

Abstract
Background
Long non-coding RNAs (lncRNAs) are important in various biological processes, but very few studies on lncRNA have been conducted in birds. To identify IncRNAs expressed during feather development, we analyzed single-stranded RNA-seq (ssRNA-seq) data from the anterior and posterior dorsal regions during zebra finch (Taeniopygia guttata) embryonic development. Using published transcriptomic data, we further analyzed the evolutionary conservation of IncRNAs in birds and amniotes.
Results
A total of 1,081 lncRNAs, including 965 intergenic lncRNAs (lincRNAs), 59 intronic lncRNAs, and 57 antisense lncRNAs (lncNATs), were identified using our newly developed pipeline. These avian IncRNAs share similar characteristics with lncRNAs in mammals, such as shorter transcript length, lower exon number, lower average expression level and less sequence conservation than mRNAs. However, the proportion of lncRNAs overlapping with transposable elements in birds is much lower than that in mammals. We predicted the functions of IncRNAs based on the enriched functions of co-expressed protein-coding genes. Clusters of lncRNAs associated with natal down development were identified. The sequences and expression levels of candidate lncRNAs that shared conserved sequences among birds were validated by qPCR in both zebra finch and chicken. Finally, we identified three highly conserved lncRNAs that may be associated with natal down development.
Conclusions
Our study provides the first systematical identification of avian lncRNAs using ssRNA-seq analysis and offers a resource of embryonically expressed lncRNAs in zebra finch. We also predicted the biological function of identified lncRNAs.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3506-z) contains supplementary material, which is available to authorized users.
Keywords: Feather development, lncRNA, Zebra finch, ssRNA-seq
Background
A large portion of the eukaryotic genome is transcribed in the form of non-coding RNAs (ncRNAs) [1–3]. NcRNAs longer than 200 nucleotides are classified as long ncRNAs (lncRNAs), which are further divided into lincRNAs (long intergenic non-coding RNAs), intronic lncRNAs (transcribed within the introns of protein-coding genes), and lncNATs (long non-coding natural antisense transcripts, which are transcribed in the opposite strand of the protein-coding sequences) [4–7]. In general, lncRNAs show fewer exons, shorter transcript length and more diverse expression levels than protein-coding mRNAs [8, 9]. Furthermore, lncRNAs are usually evolutionarily less conserved in sequence than small/short ncRNAs and protein-coding mRNAs [8–10].
LncRNAs have been found to play regulatory and structural roles in diverse biological processes. For example, X-inactive specific transcript (XIST), a X-link lncRNA, mediates chromosome inactivation [11, 12], and KCNQ1 overlapping transcript 1 (KCNQ1OT1), a paternally expressed lncRNA, regulates the establishment of genomic imprinting [13–15]. LncRNAs can work in cis- or trans-regulation. For example, HOXA transcript at the distal tip (HOTTIP) is the lncRNA produced from the 5' end of the HOXA locus that coordinates the activation of several 5' HOXA genes [16], while HOX transcription antisense RNA (HOTAIR) is the trans-acting lncRNA that is transcribed from the HOXC gene cluster but acts as the repressor on the HOXD gene cluster [17].
Mammal hair and avian feather have had evolved independently, but their developments share many signaling pathways [18, 19]. In hair formation, dermal papilla cells can be the source of dermal-derived signaling molecules and play crucial roles in hair follicle development and postnatal hair cycle. Several lncRNAs were predicted to interact with the Wnt signaling pathway during dermal papilla cell development [20]. Whether avian feather development is also regulated by lncRNAs is therefore an interesting question. A few studies on avian lncRNAs have been made [21–23] and Gardner et al. [21–23] have studied the conservation and losses of non-coding RNAs in avian genomes.
Natal down is the downy plumage in avian hatchlings. Natal down development starts with a series of reciprocal epithelio-mesenchymal molecular interactions between the dermis and the overlying epidermis to form the primordia. The signaling crosstalk between epidermis and dermis coordinates the spatial arrangement and regular outgrowth of feathers [24–26]. Our previous study investigated the natal down formation divergence in zebra finch (Taeniopygia guttata) hatchlings, using single stranded RNA-seq (ssRNA-seq) data from both the anterior and the posterior dorsal region of zebra finch embryos at developmental stages E8, E9 and E12 (Additional file 1: Figure S1) [27].
The purpose of this study was to identify lncRNAs in zebra finch, predict their function and study their evolutionary conservation in birds and amniotes. First, we designed a set of criteria to identified lncRNAs using the ssRNA-seq data of our previous study [27]. Second, we classified IncRNAs into lincRNAs, intronic lncRNAs and lncNATs and compared the genomic and expression features of the predicted lncRNAs with protein-coding genes and between zebra finch and mammals. Third, we predicted the functions of the IncRNAs in natal down development. Finally, we validated the expressions of candidate lncRNAs involved in natal down development by qPCR and studied the sequence conservation in amniotes.
Results
Identified lncRNAs
To identify lncRNAs in zebra finch, six ssRNA-seq datasets (E8A, E8P, E9A, E9P, E12A and E12P, Additional file 1: Figure S1 [26]) from anterior dorsal (AD) and posterior dorsal (PD) skins in three embryonic incubation days (E8, E9 and E12) were re-analyzed. To infer the consensus mapping locations of RNA-seq reads, the concatenated paired-end reads were aligned onto the zebra finch genome by TopHat and only properly paired reads were retained, resulting in the mapping rates of 77 to 79% for the libraries (Additional file 2: Table S1). The new annotation file (General Transfer Format, GTF file) generated by Cufflinks was used for the subsequently analyses (Fig. 1).
Fig. 1.
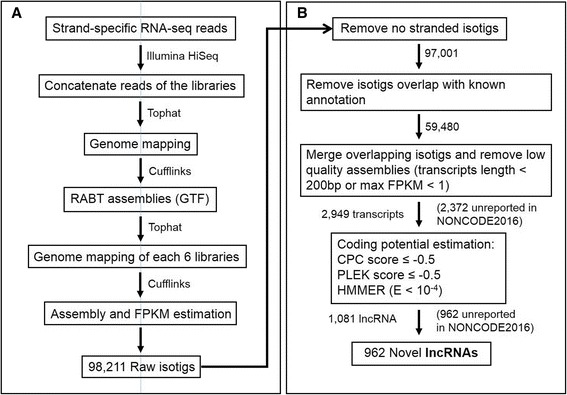
Overview of the ssRNA-seq transcriptome assembly and lncRNA identification pipeline. a Overview of the ssRNA-seq-based transcript reconstruction pipeline that was used to identify expressed transcripts in sequencing libraries. b The integrative pipeline for the stringent identification of lncRNAs in zebra finch dorsal skins. CPC: coding potential calculator; PLEK: predictor of long non-coding RNAs and messenger RNAs based on an improved k-mer scheme; HMMER (HMMER-3): Profiling protein sequence data using hidden Markov models
The strand specificities of the mapped reads were 86 to 92% for each library (Additional file 2: Table S1) [28], and the total number of the raw isotigs reconstructed using Cufflinks was 98,211 (Fig. 1). Raw isotigs without strand information (~1.3%) were removed and the remaining isotigs were separated to Ensembl annotated genes (Additional file 3: Table S2) and isotigs (59,480) that showed no overlap with any annotated genes (Fig. 1). We further merged the overlapping isotigs into raw transcripts (10,383). After removing the low quality assemblies as those with a small fragment (<200 bp) or low expression (max FPKM < 1 among all six libraries), we identified 2,949 unannotated transcripts, including 577 lncRNAs recorded in the NONCODE2016 database and 2,372 novel transcripts (Fig. 1; Additional file 4: Table S3) [29].
To identify lncRNAs, we focused on the unannotated transcripts. We first applied the coding potential calculator (CPC) to assess coding potential by considering the quality of predicted ORFs, and the homology with known proteins [30, 31]. In the 2,949 unannotated transcripts, 1,673 were identified as putative noncoding transcripts (Additional file 4: Table S3) by a cutoff score of −0.5 [8].
Although CPC has been widely used to analyze the coding potential, it only utilizes UniRef90 as the reference database [30, 32]. As the annotation of protein coding genes in the current bird genomes is not as complete as that in model mammals, it may include false positives in discovering lncRNAs. Our second approach was to use a newly developed classifier, known as the predictor of long non-coding RNAs and messenger RNAs based on an improved k-mer scheme (PLEK) [33] to estimate the coding potential of the transcripts, according to a training dataset generated from known coding and noncoding genes of chicken and zebra finch. We set the cutoff value to be −0.5 to reduce the possible bias in coding and noncoding gene classification. We identified 2,176 putative non-coding transcripts from the 2,949 unannotated transcripts (Additional file 4: Table S3).
The third approach was to eliminate the putative noncoding transcripts with similar reading frames with the Pfam protein domain database by HMMER3 (E-value < 10−4) [34]. Among the 2,949 unannotated transcripts, 2,265 transcripts remained and were taken as putative IncRNAs (Additional file 4: Table S3).
From the overlaps of the results of the three approaches, we identified 1,081 putative lncRNAs, including 119 lncRNAs annotated in NONCODE2016 [29] and 962 putative novel lncRNAs (Additional file 4: Table S3). The 1,081 lncRNAs could be classified into 965 lincRNAs, 59 intronic lncRNAs, and 57 lncNATs (Additional file 4: Table S3).
To evaluate our pipeline of coding potential estimation, we mapped the 1,081 putative lncRNAs and the remaining 1,868 unannotated transcripts to the zebra finch chromosomes (Additional file 5: Figure S2A). The 1,868 unannotated transcripts showed highest distribution in chromosome 25 and 27, while the 1,081 putative lncRNAs were distributed across all the chromosomes. Most α- and β- keratin genes were clustered in chromosomes 25 and 27 [35]. Keratin genes, especially β- keratin genes, are tandem duplicated genes with similar sequences. They are difficult to be annotated on the reference genome precisely and therefore many of them were included in our unannotated transcript pool. We mapped α- and β- keratin gene transcripts, unannotated transcripts (without lncRNAs), and lncRNAs to chromosomes 25 and 27 (Additional file 5: Figure S2B). In chromosome 25, the unannotated transcripts mainly overlapped with β- keratin genes, while in chromosome 27, the unannotated transcripts mainly overlapped with α- keratin genes. However, the overlap between lncRNAs and keratin genes was lower than that between unannotated transcripts and keratin genes (Additional file 5: Figure S2A), suggesting that our pipeline for lncRNAs identification could effectively exclude keratin-like transcripts. Chromosomes 25 is short (Chr. 25: 1.28 Mb; Chr. 26: 4.91 Mb; Chr. 27: 4.62 Mb) and therefore the values of “Transcript number/Chromosome size (Mb)” are very high for Chr. 25 (Additional file 5: Figure S2A).
The distribution range of the putative lncRNAs is from 0.40 to 3.91 lncRNAs per chromosome. We mapped the previous identified lncRNAs expressed in human skin to human chromosomes (except the Y chromosome) and found that the distribution range of the lncRNAs across the chromosomes is from 0.56 to 2.99 lncRNAs per chromosome [36], which is close to the distribution range of zebra finch skin lncRNAs we identified.
Genomic and expression features of the putative lncRNAs
We compared the transcript lengths, exon counts and sequence conservation of the 1081 putative lncRNAs with the protein-coding mRNAs. In agreement with previous studies in mammals [6, 8, 36, 37], the length distribution of the identified lncRNAs (median 0.75 kb; average 1.32 kb) is shorter than that of the mRNAs (median 1.09 kb; average 1.47 kb; p < 10−8, Student’s t-test), while the length distribution shows no significant differences between lincRNA, intronic lncRNA, and lncNAT (Fig. 2a). The exon counts of the putative lncRNAs (average 1.9 exons per transcript) is also less than that of the mRNAs (average 10.3 exons per transcript; p < 0.0001, Student’s t-test), while the exon counts of the three kinds of lncRNA show no differences (Fig. 2b). The sequences are less evolutionarily conserved in the putative lncRNAs than in protein-coding mRNAs (Fig. 2c). Finally, the proportions of overlapping lncRNAs and TEs in birds (zebra finch 39.6%; Chicken 10.3%) are much lower than those in mammals ((human 89.8%; bovine 96.4%, Fig. 2d; Additional file 6: Table S4), suggesting that TEs are not a major origin of avian lncRNAs.
Fig. 2.
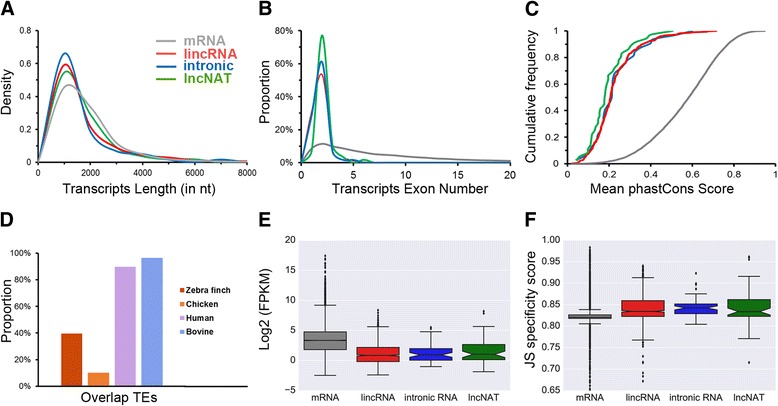
Genomic and expression features of the predicted zebra finch lncRNAs. Genomic features of (a) transcripts length, (b) exon number, and (c) mean phastCons score of zebra finch mRNA, lincRNA, intronic lncRNA, and lncNAT were compared. d The fraction of lncRNAs overlapping with at least one base of a TE (transposable element) in zebra finch, chicken, human, and bovine. Expression features of (e) expression levels and (f) JS scores of zebra finch mRNA, lincRNA, intronic lncRNA, and lncNAT were compared
We also compared the expression levels and the tissue specificities of the putative lncRNAs with those of the protein-coding mRNAs. The average expression levels of the putative lncRNAs (median 1.7; average 6.3 FPKM) tend to be lower than those of the mRNAs (median 9.6; average 114.7 FPKM; p < 0.0001, Student’s t-test; Fig. 2e). To quantify the tissue specificity of the transcripts of mRNA, lincRNA, intronic lncRNA, and lncNAT, we compared the JS scores [38] of the expressed transcripts between different skin regions and between different developmental stages. The results showed that the regional specificity is significantly different between the mRNAs and the lncRNAs (p < 0.0001, Student’s t-test; Fig. 2f), but no significant difference could be detected between different types of lncRNAs. Furthermore, no significant difference was detected between different types of lncRNAs in the three developmental stages analyzed (Additional file 7: Figure S3; also see Methods of [26]).
Co-expression analysis
Most lncRNAs lack annotated features and functional predictions for the lncRNAs have often been based on “guilt-by-association” analysis [38–40]. We clustered the lncRNAs along with the Ensembl functional annotated genes according to their expression profiles, and analyzed the GO categories enriched in each cluster. The expressed genes were classified into 12 expression clusters (A-L) (Fig. 3; Additional file 3: Table S2 and Additional file 4: Table S3). Then, we utilized the website based software g: Profiler to analyze the gene set enrichment of each cluster and excluded the clusters that may not be associated with natal down development by a series of filters; the detail of the filtering is described in Additional file 8: Supplementary Results. Only Clusters F, G, and L passed our criteria and were potentially associated with feather formation. To confirm the functional categories of these clusters, we further conducted Fisher’s exact test to gain the enrichments of GO terms and protein domains (collected from zebra finch protein domain databases: Pfam, Interpro, SMART, and SUPERFAMILY) in the three clusters. Only the GO categories with a p value < 0.01 and FDR < 0.05 were analyzed further.
Fig. 3.
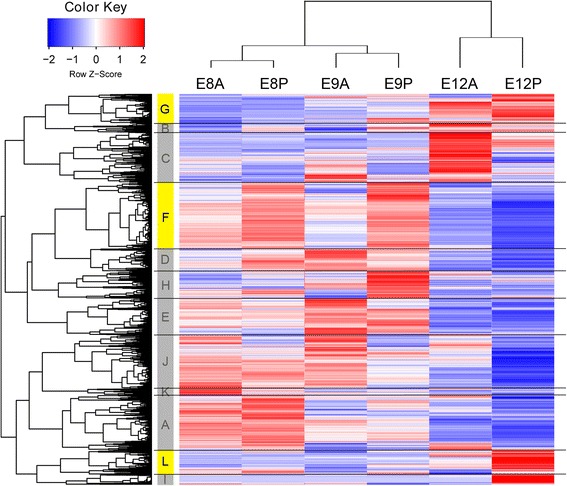
Clustering analysis of the expressed genes and the expression heat map. Hierarchical clustering analysis clustered the 13,362 expressed annotated genes and 2,949 unannotated transcripts into 12 clusters (A-L, see Additional files tables for details). The expression levels of each gene are shown as the scaled FPKM values across the six transcriptomes (scaled z-score: red = up-regulation, blue = down-regulation). Three clades (F, G, and L) used for further analysis were labeled in yellow
Genes in Cluster F were enriched in transcription factors (PF00076), mRNA metabolic process (GO:0016071), cell cycle process (GO:0022402), and DNA replication (GO:0006260) (Additional file 9: Table S5, Additional file 10: Table S6 and Additional file 11: Table S7), suggesting that lncRNAs in this cluster may be associated with cell proliferation. A previously identified feather bud growth promoter, sonic hedgehog (SHH), was in this cluster and expressed higher in downy dorsal skin than in naked dorsal skin [27]. Genes in Cluster G were enriched in the Claudin family (PF00822), the Rho protein signaling pathway (GO:0051056, GO:0046578, and PF00621), skin development (GO:0043588), keratinocyte differentiation (GO:0030216), and epithelial cell differentiation (GO:0030855) (Additional file 9: Table S5, Additional file 10: Table S6 and Additional file 11: Table S7). Claudins are the main component of tight junctions and Rho family GTPases are known to regulate the tight junctions [41]. A previous study showed that tight junctions are associated with the formation of feather branches, suggesting that lncRNAs in this cluster may regulate feather morphogenesis [42]. In Cluster L, genes showed enrichment in α-keratin domain (intermediate filament protein, PF00038) (Additional file 9: Table S5, Additional file 10: Table S6 and Additional file 11: Table S7). Although the FDR value of the protein domain enrichment exceeded 0.05, we still considered this result significant because α- keratin domains were trained based on mammalian data, so the calculation of FDR in avian α- keratin domains might be overestimated. [35]. Several β-keratins were also clustered in this cluster (Additional file 3: Table S2). It is possible that the lncRNAs in this cluster are involved in feather formation.
Validation and sequence analysis of the candidate lncRNAs associated with natal down development
To find the lncRNAs associated with natal down development in birds, we focused only on the lncRNAs that satisfied the following criteria: First, the lncRNAs were clustered in Cluster F, G, or L. Second, the lncRNAs were differentially expressed between the AD and PD skin regions (Additional file 4: Table S3). Third, the lncRNAs shared similar sequences in the same chromosomes between zebra finch and chicken. Three candidate lncRNAs, CUFF.19772.1 (in Cluster F), CUFF.6222.3 (in Cluster G), and CUFF.14902.2 (in Cluster L), were selected for further analysis. The sequence of CUFF.19772.1 is recorded in the NONCODE lncRNA database (ID: NONBTAT021324 and NONMMUT059481, found in bovine and mouse, respectively). CUFF.6222.3 and CUFF.14902.2 were putative novel lncRNAs.
The expression levels of the predicted lncRNAs were too low to be detected by whole mount in situ hybridization. To confirm the role of the three selected putative lncRNAs, we compared their expression levels in the AD and PD skins of different individuals of zebra finch and chicken by quantitative PCR. All three lncRNAs were expressed in both zebra finch and chicken. Moreover, in zebra finch, those lncRNAs were expressed more highly in the PD region than in the AD region, but no expression differences could be detected between the AD and PD skin regions in chicken (Fig. 4). Zebra finch has two types of natal down formation in dorsal skins, but chicken only has one type (Additional file 1: Figure S1). Our previous study had found that most feather formation genes were differentially expressed between the AD and PD skin regions in zebra finch, but not in chicken [27]. Therefore, these three lncRNAs might be involved in natal down development.
Fig. 4.
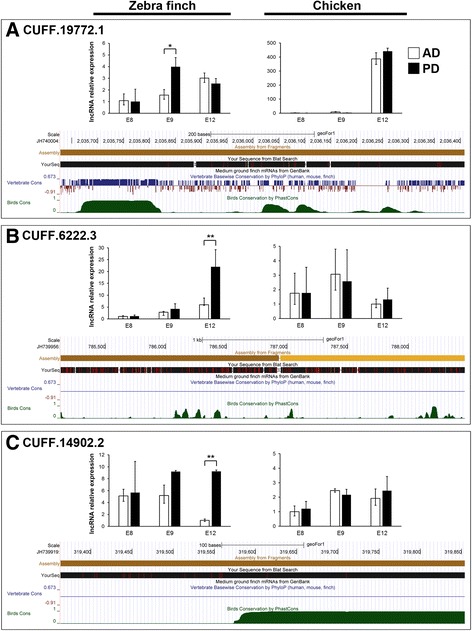
Quantitative PCR and sequence conservation analysis of the three identified lncRNAs. a Expression profiles of lncRNA CUFF.19772.1 in E8, E9, and E12 of zebra finch and chicken AD and PD skins. b Expression profiles of lncRNA CUFF.6222.3 in E8, E9, and E12 of zebra finch and chicken AD and PD skins. c Expression profiles of lncRNA CUFF.14902.2 in E8, E9, and E12 of zebra finch and chicken AD and PD skins. Sequence conservations are shown below the quantitative PCR in blue (conservation among amniotes) and green (conservation among birds)
We studied the sequence conservation of these three lncRNAs between birds and between amniotes. The multiple genome alignment of the medium ground finch in the UCSC Genome Browser provided the sequence conservation scores across birds (zebra finch, chicken, turkey, and budgerigar) and across amniotes (birds, human, and mouse) [43]. We used the UCSC BLAT algorithm to map our lncRNA sequences to the genome of medium ground finch to evaluate the sequence conservation (Fig. 4). In CUFF.19772.1, the sequence was conserved in both birds and amniotes (Fig. 4a), suggesting a function shared by amniotes. In CUFF.6222.3, the sequence has been only partially conserved in birds (Fig. 4b). In CUFF.14902.2, the sequence has been highly conserved only in birds (Fig. 4c). Interestingly, we found that CUFF.19772.1 is similar in sequence with the 3’ UTR of human BHLHE41 (the basic helix-loop-helix family, member e41, Additional file 12: Figure S4). BHLHE41 is a transcription factor and known to be the upstream signal of c-Myc [44], and c-Myc could promote the epithelium cell proliferation in feather bud elongation [45]. In our transcriptomes, the expression profiles of BHLHE41 and MYC belong to the same cluster with CUFF.19772.1 (Cluster F, Additional file 3: Table S2). Taken together, these results suggest that through the c-Myc signaling, CUFF.19772.1 promotes feather bud elongation.
Discussion
In this study, we developed a pipeline to identify zebra finch lncRNAs from the published ssRNA-seq data. We analyzed the genomic and expression features of the identified lncRNAs and compared the features with that in other vertebrates. We constructed a weighted gene co-expression network and predicted the functions of the lncRNAs based on their correlation with known protein-coding genes.
To find candidate lncRNAs in natal down formation, we compared the zebra finch lncRNA from AD and PD skins. Then, we compared the expression profiles of the candidate lncRNAs in zebra finch with those in chicken to identify avian conserved lncRNAs, which may be involved in natal down development. Feathers play important roles in heat conservation, mate attraction, physical protection, and flight. Many signaling molecules of these processes are well established in chicken [45–52]. However, as most previous studies focused on protein-coding genes, the role of non-coding RNAs (ncRNAs) in feather development is unclear.
In agreement with the previous studies in various eukaryotes [6–8, 53], our identified lncRNAs have shorter transcript length, lower exon number, lower sequence conservation, less average expression, and higher tissue specific expression than protein-coding transcripts. However, we found the overlapping proportions between lncRNAs and TEs are much lower in birds than in mammals. Previous studies proposed that TEs are one of the major origins of lncRNAs in vertebrates, and TEs embedded in lncRNAs are subjected to RNA editing or secondary structure formation [54, 55]. However, these studies did not include avian lncRNAs. Birds are known to have lower percentages of TEs in their genomes than most other vertebrates [56]. Thus, it seems that TEs have a lower contribution to lncRNAs in birds than in mammals. Although several lncRNAs play an essential role in cellular differentiation, cell lineage choice, organogenesis and tissue homeostasis, the function of most identified lncRNAs is unknown [57]. In our tissue specificity analysis, we found differential expression of lncRNAs among skin regions but not among developmental stages. Thus, our identified lncRNAs may play a role in skin or skin appendage differentiation, although probably not in skin or skin appendage growth.
In general, most lncRNAs show low primary sequence conservation between species despite having similar functions. In our study, one putative natal down development associated lncRNAs showed sequence conservation among amniotes. This is an interesting observation because feather and hair share many molecules at the start of their development, although hair and feather utilize different molecules for morphogenesis and cornification. LncRNA CUFF.19772.1 showed high sequence conservation among human, mouse, and birds. Moreover, the co-expressed SHH and MYC are important molecules that promote cell proliferations for both feather and hair formation [58–60]. Although the function of the host gene BHLHE41 in hair formation is not known, we speculate that CUFF.19772.1 is important for early stages of both feather and hair formation. Through c-Myc signaling, CUFF.19772.1 might interact with or function like SHH to promote feather bud elongation [27, 60]. In contrast, lncRNA CUFF.6222.3 and CUFF.14902.2 are co-expressed with feather morphogenesis and cornification factors, such as Claudins, Rho proteins, and α- and β-keratins, and their sequences have been conserved only in birds. CUFF.14902.2 showed high sequence conservation in birds and is located in chromosome 17. Most feather cornification factors, such as α- and β-keratins, are not located in chromosome 17, but are clustered in chromosomes 2, 25, 27, and 33 in both zebra finch and chicken [35, 61]. Therefore, we propose that CUFF.14902.2 may be associated with feather cornification in trans-regulation. Furthermore, all the three conserved lncRNAs we found do not overlap with any of the previously identified well conserved lncRNAs [23].
Several concerns arise from the analysis of this study. First, previous pipelines for lncRNA predictions in mammals excluded single-exon transcripts [19, 21]. However, compared to mammals, bird genomes are more compact with shorter introns and intergenic regions [22, 62, 63]. Therefore, we retained single exon transcripts in our lncRNA pool. Second, we used zebra finch as the model animal in this study because its unique natal down growth feature enabled us to find candidate regulators for natal down formation. However, the average protein-coding transcript length is much longer in chicken (2.3 kb) than that in zebra finch (1.47 kb), and as 1/6 of the sequences are unassigned to chromosomes, the assembly quality of the zebra finch genome is not as good as those of other model animals, and so some lncRNAs may have been missed in our data. The fast growing avian genome sequencing data may help to remove these concerns in the future [22].
Conclusion
Previous lncRNA studies covered many organisms, but less include birds. In this study, we employed ssRNA-seq to identify zebra finch lncRNAs and predicted the function of the identified lncRNAs. We identified 962 novel lncRNAs, which greatly expanded the repertoire of lncRNAs. In genomic feature analysis of the identified lncRNAs, we found that TEs are not a major origin of avian lncRNAs. Moreover, by comparing the expression profiles between zebra finch and chicken, and by examining the sequence conservation among amniotes, three lncRNAs were found to have been highly conserved and were predicted to be associated with natal down development.
Methods
RNA isolation
The zebra finch and chicken embryonic skin tissues were dissected as described in Additional file 1: Figure S1 (red dash boxes, AD: anterior dorsal skin; PD: posterior dorsal skin). Tissue total RNA was isolated and quality assessed as described in Chen et al. [27].
Data processing, reads mapping and assembly
Sequencing reads of the six libraries were described in Chen et al. [27] and summarized in Additional file 1: Figure S1 and Additional file 2: Table S1. This study used the new versions of Tophat (version 2.0.14) and Cufflinks (version 2.2.1) to process the reads. The zebra finch genome (version Taeniopygia_guttata.taeGut3.2.4) and its gene annotation were downloaded from Ensembl. The processed sequencing reads were then mapped to the genome using Tophat [64], and its embedded aligner Bowtie (version 2.1.0) [65] by the following parameters: −r 116 --mate-std-dev 100 --library-type fr-firststrand -g 2. The normalized expression levels of genes, represented by fragments per kilobase of exon per million fragments mapped (FPKMs) [66], were generated by Cufflinks [67] by the following parameters: −−library-type fr-firststrand --max-bundle-frags 1012.
Identification of novel transcripts
The pipeline for exploring novel transcripts is shown in Fig. 1. Raw transcripts generated from our mapping and assembly were filtered by the following criteria to detect putative novel transcripts: 1. Transcripts that have no strand information were removed. 2. Transcripts that overlap with the locations of the annotated genes in the Ensemble and UCSC databases were removed. 3. Transcripts with length less than 200 bp or an FPKM value lower than 1 in all the libraries were removed. 4. Transcripts not recorded in the NONCODE2016 database were retained [29].
Coding potential analysis
The coding potential calculator (CPC) is a SVM-based classifier based on the presence and integrity of the ORF in a transcript and on the Blastx-computed similarity scores between transcript ORFs and the known protein databases [30, 31]. UniRef90 [32] was used as the protein reference for the analysis and we set the cutoff score of −0.5 to distinguish noncoding RNAs from coding RNAs.
The predictor of long non-coding RNAs and messenger RNAs based on an improved k-mer scheme (PLEK) is a newly developed classifier based on the improved k-mer scheme and a SVM algorithm [33]. We used Ensembl known coding-genes of zebra finch (Taeniopygia_guttata.taeGut3.2.4.cds.all.fa) and known noncoding genes from the combination of chicken and zebra finch (Taeniopygia_guttata.taeGut3.2.4.ncrna.fa and Gallus_gallus.Galgal4.ncrna.fa) as the training dataset to score the novel transcripts. We stringently set the cutoff value to be −0.5 for the coding and noncoding genes discrimination.
Genomic and expression features of the identified lncRNAs
We analyzed several commonly characterized genomic and expression features of the identified lncRNAs according to the previous studies [6, 8, 36]. The identified 1,081 lncRNAs and the 16,869 protein-coding mRNA were used in the analysis (Additional file 3: Table S2; Additional file 4: Table S3).
Conservation analysis
We generated the three birds multiple genome alignment. Zebra finch (Taeniopygia_guttata.taeGut3.2.4) was used as the target, and chicken (Gallus_gallus.Galgal4) and flycatcher (Ficedula_albicollis.FicAlb_1.4) were used as the queries. Briefly, we downloaded the homologous genes between the species from the Ensembl database. These homologous genes were used as the anchors to construct the multi-species genomic synteny blocks. These syntenic blocks were aligned by Multiz-TBA (threaded blockset aligner) software to generate three species multiple genome alignment [68]. The average phastCon score of the location of the predicted lncRNAs and protein-coding genes were calculated by phastCons software [69]. Nucleotides which have no phastCon score were ignored.
Transposable element overlapping analysis
We analyzed the TEs and lncRNAs of human, bovine, zebra finch, and chicken. The locations of SINE, LINE, LTR, and DNA transposable elements generated by RepeatMasker were downloaded from the UCSC table browser. To reduce the possible bias from the tissue specificity of the lncRNAs, we collected published lncRNAs from similar tissues in different species. The genome version and the lncRNAs datasets were based on the previous studies in human skin [36], bovine muscle [6], and chicken muscle [21] (Additional file 6: Table S4).
Evaluation of tissue specificity
We estimated the tissue specificity of an expressed gene based on the JS (Jensen-Shannon) score. A higher JS score indicates a higher degree of tissue specific expression under that condition. We used the maximum JS score among the libraries of a transcript to represent the expression specificity of the transcript. Regional and developmental stage specificities are the two conditions used in our analysis.
Clustering analysis and differentially expressed genes (DEGs) identification
In the clustering analysis, we first defined an expressed gene as having a FPKM value > 1 in at least one library. All the expressed known genes and the identified 2,949 transcripts (1,868 unannotated protein-coding transcripts and 1,081 lncRNAs) were hierarchically clustered by the WPGMA (Weighted Pair-Group Method with Arithmetic mean) method by the R script. Heatmap of the clusters was generated by Heatmap.2. The cut-off for the cluster analysis was 0.69.
We identified the DEGs (differentially expressed genes) through several sets of comparisons. To identify the candidate genes (protein-coding gene and lncRNAs) involved in natal down developments, we compared the regional gene expression differences between the AD and PD skin regions in the three embryonic incubation days. To increase the power of detecting the DEGs with low expression, the libraries of AD skins were used as the AD replicate, while the libraries of PD skins were used as the PD replicate. The two replicates were further compared (E8A + E9A versus E8P + E9P, and E9A + E12A versus E9P + E12P). To identify the candidate genes (protein-coding gene and lncRNAs) for skin development, we compared the temporal gene expression differences between different embryonic incubation days in AD or PD skin regions. The DEGs from the comparisons were estimated by NOISeq [70]. Only the genes with q > 0.7 were defined as differentially expressed [71]. All DEGs were labeled in Additional file 4: Table S3.
Gene set enrichment and pathway analysis
To search the possible pathways involved in natal down development, the Ensemble gene ID of the expressed genes were converted to the ID of their chicken homologs and input into g:Profiler, a web-based toolset for functional profiling of gene lists from large-scale experiments. The p-value of the gene enrichment was corrected by Benjamini-Hochberg FDR (false discovery rate). Only the gene ontology with the corrected p-value < 0.05 was used in further analyses.
Quantitative PCR
To quantify the candidate lncRNA gene expression levels, the cDNAs were synthesized from the total RNAs by QuaniTect Reverse Transcription kit (Qiagen). Each cDNA sample containing SYBR green (KAPA SYBR FAST qPCR kit) was run on LightCycler 480 (Roche) under the appropriate conditions. Quantification of the TATA box binding protein (TBP) RNA was used to normalize target gene expression levels. All the PCR primers are listed in Additional file 13: Table S8.
Acknowledgments
We thank Drs. Chih-Feng Chen and Joyraj Bhattaharjee for their advice on this study. We also thank the NGS Core Facility, Academia Sinica, for transcriptome sequencing. We would like to thank National Center for High-performance Computing (NCHC) of National Applied Research Laboratories (NARLabs) of Taiwan and National Research Program for Biopharmaceuticals (NRPB, MOST 104-2325-B-492-001) for providing computational biology platform.
Funding
This study was supported by MOST 104-2621-B-001-003-MY3 to W.H.L. and MOST 105-2311-B-007-008-MY2 to C.S.N..
Availability of data and materials
The RNA-Seq data were downloaded from in the National Center for Biotechnology Information (NCBI) under the accession code PRJNA296752.
Authors’ contributions
CKC, CSN, and WHL conceived this study. CKC, CPY, CSN, CTT, and WHL designed the experiments. CKC collected all the animal samples. SCL developed the lncRNA prediction pipeline. CKC and CPY carried out the bioinformatics analysis. CKC and SMW performed the experimental validations. MYJL, YHC and DRC conducted the transcriptome sequencing. CKC, CPY and SMW generated all images. CKC, CPY, CSN, CTT and WHL wrote the manuscript. All authors read and revised the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
Unless stated otherwise below, all the animal experiments were conducted in our previous study [27] according to the protocol approved by the Institutional Animal Care and Use Committees of National Chung Hsing University (Taichung, Taiwan).
Abbreviations
- AD
Anterior dorsal skin region
- E12A
AD skin of embryo day 12
- E12P
PD skin of embryo day 12
- E8A
AD skin of embryo day 8
- E8P
PD skin of embryo day 8
- E9A
AD skin of embryo day 9
- E9P
PD skin of embryo day 9
- PD
Posterior dorsal skin region
- ssRNA-seq
Single-stranded RNA-seq
Additional files
Schematic presentation of natal down development in zebra finch and chicken and the six ssRNA-seq datasets used in this study. (A) Zebra finch embryos show two types of feather formation. The anterior dorsal (AD) tract and its two flanks show Type I feather formation (open circles) in which the feather buds do not develop into feather. On the other hand, the middle stripe of the posterior dorsal (PD) tract and the other regions shown in black circles show Type II feather formation in which the feather buds develop into down feathers, which are later replaced by contour feathers. In contrast, both the AD and the PD region of chicken embryos show Type II feather formation. (B) The skin regions and the six ssRNA-seq datasets used in this study. To reduce the complexity of skin regional specificity, only the dorsal skins (red dash boxes) were dissected and analyzed for the gene expressions. E8A: AD skin of embryo day 8; E8P: PD skin of embryo day 8; E9A: AD skin of embryo day 9; E9P: PD skin of embryo day 9; E12A: AD skin of embryo day 12; E12P: PD skin of embryo day 12; D7: 7 days post-hatch. Scale bar: 0.1 cm. The figure was modified from our previous study [27]. (TIF 20328 kb)
Read count statistics of the ssRNA seq. (DOCX 13 kb)
Information of the annotated genes. (XLSX 1440 kb)
2,949 identified transcripts information. (XLSX 517 kb)
Chromosomal distribution of the expressed unannotated transcripts. (A) Chromosomal distribution of the expressed 2949 transcripts and 1080 lncRNAs. The Y axis represents the transcript number per million bases of the chromosome. (B) The distributions of α- keratin gene transcripts, β- keratin gene transcripts, unannotated transcripts (without lncRNAs), and lncRNAs in chromosomes 25 and 27. Purple line: α- keratin gene transcripts; green line: β- keratin gene transcripts; orange line: unannotated transcripts (without lncRNAs); blue line: lncRNAs. P-value < 0.05 is indicated by dashed red line (chi-square test with one-tailed). (TIF 17522 kb)
Tables of the collected human, chicken, bovine lncRNAs. (XLSX 90 kb)
The distribution of Maximal JS stage specificity score. The JS score distributions for mRNA (grey box), lincRNA (red box), intronic lncRNA (blue box), and lncNAT (green box). A higher score value indicates a higher degree of stage specificity expression. (TIF 145 kb)
Supplementary Results. (DOCX 22 kb)
Gene set enrichment analysis using gProfiler. (XLSX 743 kb)
Gene set enrichment analysis using Fisher’s exact test. (XLSX 1040 kb)
Protein domains enriched in clades F, G and L. (XLSX 442 kb)
The location of the aligned CUFF.19772.1 (black bar) and BHLHE41 in human genome. CUFF.19772.1 was aligned to the 3’ UTR of human gene BHLHE41. (TIF 234 kb)
Primer pair sequences used in this study. (DOCX 15 kb)
Contributor Information
Chih-Kuan Chen, Email: r93b42016@gmail.com.
Chun-Ping Yu, Email: chpngyu@gmail.com.
Sung-Chou Li, Email: raymond.pinus@gmail.com.
Siao-Man Wu, Email: u941671@oz.nthu.edu.tw.
Mei-Yeh Jade Lu, Email: meiyehlu@gate.sinica.edu.tw.
Yi-Hua Chen, Email: yihua5314@gmail.com.
Di-Rong Chen, Email: cherrychen2835@gmail.com.
Chen Siang Ng, Email: gcsng@life.nthu.edu.tw.
Chau-Ti Ting, Email: ctting@ntu.edu.tw.
Wen-Hsiung Li, Email: whli@sinica.edu.tw.
References
- 1.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermuller J, Hofacker IL, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316(5830):1484–1488. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- 3.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peschansky VJ, Wahlestedt C. Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics. 2014;9(1):3–12. doi: 10.4161/epi.27473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mattick JS, Rinn JL. Discovery and annotation of long noncoding RNAs. Nat Struct Mol Biol. 2015;22(1):5–7. doi: 10.1038/nsmb.2942. [DOI] [PubMed] [Google Scholar]
- 6.Billerey C, Boussaha M, Esquerre D, Rebours E, Djari A, Meersseman C, Klopp C, Gautheret D, Rocha D. Identification of large intergenic non-coding RNAs in bovine muscle using next-generation transcriptomic sequencing. BMC Genomics. 2014;15:499. doi: 10.1186/1471-2164-15-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang YC, Liao JY, Li ZY, Yu Y, Zhang JP, Li QF, Qu LH, Shu WS, Chen YQ. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014;15(12):512. [DOI] [PMC free article] [PubMed]
- 8.Zhang K, Huang K, Luo Y, Li S. Identification and functional analysis of long non-coding RNAs in mouse cleavage stage embryonic development based on single cell transcriptome data. BMC Genomics. 2014;15:845. doi: 10.1186/1471-2164-15-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui W, Qian Y, Zhou X, Lin Y, Jiang J, Chen J, Zhao Z, Shen B. Discovery and characterization of long intergenic non-coding RNAs (lincRNA) module biomarkers in prostate cancer: an integrative analysis of RNA-Seq data. BMC Genomics. 2015;16(Suppl 7):S3. [DOI] [PMC free article] [PubMed]
- 10.St Laurent G, Wahlestedt C, Kapranov P. The Landscape of long noncoding RNA classification. Trends Genet. 2015;31(5):239–251. doi: 10.1016/j.tig.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown CJ, Hendrich BD, Rupert JL, Lafreniere RG, Xing Y, Lawrence J, Willard HF. The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell. 1992;71(3):527–542. doi: 10.1016/0092-8674(92)90520-M. [DOI] [PubMed] [Google Scholar]
- 12.Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N. Requirement for Xist in X chromosome inactivation. Nature. 1996;379(6561):131–137. doi: 10.1038/379131a0. [DOI] [PubMed] [Google Scholar]
- 13.Fitzpatrick GV, Soloway PD, Higgins MJ. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet. 2002;32(3):426–431. doi: 10.1038/ng988. [DOI] [PubMed] [Google Scholar]
- 14.Lewis A, Green K, Dawson C, Redrup L, Huynh KD, Lee JT, Hemberger M, Reik W. Epigenetic dynamics of the Kcnq1 imprinted domain in the early embryo. Development. 2006;133(21):4203–4210. doi: 10.1242/dev.02612. [DOI] [PubMed] [Google Scholar]
- 15.Mancini-Dinardo D, Steele SJ, Levorse JM, Ingram RS, Tilghman SM. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 2006;20(10):1268–1282. doi: 10.1101/gad.1416906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472(7341):120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129(7):1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhouailly D. A new scenario for the evolutionary origin of hair, feather, and avian scales. J Anat. 2009;214(4):587–606. doi: 10.1111/j.1469-7580.2008.01041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chuong CM, Yeh CY, Jiang TX, Widelitz R. Module-based complexity formation: periodic patterning in feathers and hairs. Wiley Interdiscip Rev Dev Biol. 2013;2(1):97–112. doi: 10.1002/wdev.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin CM, Liu Y, Huang K, Chen XC, Cai BZ, Li HH, Yuan YP, Zhang H, Li Y. Long noncoding RNA expression in dermal papilla cells contributes to hairy gene regulation. Biochem Biophys Res Commun. 2014;453(3):508–514. doi: 10.1016/j.bbrc.2014.09.119. [DOI] [PubMed] [Google Scholar]
- 21.Li T, Wang S, Wu R, Zhou X, Zhu D, Zhang Y. Identification of long non-protein coding RNAs in chicken skeletal muscle using next generation sequencing. Genomics. 2012;99(5):292–298. doi: 10.1016/j.ygeno.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Zhang GJ, Li C, Li QY, Li B, Larkin DM, Lee C, Storz JF, Antunes A, Greenwold MJ, Meredith RW, et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science. 2014;346(6215):1311–1320. doi: 10.1126/science.1251385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gardner PP, Fasold M, Burge SW, Ninova M, Hertel J, Kehr S, Steeves TE, Griffiths-Jones S, Stadler PF. Conservation and losses of non-coding RNAs in avian genomes. PLoS One. 2015;10(3):e0121797. doi: 10.1371/journal.pone.0121797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mou C, Pitel F, Gourichon D, Vignoles F, Tzika A, Tato P, Yu L, Burt DW, Bed'hom B, Tixier-Boichard M, et al. Cryptic patterning of avian skin confers a developmental facility for loss of neck feathering. PLoS Biol. 2011;9(3):e1001028. doi: 10.1371/journal.pbio.1001028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wells KL, Hadad Y, Ben-Avraham D, Hillel J, Cahaner A, Headon DJ. Genome-wide SNP scan of pooled DNA reveals nonsense mutation in FGF20 in the scaleless line of featherless chickens. BMC Genomics. 2012;13:257. doi: 10.1186/1471-2164-13-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hornik C, Krishan K, Yusuf F, Scaal M, Brand-Saberi B. cDermo-1 misexpression induces dense dermis, feathers, and scales. Dev Biol. 2005;277(1):42–50. doi: 10.1016/j.ydbio.2004.08.050. [DOI] [PubMed] [Google Scholar]
- 27.Chen CK, Ng CS, Wu SM, Chen JJ, Cheng PL, Wu P, Lu MJ, Chen DR, Chuong CM, Cheng HC et al. Regulatory Differences in Natal Down Development between Altricial Zebra Finch and Precocial Chicken. Mol Biol Evol. 2016;33(8):2030–43. [DOI] [PMC free article] [PubMed]
- 28.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Y, Li H, Fang S, Kang Y, Wu W, Hao Y, Li Z, Bu D, Sun N, Zhang MQ, et al. NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2016;44(D1):D203–208. doi: 10.1093/nar/gkv1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35(Web Server issue):W345–349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Housman G, Ulitsky I. Methods for distinguishing between protein-coding and long noncoding RNAs and the elusive biological purpose of translation of long noncoding RNAs. Biochim Biophys Acta. 2016;1859(1):31–40. doi: 10.1016/j.bbagrm.2015.07.017. [DOI] [PubMed] [Google Scholar]
- 32.Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH, UniProt C. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics. 2015;31(6):926–932. doi: 10.1093/bioinformatics/btu739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li A, Zhang J, Zhou Z. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC bioinformatics. 2014;15:311. doi: 10.1186/1471-2105-15-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finn RD, Clements J, Eddy SR: HMMER web server: interactive sequence similarity searching. Nucleic acids research 2011, 39(Web Server issue):W29-37 [DOI] [PMC free article] [PubMed]
- 35.Ng CS, Wu P, Fan WL, Yan J, Chen CK, Lai YT, Wu SM, Mao CT, Chen JJ, Lu MY, et al. Genomic organization, transcriptomic analysis, and functional characterization of avian alpha- and beta-keratins in diverse feather forms. Genome Biol. Evol. 2014;6(9):2258–2273. doi: 10.1093/gbe/evu181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsoi LC, Iyer MK, Stuart PE, Swindell WR, Gudjonsson JE, Tejasvi T, Sarkar MK, Li B, Ding J, Voorhees JJ, et al. Analysis of long non-coding RNAs highlights tissue-specific expression patterns and epigenetic profiles in normal and psoriatic skin. Genome Biol. 2015;16:24. doi: 10.1186/s13059-014-0570-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ren H, Wang G, Chen L, Jiang J, Liu L, Li N, Zhao J, Sun X, Zhou P. Genome-wide analysis of long non-coding RNAs at early stage of skin pigmentation in goats (Capra hircus) BMC Genomics. 2016;17:67. doi: 10.1186/s12864-016-2365-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25(18):1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dinger ME, Amaral PP, Mercer TR, Pang KC, Bruce SJ, Gardiner BB, Askarian-Amiri ME, Ru K, Solda G, Simons C, et al. Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res. 2008;18(9):1433–1445. doi: 10.1101/gr.078378.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458(7235):223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonzalez-Mariscal L, Tapia R, Chamorro D. Crosstalk of tight junction components with signaling pathways. Biochim Biophys Acta. 2008;1778(3):729–756. doi: 10.1016/j.bbamem.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 42.Alibardi L. Gap and tight junctions in the formation of feather branches: A descriptive ultrastructural study. Ann Anat. 2010;192(4):251–258. doi: 10.1016/j.aanat.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 43.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/gr.229102.ArticlepublishedonlinebeforeprintinMay2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Desbiens X, Queva C, Jaffredo T, Stehelin D, Vandenbunder B. The relationship between cell proliferation and the transcription of the nuclear oncogenes c-myc, c-myb and c-ets-1 during feather morphogenesis in the chick embryo. Development. 1991;111(3):699–713. doi: 10.1242/dev.111.3.699. [DOI] [PubMed] [Google Scholar]
- 45.Widelitz RB, Jiang TX, Yu M, Shen T, Shen JY, Wu P, Yu Z, Chuong CM. Molecular biology of feather morphogenesis: a testable model for evo-devo research. J Exp Zool B Mol Dev Evol. 2003;298(1):109–122. doi: 10.1002/jez.b.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Michon F, Forest L, Collomb E, Demongeot J, Dhouailly D. BMP2 and BMP7 play antagonistic roles in feather induction. Development. 2008;135(16):2797–2805. doi: 10.1242/dev.018341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Widelitz RB, Veltmaat JM, Mayer JA, Foley J, Chuong CM. Mammary glands and feathers: comparing two skin appendages which help define novel classes during vertebrate evolution. Semin Cell Dev Biol. 2007;18(2):255–266. doi: 10.1016/j.semcdb.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin CM, Jiang TX, Widelitz RB, Chuong CM. Molecular signaling in feather morphogenesis. Curr Opin Cell Biol. 2006;18(6):730–741. doi: 10.1016/j.ceb.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu P, Hou L, Plikus M, Hughes M, Scehnet J, Suksaweang S, Widelitz R, Jiang TX, Chuong CM. Evo-Devo of amniote integuments and appendages. Int J Dev Biol. 2004;48(2–3):249–270. doi: 10.1387/ijdb.15272390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prum RO, Dyck J. A hierarchical model of plumage: morphology, development, and evolution. J Exp Zool B Mol Dev Evol. 2003;298(1):73–90. doi: 10.1002/jez.b.27. [DOI] [PubMed] [Google Scholar]
- 51.Chen CF, Foley J, Tang PC, Li A, Jiang TX, Wu P, Widelitz RB, Chuong CM. Development, regeneration, and evolution of feathers. Annu Rev Anim Biosci. 2015;3:169–195. doi: 10.1146/annurev-animal-022513-114127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin SJ, Wideliz RB, Yue Z, Li A, Wu X, Jiang TX, Wu P, Chuong CM. Feather regeneration as a model for organogenesis. Dev Growth Differ. 2013;55(1):139–148. doi: 10.1111/dgd.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pauli A, Valen E, Lin MF, Garber M, Vastenhouw NL, Levin JZ, Fan L, Sandelin A, Rinn JL, Regev A, et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012;22(3):577–591. doi: 10.1101/gr.133009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kapusta A, Kronenberg Z, Lynch VJ, Zhuo X, Ramsay L, Bourque G, Yandell M, Feschotte C. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 2013;9(4):e1003470. doi: 10.1371/journal.pgen.1003470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kapusta A, Feschotte C. Volatile evolution of long noncoding RNA repertoires: mechanisms and biological implications. Trends Genet. 2014;30(10):439–452. doi: 10.1016/j.tig.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chalopin D, Naville M, Plard F, Galiana D, Volff JN. Comparative analysis of transposable elements highlights mobilome diversity and evolution in vertebrates. Genome Biol. Evol. 2015;7(2):567–580. doi: 10.1093/gbe/evv005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmitz SU, Grote P, Herrmann BG. Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci. 2016;73(13):2491–2509. doi: 10.1007/s00018-016-2174-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mill P, Mo R, Hu MC, Dagnino L, Rosenblum ND, Hui CC. Shh controls epithelial proliferation via independent pathways that converge on N-Myc. Dev Cell. 2005;9(2):293–303. doi: 10.1016/j.devcel.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 59.Wang N, Yang T, Li J, Lei M, Shi J, Qiu W, Lian X. The expression and role of c-Myc in mouse hair follicle morphogenesis and cycling. Acta Histochem. 2012;114(3):199–206. doi: 10.1016/j.acthis.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 60.McKinnell IW, Turmaine M, Patel K. Sonic Hedgehog functions by localizing the region of proliferation in early developing feather buds. Dev Biol. 2004;272(1):76–88. doi: 10.1016/j.ydbio.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 61.Greenwold MJ, Sawyer RH. Genomic organization and molecular phylogenies of the beta (beta) keratin multigene family in the chicken (Gallus gallus) and zebra finch (Taeniopygia guttata): implications for feather evolution. BMC Evol Biol. 2010;10:148. doi: 10.1186/1471-2148-10-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.International Chicken Genome Sequencing C Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432(7018):695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- 63.Nie H, Crooijmans RP, Lammers A, van Schothorst EM, Keijer J, Neerincx PB, Leunissen JA, Megens HJ, Groenen MA. Gene expression in chicken reveals correlation with structural genomic features and conserved patterns of transcription in the terrestrial vertebrates. PLoS One. 2010;5(8):e11990. doi: 10.1371/journal.pone.0011990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 67.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31(1):46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14(4):708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Margulies EH, Blanchette M, Program NCS, Haussler D, Green ED. Identification and characterization of multi-species conserved sequences. Genome Res. 2003;13(12):2507–2518. doi: 10.1101/gr.1602203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: a matter of depth. Genome Res. 2011;21(12):2213–2223. doi: 10.1101/gr.124321.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu WY, Chang YM, Chen SC, Lu CH, Wu YH, Lu MY, Chen DR, Shih AC, Sheue CR, Huang HC, et al. Anatomical and transcriptional dynamics of maize embryonic leaves during seed germination. Proc Natl Acad Sci U S A. 2013;110(10):3979–3984. doi: 10.1073/pnas.1301009110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The RNA-Seq data were downloaded from in the National Center for Biotechnology Information (NCBI) under the accession code PRJNA296752.