

Abstract
The design, synthesis and assessment of β-carboline core-based compounds as potential multifunctional agents against several processes that are believed to play a significant role in Alzheimer’s disease (AD) pathology, are described. The activity of the compounds was determined in Aβ self-assembly (fibril and oligomer formation) and cholinesterase (AChE, BuChE) activity inhibition, and their antioxidant properties were also assessed. To obtain insight into the mode of action of the compounds, HR-MS studies were carried out on the inhibitor-Aβ complex formation and molecular docking was performed on inhibitor-BuChE interactions. While several compounds exhibited strong activities in individual assays, compound 14 emerged as a promising multi-target lead for the further structure-activity relationship studies.
Keywords: Alzheimer’s disease, Amyloid beta, Cholinesterase inhibition, Antioxidant, β-Carbolines
Due to the rapidly aging population and the ever greater occurrence of Alzheimer’s disease (AD) a multitude of therapeutic approaches have been investigated.1-7 The most common ones that resulted in clinical effects are based on the cholinergic8 and amyloid cascade hypotheses.9 The first strategy provides a symptomatic treatment via the inhibition of cholinesterase enzymes that successfully addressed the low level of acetylcholine neurotransmitter.10 In fact, the currently available AD drugs are mostly based on this approach, namely the inhibition of the acetylcholinesterase enzyme (AChE).11 Similarly to AChE the activity of butyrylcholinesterase (BuChE) also has a negative effect on the abundance of neurotransmitters.12 However, it is widely accepted that the accumulation of neurotoxic protein assemblies of the amyloid β peptide (Aβ) in the form of soluble oligomers and insoluble fibrillar deposits are among the significant instigators of AD.13 This initiated extensive efforts on the development of Aβ self-assembly inhibitors.14 The most recent potential therapeutic tried for AD therapy, the antibody Aducanumab, targets the Aβ deposits resulting in their clearance and possible cognitive benefits.15 As recent studies suggest, there may be a connection between the cholinergic and Aβ targets. It was proposed that AChE peripheral binding site may initiate the Aβ self-assembly.16 To further highlight the complex nature of AD, metal ions and oxidative stress were also suggested to contribute to the progression of AD.17 The network of symptoms and potential causative sources of the disease suggest that the development of compounds that target multiple areas of the pathogenesis should be more effective than drug candidates that would only alter single pathogenic contributors.18
Building upon our recent efforts19-27 potential multitarget inhibitors were designed based on the β-carboline core structure. Herein, we describe the synthesis, and biochemical evaluation of these compounds. The β-carboline core or sub-unit appears in a large number of biologically active compounds. Such compounds possess a variety of biological effects including anticancer28, antiprotozoal29, or anti-leishmanial30 activities. Compounds with the β-carboline core structure have also been described targeting a variety of neurological disorders and neurodegenerative diseases and being tau phosphorylation inhibitors31, channel blockers32 cholinesterase inhibitors33 GABAA receptor modulators34, or antioxidants.35 Based on these preliminaries an initial set of β-carbolines have been designed and synthesized with the aim of incorporating structural features that could present the opportunity to apply these compounds as multitarget modulators of several processes thought to play significant roles in the development and progression of AD.
The design of the structures was based on the following major factors. The structural analysis of a large set of Aβ self-assembly inhibitors14 indicated the importance of the aromatic structure as well as the presence of H-donor and H-acceptor units. The basic β-carboline skeleton fulfills this criterion. Analyzing the structures of cholinesterase inhibitors it appears paramount to have a relatively extended structure that is able to span the active center of the cholinesterases involving a variety of hydrophobic units. For this reason the original three ring system has been extended with an additional aromatic ring either directly or via a carbonyl linker to test the role of molecular flexibility on the efficacy of anti-cholinesterase activity. The extended aromatic structures are also expected to contribute to the potential antioxidant activity. The compounds that were designed using the above principals have been synthesized using our previously developed environmentally benign method.36 The synthetic procedure and preliminary set of β-carbolines are shown in Fig. 1.
Fig. 1.
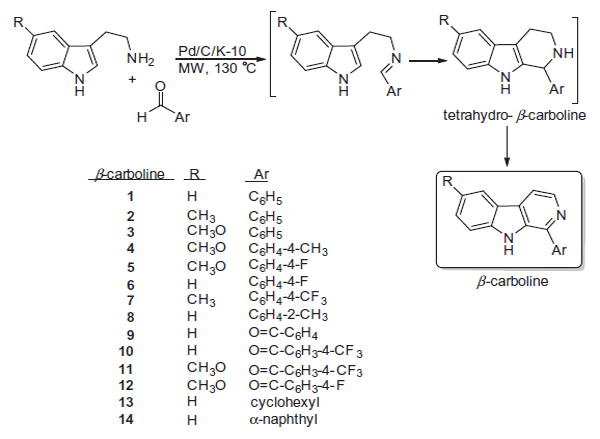
Synthesis and structure of the designed β-carboline derivatives.
The compounds were synthesized by a 3-step-one pot domino reaction by using a special mixture of commercially available 5% Pd/C and K-10 montmorillonite as a bifunctional catalyst. The first condensation step between the aldehyde and tryptamine was catalyzed by the solid acid K-10. The imine formed immediately underwent a cyclization also by K-10 catalysis. The resulting tetrahydro-β-carbolines were dehydrogenated by the Pd metal to provide the aromatic β-carbolines. Each product was characterized by 1H and 13C NMR spectroscopy and mass spectrometry (GC–MS). The spectroscopic characterization of the compounds was in agreement with their structures and the literature data.
After the completion of the synthesis the compounds were evaluated in several assays to test the design hypothesis. Assays included the inhibition of Aβ fibrillogenesis and oligomer formation, modulation of cholinesterase activity of AChE and BuChE enzymes, the determination of the antioxidant properties, high resolution mass spectrometry and molecular docking.
To determine the activity profile of the compounds they were first subjected to Aβ fibrillogenesis assays. The quantitative Thioflavin-T (THT) fluorescence assay was applied to determine the antifibrillogenic potency of the compounds.37 The data were compared to the fluorescence of the inhibitor-free control (Icontrol) and the observed decrease in fluorescence in the presence of the inhibitors was normalized to a scale of 0–100% and was tabulated as % inhibition in Table 1.
Table 1.
Effect of β-carbolines on the self-assembly of Ab and the activity of the AChE and BuChE enzymes.
| Compound | Aβ fibril inhibition (%)a | Aβ oligomer inhibition (%)b | AChE inhibition (%)c | BuChE inhibition (%)c | BuChE IC50 (μM) |
|---|---|---|---|---|---|
| 1 | −4 | 79 | −12 | 46 | >10 |
| 2 | −7 | 6 | −9 | 54 | 3.06 ± 1.27 |
| 3 | 19 | 58 | −10 | 64 | 4.48 ± 0.27 |
| 4 | 10 | 63 | −15 | 32 | >10 |
| 5 | 12 | – | −7 | 34 | >10 |
| 6 | 37 | 22 | −3 | 79 | 4.27 ± 1.30 |
| 7 | 34 | 82 | - | - | - |
| 8 | 17 | 45 | −9 | 7 | >10 |
| 9 | 20 | - | −7 | 11 | >10 |
| 10 | 11 | - | −6 | 69 | 1.29 ± 0.25 |
| 11 | −5 | 84 | −9 | 46 | 1.42 ± 0.73 |
| 12 | 6 | 82 | −9 | 1 | >10 |
| 13 | −12 | 30 | −10 | - | - |
| 14 | 39 | 58 | −11 | 95 | 0.22 ± 0.03 |
| GAL | - | - | 50 | 29 | 10 |
Aβ:inhibitor = 1:1 ratio at 100 μM Aβ concentration.
Aβ:inhibitor = 0.0002 ratio at 0.01 μM Aβ concentration.
β-carbolines were tested at 2 μM concentration for AChE and 10 μM concentration for BuChE; GAL – galantamine.
The data indicate that the compounds had a moderate effect on the fibril formation of Aβ. While based on the limited number of compounds a clear structure-activity relationship cannot be drawn, it appears that the larger structures (6–10, 14) act as better inhibitors. The best performance was shown by 6 (39%) and 14 (40%), which has an approximately 110 μM EC50 value.
As soluble Aβ oligomers are more neurotoxic than their insoluble fibril counterparts, the activity of the compounds in the inhibition of oligomer formation was also assessed by the biotinyl-Aβ single-site streptavidin-based assay.38 Similarly to anti-fibrillogenesis assays, the intensity of the inhibited samples was normalized to the inhibitor-free control sample and the inhibition was expressed in % compared to the uninhibited sample. The data are summarized in Table 1.
As the data show the compounds were highly active in preventing the formation of soluble Aβ oligomers. A majority of the compounds produced more than 50% inhibition (1, 3, 4, 7, 11, 12 and 14). The activity comparison of the compounds in the Aβ fibril and oligomer formation inhibition assays are in agreement with previous findings namely that a compound is either a fibril inhibitor or an oligomer inhibitor as shown by the respective behaviors of 1, 11 or 12.39 However, compounds 7 and 14 appear to provide a reasonable protection against both forms of self-assembled Aβ.
High resolution mass spectrometry data reveal a convincing evidence that 14, which is able to block both fibril and oligomer formation of Aβ, forms a complex with the peptide in the solution (Fig. 2). The most typical complex appears to be the 1:1 ratio of Aβ:14, although another complex with somewhat higher ratio (1:2) can be observed. The spectrum, however, shows that Aβ is still overwhelmingly in an uncomplexed form, indicating that a limited amount of inhibitor could modify the self-assembly process by partially complexing/blocking the peptide.
Fig. 2.
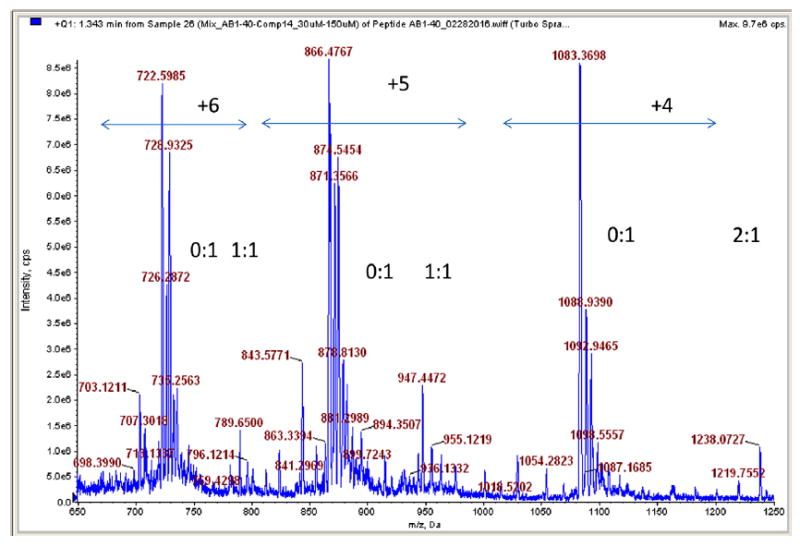
High resolution mass spectrum of the Aβ-peptide-14 mixture (30 μM to 150 μM). The relevant signals indicate 1:1 and 1:2 complex formation between the peptide and inhibitor compound. The intervals highlight the relevant peaks of the charged Aβ carrying 4–6 positive charges.
In order to observe the potential multifunctional behavior of the compounds, the β-carbolines were tested as cholinesterase inhibitors as well. The compounds were subjected to the Ellman assay using both AChE and BuChE, respectively (Table 1). The compounds were assayed at the respective IC50 of galantamine that was used as a reference compound (2 μM in AChE and 10 μM in BuChE inhibition).40,41
As shown the β-carbolines have negligible effect on the activity of AChE. In contrast, the compounds were highly active in the inhibition of BuChE; over 60% of the studied compounds appear to be a better inhibitor of BuChE than galanthamine. Several of them (e.g. 6, 14) show above 80% inhibition of the enzyme at 10 μM concentration.
In order to compare the potency of the compounds to others in the literature the IC50 values of compounds that showed >50% inhibition at 10 μM concentration were determined. The following IC50 values were obtained: 2–3.06 ± 1.27 μM, 3–4.48 ± 0.27 μM, 6–4.27 ± 1.30 μM, 10–1.29 ± 0.25 μM, 11–1.42 ± 0.73 μM, 14–0.225 ± 0.03 μM. The data show that, as expected, these compounds in fact possess a better IC50 than the reference compound. Compound 14 was found to be the best inhibitor of BuChE; its 225 nM value is of practical importance for further lead development. While at this level of the research it is difficult to make structure-activity relationship predictions it appears that the presence of an electron-donating substituent (Me, OMe) on the β-carboline ring positively affects the BuChe inhibition. In addition, considering the lower, additional ring, the bulkier the group, the better the effect. Compound 14 with the quite large naphthyl group was found to be by far the most effective inhibitor.
With the aim of understanding the butyrylcholinesterase inhibition property of these molecules, two among the best compounds (11 and 14) were docked in the active site of BuChE (PDB code: 1P0I42) using the Glide module of the Schrodinger package.43 The superimposition of compound 11 and 14 with donepezil and galantamine (known BuChE inhibitors) in the active site of the enzyme is shown in Figs. 3 and 4.
Fig. 3.
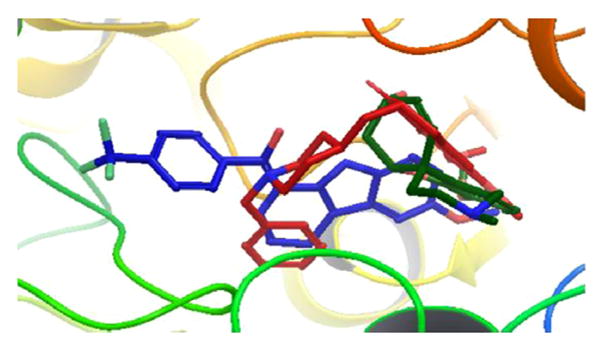
Superimposition of molecule 11 (blue) with donepezil (red) and galantamine (dark green) in the active site of huBChE (PDB ID: 1P0I). (hydrogens are concealed for clarity.)
Fig. 4.
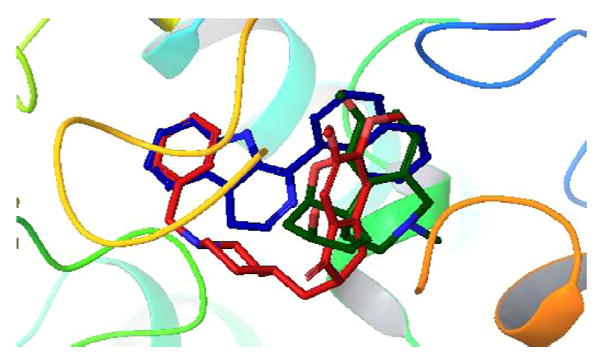
Superimposition of molecule 14 (blue) with donepezil (red) and galantamine (dark green) in the active site of huBChE (PDB ID: 1P0I). (hydrogens are concealed for clarity.)
The analysis of the docked structures revealed that while 11 is extended through the active site of BuChE the reference compounds galantamine and donepezil appear to bind in the right side of the active site in 1P0I (Figs. 3 and 4). The indole −NH of 11 shows a hydrogen bonding interaction with the Pro285 and Thr120 residues, respectively. In addition to that, the methoxy oxygen of 11 interacts with the Glu197 residue of the active site. In contrast, compound 14 appears to bind o the enzyme very similarly to donepezil, which is another known inhibitor of AChE as well as BuChE. The binding of 14 stretched through the active site explains the IC50 that is an order of magnitude lower than that of 11. The indole −NH of 14 shows hydrogen bonded interaction with the hydroxyl group of residue Ser198. In addition to that two π-π stacking interactions were observed, first between the phenyl ring of indole and residue Trp231 and the second one between the naphthalene ring of compound 14 and residue His438. Likewise, a cation-π interaction was noticed between quaternary nitrogen of 14 and residue Trp231. Since the compounds appear to act as selective BuChE inhibitors a docking study was carried out with 12 and AChE to observe whether the compound-enzyme interaction would reveal the reasons (Fig. 5).
Figure 5.
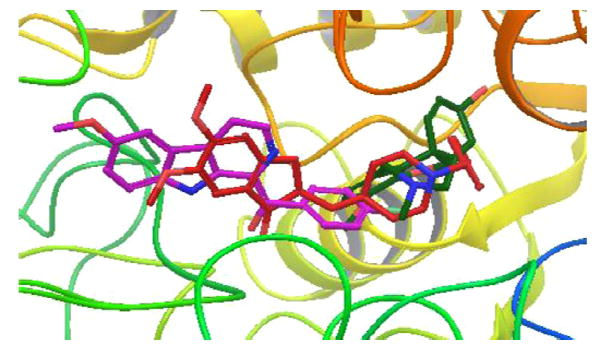
Superimposition of molecule 12 (purple) with donepezil (red) and galanthamine (dark green) in the active site of huAChE (PDB ID-4EY7) (hydrogen’s are concealed for clarity.)
Fig. 5 shows that the orientation of 12 is significantly different from those of donepezil and galantamine. The molecule streches completely through the active site while galantamine only occupies the right side of the pocket and donepezil also appears on the right side and turns back to the center. Although 12 shows hydrogen bonding interaction with residues Phe295, Tyr124 as well as π-π interaction with Trp286, The338 and Tyr337 residues, it is likely, that several of these interactions do not block residues that possess a role in the catalytic action.
Oxidative stress caused by free radicals also plays an important role in the development of AD. Thus, the potential antioxidant character of the compounds was assessed in three assays including the scavenging of the 2,2-diphenyl-1-picrylhydrazyl radical (DPPH assay), 2,2’-azino-bis(3-ethylbenzo-thiazoline-6-sulphonic acid) (ABTS assay) and the peroxyl (ORAC assay) free radicals.44 The data are compared to those obtained with reference compounds ascorbic acid45, resveratrol,46 and trolox,47 all of which are well-known antioxidants (Table 2). The data show that while the compounds have negligible effect in scavenging the large stable DPPH radical, they exhibited low to moderate scavenging activity against the also large ABTS radical.
Table 2.
Radical scavenging activity of β-carbolines (10 μM) in the DPPH, ABTS and ORAC antioxidant assays. Ascorbic acid, resveratrol and trolox that are well-known antioxidants were used as reference.
| Compound | % radical scavenging
|
||
|---|---|---|---|
| DPPH | ABTS | ORAC | |
| 1 | −7 | 12 | 10 |
| 2 | 2 | 10 | 10 |
| 3 | −6 | 16 | 35 |
| 4 | −12 | 13 | 28 |
| 5 | −13 | 15 | 41 |
| 6 | −3 | 22 | 54 |
| 7 | - | - | - |
| 8 | 0 | 15 | 58 |
| 9 | −3 | 6 | 6 |
| 10 | −1 | 7 | −10 |
| 11 | 2 | 3 | 5 |
| 12 | −4 | 5 | 5 |
| 13 | - | - | - |
| 14 | −6 | 14 | 0 |
| Ascorbic acid | 15 | 28 | 15 |
| Resveratrol | 28 | 88 | 91 |
| Trolox | 23 | 26 | 90 |
– Data not measured due to solubility problems.
Several compounds showed comparable activity to the reference compounds ascorbic acid and trolox. The β-carbolines were most active against the much smaller peroxyl radical used in the ORAC assay. Since this radical is one of the naturally occurring reactive oxygen species, these data are encouraging.
The analysis of the above data reveals that several of the synthesized substituted β-carbolines show promising properties in the AD related assays. Compounds 6, 7, 14 were able to inhibit Aβ fibril formation to a meaningful extent, while the majority of the molecules (1, 3, 4, 7, 8, 11, 12, 14) had a much stronger effect in the inhibition of Ab oligomer formation. This is in line with our earlier observations: a compound group is either a strong fibril or oligomer formation inhibitor.37 Although the compounds were inactive in AChE inhibition, they exhibited a highly selective and efficient inhibition of the BuChE enzyme. As shown, compounds 10, 11 and 14 exhibited the highest efficiency, 14 having one order of magnitude lower IC50 (225 nM) than the other compounds. The structural comparisons indicate that the added Ar part (Fig. 1.) appears highly important; the large Ar groups, such as naphthyl in 14, result in significant activity in the assays except the antioxidant tests.
In conclusion, a variety of β-carbolines with an extended aromatic ring system were synthesized and tested with the aim of identifying potential multitarget agents, that can interfere with Aβ self-assembly and cholinesterase activity while exhibiting promising antioxidant properties, for AD treatment. Based on the analysis of the data compound 14 emerged as a potential lead compound for further structure activity relationship studies. This molecule exhibited moderate to high activity in a range of assays suggesting that further modification of its basic ring system could yield a truly efficient candidate to develop effective drugs for disease management. To improve the drug-like properties of the compound the introduction of hydrophilic units such as NH (in the form of primary or secondary amines) or OH are proposed as the presence of these groups would improve water solubility (increased polarity), and antioxidant activity (presence of X-H bond) and likely would initiate further interactions with the cholinesterases that could improve the inhibition.
Acknowledgments
Financial support provided by the University of Massachusetts Boston through the 2013 Joseph P. Healey Research grant and National Institute of Health (R21AG028816-01 to H.L.) is gratefully acknowledged. Thanks are due to Alnylam Pharmaceutical Inc. for their help with the HR-MS measurements.
References and notes
- 1.Kelley BJ, Petersen RC. Neur Clinics. 2007;25:577. doi: 10.1016/j.ncl.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Lancet. 2011;377:1019. doi: 10.1016/S0140-6736(10)61349-9. [DOI] [PubMed] [Google Scholar]
- 3.Chen X, Tikhonova IG, Decker M. Bioorg Med Chem. 2011;19:1222. doi: 10.1016/j.bmc.2010.12.034. [DOI] [PubMed] [Google Scholar]
- 4.Jakob-Roetne R, Jacobsen H. Angew Chem. 2009;121:3074. doi: 10.1002/anie.200802808. [DOI] [PubMed] [Google Scholar]
- 5.Neugroschl J, Sano M. Curr Neurol Neurosci Rep. 2009;5:368. doi: 10.1007/s11910-009-0054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bolognesi ML, Matera R, Minarini A, Rosini M, Melchiorre C. Curr Opinion Chem Biol. 2009;13:303. doi: 10.1016/j.cbpa.2009.04.619. [DOI] [PubMed] [Google Scholar]; Relkin NR. Expert Rev Neurother. 2007;7:735. doi: 10.1586/14737175.7.6.735. [DOI] [PubMed] [Google Scholar]; Findeis MA. Biochim Biophys Acta. 2000;1502:76. doi: 10.1016/s0925-4439(00)00034-x. [DOI] [PubMed] [Google Scholar]
- 6.Ballard CG, Greig NH, Guillozet-Bongaarts AL, Enz A, Darvesh S. Curr Alz Res. 2005;2:307. doi: 10.2174/1567205054367838. [DOI] [PubMed] [Google Scholar]
- 7.Darvesh S, Hopkin DA, Geula C. Nat Rev Neurosci. 2003;4:131. doi: 10.1038/nrn1035. [DOI] [PubMed] [Google Scholar]
- 8.Bartus RT, Dean RL, III, Beer B, Lippa AS. Science. 1982;217:408. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]; Silman I, Sussman JL. Curr Opin Pharmacol. 2005;5:293. doi: 10.1016/j.coph.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 9.Belinson H, Kariv-Inbal Z, Kayed R, Masliah E, Michaelson DM. J Alz Dis. 2010;22:959. doi: 10.3233/JAD-2010-101008. [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu L, Murphy RM. Biochemistry. 2006;45:15702. doi: 10.1021/bi0618520. [DOI] [PubMed] [Google Scholar]; Selkoe DJ. Nature. 2003;426:900. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 10.DeKosky ST, Scheff SW. Ann Neurol. 1990;27:457. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 11.Larner AJ. Expert Rev Neurother. 2010;10:1699. doi: 10.1586/ern.10.105. [DOI] [PubMed] [Google Scholar]; Wollen KA. Alt Med Rev. 2010;15:223. [PubMed] [Google Scholar]; Francis PT, Ramirez MJ, Lai MK. Neuropharmacology. 2010;59:221. doi: 10.1016/j.neuropharm.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 12.Darvesh H, Hopkins DA, Geula C. Nat Rev Neurosci. 2003;4:131. doi: 10.1038/nrn1035. [DOI] [PubMed] [Google Scholar]
- 13.Kayed R, Canto I, Breydo L, et al. Mol Neurodegeneration. 2010;557 doi: 10.1186/1750-1326-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhao WQ, Santini F, Breese R, et al. J Biol Chem. 2010;285:7619. doi: 10.1074/jbc.M109.057182. [DOI] [PMC free article] [PubMed] [Google Scholar]; Shankar GM, Leissring MA, Adame A, et al. Neurobiol Dis. 2009;36:293. doi: 10.1016/j.nbd.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kayed R, Head E, Thompson JL, et al. Science. 2003;300:486. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 14.Török B, Dasgupta S, Török M. Curr Bioact Comp. 2008;4:159. [Google Scholar]; Török B, Bag S, Sarkar M, Dasgupta S, Török M. Curr Bioact Comp. 2013;9:37. [Google Scholar]
- 15. [09/25/2016]; http://www.alzforum.org/therapeutics/aducanumab.
- 16.Alvarez A, Alarcon R, Opazo C, et al. J Neurosci. 1998;18:3213. doi: 10.1523/JNEUROSCI.18-09-03213.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]; De Ferrari GV, Canales MA, Shin I, Weiner LM, Silman I, Inestrosa NC. Biochemistry. 2001;40:10447. doi: 10.1021/bi0101392. [DOI] [PubMed] [Google Scholar]; Bartolini M, Bertucci C, Cavrini V, Andrisano V. Biochem Pharmacol. 2003;65:407. doi: 10.1016/s0006-2952(02)01514-9. [DOI] [PubMed] [Google Scholar]
- 17.Huang X, Moir RD, Tanzi RE, Bush AI, Rogers JT. Ann N Y Acad Sci USA. 2004;1012:153. doi: 10.1196/annals.1306.012. [DOI] [PubMed] [Google Scholar]; Barnham KJ, Masters CL, Bush AI. Nat Rev. 2004;3:205. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- 18.Bolognesi ML, Simoni E, Rossini M, Minarini A, Tumiatti V, Melchiore C. Curr Top Med Chem. 2011;11:2797. doi: 10.2174/156802611798184373. [DOI] [PubMed] [Google Scholar]
- 19.Török M, Milton S, Kayed R, et al. J Biol Chem. 2002;277:40810. doi: 10.1074/jbc.M205659200. [DOI] [PubMed] [Google Scholar]
- 20.Török M, Abid M, Mhadgut SC, Török B. Biochemistry. 2006;45:5377. doi: 10.1021/bi0601104. [DOI] [PubMed] [Google Scholar]
- 21.Sood A, Abid M, Hailemichael S, Foster M, Török B. Bioorg Med Chem Lett. 2009;19:6931. doi: 10.1016/j.bmcl.2009.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sood A, Abid M, Sauer C, et al. Bioorg Med Chem Lett. 2011;21:2044. doi: 10.1016/j.bmcl.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borkin D, Morzhina E, Datta S, et al. Org Biomol Chem. 2011;9:1394. doi: 10.1039/c0ob00638f. [DOI] [PubMed] [Google Scholar]
- 24.Török B, Sood A, Bag S, et al. Biochemistry. 2013;52:1137. doi: 10.1021/bi3012059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bag S, Ghosh S, Tulsan R, et al. Bioorg Med Chem Lett. 2013;23:2614. doi: 10.1016/j.bmcl.2013.02.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bag S, Tulsan R, Sood A, et al. Bioorg Med Chem Lett. 2015;25:626. doi: 10.1016/j.bmcl.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 27.Rudnitskaya A, Török B, Török M. Biochem Mol Biol Ed. 2010;38:261. doi: 10.1002/bmb.20392. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y-F, Lin Y-C, Chen J-P, et al. Bioorg Med Chem Lett. 2015;25:3873. doi: 10.1016/j.bmcl.2015.07.058. [DOI] [PubMed] [Google Scholar]; Chen Hao, Gao Pengchao, Zhang Meng, Liao Wei, Zhang Jianwei. New J Chem. 2014;38:4155. [Google Scholar]
- 29.Gellis A, Dumetre A, Lanzada G, et al. Biomed Pharmacother. 2012;66:339. doi: 10.1016/j.biopha.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 30.Gohil VM, Brahmbhatt KG, Loiseau PM, Bhutani KK. Bioorg Med Chem Lett. 2012;22:3905. doi: 10.1016/j.bmcl.2012.04.115. [DOI] [PubMed] [Google Scholar]
- 31.Dunckley Travis. WO 2012024433 A2. PCT Int Appl. 2012 Feb 23;
- 32.Espinoza-Moraga M, Caballero J, Gaube F, Winckler T, Santos LS. Chem Biol Drug Des. 2012;79:594. doi: 10.1111/j.1747-0285.2012.01317.x. [DOI] [PubMed] [Google Scholar]
- 33.Winckler T, Fleck C, Lehmann J, Otto R, Appenroth D, Gaube F. DE 102012003065 A1. Ger Offen. 2013 Aug 14;
- 34.May AC, Fleischer W, Kletke O, Haas HL, Sergeeva OA, British J. Pharmacology. 2013;170:222. doi: 10.1111/bph.12280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Francik R, Kazek G, Cegla M, Stepniewski M. Acta Pol Pharma. 2011;68:185. [PubMed] [Google Scholar]
- 36.Kulkarni A, Abid M, Török B, Huang X. Tet Lett. 2009;50:1791. [Google Scholar]
- 37.Naiki H, Higuchi K, Hosokawa M, Takeda T. Anal Biochem. 1989;177:244. doi: 10.1016/0003-2697(89)90046-8. [DOI] [PubMed] [Google Scholar]; LeVine H., III Protein Sci. 1993;2:404. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nilsson MR. Methods. 2004;34:151. doi: 10.1016/j.ymeth.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 38.LeVine H., III Anal Biochem. 2006;356:265. doi: 10.1016/j.ab.2006.04.036. [DOI] [PubMed] [Google Scholar]; LeVine H, III, Ding Q, Walker JA, Voss RS, Augelli-Szafran CE. Neurosci Lett. 2009;465:99. doi: 10.1016/j.neulet.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Necula M, Kayed R, Milton S, Glabe CG. J Biol Chem. 2007;282:10311. doi: 10.1074/jbc.M608207200. [DOI] [PubMed] [Google Scholar]
- 40.Greenblatt H, Kryger G, Lewis T, Silman I, Sussman JL. FEBS Lett. 1999;463:321. doi: 10.1016/s0014-5793(99)01637-3. [DOI] [PubMed] [Google Scholar]
- 41.Sussman JL, Harel M, Frolow F, et al. Science. 1999;253:872. doi: 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]; Zhou X, Wang X, Wang T, Kong L. Bioorg Med Chem. 2008;16:8011. doi: 10.1016/j.bmc.2008.07.068. [DOI] [PubMed] [Google Scholar]
- 42.Nicolet Y, Lockridge O, Masson P, Fontecilla-Camps JC, Nachon F. J Biol Chem. 2003;278:41141. doi: 10.1074/jbc.M210241200. [DOI] [PubMed] [Google Scholar]
- 43.Glide version 6.8. New York, NY: Schrödinger, LLC; 2015. [Google Scholar]
- 44.Magalhães LM, Segundo MA, Reis S, Lima JLFC. Anal Chim Acta. 2008;613:1. doi: 10.1016/j.aca.2008.02.047. [DOI] [PubMed] [Google Scholar]; Cao G, Alessio HM, Cutler RG. Free Radic Biol Med. 1993;14:303. doi: 10.1016/0891-5849(93)90027-r. [DOI] [PubMed] [Google Scholar]; Peerannawar S, Horton W, Kokel A, Török F, Török M, Török B. Struct Chem. 2017 in press. [Google Scholar]
- 45.Balsano C, Alisi A. Curr Pharm Des. 2009;15:3063. doi: 10.2174/138161209789058084. [DOI] [PubMed] [Google Scholar]
- 46.Ono K, Condron MM, Ho L, et al. J Biol Chem. 2008;283:32176. doi: 10.1074/jbc.M806154200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berg R, Haenen G, Berg H, Bast A. Food Chem. 1999;66:511. [Google Scholar]