutes of rest in the cage. The animals were tested until the recordings match the standards described above, for a maximum of 3 days, with a maximum of three sessions per day.
The acquired movies were pre‐processed, postprocessed, and edited using the DigiGait™ Analysis software (version 14.0) and basic gait metrics calculated. Values for fore‐ and hind limbs were calculated separately with values for right and left limb pooled.
Statistical analysis using multiple regression modeling in R
The DigiGait™ analysis results were analyzed using partial least squares (PLS) regression with Orthogonal Signal Correction PLS (OSC‐PLS). We modified an existing R script (Grapov, 2014; R Development Core Team, 2011) and applied the OSC‐PLS method to the complete, first standardized, dataset in order to define and optimize a 2‐component model. The model was compared to 100 randomly permuted models according to the leave‐one‐out method. We conducted training and testing validations by randomly splitting our original data into 2/3 training set to fit the model, and then using this to predict group memberships on the remaining 1/3 testing set.
Our model's fit and predictability to the training data was defined by its coefficient of determination, Q2, and the root mean square error of prediction, RMSEP (Wehrens, 2011). The results of the two‐sample Student's t‐test used for the comparisons indicated a < 0.1% probability of achieving a performance similar to our model by random chance.
Statistical analysis
GraphPad PRISM was used to perform all statistical analysis unless indicated otherwise using ANOVA (Tukey post hoc) or Student's t‐test, paired or unpaired.
Author contributions
SV and DB designed and performed experiments and co‐wrote the manuscript; CL, LT, GD, CP, NS‐D, and SJ performed experiments; MM wrote the FIJI macro, and SG and K‐AN contributed mouse lines; JBS, TM, and PL contributed analyses tools; and JS designed the study and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
This work was supported by the Michael J. Fox Foundation for Parkinson's Research: Target Validation Program 2013 (JS), the Max Planck Society (JS), the GGNB Excellence Stipend (SV), the GGNB Junior Research Group Stipend (JS), and START (RWTH Aachen, JS). CL and TM were supported by European Research Council (ERC) Grant Agreement 311710‐MU TUNING of the European Union's Seventh Framework Programme (FP/2007‐2013). The Proteomics Facility is supported by a grant from the Interdisciplinary Center for Clinical Research within the faculty of Medicine at the RWTH Aachen. We thank Benoît Lancelin (Maïsadour Semences, France) for help with the statistical analyses of the DigiGait data, Bekir Altas, and Hiroshi Kawabe (MPI of Experimental Medicine, Göttingen) for providing reagents and both Ekrem Dere and Hannelore Ehrenreich (MPI of Experimental Medicine, Göttingen) for advice on behavioral analyses. Aaron Voigt (UK Aachen, Germany) is thanked for critical reading of the manuscript and Björn Falkenburger (UK Aachen, Germany) for helpful discussions.
The EMBO Journal (2016) 35: 2008–2025
References
- Abeliovich A, Schmitz Y, Farinas I, Choi‐Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A (2000) Mice lacking alpha‐synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25: 239–252 [DOI] [PubMed] [Google Scholar]
- Amende I, Kale A, McCue S, Glazier S, Morgan JP, Hampton TG (2005) Gait dynamics in mouse models of Parkinson's disease and Huntington's disease. J Neuroeng Rehabil 2: 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres‐Mateos E, Mejias R, Sasaki M, Li X, Lin BM, Biskup S, Zhang L, Banerjee R, Thomas B, Yang L, Liu G, Beal MF, Huso DL, Dawson TM, Dawson VL (2009) Unexpected lack of hypersensitivity in LRRK2 knock‐out mice to MPTP (1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine). J Neurosci 29: 15846–15850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford L, Hay D, Devoy A, Paine S, Powe DG, Seth R, Gray T, Topham I, Fone K, Rezvani N, Mee M, Soane T, Layfield R, Sheppard PW, Ebendal T, Usoskin D, Lowe J, Mayer RJ (2008) Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy‐like inclusions resembling human pale bodies. J Neurosci 28: 8189–8198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet‐Dubouch MP, Baudelet E, Guerin F, Matondo M, Uttenweiler‐Joseph S, Burlet‐Schiltz O, Monsarrat B (2009) Affinity purification strategy to capture human endogenous proteasome complexes diversity and to identify proteasome‐interacting proteins. Mol Cell Proteomics 8: 1150–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchell VS, Nelson DE, Sanchez‐Martinez A, Delgado‐Camprubi M, Ivatt RM, Pogson JH, Randle SJ, Wray S, Lewis PA, Houlden H, Abramov AY, Hardy J, Wood NW, Whitworth AJ, Laman H, Plun‐Favreau H (2013) The Parkinson's disease‐linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat Neurosci 16: 1257–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo T, Pagano M (2004) The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol 5: 739–751 [DOI] [PubMed] [Google Scholar]
- Chan CH, Li CF, Yang WL, Gao Y, Lee SW, Feng Z, Huang HY, Tsai KK, Flores LG, Shao Y, Hazle JD, Yu D, Wei W, Sarbassov D, Hung MC, Nakayama KI, Lin HK (2012) The Skp2‐SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 149: 1098–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Orian A, Schwartz AL (2000) Ubiquitin‐mediated proteolysis: biological regulation via destruction. BioEssays 22: 442–451 [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- Di Fonzo A, Dekker MC, Montagna P, Baruzzi A, Yonova EH, Correia Guedes L, Szczerbinska A, Zhao T, Dubbel‐Hulsman LO, Wouters CH, de Graaff E, Oyen WJ, Simons EJ, Breedveld GJ, Oostra BA, Horstink MW, Bonifati V (2009) FBXO7 mutations cause autosomal recessive, early‐onset parkinsonian‐pyramidal syndrome. Neurology 72: 240–245 [DOI] [PubMed] [Google Scholar]
- Fabre B, Lambour T, Garrigues L, Amalric F, Vigneron N, Menneteau T, Stella A, Monsarrat B, Van den Eynde B, Burlet‐Schiltz O, Bousquet‐Dubouch MP (2015) Deciphering preferential interactions within supramolecular protein complexes: the proteasome case. Mol Syst Biol 11: 771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S, Oakes D, Shoulson I, Kieburtz K, Rudolph A, Lang A, Olanow CW, Tanner C, Marek K, Parkinson Study Group (2004) Levodopa and the progression of Parkinson's disease. N Engl J Med 351: 2498–2508 [DOI] [PubMed] [Google Scholar]
- Finley D (2009) Recognition and processing of ubiquitin‐protein conjugates by the proteasome. Annu Rev Biochem 78: 477–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Fonseca PC, He J, Morris EP (2012) Molecular model of the human 26S proteasome. Mol Cell 46: 54–66 [DOI] [PubMed] [Google Scholar]
- Forster F, Unverdorben P, Sledz P, Baumeister W (2013) Unveiling the long‐held secrets of the 26S proteasome. Structure 21: 1551–1562 [DOI] [PubMed] [Google Scholar]
- Gautier CA, Kitada T, Shen J (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci USA 105: 11364–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA (2006) Genetic targeting of principal neurons in neocortex and hippocampus of NEX‐Cre mice. Genesis 44: 611–621 [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J (2003) Parkin‐deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278: 43628–43635 [DOI] [PubMed] [Google Scholar]
- Grapov D (2014) DeviumWeb: Dynamic Multivariate Data Analysis and Visualization Platform. v0.3.2 (25/11/14) doi: 10.5281/zenodo.12879
- Gunduz A, Eken AG, Bilgic B, Hanagasi HA, Bilguvar K, Gunel M, Basak AN, Ertan S (2014) FBXO7‐R498X mutation: phenotypic variability from chorea to early onset parkinsonism within a family. Parkinsonism Relat Disord 20: 1253–1256 [DOI] [PubMed] [Google Scholar]
- Hampton TG, Stasko MR, Kale A, Amende I, Costa AC (2004) Gait dynamics in trisomic mice: quantitative neurological traits of Down syndrome. Physiol Behav 82: 381–389 [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Lenzner C, Leneuve P, Zaoui R, Hamard G, Vaulont S, Bouc YL (2000) Cre‐mediated germline mosaicism: a method allowing rapid generation of several alleles of a target gene. Nucleic Acids Res 28: E92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW (2004) Systematic analysis and nomenclature of mammalian F‐box proteins. Genes Dev 18: 2573–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Williamson A, Banerjee S, Philipp I, Rape M (2008) Mechanism of ubiquitin‐chain formation by the human anaphase‐promoting complex. Cell 133: 653–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin G, Lee SW, Zhang X, Cai Z, Gao Y, Chou PC, Rezaeian AH, Han F, Wang CY, Yao JC, Gong Z, Chan CH, Huang CY, Tsai FJ, Tsai CH, Tu SH, Wu CH, dos Sarbassov D, Ho YS, Lin HK (2015) Skp2‐mediated RagA ubiquitination elicits a negative feedback to prevent amino‐acid‐dependent mTORC1 hyperactivation by recruiting GATOR1. Mol Cell 58: 989–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kale A, Amende I, Meyer GP, Crabbe JC, Hampton TG (2004) Ethanol's effects on gait dynamics in mice investigated by ventral plane videography. Alcohol Clin Exp Res 28: 1839–1848 [DOI] [PubMed] [Google Scholar]
- Kato H, Araki T, Imai Y, Takahashi A, Itoyama Y (2003) Protection of dopaminergic neurons with a novel astrocyte modulating agent (R)‐(‐)‐2‐propyloctanoic acid (ONO‐2506) in an MPTP‐mouse model of Parkinson's disease. J Neurol Sci 208: 9–15 [DOI] [PubMed] [Google Scholar]
- Kett LR, Stiller B, Bernath MM, Tasset I, Blesa J, Jackson‐Lewis V, Chan RB, Zhou B, Di Paolo G, Przedborski S, Cuervo AM, Dauer WT (2015) alpha‐Synuclein‐independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2. J Neurosci 35: 5724–5742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, Pownall S, Wakeham A, You‐Ten AJ, Kalia SK, Horne P, Westaway D, Lozano AM, Anisman H, Park DS, Mak TW (2005) Hypersensitivity of DJ‐1‐deficient mice to 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci USA 102: 5215–5220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, Harper JW, Gygi SP (2011) Systematic and quantitative assessment of the ubiquitin‐modified proteome. Mol Cell 44: 325–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk R, Laman H, Knowles PP, Murray‐Rust J, Lomonosov M, el Meziane K, McDonald NQ (2008) Structure of a conserved dimerization domain within the F‐box protein Fbxo7 and the PI31 proteasome inhibitor. J Biol Chem 283: 22325–22335 [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: 605–608 [DOI] [PubMed] [Google Scholar]
- Kumar KG, Barriere H, Carbone CJ, Liu J, Swaminathan G, Xu P, Li Y, Baker DP, Peng J, Lukacs GL, Fuchs SY (2007) Site‐specific ubiquitination exposes a linear motif to promote interferon‐alpha receptor endocytosis. J Cell Biol 179: 935–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laman H, Funes JM, Ye H, Henderson S, Galinanes‐Garcia L, Hara E, Knowles P, McDonald N, Boshoff C (2005) Transforming activity of Fbxo7 is mediated specifically through regulation of cyclin D/cdk6. EMBO J 24: 3104–3116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SW, Li CF, Jin G, Cai Z, Han F, Chan CH, Yang WL, Li BK, Rezaeian AH, Li HY, Huang HY, Lin HK (2015) Skp2‐dependent ubiquitination and activation of LKB1 is essential for cancer cell survival under energy stress. Mol Cell 57: 1022–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennox G, Lowe J, Morrell K, Landon M, Mayer RJ (1988) Ubiquitin is a component of neurofibrillary tangles in a variety of neurodegenerative diseases. Neurosci Lett 94: 211–217 [DOI] [PubMed] [Google Scholar]
- Li X, Thompson D, Kumar B, DeMartino GN (2014) Molecular and cellular roles of PI31 (PSMF1) protein in regulation of proteasome function. J Biol Chem 289: 17392–17405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livnat‐Levanon N, Kevei E, Kleifeld O, Krutauz D, Segref A, Rinaldi T, Erpapazoglou Z, Cohen M, Reis N, Hoppe T, Glickman MH (2014) Reversible 26S proteasome disassembly upon mitochondrial stress. Cell Rep 7: 1371–1380 [DOI] [PubMed] [Google Scholar]
- Lohmann E, Leclere L, De Anna F, Lesage S, Dubois B, Agid Y, Durr A, Brice A, French Parkinson's Disease Genetics Study Group (2009) A clinical, neuropsychological and olfactory evaluation of a large family with LRRK2 mutations. Parkinsonism Relat Disord 15: 273–276 [DOI] [PubMed] [Google Scholar]
- Lohmann E, Coquel AS, Honore A, Gurvit H, Hanagasi H, Emre M, Leutenegger AL, Drouet V, Sahbatou M, Guven G, Erginel‐Unaltuna N, Deleuze JF, Lesage S, Brice A (2015) A new F‐box protein 7 gene mutation causing typical Parkinson's disease. Mov Disord 30: 1130–1133 [DOI] [PubMed] [Google Scholar]
- Lowe J, Blanchard A, Morrell K, Lennox G, Reynolds L, Billett M, Landon M, Mayer RJ (1988) Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson's disease, Pick's disease, and Alzheimer's disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and mallory bodies in alcoholic liver disease. J Pathol 155: 9–15 [DOI] [PubMed] [Google Scholar]
- Moiseeva TN, Bottrill A, Melino G, Barlev NA (2013) DNA damage‐induced ubiquitylation of proteasome controls its proteolytic activity. Oncotarget 4: 1338–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DE, Randle SJ, Laman H (2013) Beyond ubiquitination: the atypical functions of Fbxo7 and other F‐box proteins. Open Biol 3: 130131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisan‐Ruiz C, Guevara R, Federoff M, Hanagasi H, Sina F, Elahi E, Schneider SA, Schwingenschuh P, Bajaj N, Emre M, Singleton AB, Hardy J, Bhatia KP, Brandner S, Lees AJ, Houlden H (2010) Early‐onset L‐dopa‐responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord 25: 1791–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathare GR, Nagy I, Bohn S, Unverdorben P, Hubert A, Korner R, Nickell S, Lasker K, Sali A, Tamura T, Nishioka T, Forster F, Baumeister W, Bracher A (2012) The proteasomal subunit Rpn6 is a molecular clamp holding the core and regulatory subcomplexes together. Proc Natl Acad Sci USA 109: 149–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP (2003) A proteomics approach to understanding protein ubiquitination. Nat Biotechnol 21: 921–926 [DOI] [PubMed] [Google Scholar]
- Pickart CM (2000) Ubiquitin in chains. Trends Biochem Sci 25: 544–548 [DOI] [PubMed] [Google Scholar]
- Poulopoulos M, Levy OA, Alcalay RN (2012) The neuropathology of genetic Parkinson's disease. Mov Disord 27: 831–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2011) R: a language and environment for statistical computing. Vienna: R Foudation for statistical computing; http://www.R-project.org/ [Google Scholar]
- Sakata E, Yamaguchi Y, Kurimoto E, Kikuchi J, Yokoyama S, Yamada S, Kawahara H, Yokosawa H, Hattori N, Mizuno Y, Tanaka K, Kato K (2003) Parkin binds the Rpn10 subunit of 26S proteasomes through its ubiquitin‐like domain. EMBO Rep 4: 301–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh K, Sasajima H, Nyoumura KI, Yokosawa H, Sawada H (2001) Assembly of the 26S proteasome is regulated by phosphorylation of the p45/Rpt6 ATPase subunit. Biochemistry 40: 314–319 [DOI] [PubMed] [Google Scholar]
- Saura J, Pares M, Bove J, Pezzi S, Alberch J, Marin C, Tolosa E, Marti MJ (2003) Intranigral infusion of interleukin‐1beta activates astrocytes and protects from subsequent 6‐hydroxydopamine neurotoxicity. J Neurochem 85: 651–661 [DOI] [PubMed] [Google Scholar]
- Savitt JM, Jang SS, Mu W, Dawson VL, Dawson TM (2005) Bcl‐x is required for proper development of the mouse substantia nigra. J Neurosci 25: 6721–6728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shojaee S, Sina F, Banihosseini SS, Kazemi MH, Kalhor R, Shahidi GA, Fakhrai‐Rad H, Ronaghi M, Elahi E (2008) Genome‐wide linkage analysis of a Parkinsonian‐pyramidal syndrome pedigree by 500 K SNP arrays. Am J Hum Genet 82: 1375–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sortwell CE, Daley BF, Pitzer MR, McGuire SO, Sladek JR Jr, Collier TJ (2000) Oligodendrocyte‐type 2 astrocyte‐derived trophic factors increase survival of developing dopamine neurons through the inhibition of apoptotic cell death. J Comp Neurol 426: 143–153 [PubMed] [Google Scholar]
- Sullivan PG, Dragicevic NB, Deng JH, Bai Y, Dimayuga E, Ding Q, Chen Q, Bruce‐Keller AJ, Keller JN (2004) Proteasome inhibition alters neural mitochondrial homeostasis and mitochondria turnover. J Biol Chem 279: 20699–20707 [DOI] [PubMed] [Google Scholar]
- Tian G, Park S, Lee MJ, Huck B, McAllister F, Hill CP, Gygi SP, Finley D (2011) An asymmetric interface between the regulatory and core particles of the proteasome. Nat Struct Mol Biol 18: 1259–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomko RJ Jr, Hochstrasser M (2013) Molecular architecture and assembly of the eukaryotic proteasome. Annu Rev Biochem 82: 415–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ III, Shen J (2010) Loss of leucine‐rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha‐synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci USA 107: 9879–9884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turriziani B, Garcia‐Munoz A, Pilkington R, Raso C, Kolch W, von Kriegsheim A (2014) On‐beads digestion in conjunction with data‐dependent mass spectrometry: a shortcut to quantitative and dynamic interaction proteomics. Biology (Basel) 3: 320–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens R (2011) Chemometrics with R‐multivariate data analysis in the natural sciences and life sciences. Berlin: Springer Verlag; [Google Scholar]
- Yalcin‐Cakmakli G, Olgiati S, Quadri M, Breedveld GJ, Cortelli P, Bonifati V, Elibol B (2014) A new Turkish family with homozygous FBXO7 truncating mutation and juvenile atypical parkinsonism. Parkinsonism Relat Disord 20: 1248–1252 [DOI] [PubMed] [Google Scholar]
- Zaiss DM, Standera S, Kloetzel PM, Sijts AJ (2002) PI31 is a modulator of proteasome formation and antigen processing. Proc Natl Acad Sci USA 99: 14344–14349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, De Graaff E, Breedveld GJ, Loda A, Severijnen LA, Wouters CH, Verheijen FW, Dekker MC, Montagna P, Willemsen R, Oostra BA, Bonifati V (2011) Loss of nuclear activity of the FBXO7 protein in patients with parkinsonian‐pyramidal syndrome (PARK15). PLoS One 6: e16983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, Conaway RC, Conaway JW, Harper JW, Pavletich NP (2002) Structure of the Cul1‐Rbx1‐Skp1‐F boxSkp2 SCF ubiquitin ligase complex. Nature 416: 703–709 [DOI] [PubMed] [Google Scholar]
- Zong N, Ping P, Lau E, Choi HJ, Ng DC, Meyer D, Fang C, Li H, Wang D, Zelaya IM, Yates JR III, Lam MP (2014) Lysine ubiquitination and acetylation of human cardiac 20S proteasomes. Proteomics Clin Appl 8: 590–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6