

Abstract
Pre‐B‐cell leukemia homeobox (PBX) transcription factors are known to regulate organogenesis, but their molecular targets and function in midbrain dopaminergic neurons (mDAn) as well as their role in neurodegenerative diseases are unknown. Here, we show that PBX1 controls a novel transcriptional network required for mDAn specification and survival, which is sufficient to generate mDAn from human stem cells. Mechanistically, PBX1 plays a dual role in transcription by directly repressing or activating genes, such as Onecut2 to inhibit lateral fates during embryogenesis, Pitx3 to promote mDAn development, and Nfe2l1 to protect from oxidative stress. Notably, PBX1 and NFE2L1 levels are severely reduced in dopaminergic neurons of the substantia nigra of Parkinson's disease (PD) patients and decreased NFE2L1 levels increases damage by oxidative stress in human midbrain cells. Thus, our results reveal novel roles for PBX1 and its transcriptional network in mDAn development and PD, opening the door for new therapeutic interventions.
Keywords: ChIP‐Seq, dopamine, dopaminergic differentiation, stem cells, mesencephalon
Subject Categories: Development & Differentiation, Neuroscience
Introduction
Midbrain dopaminergic neurons (mDAn) play a central role in the modulation of several brain functions, including voluntary movements, emotion, and cognition. The progressive degeneration of substantia nigra (SN) mDAn gives rise to some of the main motor features of PD (Lees et al, 2009). Current treatments for PD are symptomatic and there is a need for therapies capable of changing the course of this disease. The PBX family of transcription factors is composed of four members in mammals (PBX1‐4) (Moens & Selleri, 2006; Longobardi et al, 2013). Expression of Pbx genes has been detected in both the mouse and human midbrain as well as mDAn (Thompson et al, 2006; Yin et al, 2009; Ganat et al, 2012; Sgado et al, 2012; Veenvliet et al, 2013). However, to date only a very mild mDAn axon guidance phenotype has been described in Pbx1 null embryos (Sgado et al, 2012). Thus, the function and precise role of different PBX family members on mDAn remains largely unexplored. Moreover, the mechanism of action and molecular targets of PBX transcription factors in mDAn, as well as any role in neurodegenerative diseases, such as PD, remains unknown.
The development of mDAn is controlled by a combination of cell extrinsic and intrinsic signals (Prakash & Wurst, 2006; Ribes et al, 2010; Arenas et al, 2015). One such pathway is that formed by SHH, which regulates and is regulated by FOXA2, a forkhead/winged helix transcription factor required for mDAn development (Ferri et al, 2007). A second main pathway involves WNT1, which regulates and is regulated by LMX1A, a LIM homeodomain transcription factor required for mDAn specification (Andersson et al, 2006). Notably, LMX1A forms an autoregulatory loop together with LMX1B and WNT1, and directly regulates the expression of the orphan nuclear receptor Nurr1/Nr4a2 and the paired‐like homeodomain transcription factor 3 (Pitx3) gene (Chung et al, 2009). Both Nurr1 and Pitx3 are required for mDA neuron differentiation and survival (Zetterstrom et al, 1997; Nunes et al, 2003; Smidt et al, 2004; Maxwell et al, 2005). While LMX1A labels all the cells in the mDA lineage, from proliferating progenitors to postmitotic neuroblasts and mDAn (Andersson et al, 2006), NURR1 labels all postmitotic cells, from mDA neuroblasts to neurons. The very first mDA neuroblasts in mice are detected at embryonic day (E) 10, as LMX1A+NURR1+ cells, and mDA neurons are first detected at E10.5 by the expression of tyrosine hydroxylase (Th), as LMX1A+NURR1+TH+. PITX3 also labels NURR1+TH+ mDA neurons and a subpopulation of postmitotic NURR1+TH− mDA neuroblasts. These factors are thus sequentially expressed during development (Nurr1 followed by Pitx3 and then Th) in progressively restricted mDA postmitotic cells.
We hereby show that the Pbx1 homeobox gene is a novel intrinsic determinant important for the specification and survival of mDA neurons. PBX1 is present in a subpopulation of NURR1+ neuroblasts and in all mDAn, where it plays a dual role in transcription by directly activating genes such as Pitx3, to promote mDAn development, or repressing genes such as Onecut2, to inhibit lateral fates in the midbrain floor plate (mFP). We also found that PBX1 directly increases the expression of a neuroprotective gene activated by oxidative stress, Nfe2l1, which is known to regulate mitochondrial function and proteasomal activity. Notably, a reduction of PBX1 and NFE2L1 levels was detected in the nuclei of mDAn in the substantia nigra (SN) of PD patients. Moreover, we found that decreased levels of NFE2L1 results in increased vulnerability of human midbrain cells to oxidative stress. Thus, our results reveal novel roles of PBX1 and its transcriptional network in mDAn development and PD, opening the door for the future development of novel therapeutic strategies.
Results
PBX1A is present in the developing mDAn and type 2 neuroblasts
Transcriptome analyses (RNA‐Seq) of the mFP at E12.5, compared to adjacent anterior and posterior structures and the dorsal midbrain, revealed enriched expression of the transcription factor Pbx1 together with markers of mDAn such as Th, the rate‐limiting enzyme for the synthesis of DA, and Pitx3, a factor required for the survival of mDAn of the SN (Nunes et al, 2003; Smidt et al, 2004; Maxwell et al, 2005) (Fig 1A). In situ hybridization analyses at E12.5 confirmed that Pbx1 was highly expressed from rostral to caudal levels in the intermediate and marginal zones of the mFP, while Pbx3 transcripts were only weakly detectable in the LMX1A+ mFP and then only at the rostral level (Figs 1B and EV1). A developmental time‐course analysis revealed that the first PBX1+ cells appeared in the mFP at around E10, a few hours before the first TH+ mDAn (at E10.5), and that all TH+ cells at E12.5 in the marginal zone contained PBX1+ nuclei (Fig 1C). Examination of mDA neuroblasts characterized by the expression of Nurr1/Nr4a2, an orphan nuclear receptor required for the development of mDAn (Zetterstrom et al, 1997), revealed that only a subpopulation of NURR1+ neuroblasts become PBX1+ at E11.5 (purple nuclei, Fig 1D). PBX1 was also present in all PITX3+ mDAn, but PITX3+ labeled only a fraction of all PBX1+ cells in the mFP at E12.5 (Fig 1E). Analysis of the two main PBX1 isoforms (PBX1A and B), differing in their C‐terminus domain (Moens & Selleri, 2006), revealed that only PBX1A, the full‐length isoform, is present in mDAn during embryonic development in neuroblasts type 2 and mDAn (Fig 2A and B and Appendix Fig S1). To examine the relevance of these findings to human midbrain development, we undertook similar studies in human embryos. Unlike a previous study, which showed a broad distribution of PBX1+ cells in week 6–8 human ventral midbrain (VM) (Ganat et al, 2012), our analysis of 7‐week human embryonic VM tissue shows that all TH+ and some cells in the intermediate zone were PBX1+ (Fig 2C), suggesting a conserved role of PBX1 in mouse and human mDAn development. In agreement with the study by Ganat et al (2012), PBX1 was found in mDAn of the ventral tegmental area (VTA, A10) and SN (A9) of adult mice (Fig 2D), suggesting a possible conserved function from development through to adulthood.
Figure 1. PBX1 is present in mDAn.
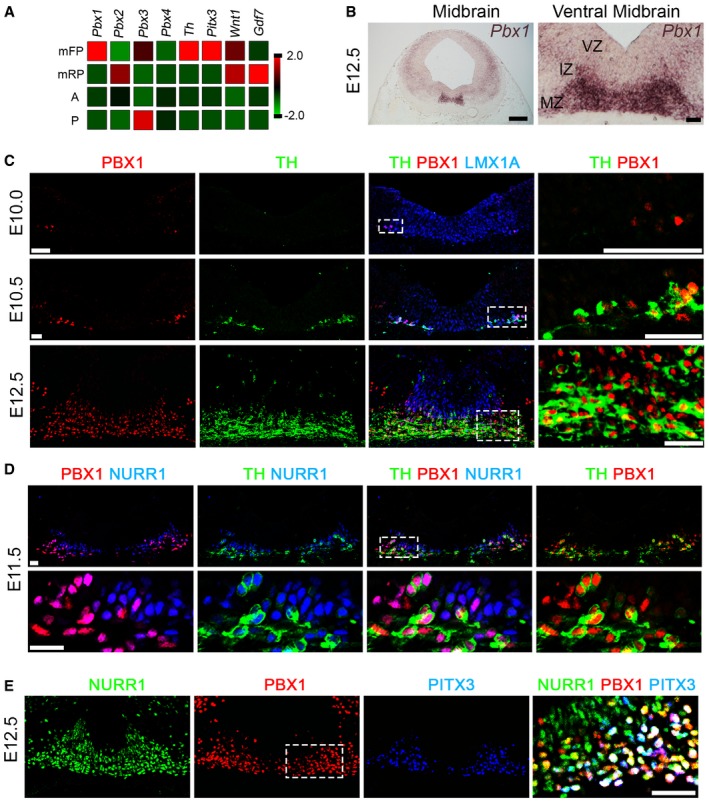
- Tru‐Seq RNA sequence analysis of E12.5 midbrain floor plate (mFP), midbrain roof‐plate (mRP), anterior (A, adjacent anterior FP), and posterior (P, adjacent posterior FP). Pbx1 is enriched in the midbrain FP, together with Th and Pitx3. Lower levels of Pbx3 are also expressed in the mFP. Gdf7 and Wnt1 are restricted to the mRP at E12.5.
- Pbx1 is expressed in the intermediate (IZ) and marginal zones (MZ), but not the ventricular zone (VZ), of the mFP at E12.5, as detected by in situ hybridization.
- PBX1 is first detected in the ventro‐lateral part of the LMX1A+ domain at E10, preceding the birth of the first (TH+) mDA neurons at E10.5. At E12.5, PBX1 is present in all mDA neurons, but not all PBX1+ cells are TH+. White boxes indicate the area shown in higher magnification (right).
- At E11.5, PBX1 protein defines a subpopulation of NURR1+ neuroblasts and labels all NURR1+TH+ mDA neurons.
- PBX1 co‐localizes with PITX3 and is also detected in a subpopulation of NURR1+PITX3− postmitotic neuroblasts at E12.5. Higher magnification revealed three different populations of postmitotic cells: primary neuroblasts (NURR1+PBX1A−PITX3− cells, green), secondary neuroblasts (NURR1+PBX1A+PITX3− cells, yellow/orange), and tertiary neuroblasts/mDA neurons (NURR1+PBX1A+PITX3+ cells, white).
Figure EV1. Pbx1 and Pbx3, but not Pbx2, are expressed in the mouse midbrain.
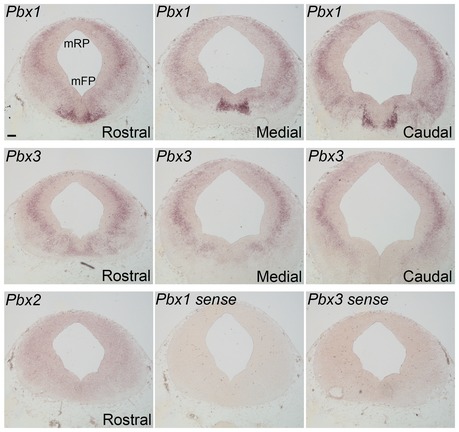
Pbx1 is expressed at high levels in the VM, at rostral, medial, and caudal levels at E12.5. Weak expression of Pbx3 is found only in rostral levels of the VM. Pbx2 was not detected in the midbrain. Pbx1 and Pbx3 sense controls show the specificity of the antisense probe. Scale bar, 80 μm.
Figure 2. PBX1A is present in mDA neuroblasts and in adulthood.
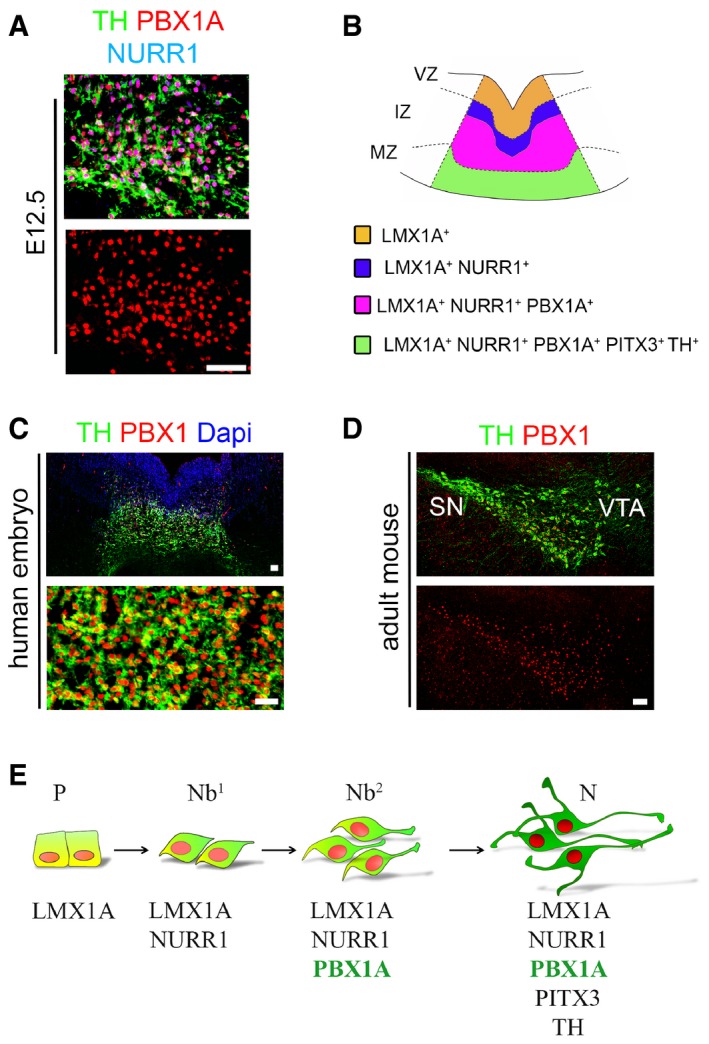
- PBX1A is the isoform detected in the TH+NURR1+ mDA neurons at E12.5.
- Scheme representing the LMX1A, NURR1, PBX1, and TH/PITX3 domains in the embryonic VM at E11.5‐12.5.
- Immunofluorescence analysis of 7‐week‐old human VM tissue showing that all mDAn are positive for PBX1, suggesting a conserved role for this factor in the development of these neurons in mice and humans.
- PBX1 is also present in TH+ adult mDA neurons of the substantia nigra (SN) and ventral tegmental area (VTA).
- Schematic representation of sequential markers expressed in the mDA lineage from progenitors (P), primary neuroblasts (Nb1) to the secondary neuroblasts (Nb2), and mDAn (N), both of which are PBX1+.
In summary therefore, our results indicate that during development, NURR1+PBX1− mDA neuroblasts (neuroblasts type 1, Nb1) mature into NURR1+PBX1+ neuroblasts type 2 (Nb2), before they become NURR1+PBX1+PITX3+TH+ mDAn (Fig 2E).
Pbx1 controls the specification and survival of mDAn
To further uncover the function of Pbx1 in mDAn development, we examined Pbx1 −/− mutant mice. Pbx1 −/− embryos do not survive past E16.5 (DiMartino et al, 2001; Selleri et al, 2001; Kim et al, 2002; Schnabel et al, 2003), and we thus focused our analysis on earlier developmental stages. Pbx1 mutant embryos showed no reduction in NURR1+ cells, but a modest reduction in the number of TH+ neurons (19%), which resulted in a 30% reduction in TH+/NURR1+ cells at E12.5 (Fig 3A and Appendix Fig S2). These results indicate that Pbx1 is required for the proper differentiation of NURR1+ neuroblasts into NURR1+TH+ mDAn. As overlapping functions and functional redundancy among Pbx family members has been previously suggested (Selleri et al, 2004; Ferretti et al, 2011), we examined the levels of other PBX members in Pbx1 −/− embryos. To our surprise, Pbx1 −/− mice exhibited high levels of PBX3 protein from rostral to caudal levels in the VM, suggesting that an up‐regulation of Pbx3 could partially compensate for the loss of Pbx1 (Fig 3B and Appendix Fig S3).
Figure 3. Pbx1 controls the specification and survival of mDAn.
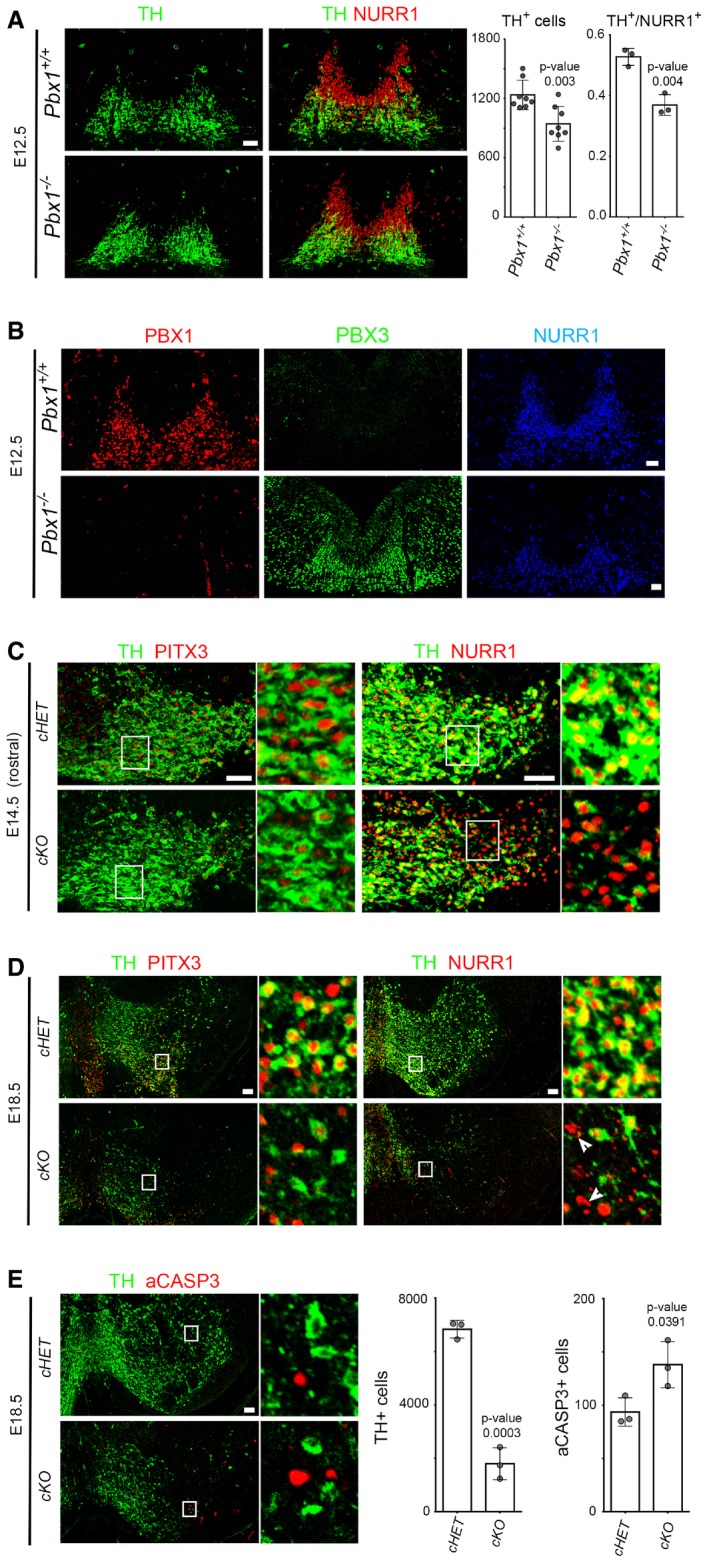
- To the left, representative immunofluorescence/confocal images showing that the number of TH+ cells is reduced in Pbx1 −/− embryos. Deletion of Pbx1 decreases the proportion of NURR1+ neuroblasts that become TH+ neurons at E12.5, indicating that Pbx1 is required for the proper maturation of NURR1+ neuroblasts. To the right, quantification of these experiments.
- Ectopic PBX3 is detected in all rostro‐caudal levels of the VM in Pbx1 −/− mice at E12.5, indicating a repressive function of PBX1 on Pbx3 expression.
- Representative immunofluorescence/confocal images and quantification showing that the conditional deletion of Pbx1 and Pbx3 (cKO, Pbx1 flox/flox ;Pbx3 −/− ;Th‐IRES‐Cre‐ERT) reduces the number of PITX3+ cells in rostral VM levels, but not of NURR1+ cells, when compared to double heterozygous at E14.5 (cHET, Pbx1 flox/+ ;Pbx3 −/+ ;Th‐IRES‐Cre‐ERT). White boxes are magnified to the right.
- At E18.5, a drastic reduction of TH+, PITX3+, and NURR1+ cells is detected from anterior to posterior levels in cKO embryos, but not in cHET. Pyknotic NURR1+ nuclei are observed in cKO embryos (arrowheads). White boxes are magnified to the right.
- The total number of TH+ mDA neurons is dramatically reduced, and there is an increase in aCASP3+ cells in cKO embryos compared to cHET mice at E18.5.
As double Pbx1 −/− ;Pbx3 −/− embryos die before E10.5, we examined Pbx1 −/− ;Pbx3 +/− embryos, which survived until E12.5. Analysis of the VM phenotype at this stage showed no alteration in the number of NURR1+ cells but a reduction of TH+ cells (Fig EV2A) comparable to that seen in the Pbx1 −/− embryos (Fig 3A). Moreover, E12.5 Pbx1 −/− ;Pbx3 +/− embryos showed no difference in the number of cells immunoreactive for neurogenin 2 (NGN2+, Fig EV2B), a transcription factor required for mDA neurogenesis (Kele et al, 2006), or in SOX6 and OTX2 (Fig EV2C), two transcription factors involved in SN vs. VTA subtype specification, respectively (Di Salvio et al, 2010; Panman et al, 2014). Together, our data indicate that there is no alteration in mDA neurogenesis or neuron subtype specification, but rather a specific defect in the differentiation of NURR1+TH− neuroblasts into NURR1+TH+ mDA neurons.
Figure EV2. Analysis of the VM phenotype of Pbx1 −/− ;Pbx3 +/− and cKO embryos.
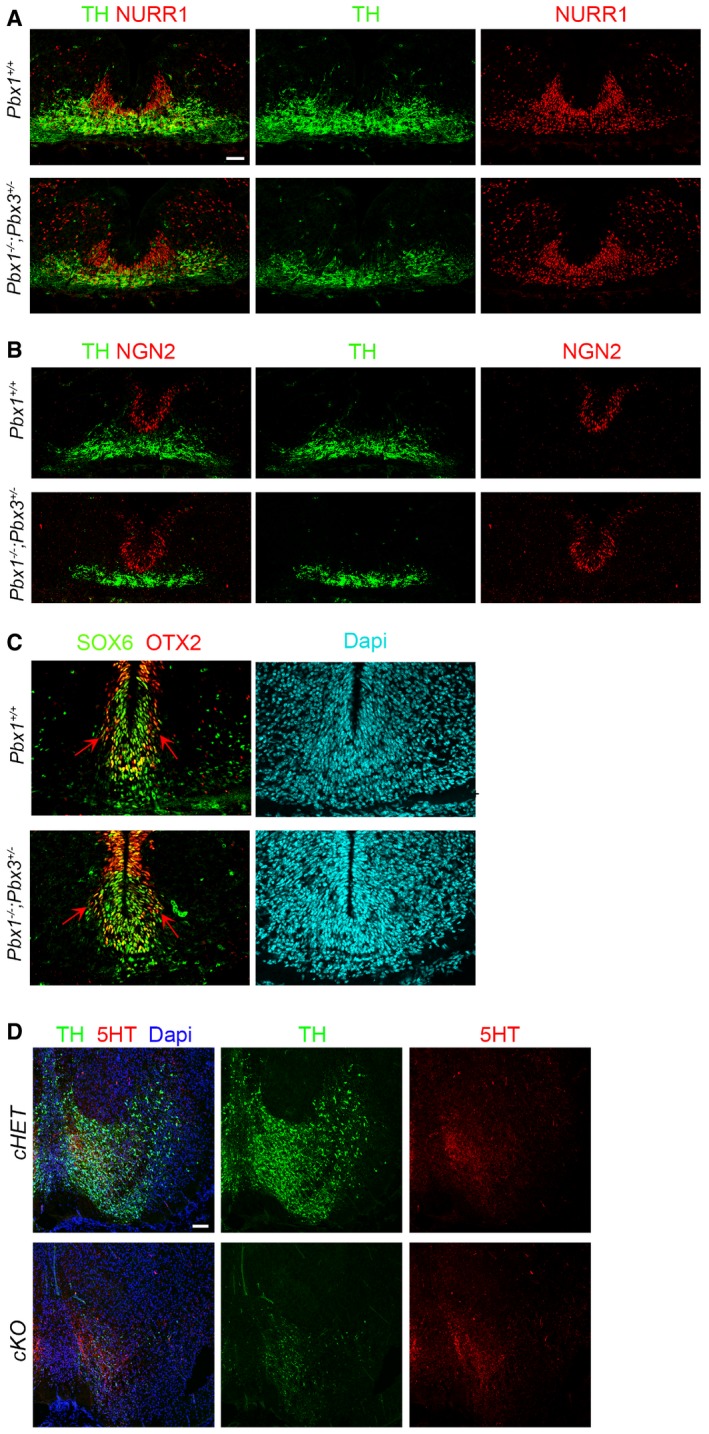
- No alteration in the number of NURR1+ cells and a reduction in TH+ cells is detected in Pbx1 −/− ;Pbx3 +/− embryos at E12.5.
- Immunostaining for NGN2, a transcription factor required for mDA neurogenesis, was not altered in Pbx1 −/− ;Pbx3 +/− embryos at E12.5.
- Immunostaining for SOX6 and OTX2, two transcription factors involved in SN vs. VTA subtype specification, was not different in Pbx1 −/− ;Pbx3 +/− compared to Pbx1 +/+ embryos at E12.5. Red arrows indicate the lateral OTX2+ domain.
- Staining for 5HT (serotoninergic neurons) showed no major differences in cKO vs. cHET embryos at E18.5, indicating that there is no misspecification of mDAn into serotoninergic neurons.
Next, we generated conditional mutant embryos with the Shh‐Cre‐ERT or TH‐IRES‐Cre‐ERT deleter strains. While Pbx3 −/− mice in which Pbx1 was conditionally deleted by Shh before E11.5 were embryonically lethal, conditional deletion by TH (cKO, Pbx1 flox/flox ;Pbx3 −/− ;Th‐IRES‐Cre‐ERT) resulted in viable embryos. Analysis of E14.5 cKO compared to conditional heterozygous (cHET, Pbx1 flox/+ ;Pbx3 +/− ;TH‐IRES‐Cre‐ERT) revealed no alteration in the number of NURR1+ cells, but a dramatic reduction of PITX3+ cells and the loss of the most lateral (and anterior) population of TH+ mDA neurons (Fig 3C). These results showed a defect in the differentiation of mDAn at E14.5, which followed the pattern of nuclear CRE and recombination (Fig EV3A and B) (Rotolo et al, 2008; Badea et al, 2009). Analysis of cKO embryos at E18.5 (Figs 3D and E, and EV3C) revealed a very severe loss of TH+, PITX3+ and NURR1+ cells, which was accompanied by the presence of NURR1+ pyknotic nuclei (Fig 3D, right, arrowheads). The loss of TH+ mDA neurons affected all rostro‐caudal levels and was accompanied by cell death, as shown by an increased number of active caspase‐3+ (aCASP3+) cells, a marker of cellular damage and apoptosis, both at E14.5 (cHet = 43 ± 8.7 vs. cKO = 71 ± 4.4, mean ± SD, P‐value 0.016) and E18.5 (Fig 3E). In addition, staining for 5HT (serotoninergic neurons) showed no major differences in cKO vs. cHET embryos at E18.5 (Fig EV2D), indicating that there is no anterior‐posterior misspecification. The loss of TH+ neurons was also reflected by a loss of TH immunoreactive fibers in the striatum of cKO at E18.5 (Fig EV3D). Since Pbx3 −/− mice did not show any detectable phenotype in mDAn (data not shown), our results indicate that Pbx1 and, in its absence, also Pbx3 are required for the correct differentiation of neuroblasts type 1 into type 2 and into mDAn, as well as for mDAn survival.
Figure EV3. CRE recombination does not take place in all mDA TH+ cells of cKO embryos.
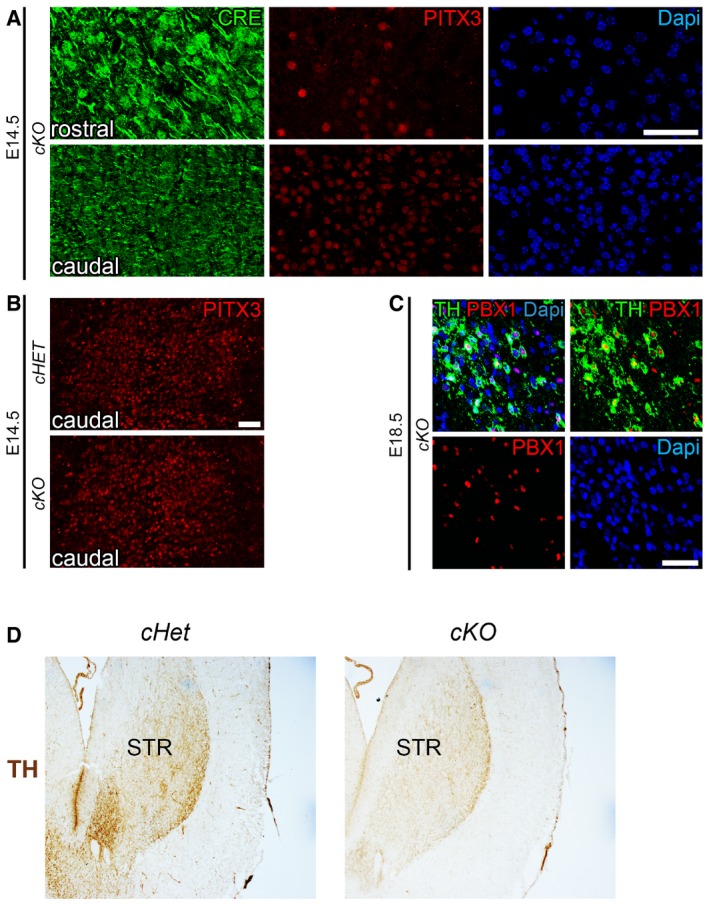
- Pbx1 flox/flox ;Pbx3 −/− ;Th‐IRES‐Cre‐ERT (cKO) embryos treated with tamoxifen at E12.5 and E13.5 showed a reduction of PITX3 at E14.5 in rostral VM levels, where a strong nuclear localization of CRE was detected, but not in caudal VM, where diffuse low levels of CRE were found.
- PITX3 levels were not affected in caudal levels of E14.5 cKO embryos compared to cHET.
- At E18.5, CRE recombination still did not occur in a few cells in the most medial and caudal aspect of the VM of cKO embryos, which remained PBX1+TH+.
- Immunohistochemistry (DAB) for TH indicates that the loss of TH+ neurons in the VM was also reflected by a loss of TH immunoreactive fibers in the striatum (STR) of cKO at E18.5, compared to cHET.
PBX1 improves the differentiation of neural progenitor cells toward a mDA fate
We next performed gain‐of‐function experiments and examined whether expression of PBX1 may contribute toward programming human stem cells into mDAn. Human long‐term neuroepithelial stem (hNES) cells derived from induced pluripotent stem cells (line AF22) (Falk et al, 2012), a source of rosette‐type neural stem cells with extensive self‐renewal and stable neurogenic capacities (Koch et al, 2009), were differentiated using a basic 8 day differentiation protocol (Fig 4A). Lentivirus‐mediated overexpression of human PBX1 was sufficient to increase the final number of TH+ neurons compared to control (Fig 4B). While PBX1 expression did not change the total number of Dapi+ cells, a remarkable 2.3‐fold increase in TH+ cells was detected (Fig 4B and C). TH+ cells in the PBX1‐overexpressing condition were also positive for mDA lineage markers such as LMX1A and PITX3 (Fig 4D). Thus, our results indicate that PBX1 can improve the generation of mDAn from hNES cells, opening the door to new future avenues to improve mDAn replacement therapies for PD (Arenas et al, 2015).
Figure 4. PBX1 improves the differentiation of neural progenitor cells toward mDA fate.
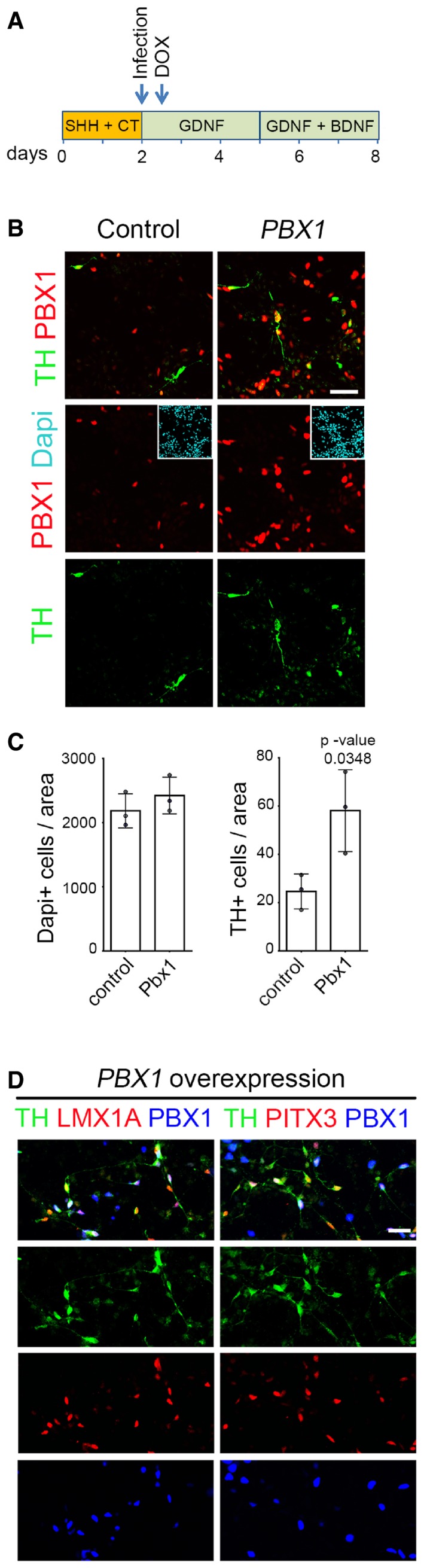
- Scheme of the differentiation protocol for hNES cells. The AF22 cells (NES cells derived from induced pluripotent stem cells) were infected at day 2 with TET‐on PBX1 lentiviral particles, and doxycycline (DOX) treatment started the day after.
- Representative immunofluorescence/confocal images showing that PBX1 overexpression increases the number of TH+ neurons after 8 days of differentiation of hNES cells (AF22 line derived from induced pluripotent stem cells).
- While the number of cells (Dapi+) in both conditions did not change, the number of TH+ cells increased 2.3‐fold in PBX1‐overexpressing cells.
- TH+ cells derived from the PBX1‐overexpressing hNES cells were also positive for mDAn markers such as LMX1A and PITX3.
Identification of direct PBX1 target genes
To identify the molecular mechanisms by which PBX1 regulates mDAn development, we performed chromatin immunoprecipitation sequencing (ChIP‐seq) on E12.5 mouse VM tissue using a PBX1 antibody validated for immunoprecipitation (Fig EV4A). Our analysis identified a PBX1 binding site composed of a 10‐nucleotide motif (A) separated by 8–11 nucleotides from a 5‐nucleotide motif (B) (Fig 5A). A similar 10‐nucleotide motif was recently reported, where the binding capacity of PBX1 was demonstrated using an electrophoretic mobility assay (Ferretti et al, 2011; Penkov et al, 2013). Most PBX1‐binding sites were in close proximity to transcription starting sites (TSS) (Fig EV4B and C). The enrichment profiles of some PBX1‐binding sites and their corresponding genomic regions are shown in Figs 5B and EV4D and E. Enriched gene categories were related to signal transduction (P‐value 10−13), RNA metabolic processes (P‐value 10−10), as well as transcription and response to stress (P‐value 10−7) (Fig EV4F). We next combined our RNA‐Seq analysis of VM tissue at E12.5 with our ChIP‐seq data in order to identify possible direct PBX1 target genes differentially expressed in the VM. Gene Set Enrichment Analysis (GSEA) showed a strong correlation between the cistrome and transcriptome (P‐value < 0.001, q‐value < 0.005). Moreover, the highly correlated genes were either enriched (green) or depleted (red) in the E12.5 VM, suggesting a possible dual role of PBX1 as both an activator and repressor of transcription in the VM, as described for other tissues (Asahara et al, 1999; Saleh et al, 2000). Most of the PBX1 loci were located in the proximity of a single gene (87.6%, 177 of 202) (Fig EV5A and B). De novo motif analysis in 200‐bp regions centered on the PBX1 peaks, to identify additional motifs, revealed slight variations of the PBX1 binding site motif (Fig EV5C, motif 1 and 2) and additional domain B motifs (Fig EV5C, motif 2 and 3), all of which were associated with activated or repressed genes, except for a repressor loci, a GC‐box (Fig EV5C, motif 5), previously identified as an SP1 binding site (Suske, 1999). Moreover, using an unbiased bioinformatics analysis of the promoter regions of PBX target genes for transcription factor binding site enrichment (Kwon et al, 2012), we found only a few binding sites exclusive for activated or repressed loci (Fig EV5D–F). The most noticeable was the presence of a binding site for NR4A2/NURR1 at a repressor locus, suggesting a possible concerted action of PBX1 and NR4A2/NURR1 to repress common target genes.
Figure EV4. PBX1‐chromatin immunoprecipitation.
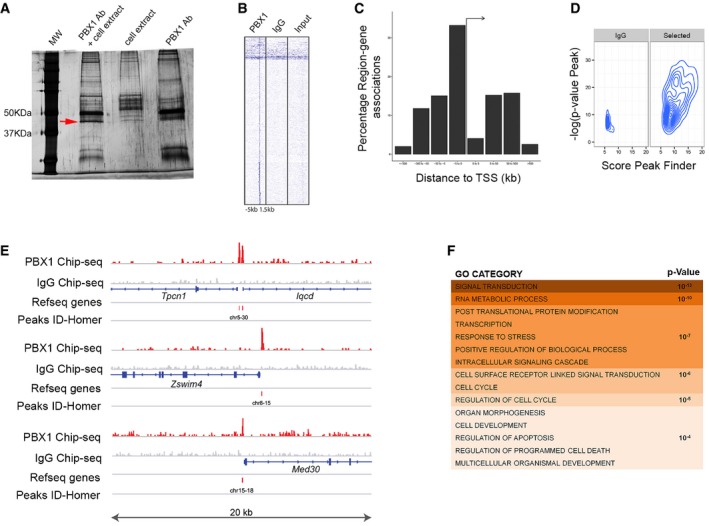
- Validation of the antibody against PBX1 (rabbit anti‐PBX1, #43428, 1:500, Cell Signaling) to immunoprecipitate PBX1 from cell extracts obtained from SN4741 cells transfected with a psg‐PBX1 plasmid. The red arrow indicates the band identified as PBX1 by mass spectrometry.
- Heatmap enrichment of the peaks found within the ChIP‐seq for PBX1 and IgG, in a window of −5 kb to +1.5 kb from the transcription starting site (TSS).
- Summary of the localization of the detected peaks according to their distance to the TSS.
- Quality representation of the peaks detected with HOMER in IgG and PBX1 (selected) ChIP‐seq when statistical significance and peak scores are plotted. The values for peak score represent adjusted number of reads per peak detected.
- Schematic of PBX1 tracks compared to control IgG ChIP‐seq on different representative loci. PBX1 peaks were identified with HOMER. Each locus represents a 20‐kb chromosome region.
- Gene Ontology terms of the enriched categories identified by GSEA.
Figure 5. Identification of direct PBX1 target genes.
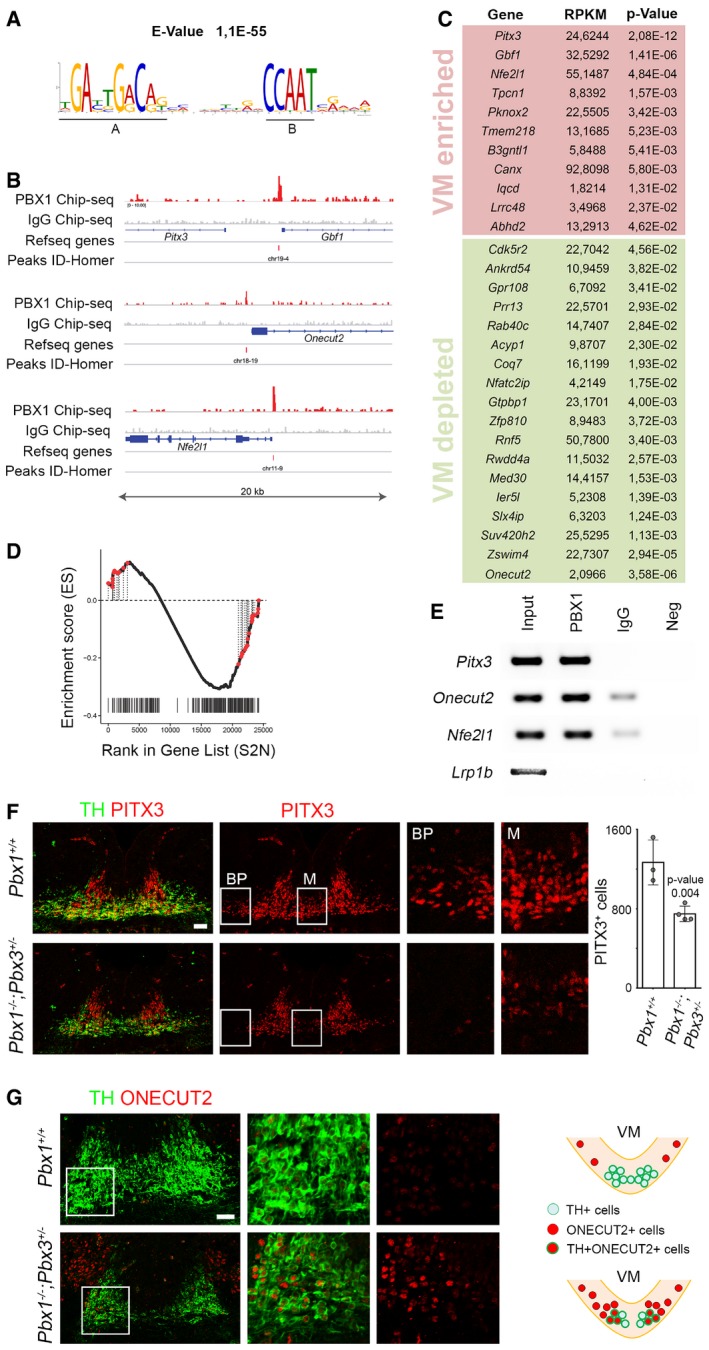
-
AChromatin immunoprecipitation sequencing (ChIP‐seq) identified a PBX1 binding core sequence motif composed of a 10‐nucleotide motif (A) and a 5‐nucleotide motif (B).
-
BSchematic of the track of PBX1 ChIP‐seq compared to IgG ChIP‐seq as a control on different loci with peaks identified by HOMER. Each locus represents a 20‐kb chromosome region.
-
CList of the 29 most regulated genes (t‐test q‐value < 0.05) identified by Gene Set Enrichment Analysis (GSEA) and ranked by P‐value. RPKM stands for reads per kilobase of transcript per million mapped reads. GSEA was performed on genes identified by RNA‐Seq as differentially expressed in the VM at E12.5 compared to adjacent brain regions and identified by PBX1 Chip‐Seq in proximity to transcription starting sites.
-
DGraphic representation of these top 29 genes (red dots) in the GSEA curve showing a high correlation between the cistrome and transcriptome (P‐value < 0.001, q‐value < 0.005).
-
EChIP followed by PCR confirmed that PBX1 binds to genomic regions in close proximity to the TSS of Pitx3 (Pitx3‐Gbf1), Onecut2, and Nfe2l1 genes. No co‐immunoprecipitation was detected for the unrelated gene, Lrp1b (low‐density lipoprotein receptor‐related protein 1B).
-
F, GThe regulation of Pitx3 and Onecut2 by PBX1 was confirmed in Pbx1 −/− ;Pbx3 +/− mutant embryos at E12.5, compared to wild‐type (Pbx1 +/+) littermates. (F) Representative immunofluorescence/confocal images showing that decreased levels of PITX3 and number of PITX3+ cells in E12.5 Pbx1 −/− ;Pbx3 +/− embryos. This reduction is more evident in the basal plate (BP) and in the midline (M), as shown at higher magnification (right). (G) Double ONECUT2+TH+ cells were detected only in the lateral part of the mFP in E12.5 Pbx1 −/− ;Pbx3 +/− embryos. Selected areas are magnified to the right. A scheme (right) shows the distribution of cells in Pbx1 +/+ and Pbx1 −/− ;Pbx3 +/− embryos.
Figure EV5. Dual role of PBX1 in transcription.
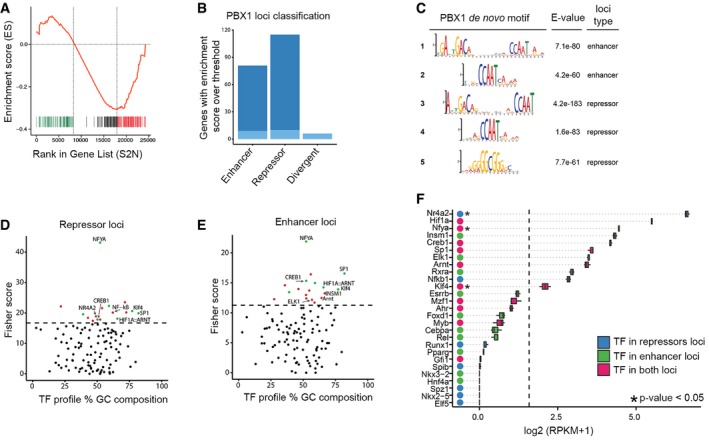
-
AGSEA curve showing genes that are enriched (green) or depleted (red) in the E12.5 VM.
-
BAnalysis of PBX1 peaks in the proximity of two neighbors TSS (as described in Materials and Methods). Dark blue indicates transcriptional regulation of only one gene of the two neighbors. Light blue indicates regulation of both genes. Peaks were classified as divergent if one gene was upregulated and the other downregulated.
-
CMotifs identified de novo on regions of 200 bp centered on PBX1 peaks.
-
D, EOverrepresented transcription factors binding sites found on neighboring loci of PBX1 using the oPOSSUM‐3 software. In green, transcription factor binding sites common in both loci. In red, transcription factor binding sites found only in one locus. Threshold of Fisher score (dashed line) is mean plus standard deviation.
-
FBoxplot of the expression value of transcriptions factors associated with the enriched TFBS at ventral midbrain at E12.5. Color of the dot indicates the type of loci in which the transcription factor is found. DEG genes in the VM at E12.5 are annotated with an asterisk. Dashed line at RPKM = 2.
Twenty‐nine genes with PBX1 binding sites in close proximity to their TSS (−5,000 to + 500 bp) were differentially up‐regulated or down‐regulated in the mFP vs. adjacent regions and midbrain roof‐plate (t‐test q‐value < 0.05) (Figs 5C and EV6). Moreover, these 29 genes were positioned in the leading edges of the curve generated by GSEA (red dots in Fig 5D), indicating a dual activator–repressor activity of PBX1. The highest enriched gene in the list was the homeobox gene Pitx3. Specific binding of PBX1 to a Pitx3‐associated region identified in our ChIP‐Seq was confirmed by ChIP‐PCR (Fig 5E). As conditional double KO embryos showed a drastic reduction of TH+ cells, we decided to analyze embryos with only one allele of Pbx3. Accordingly, Pbx1 −/− ;Pbx3 +/− embryos showed decreased levels of PITX3 and a 40% reduction in the number of PITX3+ cells at E12.5, which was more prominent in the midline and basal plate (Fig 5F and Appendix Fig S4). The most down‐regulated gene in our list was the homeobox gene Onecut2, which is broadly expressed in the central nervous system (Francius & Clotman, 2010; Espana & Clotman, 2012; Stam et al, 2012; Wu et al, 2012), but its deletion does not lead to abnormalities in mDAn development (Espana & Clotman, 2012). We found that Onecut2 is expressed in the midbrain from the basal to the roof‐plate, but not in the mFP (Appendix Fig S5). However, in Pbx1 −/− ;Pbx3 +/− embryos, the ONECUT2 domain extended into the floor plate, resulting in misspecified double TH+ONECUT2+ mDAn (Fig 5G). Thus, our results show that inhibition of alternative lateral fates does take place in postmitotic NURR1+ neuroblasts. Combined these results indicate that PBX1 controls the specification of mDA neurons by directly activating Pitx3 and by repressing lateral fate genes such as Onecut2.
Figure EV6. Multiple comparison of PBX1 binding sites located in different regulatory regions.
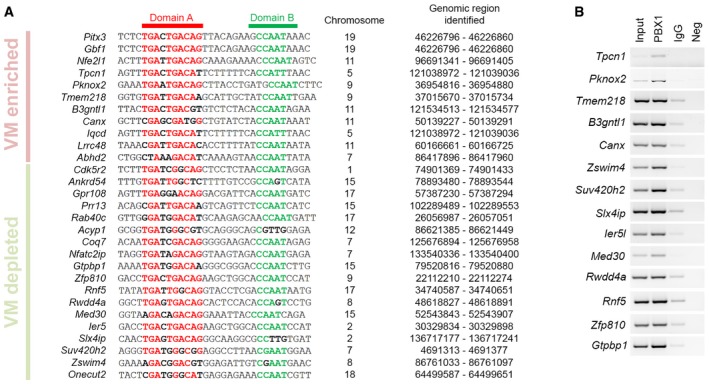
- Our analysis identified a PBX1 binding site composed of a 10‐nucleotide motif (domain A, red) separated by 8–11 nucleotides from a 5‐nucleotide motif (domain B, green). Interval numbers indicate the genomic regions identified in our ChIP experiments. Inside each domain, highly conserved amino acids are in bold red or green, and low conserved amino acids are in bold black.
- PBX1 ChIP in E12.5 mouse VM followed by PCR confirmed that PBX1 binds to multiple regions.
PBX1 promotes survival of human TH+ cells via NFE2L1
The Nfe2l1 gene (nuclear factor erythroid‐derived 2‐like 1, also known as Nrf1 and Tcf11) was the third most enriched target gene of PBX1 in the VM (Fig 5C). ChIP‐PCR confirmed that PBX1 binds to a regulatory region in close proximity to the TSS of Nfe2l1 (60 bp distance) in E12.5 mouse VM (Fig 5E). Forced expression of human PBX1 with lentiviral particles increased protein levels of NFE2L1 by 1.5‐fold, compared to the control condition, in a mouse SN mDAn cell line (SN4741 cells (Son et al, 1999)) (Appendix Fig S6A). Multiple cells in the VM, including TH+ neurons, were found to be NFE2L1+ at E12.5 (Fig 6A). To examine whether the expression of Nfe2l1 in TH+ cells is regulated by PBX1, we analyzed the VM of Pbx1 −/− ;Pbx3 +/− embryos and found a near complete loss of NFE2L1 in TH+ neurons at E12.5 (Fig 6B), but not in PBX1‐free structures such as the branchial arches (Appendix Fig S6B), underlining the specificity of the regulation of NEF2L1 by PBX1/3 in mDAn. However, the function of NFE2L1 is not limited to mDAn as deletion of Nfe2l1 impairs cell survival in other cell types and results in embryonic lethality (Farmer et al, 1997).
Figure 6. PBX1 promotes survival of human neural cells via NFE2L1, a pathway disrupted in PD patients.
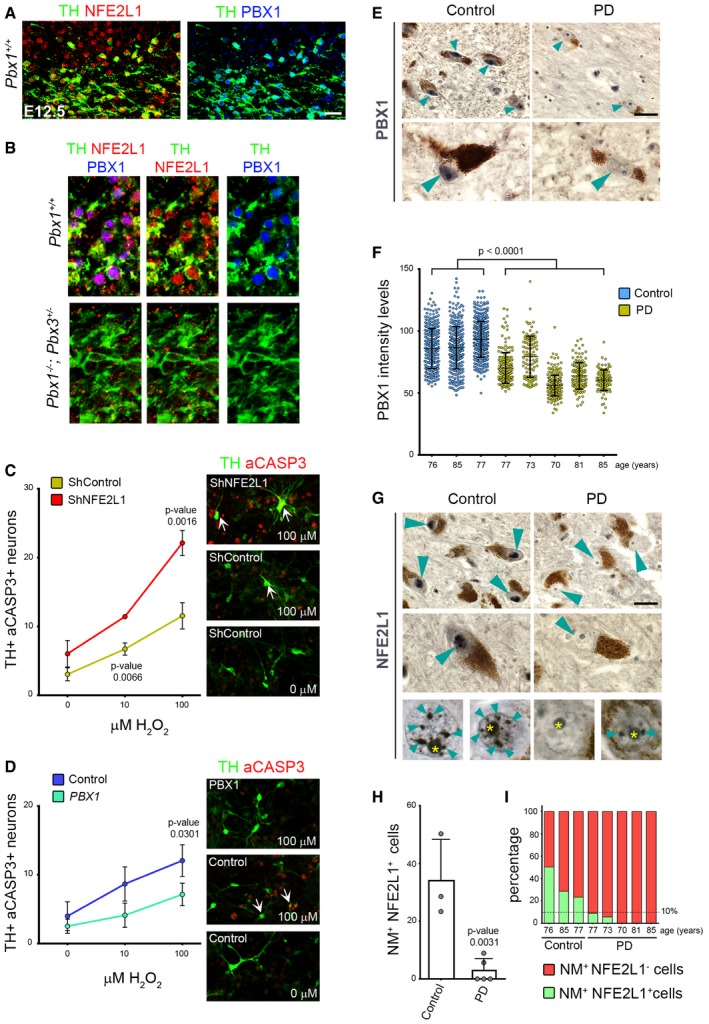
- Representative immunofluorescence/confocal images showing that NFE2L1 is detected in TH+ mDAn and neuroblasts at E12.5.
- The expression of NFE2L1 in TH+ neurons is dramatically reduced in Pbx1 −/− ;Pbx3 +/− compared to Pbx1 +/+ embryos at E12.5.
- shRNA lentiviruses against NFE2L1 increased the levels of aCASP3 in H2O2‐treated (100 μm for 12 h) TH+ neurons derived from hNES cells differentiated for 8 days (percentage). White arrows indicate double TH+aCASP3+ neurons.
- PBX1 overexpression protected TH+ neurons derived from hNES cells from the oxidative stress induced by H2O2 (100 μm for 12 h), resulting in reduced levels of aCASP3 in TH+ neurons (percentage).
- Human postmortem SN tissue of control donors exhibited PBX1 chromogenic staining (dark blue/black, indicated by arrowheads) in the nuclei of neuromelanin+ cells (NM+, cytosolic brown aggregates) of the SN. In contrast, PBX1 staining was severely reduced in PD patients.
- Graphical representation of PBX1 intensity levels in the SN of NM+ neurons in all individual controls and PD patients analyzed. Each dot in the graph represents the level of PBX1 in the nuclei of one single NM+ neuron.
- Strong immunoreactivity of the PBX1 direct target, NFE2L1, was also detected as very well‐defined nuclear punctuate structures in NM+ cells of the SN in control samples (arrowheads), but were dramatically reduced in PD patients. Yellow asterisks indicate the nucleoli.
- Percentage of NM+ neurons showing nuclear NFE2L1 staining in the SN of control donors and PD patients.
- Graphical representation of all individual controls and PD patients analyzed, showing that NM+ neurons with NFE2L1+ nuclei in the SN of PD patients are < 10% or are totally absent.
Nfe2l genes encode basic leucine zipper transcription factors that bind to antioxidant response elements to regulate the expression of cytoprotective target genes in response to oxidative stress (Jennings et al, 2012). We thus examined whether NFE2L1 plays a role in preventing oxidative stress in human mDAn. hNES cells were differentiated for 8 days, infected with lentiviral particles containing a combination of three different NFE2L1 shRNAs to block NFE2L1 expression (Appendix Fig S6C), and 4 days later were treated for 12 hours with H2O2 (Appendix Fig S6D). We found that cells exposed to NFE2L1 shRNAs showed reduced NFE2L1 levels (Appendix Fig S6C) and responded to H2O2 treatment with increased number of both TH+aCASP3+ and aCASP3+ cells, compared to control (Fig 6C, Appendix Fig S6E), indicating a role of NFE2L1 in preventing oxidation in TH+ cells. We next examined whether PBX1 can elicit the antioxidant effect of NFE2L1 on TH+ neurons. Lentiviral overexpression of PBX1 in human NES cells differentiated into mDAn significantly reduced the number of TH+aCASP3+ cells compared to controls, in cultures treated with 100 μm H2O2 (Fig 6D). Combined, our results indicate that the PBX1‐NFE2L1 axis plays a role in preventing damage by oxidative stress in human mDAn.
The PBX1‐NFE2L1 pathway is disrupted in PD patients
Nfe2l1 has been also found to regulate genomic stability (Oh et al, 2012) and increase proteasome transcription and activation in response to cell stressors such as proteasome inhibition (Steffen et al, 2010) or PARK2/Parkin‐mediated mitophagy (Furuya et al, 2014), two functions impaired in PD (Scarffe et al, 2014; Pickrell & Youle, 2015). To examine the role of NFE2L1 in vivo and its possible implication in PD, we next examined human postmortem tissue samples from both PD brains and aged‐matched controls with no other brain pathology for the presence of nuclear PBX1 and NFE2L1. We only considered sections from patients and controls in which nuclear immunoreactivity for NFE2L1 antibody was clearly detected, and used consecutive sections for PBX1 analyses. We found that the levels of PBX1 in the nuclei of neuromelanin‐positive (NM+) cells in the SN were significantly reduced in PD patients compared to controls (dark blue/black precipitate, Fig 6E and F). NFE2L1 showed strong nuclear punctuate staining in 20–50% of the nuclei of NM+ cells in the SN of control donors (Fig 6G–I). To our surprise, the number of NM+ neurons with nuclear NFE2L1 staining was dramatically reduced or completely absent in PD patients (0–10%) (Fig 6G–I). This effect was specific to the NM+ cells of the SN since other midbrain cells outside the mDA domain remained NFE2L1+ (Appendix Fig S6F). This result indicated that the PBX1‐NFE2L1 pathway is impaired in mDAn of the SN in PD patients.
Discussion
Our study identifies Pbx1 as a novel intrinsic determinant required for the specification and survival of mDAn. Notably, deletion of Pbx1 was compensated by an upregulation of Pbx3, which is otherwise only expressed at low levels in the anterior midbrain and is not required per se for mDAn development. Pbx3 upregulation partially compensated for the loss of Pbx1, as indicated by the strong phenotype in compound mutant mice. Pbx1 −/− ;Pbx3 +/− mice revealed a lateral misspecification of mDAn, as shown by the loss of PITX3 and the appearance of ONECUT2 in midbrain TH+ cells. Moreover, conditional deletion of Pbx1 in TH+ cells of Pbx3 −/− mice resulted first in reduced PITX3 levels and the loss of PITX3+ cells at E14.5. This was followed by a dramatic loss of NURR1+, PITX3+ and TH+ cells, as well as increased numbers of aCASP‐3+ cells and pyknotic NURR1+ nuclei at E18.5. Combined, our results indicate that Pbx1, and in its absence Pbx3, are required for the correct medio‐lateral specification, differentiation and survival of mDA type 2 neuroblasts and mDAn during development.
Mechanistically, we found that PBX1A binds to a DNA sequence highly conserved in vertebrates (Figs 5A and EV6A), which is formed by two motifs: a PBX1 binding site and a small CCAAT box extension, which may be bound by other transcription factors such as C/EBPs (CCAAT‐enhancer‐binding proteins), also known to control neurogenesis and neuronal differentiation (Menard et al, 2002; Cortes‐Canteli et al, 2011). Our combined ChIP‐Seq and RNA‐Sep analysis revealed that PBX1 has a dual transcriptional role in mDAn, which involves both directly repressing the expression of Onecut2 (to repress lateral fates) as well as activating the expression of both Pitx3 (to promote mDAn differentiation) and Nfe2l1 (to promote mDAn survival). Our results thus indicate that PBX1 controls a full transcriptional program essential for different aspects of mDAn development and maintenance.
Current therapies for PD focus on correcting or balancing neurotransmitter levels or function by different means, which result in symptomatic relief, but do not change the course of the disease. The design of novel strategies to treat PD is currently limited by our incomplete knowledge of the mechanisms that control the development and maintenance of mDAn as well as the pathogenic pathways driving the disease. By unraveling some of these mechanisms, our work may contribute to the development of such therapies. For instance, the finding that expression of PBX1 in human neural stem cells improves the generation of mDAn suggests a possible application of PBX1 to improve current protocols for the generation of mDAn from pluripotent stem cells (Kriks et al, 2011; Kirkeby et al, 2012), which could lead to improvements for cell replacement therapy or disease modeling of PD. We also report that PBX1 directly controls the expression of an antioxidant transcription factor, Nfe2l1, which counteracts oxidative stress, mitochondrial dysfunction, and proteasome impairment (Hirotsu et al, 2012; Furuya et al, 2014; Bugno et al, 2015). Our results show a dramatic reduction or complete absence of nuclear PBX1 and NFE2L1 in neuromelanin+ neurons of the substantia nigra of PD patients, pointing to a possible defect in the PBX1‐NFE2L1 pathway in PD. A previous postmortem study has reported that the nuclear localization of NFE2L2, unlike that of NFE2L1 (in our study), is strongly induced in PD patients (Ramsey et al, 2007), underlining a differential regulation of NFE2L1 and 2 in PD. Importantly, our results suggest that NFE2L1 serves a physiological role in healthy mDAn of the substantia nigra and that its near complete absence in the nuclei of substantia nigra neurons in PD, impairs its function. Accordingly, we found that while increased PBX1 levels promote the survival and differentiation of human mDAn, reduced levels of NFE2L1 increase vulnerability to oxidative stress. We thus suggest PBX1 and NFE2L1 as novel targets for drug discovery, aimed at increasing neuroprotection in PD.
In summary, our data highlight the important role of Pbx1 in the specification and maintenance of mDAn. Moreover, we identify that the PBX1‐NFE2L1 pathway is a novel neuroprotective pathway that is dramatically compromised in PD patients. Thus, our results open the door for the future development of novel therapies for PD designed to alter the course of disease.
Materials and Methods
Animals
Wild‐type embryos were obtained from CD1 and C57BL/6 mice strains. All mutant alleles used in this study were previously described: Pbx1 (Selleri et al, 2001); Pbx3 (Rhee et al, 2004); Pbx1 conditional mice (Ferretti et al, 2011); Th‐Gfp (Matsushita et al, 2002). As for deleter strains, Shh‐Cre‐ERT2 knock‐in mice (Harfe et al, 2004) and TH‐IRES‐Cre‐ERT2 (Rotolo et al, 2008; Badea et al, 2009) were used for Pbx1 inactivation in the VM or in mDA neurons. Mice were housed, bred, and treated in accordance with protocols approved by the local ethics committees (Stockholm's Norra Djurförsöketiska Nämnd N154/06, N88/07, N135/08, N145/09, N273/11 and, N326/12). All mutant mice were kept on a C57BL/6 background. Conditional pregnant females (between 3 and 7 months old) were treated with 4 mg tamoxifen per day (dissolved in corn oil) through oral gavage with animal feeding needles (Fisher Scientific, Waltham, MA) at 12 pm at 11.5, 12.5 and 13.5 days post‐coitum.
Fetal tissue and patient sections
Human fetal tissue collection and processing ethical approval for the use of postmortem human fetal tissue was provided by the National Research Ethics Service Committee East of England—Cambridge Central, UK (ethics number 96/085).
All brain sections were obtained from the Cambridge Brain Bank. Information about their protocols and ethical committees can be found at http://www.cuh.org.uk/for-public/cambridge-brain-bank. We only considered sections from patients and controls in which nuclear immunoreactivity for NFE2L1 antibody was clearly detected, and used consecutive sections for PBX1 analyses. We excluded material in which no NFE2L1 immunoreactivity was detected in NM+ or NM− cells.
In this study, the control group was
PT151—76 years old (yo), male, no evident pathological condition
PT190—85 yo, female, no evident pathological condition
BB137/PT69—77 yo, female, with bilateral old lacunar infarcts, and no evidence of dementia.
For the PD group,
P65—77 yo, male, with idiopathic PD, and some Alzheimer pathology, insufficient for a diagnosis in life
PT161—73 yo, female, with idiopathic PD
P67—70 yo, female, with idiopathic PD
P69— 81 yo, female, with idiopathic PD
P70—85 yo, female, with idiopathic PD
Gene and protein nomenclature
We followed the guidelines from the Human Genome Organization (HUGO) Gene Nomenclature Committee (HGNC) International Advisory Committee and the guidelines from the International Committee on Standardized Genetic Nomenclature for Mice.
For mice, gene symbols are italicized, with only the first letter in uppercase and the remaining letters in lowercase (Pbx1). Protein designations are the same as the gene symbol, but are not italicized and all are upper case (PBX1). mRNAs use the same formatting conventions as the gene symbol.
For human, gene symbols are italicized, with all letters in uppercase (PBX1). Protein designations are the same as the gene symbol, but are not italicized and all letters are in uppercase (PBX1).
Immunofluorescence, immunochemistry, and in situ hybridization
For embryo analyses, heterozygous mice of the relevant genotype were mated overnight, and at noon of the day, the plug was considered E0.5. Briefly, embryos were dissected out of the uterine horns in ice‐cold PBS, fixed in 4% paraformaldehyde (PFA) in PBS overnight, cryoprotected in 30% sucrose, and frozen in Tissue‐Tek optimum cutting temperature (OCT) compound (Sakura Fine‐Tek) on dry ice. Serial coronal 16 μm sections were used for immunohistochemistry or in situ hybridization.
For immunohistochemistry or immunofluorescence, all the sections were treated for antigen retrieval by microwave boiling in antigen retrieval solution (DAKO, S1699). Sections were washed in PBT (PBS with 0.5% Tween‐20) and blocked in PBTA (PBS, 5% donkey serum, 0.3% Triton X‐100, 1% BSA, except for human adult sections where the concentration of donkey serum was 10%). Primary antibodies were diluted in PBTA, and incubations were carried out overnight at 4°C. After washes in PBT, incubations with the appropriate secondary antibodies (Alexa, 1:500, Invitrogen, or horseradish peroxidase, The Jackson Laboratories) were carried out for 1 h at room temperature. For DAB staining with dark/blue coloration, addition of nickel and cobalt was added to the DAB solution.
Non‐radioactive in situ hybridization was performed as previously described (Kele et al, 2006). The mouse antisense RNA probes have been described previously (Ferretti et al, 2011).
Antibodies used for immunofluorescence were as follows: rabbit anti‐NURR1 (sc‐990, 1:500, Santa Cruz Biotechnology); goat anti‐NURR1 (AF2156, 1:500, R&D); rabbit anti‐TH (P40101‐0, 1:500; Pel‐Freeze); sheep anti‐TH (NB300‐110, 1:500, Novus Biologicals); mouse anti‐TH (22941, 1:500; Immunostar); rabbit anti‐LMX1A (1:1,000; gift from Mike German, University of California, San Francisco, CA); goat anti‐LMX1A (C‐17, sc‐54273, 1:50, Santa Cruz Biotechnology); mouse anti‐PBX1A (710.2, sc‐101851, 1:250, Santa Cruz Biotechnology); mouse anti‐PBX1B (41.1, sc‐101852, 1:250, Santa Cruz Biotechnology); rabbit anti‐PBX1 (#43428, 1:750, Cell Signaling); rabbit anti‐PBX1 (P‐20X, sc‐889X, Santa Cruz Biotechnology); goat anti‐PITX3 (N‐20, sc‐19307, 1:50, Santa Cruz Biotechnology); rabbit anti‐PITX3 (382850, 1:250, Invitrogen); rabbit anti‐ONECUT2 (HPA057058, 1:200, ATLAS Antibodies); rabbit anti‐cleaved caspase‐3 (D175, 1:500, Cell Signaling); rabbit anti‐5HT (S5545, 1:1,000; Sigma); and goat anti‐OTX2 (AF1979, 1:500, R&D). All secondary antibodies were Alexa‐conjugated (Invitrogen) with 405, 488, 555, or 647 fluorophores. For nuclear staining, we used Dapi (1:5,000, Sigma). Slides were mounted with fluorescent mounting media (DAKO, S3023).
Immunohistochemistry and in situ hybridization pictures were taken with a Zeiss Axioplan2 microscope. Immunofluorescence pictures were taken using a Zeiss Exciter LSM 510 or an Olympus FV1000 Confocal Microscope. Figures were assembled in Adobe Photoshop (Adobe).
Western blot
For Western blot analysis, samples were subjected to 10% SDS–PAGE, electrotransferred onto Hybond‐P membrane, immunodetected using appropriate primary and secondary antibodies, and visualized by ECL+Plus reagents according to the manufacturer's instructions (Amersham). Antibodies used were as follows: polyclonal rabbit anti‐NRF1 (NFE2L1, Santa Cruz, H‐285, sc‐13031), mouse anti‐beta‐Actin (Abcam, ab8226), and mouse anti‐GAPDH (G8795, Sigma).
Blots were analyzed using a BIO‐RAD Molecular Imager ChemiDoc XRS+ Imaging system.
Chromatin immunoprecipitation (ChIP)
Ventral midbrain E12.5 tissues were trypsinized to a single‐cell suspension using TrypLE (Invitrogen). Cells were filtered and washed using cold PBS with 1% BSA. Chromatin immunoprecipitation assays were performed following the manufacturer's instructions (Pierce, Agarose ChIP kit; 26156). The chipped fragmented DNA samples obtained from the ChIP were end repaired and A‐tailed. Then, the Illumina adapters were ligated and size selected (200–350 bp) from the gel before going to PCR amplification. The PCR samples were purified and size selected again in the gel. The final eluted samples from the gel were sequenced in Genome Analyzer IIx. Both the samples and input were loaded in a single lane (used two different barcode to track two different samples). Single‐end sequencing reads were generated from the final cDNA libraries. The final outcome sequences were aligned to the mouse genome. Peak detection was performed using the HOMER base suite v4.3 (Heinz et al, 2010) with a false discovery rate filter of 5% utilizing the read from PBX1‐ChIP‐seq, normal IgG ChIP‐seq normalized to inputs. The top 10% of the peaks were used for de novo motif finding using the MEME‐ChIP tool from MEME suite (Machanick & Bailey, 2011) (http://meme-suite.org/) with default settings and the HOMER‐based suite (Heinz et al, 2010).
The peak‐gene association was done using GREAT v2.0.2, associating a genomic region with the two nearest genes (McLean et al, 2010). Peaks heatmap was done using SeqMiner 1.3.3 (Ye et al, 2011) with default settings. Gene set enrichment analysis (GSEA) was performed as described previously (Subramanian et al, 2005), with genes scored according to their signal to noise metric of expression in the ventral midbrain vs. the other samples and custom gene sets for PBX1 ChIP‐seq results. ChIP‐seq representation of reads was made using the integrative genome browser (Robinson et al, 2011). ChIP‐seq data are deposited in the public NCBI GEO database with accession number GSE82098 and GSE82100.
Locus classification and analysis
Genes of the GSEA with a running enrichment score that was positive were listed as genes upregulated by PBX1. Genes with running enrichment scores that were after the minimum (leading edge) were classified as genes repressed by PBX1. The genes peaks were classified as enhancer if at least one of the genes were upregulated, classified as repressor if at least one was downregulated. Peaks on which one gene was upregulated and the other downregulated were classified as divergent. Transcription factor binding site enrichment was performed using the oPOSSUM‐3 single‐site analysis software with all genes as background, and the Jaspar Core profiles (Portales‐Casamar et al, 2010) with the rest of the options as default (Kwon et al, 2012). Motif comparison was made using the TOMTOM software (Gupta et al, 2007) of the MEME suit (Bailey et al, 2015).
Tru‐Seq
Different parts of embryonic (E12.5) mouse brains (Th‐Gfp strain) were dissected under the microscope. RNA was extracted from the collected tissue according to a Trizol RNA isolation protocol (Life Technology). The starting RNA material to construct cDNA libraries from each sample was 200 ng. cDNA libraries were prepared according to Illumina's Tru‐Seq DNA sample preparation guide. Ten enriched libraries (duplicate samples from five different parts of the brain) were subjected to Illumina sequencing in one lane of HiSeq 2000. The sequencing reads were sorted by barcode and then mapped into the mouse genome (mm9) using Bowtie short read aligner. The data were analyzed using Qlucore omics explorer. RNA‐seq data are deposited in the public NCBI Gene Expression Omnibus (GEO) database with accession number GSE82099 and GSE82100.
Mass spectrometry analysis
The antibody against PBX1 used for ChIP (rabbit, #43428, 1:500, Cell Signaling) was validated by mass spectrometry, identifying PBX1 protein as the main immunoprecipitated protein, using E14.5 VM extracts. Protein bands were excised manually from a silver‐stained SDS–PAGE gel and de‐stained, reduced, and trypsin‐digested in a robotic protein handling system (MassPREP station, Waters, Milford, MA). Tryptic peptides were extracted with 30 μl of 5% formic acid/2% acetonitrile and subsequently with 24 μl 2.5% formic acid/50% acetonitrile. Protein fragments were separated using a nanoAcquity HPLC system (Waters, Milford, MA) and sequenced using collision‐induced dissociation in a quadruple time‐of‐flight mass spectrometer (Q‐TOF Premier API, Waters, Milford, MA, USA) with a standard Z‐spray source. Data analysis was performed using ProteinLynx Global SERVER 2.3 software (Waters, Milford, MA). Lockmass reference of a [Glu1] fibrinopeptide B (Sigma, St. Louis, MO, USA) was used for mass scale correction when processing the data.
Cell lines
For maintenance of hNES cells (AF22 line, iPSC‐derived (Falk et al, 2012)), AF22 cells are kept in 6‐well plates coated with laminin (1:500 in water, Invitrogen, 23017‐015) and poly‐L‐ornithine (PLO, 1:5 in water; Sigma), in maintenance medium (N2 supplement (1:100, Gibco), B27 supplement (1:1,000, Gibco), FGF2 (10 ng/ml, R&D), and bFGF (10 ng/ml, R&D).
For differentiation of hNES cells, they are dissociated (TrypLE Select), seeded in 48‐well plates coated with laminin and PLO (100,000 cells per well), and cultivated for 2 days with N2 (1:100), B27 (1:1,000), SHH (200 ng/ml, R&D), and CT99021 (1 μM). After that, cells were kept on media containing N2 (1:100), B27 (1:100), GDNF (20 ng/ml, R&D), and BDNF (20 ng/ml, R&D).
SN4741 cells were maintained as previously described (Son et al, 1999).
Cell lines were routinely tested for mycoplasma contamination. The AF22 line was authenticated recently (Falk et al, 2012). SN4741 line was not authenticated by any method.
Cloning, lentivirus production, and infection
Human PBX1 cDNAs were cloned into the lentiviral backbone Tet‐O‐Fuw using the EcoRI restriction site present on the vector. The cDNAs were amplified with the following primers:
hPbx1 EcoRI Fwd: ggccGAATTCATCGACGAGCAGCCCAGGC
hPbx1 EcoRI Rev: ccggGAATTCTCAGTTGGAGGTATCAGAGTG
The Tet‐O‐Fuw‐hPBX1 and FUW‐rtTA (Addgene 20342) lentiviruses were produced in HEK293FT packaging cells (Invitrogen) cultured in DMEM with 10%FBS and penicillin/streptomycin. On the day prior to the transfection, HEK293FT cells were plated in order to reach a 70–80% confluence on the next day. On the following day, the cells were transfected using the XtremeGene9 transfection reagent (Roche) according to the manufacturer's instructions. The Tet‐O‐Fuw plasmids harboring hPBX1 and the FUW‐rtTA plasmid were individually cotransfected with the vectors pMDLg/pRRE (Addgene 12251), pRSV‐Rev (Addgene 12253), and pMd2.G (Addgene 12259) in a 4:2:1:1 ratio, respectively. The transfection mix was removed after an overnight incubation, and fresh media were supplied to the cells. The volume of the fresh media was just sufficient to cover the cells and avoid their dehydration. Thirty hours after transfection, the media of the cells were collected and centrifuged for 5 min at 435 g in order to separate the supernatant from cellular debris. The supernatants were then centrifuged for 2 h and 30 min at 60,000 g at 4°C in a Beckman Coulter Avanti‐JE Centrifuge. The resulting pellets were slowly resuspended in PBS over 2 hours, then aliquoted, and kept at −80°C.
The three viruses were subsequently titrated using the Lenti‐X Provirus Quantitation Kit (Clontech) in SVGp12 cells (ATCC) according to the manufacturer's instructions.
hNES cells were infected in their normal culture medium overnight, while the SN cells were infected in their normal culture medium supplemented with 8 μg/ml of polybrene for 24 h.
ShRNA experiments
NFE2L1 (NRF1) shRNA lentiviral particles (sc‐43575‐V, Santa Cruz Biotechnology Inc.) are a pool of concentrated, transduction ready viral particles containing 3 target‐specific constructs that encode 19–25 nucleotides (plus hairpin) shRNA designed to knock down human NFE2L1 gene expression. The volume selected to treat 1 single well of a 48‐well plate was 10 μl, corresponding to 50,000 infection units of virus (each vial contains 1.0 × 106 infectious units of virus in 200 μl). As a negative control for experiments, we used the shRNA lentiviral particles (sc‐108080, Santa Cruz Biotechnology Inc.), which encodes a scrambled shRNA sequence that does not lead to the specific degradation of any known cellular mRNA.
RT–PCR
All the reagents were purchased from Invitrogen. The following oligonucleotides (Eurofins MWG Operon and/or SIGMA) were used to amplify the genomic regulatory region identified in our ChIP experiments:
| Pitx3 | Forward: 5′‐AGGGAAAGAAGAGTAGGTAGGG‐3′ |
| Reverse: 5′‐TGGTAGGATGAAGTTGGGGG‐3′ | |
| Onecut2 | Forward: 5′‐CCGCCCCTTCTATAGCCATT‐3′ |
| Reverse: 5′‐GGAGCAGCCAACCTATCCTC‐3′ | |
| Forward: 5′‐TCAGAGAAGGTCTGCCTTTCG‐3′ | |
| Reverse: 5′‐GGGAGCAGCCAACCTATCCT‐3′ | |
| Nfe2l1 | Forward: 5′‐GGTCCAATCAGATAGCCGGG‐3′ |
| Reverse: 5′‐TGGGCAGACACAAACTCGAC‐3′ | |
| Forward: 5′‐CCACCCCAGAAACAGTTGGT‐3′ | |
| Reverse: 5′‐TCTCTCCACGTCAGTCTCGG‐3′ | |
| Tpcn1 | Forward: 5′‐TCTGACGTTTCGCCAATGGA‐3′ |
| Reverse: 5′‐CATCAGAGGGGTCGCAATGA‐3′ | |
| Pknox2 | Forward: 5′‐GGGAGACACTCTAGCAGCTC‐3′ |
| Reverse: 5′‐ACAGCCTATACTTCCCAGCAC‐3′ | |
| Tmem218 | Forward: 5′‐CCCTTCATCCAGGCTTTGGT‐3′ |
| Reverse: 5′‐AACGGTCTTCCCGTTGTTGT‐3′ | |
| B3gntl1 | Forward: 5′‐GAAGGACCGCCTTCCCTTAC‐3′ |
| Reverse: 5′‐CTGCCCTTTTCCTGCAATCG‐3′ | |
| Canx | Forward: 5′‐TTCCTGCCTCGCTATTGACC‐3′ |
| Reverse: 5′‐TCACGTGCCGCAGAACTAAG‐3′ | |
| Zswim4 | Forward: 5′‐GAAGCGACCCTCCGTAAACA‐3′ |
| Reverse: 5′‐CAAAGACCCGCCTCCAAGAT‐3′ | |
| Suv420h2 | Forward: 5′‐CTTAAAGAGCCGTCGCTCCA‐3′ |
| Reverse: 5′‐CTCAGGGATTTTGCCCCACT‐3′ | |
| Slx4ip | Forward: 5′‐GACAACTGAGTGACAGGGCA‐3′ |
| Reverse: 5′‐ACCGGAAATCTTACGGGCAG‐3′ | |
| Ierl5 | Forward: 5′‐GCTACGAATTTCGACGTGGC‐3′ |
| Reverse: 5′‐ACTGGCCAATCAGACCAAGG‐3′ | |
| Med30 | Forward: 5′‐CCACTCGGCCGATTTCAAAAA‐3′ |
| Reverse: 5′‐AGCCCACCCCATCTATCACT‐3′ | |
| Rwdd4a | Forward: 5′‐ACCCAGGATCGCGTTATGTG‐3′ |
| Reverse: 5′‐TGAGTGACAGCACTCCACAC‐3′ | |
| Rnf5 | Forward: 5′‐GCTTATGGTGGGCGAGTTCT‐3′ |
| Reverse: 5′‐GAAAAAGGTGTGTGTGGGGC‐3′ | |
| Zfp810 | Forward: 5′‐TCGATCCAGACCCTAGGACG‐3′ |
| Reverse: 5′‐TGACTGACAGAAGCTGGCAC‐3′ | |
| Gtpbp1 | Forward: 5′‐CGGGACCCAATCTTGTCCTC‐3′ |
| Reverse: 5′‐CAGTTGCTCTCCGAGGAGTC‐3′ |
Quantification of chromogen intensity in PBX1 immunohistochemistry
For quantification of PBX1 intensity of a chromogen stain under bright field microscopy, we used a method previously described (Nguyen et al, 2013). Briefly, we considered that standard bright field red‐green‐blue color images have a maximum intensity of value 250 (intensity function in ImageJ software). Darker areas have lower intensity values. This results in an inverse correlation between the amount of antigen and its numerical value. The intensity of a stained region in an RGB image analyzed in ImageJ can be subtracted from 250, obtaining a value that is directly proportional to the amount of chromogen present. We selected a round area of 328 square pixels and placed, when possible, in the nucleus of each NM+ cell. The value obtained from ImageJ (mean) was used to calculate the new value (250‐mean) for each cell, proportional to the amount of chromogen present in the nucleus.
Statistics
Sample size for embryonic analyses was chosen according to previous studies in the same area of research. A minimum of 3 embryos was analyzed for each genotype for comparison. The only exclusion criterion was an evident developmental delay between embryos of different genotypes. The exclusion criterion was pre‐established. No randomization or blinding procedure was used for animal studies. Although we cannot be certain that our data in the population has a normal distribution, we considered our data as being close to Gaussian distribution and applied two‐tailed unpaired t‐tests for comparison between two groups of samples, without assuming similar variance. This is a common standard in the field, and a t‐test is commonly used in other similar studies. No special steps were taken to minimize the effects of subjective bias when animals or cells were under treatment. Two‐tailed unpaired t‐test statistical analyses were performed using GraphPad Prism 6. Bars represent mean ± standard deviation. Biological replicates (dot) per condition and P‐value of t‐test are indicated in each graph.
Author contributions
JCV designed and performed experiments, analyzed data, prepared all figures, and wrote the manuscript; BL, SY, PRdVC, EMT, and KK designed and performed experiments; SRWS, DG, ML, SI, and RLG performed experiments; AF contributed with cell line AF22; PL provided bioinformatic support; TB and RAB designed experiments and contribute to the editing of the manuscript; SL designed experiments and discussed the results; LS discussed results and provided genetically engineered mouse strains, embryos, and contributed to editing of the manuscript; EA designed experiments, analyzed data, and wrote the manuscript. All authors approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Patrik Ernfors, Francesco Blasi, and Dmitry Penkov for critical reading of the manuscript; Patrik Ernfors and Marina C. Franck for the deleter TH‐IRES‐CRE‐ERT2 mouse line; Nadia Wänn for maintenance of mice colonies; the members of the Arenas laboratory for their help and suggestions; and Johnny Söderlund and Alessandra Nanni for technical and secretarial assistance. Financial support was obtained from Swedish Research Council (VR projects: DBRM, 2008:2811, 2011–3116, and 2011–3318), Swedish Foundation for Strategic Research (SRL program), European Commission (NeuroStemcellRepair and DDPD‐Genes) and Karolinska Institutet (SFO Thematic Center in Stem cells and Regenerative Medicine) to EA, Karolinska Institutet's Research Foundation and FEBS Long‐Term Fellowship to JCV, Czech Ministry of Education and European Regional Development Fund (KI‐MU; CZ.1.07/2.3.00/20.0180) to JCV and KK, and the NIH Research Project Grants 2RO1 HD043997 and RO1 DE024745 to L.S. SRWS is funded by European Commission (DDPD‐Genes) and RAB is funded by the NIHR Biomedical Research Centre which also supports the Cambridge Brain Bank. LS would like to dedicate this work to her father Giuseppe, who died with Parkinson's disease.
The EMBO Journal (2016) 35: 1963–1978
See also: DS Castro (September 2016)
References
- Andersson E, Tryggvason U, Deng Q, Friling S, Alekseenko Z, Robert B, Perlmann T, Ericson J (2006) Identification of intrinsic determinants of midbrain dopamine neurons. Cell 124: 393–405 [DOI] [PubMed] [Google Scholar]
- Arenas E, Denham M, Villaescusa JC (2015) How to make a midbrain dopaminergic neuron. Development 142: 1918–1936 [DOI] [PubMed] [Google Scholar]
- Asahara H, Dutta S, Kao HY, Evans RM, Montminy M (1999) Pbx‐Hox heterodimers recruit coactivator‐corepressor complexes in an isoform‐specific manner. Mol Cell Biol 19: 8219–8225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badea TC, Hua ZL, Smallwood PM, Williams J, Rotolo T, Ye X, Nathans J (2009) New mouse lines for the analysis of neuronal morphology using CreER(T)/loxP‐directed sparse labeling. PLoS ONE 4: e7859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Johnson J, Grant CE, Noble WS (2015) The MEME Suite. Nucleic Acids Res 43: W39–W49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugno M, Daniel M, Chepelev NL, Willmore WG (2015) Changing gears in Nrf1 research, from mechanisms of regulation to its role in disease and prevention. Biochim Biophys Acta 1849: 1260–1276 [DOI] [PubMed] [Google Scholar]
- Chung S, Leung A, Han BS, Chang MY, Moon JI, Kim CH, Hong S, Pruszak J, Isacson O, Kim KS (2009) Wnt1‐lmx1a forms a novel autoregulatory loop and controls midbrain dopaminergic differentiation synergistically with the SHH‐FoxA2 pathway. Cell Stem Cell 5: 646–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes‐Canteli M, Aguilar‐Morante D, Sanz‐Sancristobal M, Megias D, Santos A, Perez‐Castillo A (2011) Role of C/EBPbeta transcription factor in adult hippocampal neurogenesis. PLoS ONE 6: e24842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Salvio M, Di Giovannantonio LG, Acampora D, Prosperi R, Omodei D, Prakash N, Wurst W, Simeone A (2010) Otx2 controls neuron subtype identity in ventral tegmental area and antagonizes vulnerability to MPTP. Nat Neurosci 13: 1481–1488 [DOI] [PubMed] [Google Scholar]
- DiMartino JF, Selleri L, Traver D, Firpo MT, Rhee J, Warnke R, O'Gorman S, Weissman IL, Cleary ML (2001) The Hox cofactor and proto‐oncogene Pbx1 is required for maintenance of definitive hematopoiesis in the fetal liver. Blood 98: 618–626 [DOI] [PubMed] [Google Scholar]
- Espana A, Clotman F (2012) Onecut transcription factors are required for the second phase of development of the A13 dopaminergic nucleus in the mouse. J Comp Neurol 520: 1424–1441 [DOI] [PubMed] [Google Scholar]
- Falk A, Koch P, Kesavan J, Takashima Y, Ladewig J, Alexander M, Wiskow O, Tailor J, Trotter M, Pollard S, Smith A, Brustle O (2012) Capture of neuroepithelial‐like stem cells from pluripotent stem cells provides a versatile system for in vitro production of human neurons. PLoS One 7: e29597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer SC, Sun CW, Winnier GE, Hogan BL, Townes TM (1997) The bZIP transcription factor LCR‐F1 is essential for mesoderm formation in mouse development. Genes Dev 11: 786–798 [DOI] [PubMed] [Google Scholar]
- Ferretti E, Li B, Zewdu R, Wells V, Hebert JM, Karner C, Anderson MJ, Williams T, Dixon J, Dixon MJ, Depew MJ, Selleri L (2011) A conserved Pbx‐Wnt‐p63‐Irf6 regulatory module controls face morphogenesis by promoting epithelial apoptosis. Dev Cell 21: 627–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri AL, Lin W, Mavromatakis YE, Wang JC, Sasaki H, Whitsett JA, Ang SL (2007) Foxa1 and Foxa2 regulate multiple phases of midbrain dopaminergic neuron development in a dosage‐dependent manner. Development 134: 2761–2769 [DOI] [PubMed] [Google Scholar]
- Francius C, Clotman F (2010) Dynamic expression of the Onecut transcription factors HNF‐6, OC‐2 and OC‐3 during spinal motor neuron development. Neuroscience 165: 116–129 [DOI] [PubMed] [Google Scholar]
- Furuya N, Ikeda S, Sato S, Soma S, Ezaki J, Oliva Trejo JA, Takeda‐Ezaki M, Fujimura T, Arikawa‐Hirasawa E, Tada N, Komatsu M, Tanaka K, Kominami E, Hattori N, Ueno T (2014) PARK2/Parkin‐mediated mitochondrial clearance contributes to proteasome activation during slow‐twitch muscle atrophy via NFE2L1 nuclear translocation. Autophagy 10: 631–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganat YM, Calder EL, Kriks S, Nelander J, Tu EY, Jia F, Battista D, Harrison N, Parmar M, Tomishima MJ, Rutishauser U, Studer L (2012) Identification of embryonic stem cell‐derived midbrain dopaminergic neurons for engraftment. J Clin Invest 122: 2928–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS (2007) Quantifying similarity between motifs. Genome Biol 8: R24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe BD, Scherz PJ, Nissim S, Tian H, McMahon AP, Tabin CJ (2004) Evidence for an expansion‐based temporal Shh gradient in specifying vertebrate digit identities. Cell 118: 517–528 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirotsu Y, Hataya N, Katsuoka F, Yamamoto M (2012) NF‐E2‐related factor 1 (Nrf1) serves as a novel regulator of hepatic lipid metabolism through regulation of the Lipin1 and PGC‐1beta genes. Mol Cell Biol 32: 2760–2770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings P, Limonciel A, Felice L, Leonard MO (2012) An overview of transcriptional regulation in response to toxicological insult. Arch Toxicol 87: 49–72 [DOI] [PubMed] [Google Scholar]
- Kele J, Simplicio N, Ferri AL, Mira H, Guillemot F, Arenas E, Ang SL (2006) Neurogenin 2 is required for the development of ventral midbrain dopaminergic neurons. Development 133: 495–505 [DOI] [PubMed] [Google Scholar]
- Kim SK, Selleri L, Lee JS, Zhang AY, Gu X, Jacobs Y, Cleary ML (2002) Pbx1 inactivation disrupts pancreas development and in Ipf1‐deficient mice promotes diabetes mellitus. Nat Genet 30: 430–435 [DOI] [PubMed] [Google Scholar]
- Kirkeby A, Grealish S, Wolf DA, Nelander J, Wood J, Lundblad M, Lindvall O, Parmar M (2012) Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep 1: 703–714 [DOI] [PubMed] [Google Scholar]
- Koch P, Opitz T, Steinbeck JA, Ladewig J, Brustle O (2009) A rosette‐type, self‐renewing human ES cell‐derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc Natl Acad Sci U S A 106: 3225–3230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo‐Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L (2011) Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature 480: 547–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon AT, Arenillas DJ, Worsley Hunt R, Wasserman WW (2012) oPOSSUM‐3: advanced analysis of regulatory motif over‐representation across genes or ChIP‐Seq datasets. G3 2: 987–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees AJ, Hardy J, Revesz T (2009) Parkinson's disease. Lancet 373: 2055–2066 [DOI] [PubMed] [Google Scholar]
- Longobardi E, Penkov D, Mateos D, De Florian G, Torres M, Blasi F (2013) Biochemistry of the tale transcription factors PREP, MEIS, and PBX in vertebrates. Dev Dyn 243: 59–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machanick P, Bailey TL (2011) MEME‐ChIP: motif analysis of large DNA datasets. Bioinformatics 27: 1696–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita N, Okada H, Yasoshima Y, Takahashi K, Kiuchi K, Kobayashi K (2002) Dynamics of tyrosine hydroxylase promoter activity during midbrain dopaminergic neuron development. J Neurochem 82: 295–304 [DOI] [PubMed] [Google Scholar]
- Maxwell SL, Ho HY, Kuehner E, Zhao S, Li M (2005) Pitx3 regulates tyrosine hydroxylase expression in the substantia nigra and identifies a subgroup of mesencephalic dopaminergic progenitor neurons during mouse development. Dev Biol 282: 467–479 [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G (2010) GREAT improves functional interpretation of cis‐regulatory regions. Nat Biotechnol 28: 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard C, Hein P, Paquin A, Savelson A, Yang XM, Lederfein D, Barnabe‐Heider F, Mir AA, Sterneck E, Peterson AC, Johnson PF, Vinson C, Miller FD (2002) An essential role for a MEK‐C/EBP pathway during growth factor‐regulated cortical neurogenesis. Neuron 36: 597–610 [DOI] [PubMed] [Google Scholar]
- Moens CB, Selleri L (2006) Hox cofactors in vertebrate development. Dev Biol 291: 193–206 [DOI] [PubMed] [Google Scholar]
- Nguyen DH, Zhou T, Shu J, Mao JH (2013) Quantifying chromogen intensity in immunohistochemistry via reciprocal intensity. Cancer InCytes 2: e [Google Scholar]
- Nunes I, Tovmasian LT, Silva RM, Burke RE, Goff SP (2003) Pitx3 is required for development of substantia nigra dopaminergic neurons. Proc Natl Acad Sci U S A 100: 4245–4250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DH, Rigas D, Cho A, Chan JY (2012) Deficiency in the nuclear‐related factor erythroid 2 transcription factor (Nrf1) leads to genetic instability. FEBS J 279: 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panman L, Papathanou M, Laguna A, Oosterveen T, Volakakis N, Acampora D, Kurtsdotter I, Yoshitake T, Kehr J, Joodmardi E, Muhr J, Simeone A, Ericson J, Perlmann T (2014) Sox6 and Otx2 control the specification of substantia nigra and ventral tegmental area dopamine neurons. Cell Rep 8: 1018–1025 [DOI] [PubMed] [Google Scholar]
- Penkov D, Mateos San Martin D, Fernandez‐Diaz LC, Rossello CA, Torroja C, Sanchez‐Cabo F, Warnatz HJ, Sultan M, Yaspo ML, Gabrieli A, Tkachuk V, Brendolan A, Blasi F, Torres M (2013) Analysis of the DNA‐binding profile and function of TALE homeoproteins reveals their specialization and specific interactions with Hox genes/proteins. Cell Rep 3: 1321–1333 [DOI] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85: 257–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portales‐Casamar E, Thongjuea S, Kwon AT, Arenillas D, Zhao X, Valen E, Yusuf D, Lenhard B, Wasserman WW, Sandelin A (2010) JASPAR 2010: the greatly expanded open‐access database of transcription factor binding profiles. Nucleic Acids Res 38: D105–D110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash N, Wurst W (2006) Genetic networks controlling the development of midbrain dopaminergic neurons. J Physiol 575: 403–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, Hamilton RL, Chu CT, Jordan‐Sciutto KL (2007) Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol 66: 75–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee JW, Arata A, Selleri L, Jacobs Y, Arata S, Onimaru H, Cleary ML (2004) Pbx3 deficiency results in central hypoventilation. Am J Pathol 165: 1343–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribes V, Balaskas N, Sasai N, Cruz C, Dessaud E, Cayuso J, Tozer S, Yang LL, Novitch B, Marti E, Briscoe J (2010) Distinct Sonic Hedgehog signaling dynamics specify floor plate and ventral neuronal progenitors in the vertebrate neural tube. Genes Dev 24: 1186–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotolo T, Smallwood PM, Williams J, Nathans J (2008) Genetically‐directed, cell type‐specific sparse labeling for the analysis of neuronal morphology. PLoS ONE 3: e4099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh M, Rambaldi I, Yang XJ, Featherstone MS (2000) Cell signaling switches HOX‐PBX complexes from repressors to activators of transcription mediated by histone deacetylases and histone acetyltransferases. Mol Cell Biol 20: 8623–8633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarffe LA, Stevens DA, Dawson VL, Dawson TM (2014) Parkin and PINK1: much more than mitophagy. Trends Neurosci 37: 315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel CA, Selleri L, Cleary ML (2003) Pbx1 is essential for adrenal development and urogenital differentiation. Genesis 37: 123–130 [DOI] [PubMed] [Google Scholar]
- Selleri L, Depew MJ, Jacobs Y, Chanda SK, Tsang KY, Cheah KS, Rubenstein JL, O'Gorman S, Cleary ML (2001) Requirement for Pbx1 in skeletal patterning and programming chondrocyte proliferation and differentiation. Development 128: 3543–3557 [DOI] [PubMed] [Google Scholar]
- Selleri L, DiMartino J, van Deursen J, Brendolan A, Sanyal M, Boon E, Capellini T, Smith KS, Rhee J, Popperl H, Grosveld G, Cleary ML (2004) The TALE homeodomain protein Pbx2 is not essential for development and long‐term survival. Mol Cell Biol 24: 5324–5331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgado P, Ferretti E, Grbec D, Bozzi Y, Simon HH (2012) The atypical homeoprotein Pbx1a participates in the axonal pathfinding of mesencephalic dopaminergic neurons. Neural Dev 7: 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smidt MP, Smits SM, Bouwmeester H, Hamers FP, van der Linden AJ, Hellemons AJ, Graw J, Burbach JP (2004) Early developmental failure of substantia nigra dopamine neurons in mice lacking the homeodomain gene Pitx3. Development 131: 1145–1155 [DOI] [PubMed] [Google Scholar]
- Son JH, Chun HS, Joh TH, Cho S, Conti B, Lee JW (1999) Neuroprotection and neuronal differentiation studies using substantia nigra dopaminergic cells derived from transgenic mouse embryos. J Neurosci 19: 10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam FJ, Hendricks TJ, Zhang J, Geiman EJ, Francius C, Labosky PA, Clotman F, Goulding M (2012) Renshaw cell interneuron specialization is controlled by a temporally restricted transcription factor program. Development 139: 179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen J, Seeger M, Koch A, Kruger E (2010) Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD‐dependent feedback loop. Mol Cell 40: 147–158 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suske G (1999) The Sp‐family of transcription factors. Gene 238: 291–300 [DOI] [PubMed] [Google Scholar]
- Thompson LH, Andersson E, Jensen JB, Barraud P, Guillemot F, Parmar M, Bjorklund A (2006) Neurogenin2 identifies a transplantable dopamine neuron precursor in the developing ventral mesencephalon. Exp Neurol 198: 183–198 [DOI] [PubMed] [Google Scholar]
- Veenvliet JV, Dos Santos MT, Kouwenhoven WM, von Oerthel L, Lim JL, van der Linden AJ, Koerkamp MJ, Holstege FC, Smidt MP (2013) Specification of dopaminergic subsets involves interplay of En1 and Pitx3. Development 140: 3373–3384 [DOI] [PubMed] [Google Scholar]
- Wu F, Sapkota D, Li R, Mu X (2012) Onecut 1 and Onecut 2 are potential regulators of mouse retinal development. J Comp Neurol 520: 952–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye T, Krebs AR, Choukrallah MA, Keime C, Plewniak F, Davidson I, Tora L (2011) seqMINER: an integrated ChIP‐seq data interpretation platform. Nucleic Acids Res 39: e35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin M, Liu S, Yin Y, Li S, Li Z, Wu X, Zhang B, Ang SL, Ding Y, Zhou J (2009) Ventral mesencephalon‐enriched genes that regulate the development of dopaminergic neurons in vivo. J Neurosci 29: 5170–5182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterstrom RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T (1997) Dopamine neuron agenesis in Nurr1‐deficient mice. Science 276: 248–250 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File