

Abstract
The genomes of four strains (MB11, MB14, MB30, and MB66) of the species Corynebacterium pseudotuberculosis biovar equi were sequenced on the Ion Torrent PGM platform, completely assembled, and their gene content and structure were analyzed. The strains were isolated from horses with distinct signs of infection, including ulcerative lymphangitis, external abscesses on the chest, or internal abscesses on the liver, kidneys, and lungs. The average size of the genomes was 2.3 Mbp, with 2169 (Strain MB11) to 2235 (Strain MB14) predicted coding sequences (CDSs). An optical map of the MB11 strain generated using the KpnI restriction enzyme showed that the approach used to assemble the genome was satisfactory, producing good alignment between the sequence observed in vitro and that obtained in silico. In the resulting Neighbor-Joining dendrogram, the C. pseudotuberculosis strains sequenced in this study were clustered into a single clade supported by a high bootstrap value. The structural analysis showed that the genomes of the MB11 and MB14 strains were very similar, while the MB30 and MB66 strains had several inverted regions. The observed genomic characteristics were similar to those described for other strains of the same species, despite the number of inversions found. These genomes will serve as a basis for determining the relationship between the genotype of the pathogen and the type of infection that it causes.
Keywords: C. pseudotuberculosis, Biovar equi, Ulcerative lymphangitis, Horse, Genomic
Introduction
As of February 2016, thirty-three genomes of the species Corynebacterium pseudotuberculosis had been deposited into the National Center for Biotechnology Information database. This species is an animal pathogen that infects goats and sheep, causing caseous lymphadenitis, as well as horses, which can show distinct signs and symptoms. C. pseudotuberculosis can be classified into two biovars based on its ability to reduce nitrate to nitrite [1]. Non-reducing, i.e., nitrate-negative, strains are grouped into the ovis biovar and are responsible for CL. The reducing, i.e., nitrate-positive, strains are grouped into the equi biovar and mainly infect horses.
Recent increases in the number of infections in horses have led to C. pseudotuberculosis bv. equi being classified as a re-emerging pathogen. In Texas, USA, the number of cases increased 10-fold between 2005 and 2011, with a cumulative increase in annual incidence from 9.3 to 99.5 infections per 100,000 horses over the same period [2]. Kilcoyne et al. [3] analyzed the number of cultures positive for C. pseudotuberculosis in samples isolated from infected horses in 23 states in the USA. The proportion of positive cultures was higher for the most recent years, 2011 and 2012 (54% of the total number of samples), than for the period spanning 2003 to 2010 (46% of the total number of samples). These current data show the growing numbers of infections caused by this bacterium and emphasize the need for new studies on the genotypic characteristics of the biovar.
C. pseudotuberculosis bv. equi infection is commonly known as “pigeon fever” because it leads to the formation of external abscesses on the chest of the animal, making it expand, similar to a pigeon breast. Despite its common name, the bacteria can also cause other types of infections with distinct signs and symptoms, such as the formation of internal abscesses or ulcerative lymphangitis, which is characterized by the infection of limbs and compromises the lymphatic system [4]. It is currently believed that the major vectors of the disease are domestic flies of the species Haematobia irritans, Stomoxys calcitrans , and Musca domestica [5].
The pathogenesis of C. pseudotuberculosis is intrinsically linked to its genetic content. Several virulence factors have previously been described in the literature that strongly influence the ability of the bacteria to interact with the host, causing infection. Phospholipase D, the iron uptake system, and pili proteins are examples of these factors [6]. Characterization of these and novel virulence factors depends on the sequencing of new genomes from the biovar, as the vast majority of the genomes in databases belong to the ovis biovar. Therefore, to generate data that allows for a more robust genotypic analysis of the equi biovar, four genomes from strains isolated from horses with distinct signs of infection by C. pseudotuberculosis were sequenced using the next-generation Ion Torrent PGM platform.
Organism information
Classification and features
C. pseudotuberculosis bv. equi is a facultative intracellular, beta-hemolytic, pleomorphic (Fig. 1), non-sporulating, unencapsulated, non-mobile, facultative anaerobic, Gram-positive pathogen. [6]. The main characteristics of the species are shown in Table 1. C. pseudotuberculosis is taxonomically classified in the phylum Actinobacteria, class Actinobacteria, order Corynebacteriales, family Corynebacteriaceae, and genus Corynebacteria. The strains included in this study were isolated from horses in the state of California, USA. The animals had distinct signs and symptoms of infection. Strain MB11 was isolated from a 6-month-old American Paint horse with ulcerative lymphangitis. Strain MB14 was isolated from an Arab/Saddle horse with abscess formation in internal organs (liver and kidney). The animal also presented hepatic lipidosis and myocardial fibroses. Strain MB30 was isolated from the pectoral abscess of a 2-year-old Quarter horse. Finally, strain MB66 was isolated from a 20-year-old Polish Arab mare with metastatic melanoma and multiple external and internal abscesses. These distinct signs, such as pectoral abscesses (“pigeon fever”), abscesses on the internal organs, or abscesses on the limbs (ulcerative lymphangitis), suggest that the equi biovar can interact in several ways with the host animal to cause infection. All strains were isolated over the period of October-1996 up to June-2002.
Fig. 1.

Transmission Electron Micrograph of three strains sequenced in this study. The electron micrographs of a MB11, b MB30 and c MB66, demonstrate the pleomorphic morphology of the species
Table 1.
Classification and general features of the species strain designationT [cite MIGS reference]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain: Bacteria | TAS [22] | |
| Phylum: Actinobacteria | TAS [23] | ||
| Class: Actinobacteria | TAS [24] | ||
| Order: Corynebacteriales | TAS [25, 26] | ||
| Family: Corynebacteriaceae | TAS [27, 28] | ||
| Genus: Corynebacterium | TAS [28, 29] | ||
| Species: C. pseudotuberculosis | TAS [28, 30] | ||
| strain: MB11, MB14, MB30 and MB66 | IDA | ||
| Gram stain | Positive | TAS [31] | |
| Cell shape | Pleomorphic | TAS [31] | |
| Motility | Non-motile | TAS [31] | |
| Sporulation | Non-sporulated | TAS [31] | |
| Temperature range | Mesophilic | TAS [32] | |
| Optimum temperature | 37 °C | TAS [32] | |
| pH range; optimum | 7.0–7.2 | TAS [32] | |
| Carbon source | Glucose, fructose, maltose, mannose, and sucrose | TAS [6] | |
| MIGS-6 | Habitat | Soil and animal pathogens | TAS [4, 33] |
| MIGS-6.3 | Salinity | Up to 2 M NaCl | TAS [32] |
| MIGS-22 | Oxygen requirement | Facultative anaerobe | TAS [6] |
| MIGS-15 | Biotic relationship | Intracellular facultative pathogen | TAS [6] |
| MIGS-14 | Pathogenicity | Equus caballus | TAS [4] |
| MIGS-4 | Geographic location | California, USA | IDA |
| MIGS-5 | Sample collection | MB11: Oct-96 MB14: Dec-96 MB30: Nov-00 MB66: Jun-02 |
IDA |
| MIGS-4.1 | Latitude | MB11 - 38°21′23″ MB14 - 37°00′20″ MB30 - 39°39′32″ MB66 - 38°32′41″ |
IDA |
| MIGS-4.2 | Longitude | MB11 - 121°59′15″ MB14 - 121°34′05″ MB30 - 121°37′52″ MB66 - 121°44′25″ |
IDA |
| MIGS-4.4 | Altitude | MB11 - 180 ft MB14 - 196 ft MB30 - 351 ft MB66 - 55 ft |
IDA |
aEvidence codes - IDA Inferred from Direct Assay, TAS Traceable Author Statement (i.e., a direct report exists in the literature), NAS Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property of the species or anecdotal evidence). These evidence codes are from the Gene Ontology project [cite this reference]
A dendrogram was calculated with the Neighbor-joining statistical method using a bootstrap analysis with 1000 replicates. The rpoB gene, which codes for the beta subunit of the RNA polymerase enzyme, was used as a marker when constructing the dendrogram. The analysis was performed using the NCBI reference sequence for the species, retrieving from the database at least one representative from each genus in the Corynebacterium, Mycobacterium, Nocardia , and Rhodococcus group (Fig. 2). This group is composed of species that share cellular characteristics, such as a cell wall composed of peptidoglycan, arabinogalactan, and mycolic acids, as well as a genome with a high GC content [6]. The first phylogenetic studies on the CMNR group used the 16S rRNA gene as a marker. These studies demonstrated that the genera in the family Corynebacteriaceae form a monophyletic clade composed of four groups, in which C. pseudotuberculosis is phylogenetically closest to the species C. ulcerans and C. diphtheriae [7]. Recently, Khamis et al. [8] proposed that the gene rpoB could be used as a marker to identify clinical isolates of the genus Corynebacterium. The positive results for identification using the rpoB gene were greater than those of the 16S rRNA gene, indicating that rpoB is useful for taxonomic classification the family Corynebacteriaceae [8]. The dendrogram in Fig. 2 shows the phylogenetic proximity between the sequenced biovars of the species C. pseudotuberculosis. In addition, it corroborates the analyses performed with the 16S rRNA gene, which designated C. diphtheriae as the species most closely related to C. pseudotuberculosis. The results show that each genus in the CMNR group is divided into clades supported by high bootstrap values.
Fig. 2.

Dendrogram of the representative genomes in the CMNR group. The analysis was performed using MEGA 5.10. Only bootstraps greater than 50% are shown in the branches of the dendrogram. The accession numbers for the sequences used in the analysis are: C. pseudotuberculosis MB11 (CP013260), C. pseudotuberculosis MB14 (CP013261), C. pseudotuberculosis MB30 (CP013262), C. pseudotuberculosis MB66 (CP013263), C. pseudotuberculosis 316 (CP003077), C. pseudotuberculosis 258 (CP003540), C. pseudotuberculosis 1002 (CP001809), C. pseudotuberculosis C231 (CP001829), C. diphtheriae NCTC 13129 (BX248353), C. glutamicum ATCC 13032 (BA000036), C. striatum ATCC 6940 (GCA_000159135), C. accolens ATCC 49725 (GCA_000159115), C. pseudogenitalium ATCC 33035 (NZ_ABYQ00000000), C. jeikeium K411 (NC_007164), N. brasiliensis ATCC 700358 (CP003876), N. farcinica IFM 10152 (NC_006361), M. bovis AF2122/97 (BX248333), M. ulcerans Agy99 (CP000325), M. smegmatis MC2 155 (CP000480), R. equi 103S (FN563149), R. fascians NBRC 12155 (GCA_001894785), R. erythropolis PR4 (NC_012490), R. jostii RHA1 (NC_008268)
Genome sequencing information
Genome project history
The four C. pseudotuberculosis genomes in this short report are part of a collaboration between the University of California, Davis, USA, and the Federal Universities of Minas Gerais and Pará, Brazil. The project seeks to determine the genomic characteristics of 12 strains of the equi biovar isolated from horses in California showing distinct signs and symptoms of infection. Isolation was performed over several years from different horse breeds (Table 2). One of the major aims of the project is to determine if a relationship exists between the genetic content of the strains and the type of infection that it causes (i.e., ulcerative lymphangitis, external abscesses, or internal abscesses). In parallel, the project seeks to increase the amount of genomic data for the species C. pseudotuberculosis in databases, which will form the basis for broader functional studies. The genomes obtained in this study have been deposited into the NCBI database under accession number CP013260, CP013261, CP013262, CP013263. The project information is also presented in Table 2.
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS 31 | Finishing quality | Completed |
| MIGS-28 | Libraries used | Fragments library |
| MIGS 29 | Sequencing platforms | Ion Torrent PGM |
| MIGS 31.2 | Fold coverage | 842x (MB11); 867x (MB14); 309x (MB30); 658x (MB66). |
| MIGS 30 | Assemblers | MIRA4, Lasergene (DNASTAR), GapBlaster. |
| MIGS 32 | Gene calling method | Pannotator (FgenesB; Glimmer; tRNAscan; RNAmer) |
| Locus Tag | ATN02_ (MB11); ATN03_ (MB14); ATN04_ (MB30); ATN05_ (MB66) | |
| GenBank ID | CP013260 (MB11); CP013261 (MB14); CP013262 (MB30); CP013263 (MB66). | |
| GenBank Date of Release | 2016-03-01 | |
| GOLD ID | Gp0131493 (MB11); Gp0131495 (MB14); Gp0131496 (MB30); Gp0131497 (MB66). | |
| BIOPROJECT | PRJNA256958 | |
| MIGS 13 | Source Material Identifier | Isolated directly from the infected animal |
| Project relevance | Animal pathogen |
Growth conditions and genomic DNA preparation
After isolation, the bacteria were maintained in 25% glycerol at −80 °C, and the medium was refreshed routinely. To extract genomic DNA, the bacteria were first cultured in liquid brain heart infusion (BHI) medium at 37 °C with shaking. DNA was extracted during the log-phase of cell growth according to the protocol described by Pacheco et al. [9] for clinical isolates. The extracted DNA was subjected to electrophoresis on a 1% agarose gel to determine the quality of the material.
Genome sequencing and assembly
Genomic DNA was sequenced on the Ion Torrent PGM (Thermo Scientific) platform using the 318 chip v2 in accordance with the manufacturer’s instructions. The quality of the reads was analyzed using FastQC software [10]. The reads were then trimmed and filtered to remove those with a phred-scaled quality score less than 20. Next, the reads were assembled using Mira 4 software [11]. Redundancy within the assembled contigs was eliminated using the SeqMan Pro tool in the Lasergene software package (DNASTAR). The few remaining gaps after redundancy removal were manually closed using local BLAST or a program developed by our research group called GapBlaster [12], which uses a reference genome to assemble similar sequences to close the gap using the sequencing reads. For this analysis, we used C. pseudotuberculosis biovar equi strain 316 as a reference. An optical map using KpnI restriction sites was generated to evaluate the quality of the genome assembly for the MB11 strain (Fig. 3). The optical map was analyzed using MapSolver v.3.2.0 (OpGen). Figure 3 shows that the in silico assembly for strain MB11 was very satisfactory; the positions of the restriction sites were corroborated by the optical map analysis.
Fig. 3.

Optical map of Corynebacterium pseudotuberculosis MB11. The figure shows the alignment of the KpnI sites observed in the optical map (bottom scale bar) with those predicted by the in silico assembly (top scale bar). Vertical lines connect identical restriction sites observed in the optical map and those predicted by the assembly, demonstrating that the genome was assembled in the correct order
Genome annotation
An automatic annotation was first conducted using the online software Pannotator [13], which provided the .fasta files for the assembled genomes and a reference .embl file for C. pseudotuberculosis 316. The results were then manually curated to meet the gene annotation standards set by UniProt [14] using Artemis software [15] to visualize the coding sequences. Next, pseudogenes were also manually curated to resolve mismatches using CLC Genomics Workbench 5 (CLC Bio) and Artemis. Predicted genes for the four genomes were classified by the clusters of orthologous groups functional category, as shown in Table 3.
Table 3.
Number of genes associated with general COG functional categories
| Code | MB11 | MB14 | MB30 | MB66 | Description | ||||
|---|---|---|---|---|---|---|---|---|---|
| Value | %age | Value | %age | Value | %age | Value | %age | ||
| J | 127 | 5.83 | 148 | 6.62 | 123 | 5.64 | 122 | 5.54 | Translation, ribosomal structure, and biogenesis |
| A | 1 | 0.05 | 1 | 0.04 | 1 | 0.05 | 1 | 0.05 | RNA processing and modification |
| K | 55 | 2.52 | 90 | 4.03 | 55 | 2.52 | 54 | 2.45 | Transcription |
| L | 63 | 2.89 | 96 | 4.29 | 67 | 3.07 | 66 | 3.00 | Replication, recombination, and repair |
| B | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | Chromatin structure and dynamics |
| D | 16 | 0.73 | 25 | 1.12 | 16 | 0.73 | 16 | 0.73 | Cell cycle control, cell division, and chromosome partitioning |
| V | 13 | 0.60 | 23 | 1.03 | 13 | 0.60 | 13 | 0.59 | Defense mechanisms |
| T | 17 | 0.78 | 55 | 2.46 | 17 | 0.78 | 16 | 0.73 | Signal transduction mechanisms |
| M | 55 | 2.52 | 82 | 3.67 | 55 | 2.52 | 54 | 2.45 | Cell wall/membrane biogenesis |
| N | 1 | 0.05 | 14 | 0.63 | 1 | 0.05 | 1 | 0.05 | Cell motility |
| U | 17 | 0.78 | 21 | 0.94 | 17 | 0.78 | 17 | 0.77 | Intracellular trafficking and secretion |
| O | 53 | 2.43 | 79 | 3.53 | 55 | 2.52 | 53 | 2.41 | Posttranslational modification, protein turnover, and chaperones |
| C | 73 | 3.35 | 121 | 5.41 | 74 | 3.40 | 73 | 3.32 | Energy production and conversion |
| G | 73 | 3.35 | 100 | 4.47 | 74 | 3.40 | 72 | 3.27 | Carbohydrate transport and metabolism |
| E | 122 | 5.60 | 180 | 8.05 | 122 | 5.60 | 122 | 5.54 | Amino acid transport and metabolism |
| F | 58 | 2.66 | 74 | 3.31 | 57 | 2.62 | 57 | 2.59 | Nucleotide transport and metabolism |
| H | 83 | 3.81 | 113 | 5.05 | 83 | 3.81 | 83 | 3.77 | Coenzyme transport and metabolism |
| I | 36 | 1.65 | 51 | 2.28 | 36 | 1.65 | 35 | 1.59 | Lipid transport and metabolism |
| P | 68 | 3.12 | 118 | 5.28 | 67 | 3.07 | 67 | 3.04 | Inorganic ion transport and metabolism |
| Q | 13 | 0.60 | 28 | 1.25 | 13 | 0.60 | 12 | 0.55 | Secondary metabolite biosynthesis, transport, and catabolism |
| R | 113 | 5.19 | 275 | 12.30 | 111 | 5.09 | 110 | 5.00 | General function prediction only |
| S | 112 | 5.14 | 153 | 6.84 | 112 | 5.14 | 113 | 5.13 | Function unknown |
| - | 1010 | 46.35 | 389 | 17.40 | 1056 | 46.35 | 1044 | 47.43 | Not in COGs |
The total is based on the total number of protein coding genes in the genome
Genome properties
All of the genomes were completely closed, resulting in a size of 2,363,423 bp for strain MB11, 2,370,761 bp for MB14, 2,364,377 for MB30, and 2,372,202 bp for MB66. The approximately 2.3 Mbp size is similar to other previously studied and published equi strains [16–18]. Four ribosomal RNA clusters were observed in all of the genomes. The strains had an average GC content of 52% and a total of 51 tRNAs predicted by tRNAscan-SE for each strain [19]. MB11 had a total of 2179 CDSs and 37 pseudogenes after manual curation. MB14 had 2235 CDSs and 20 pseudogenes, while MB30 had 2225 CDSs and six pseudogenes, and finally, MB66 had 2201 CDSs and 54 pseudogenes. A more detailed description of the genomic statistics is presented in Table 4.
Table 4.
Genome statistics
| Attribute | MB11 | MB14 | MB30 | MB66 | ||||
|---|---|---|---|---|---|---|---|---|
| Value | % of Total | Value | % of Total | Value | % of Total | Value | % of Total | |
| Genome size (bp) | 2,363,423 | 100.0 | 2,370,761 | 100.0 | 2,364,377 | 100.0 | 2,372,202 | 100.0 |
| DNA coding (bp) | 2,021,172 | 85.52 | 2,052,709 | 86.58 | 2,066,802 | 87.41 | 2,006,473 | 84.58 |
| DNA G + C (bp) | 1,067,329 | 52.09 | 1,235,085 | 52.1 | 1,231,731 | 52.09 | 1,235,856 | 52.1 |
| DNA scaffolds | 1 | 100.0 | 1 | 100.0 | 1 | 100.0 | 1 | 100.0 |
| Total genes | 2,260 | 100.0 | 2,317 | 100.0 | 2,237 | 100.0 | 2,334 | 100.0 |
| Protein coding genes | 2,179 | 96.41 | 2,235 | 96.46 | 2,225 | 99.46 | 2,201 | 94.30 |
| RNA genes | 63 | 2.79 | 63 | 2.78 | 63 | 2.82 | 63 | 2.70 |
| Pseudo genes | 37 | 1.64 | 20 | 0.86 | 6 | 0.27 | 54 | 2.31 |
| Genes in internal clusters | 775 | 34.29 | 785 | 33.88 | 779 | 34.82 | 774 | 33.16 |
| Genes with function prediction | 1,526 | 67.52 | 1,576 | 68.02 | 1,577 | 70.50 | 1,550 | 66.41 |
| Genes assigned to COGs | 1,169 | 51.72 | 1,847 | 79.71 | 1,169 | 52.26 | 1,157 | 49.57 |
| Genes with Pfam domains | 1,722 | 76.19 | 1,819 | 80.49 | 1,823 | 78.68 | 1,797 | 76.99 |
| Genes with signal peptides | 88 | 3.89 | 92 | 3.97 | 93 | 4.16 | 86 | 3.68 |
| Genes with transmembrane helices | 589 | 26.06 | 607 | 26.20 | 604 | 27.00 | 583 | 24.98 |
| CRISPR repeats | 3 | 0.01 | 3 | 0.01 | 3 | 0.01 | 2 | 0.01 |
A circular map was generated using the CGView web tool [20] that shows the relationship of the predicted proteins in the MB14, MB30, and MB66 genomes compared to strain MB11, in which the in silico assembly was corroborated by the optical map (Fig. 4). All of the genomes had similar sizes and a similar number of CDSs, with few differences between the coding regions of the genomes. Structural analyses were conducted by comparing the four genomes with a local database using blastn, and the results were analyzed using the Artemis Comparison Tool [21]. The MB11 and MB14 strains showed extensive structural similarity, while MB30 had a large inversion of approximately 1.2 Mbp compared to MB14 (Fig. 5). However, MB66 had the largest number of structural rearrangements (Fig. 5). It is worth noting that two strains with distinct infection phenotypes (MB11 and MB14) that were isolated eight years apart had largely similar genomic structures, which did not occur in the other analyzed strains.
Fig. 4.

Circular map of the genome for the sequenced Corynebacterium pseudotuberculosis strains. The outermost ring in blue shows the features extracted from the MB11 genome using a .gbk file. The next ring shows the CDSs predicted on the forward strand of MB11 in red, followed by the CDSs on the reverse strand with their features in blue. The other three rings in red, green, and blue show proteins predicted by blastx for the MB14, MB30, and MB66 genomes, respectively, compared to the MB11 genome. The two innermost rings show the GC content and GC skew, followed by the size of the genome in base pairs
Fig. 5.
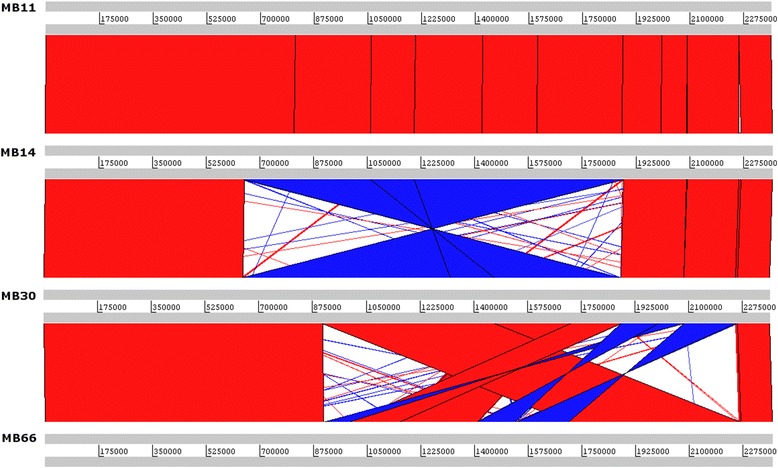
Comparison of C. pseudotuberculosis genome structures using blastn. The names of the strains are indicated at the side of the gray bars showing the size of each genome. Red bars show conserved regions between two genomes using an e-value of 1-e05, while blue bars show inverted regions
Conclusions
Because of the large number of infections reported for C. pseudotuberculosis biovar equi in recent years, sequencing and analyzing genomes for this biovar is an essential step towards new perspectives that will improve our understanding of pathogen-host interactions and facilitate the development of vaccines to eradicate the disease. The four genomes presented in this study showed structural differences, except for strains MB11 and MB14. The phylogenetic relationship is closer to other strains of the equi biovar, and other genomic characteristics, such as the GC content, number of CDSs, and tRNA and rRNA clusters, are similar to those described for other strains of the same species. Virulence factors that were previously described in the literature were identified in the analyzed genomes. In addition, in silico assembly of the MB11 genome was validated by an optical map of the KpnI restriction sites.
These initial data suggest that differences between types of infection should be analyzed using a reductionist approach, taking into account factors such as pathogenicity islands in each strain, the transmission method, and the entry point of the pathogen for each case, as well as expression levels and use of virulence factors specific to the bacteria, among other factors. Phylogenetic studies and the detection of small genetic changes such as SNPs and INDELs should then be performed because the bacteria have a very high gene density, and therefore, point mutations can strongly affect the biological response of the pathogen.
Acknowledgements
The authors are thankful for the financial support granted by CNPq and CAPES. The authors also thank the Pró-Reitoria de Pesquisa e Pós-Graduação of Universidade Federal do Pará for the financial support for the publication of the article.
Funding
This study was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior.
Authors’ contributions
RAB, RTJR, PHCGS, AAOV, LCG, DAG and ARC conducted the bioinformatics analyses, evaluated the results, and wrote the manuscript. SJS and JJE isolated the strains and designed the project together with VA and AS, in addition to helping to write the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- BHI
Brain heart infusion
- CDS
Coding DNA sequence
- CL
Caseous lymphadenitis
- CMNR
Corynebacterium Mycobacterium, Nocardia, Rhodococcus
- INDEL
Insertion/Deletion
- Ion Torrent PGM
Ion torrent personal genome machine
- PLD
Phospholipase D
- SNP
Single nucleotide polymorphism
References
- 1.Biberstein EL, Knight HD, Jang S. Two biotypes of Corynebacterium pseudotuberculosis. Vet Rec. 1971;89:691–2. doi: 10.1136/vr.89.26.691. [DOI] [PubMed] [Google Scholar]
- 2.Szonyi B, Swinford A, Clavijo A, Ivanek R. Re-emergence of pigeon fever (Corynebacterium pseudotuberculosis) infection in texas horses: epidemiologic investigation of laboratory-diagnosed cases. J Equine Vet Sci. 2014;34(2):281–7. doi: 10.1016/j.jevs.2013.06.006. [DOI] [Google Scholar]
- 3.Kilcoyne I, Spier SJ, Carter CN, Smith JL, Swinford AK, Cohen ND. Frequency of Corynebacterium pseudotuberculosis infection in horses across the United States during a 10-year period. J Am Vet Med Assoc. 2014;245(3):309–14. doi: 10.2460/javma.245.3.309. [DOI] [PubMed] [Google Scholar]
- 4.Aleman M, Spier SJ, Wilson WD, Doherr M. Retrospective study of Corynebacterium pseudotuberculosis infection in horses: 538 cases. J Am Vet Med Ass. 1996;209:804–9. [PubMed] [Google Scholar]
- 5.Spier SJ, Leutenegger CM, Carroll SP, Loye JE, Pusterla JB, Carpenter TE, et al. Use of a real-time polymerase chain reaction-based fluorogenic5′ nuclease assay to evaluate insect vectors of Corynebacterium pseudotuberculosis infections in horses. Am J Vet Res. 2004;65:829–34. doi: 10.2460/ajvr.2004.65.829. [DOI] [PubMed] [Google Scholar]
- 6.Dorella FA, Pacheco LGC, Oliveira SC, Miyoshi A, Azevedo V. Corynebacterium pseudotuberculosis: microbiology, biochemical properties, pathogenesis and molecular studies of virulence. Vet Res. 2006;37:201–18. doi: 10.1051/vetres:2005056. [DOI] [PubMed] [Google Scholar]
- 7.Ruimy R, Riegel P, Boiron P, Monteil H, Christen R. Phylogeny of the genus Corynebacterium deduced from analyses of small-subunit ribosomal DNA sequences. Int J Syst Evol Microbiol. 1995;45:740–6. doi: 10.1099/00207713-45-4-740. [DOI] [PubMed] [Google Scholar]
- 8.Khamis A, Raoult D, La Scola B. Comparison between rpoB and 16S rRNA gene sequencing for molecular identification of 168 clinical isolates of Corynebacterium. J Clin Microbiol. 2005;43:1934–6. doi: 10.1128/JCM.43.4.1934-1936.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pacheco LGC, Pena RR, Castro TLP, Dorella FA, Bahia RC, Carminati R, Frota MNL, Oliveira SC, Meyer R, Alves FSF, Miyoshi A, Azevedo V. Multiplex PCR assay for identification of Corynebacterium pseudotuberculosis from pure cultures and for rapid detection of this pathogen in clinical samples. J Med Microbiol. 2007;56:480–6. doi: 10.1099/jmm.0.46997-0. [DOI] [PubMed] [Google Scholar]
- 10.Babraham Bioinformatics: FastQC. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 18 Nov 2015.
- 11.Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WEG, Wetter T, Suhai S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004;14:1147–59. doi: 10.1101/gr.1917404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Sá PHCG, Miranda F, Veras A, de Melo DM, Soares S, Pinheiro K, Guimarães L, Azevedo V, Silva A, Ramos RTJ. GapBlaster – a graphical gap filler for prokaryotes genomes. PLoS One. 2016;11(5):e0155327. doi: 10.1371/journal.pone.0155327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santos AR, Barbosa E, Fiaux K, Zurita-Turk M, Chaitankar V, Kamapantula B, Abdelzaher A, Ghosh P, Tiwari S, Barve N, Jain N, Barh D, Silva A, Miyoshi A, Azevedo V. Pannotator: an automated tool for annotation of pan-genomes. Genet Mol Res. 2013;12(3):2982–9. doi: 10.4238/2013.August.16.2. [DOI] [PubMed] [Google Scholar]
- 14.The UniProt Consortium The universal protein resource (UniProt) Nucl Acids Res. 2008;36:D190–5. doi: 10.1093/nar/gkm895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16(10):944–5. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 16.Ramos RTJ, Carneiro AR, Soares SC, Santos AR, Almeida S, Guimarães L, Figueira F, Barbosa E, Tauch A, Azevedo V, Silva A. Tips and tricks for the assembly of a Corynebacterium pseudotuberculosis genome using a semiconductor sequencer. Microb Biotechnol. 2013;6(2):150–6. doi: 10.1111/1751-7915.12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soares SC, Trost E, Ramos RTJ, Carneiro AR, Santos AR, Pinto AC, Barbosa E, Aburjaile F, Ali A, Diniz CAA, Hassan SS, Fiaux K, Guimarães LC, Bakhtiar SM, Pereira U, Almeida SS, Abreu VAC, Rocha FS, Dorella FA, Miyoshi A, Silva A, Azevedo V, Tauch A. Genome sequence of Corynebacterium pseudotuberculosis biovar equi strain 258 and prediction of antigenic targets to improve biotechnological vaccine production. J Biotechnol. 2013;167(2):135–41. doi: 10.1016/j.jbiotec.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 18.Baraúna RA, Guimarães LC, Veras AAO, de Sá PHCG, Graças DA, Pinheiro KP, Silva ASS, Folador EL, Benevides LJ, Viana MVC, Carneiro AR, Schneider MPC, Spier SJ, Edman JM, Ramos RTJ, Azevedo V, Silva A. Genome sequence of Corynebacterium pseudotuberculosis MB20 bv. equi isolated from a pectoral abscess of an Oldenburg horse in California. Genome Announc. 2014;2(6):e00977–14. doi: 10.1128/genomeA.00977-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucl Acids Res. 1997;25(5):955–64. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grant JR, Stothard P. The CGView server: a comparative genomics tool for circular genomes. Nucl Acids Res. 2008;36(2):W181–4. doi: 10.1093/nar/gkn179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: Artemis Comparison Tool. Bioinformatics. 2005;21(16):3422–3. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- 22.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–9. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodfellow M. Phylum XXVI. Actinobacteria phyl. nov. In: Goodfellow M, Kämpfer P, Busse H-J, Trujillo ME, Suzuki K-I, Ludwig W, Whitman WB, editors. Bergey’s manual of systematic bacteriology. 2. New York: Springer; 2001. pp. 119–169. [Google Scholar]
- 24.Stackebrandt E, Rainey FA, Ward-Rainey NL. Proposal for a new hierarchic classification system, Actinobacteria classis nov. Int J Syst Bacteriol. 1997;47:479–91. doi: 10.1099/00207713-47-2-479. [DOI] [Google Scholar]
- 25.Goodfellow M, Jones AL, Order V. Corynebacteriales ord. nov. In: Goodfellow M, Kämpfer P, Busse H-J, Trujillo ME, Suzuki K-I, Ludwing W, Whitman WB, editors. Bergey’s manual of systematic bacteriology. 2. New York: Springer; 2012. pp. 235–43. [Google Scholar]
- 26.Oren A, Garrity GM. Validation List No. 164. Listo f new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2015;65:2017–25. doi: 10.1099/ijs.0.000317. [DOI] [PubMed] [Google Scholar]
- 27.Lehmann KB, Neumann R. Lehmann’s Medizin, Handatlanten. X Atlas und Grundriss der Bakteriologie und Lehrbuch der speziellen bakteriologischen Diagnostik. 4. München: J.F. Lehmann; 1907. [Google Scholar]
- 28.Skerman VBD, McGowan V, Sneath PHA. Approved lists of bacterial names. Int J Sys Bacteriol. 1980;30:225–420. doi: 10.1099/00207713-30-1-225. [DOI] [PubMed] [Google Scholar]
- 29.Lehmann KB, Neumann R. Atlas und Grundriss der Bakteriologie und Lehrbuch der speziellen bakteriologischen Diagnostik. 1. München: J.F. Lehmann; 1986. pp. 1–448. [Google Scholar]
- 30.Eberson F. A bacteriologic study of the diphtheroid organisms with special reference to Hodgkin’s disease. J Infect Dis. 1918;23:1–42. doi: 10.1086/infdis/23.1.1. [DOI] [Google Scholar]
- 31.Moura-Costa LF, Bahia RC, Carminati R, Vale VLC, Paule BJA, Portela RW, Freire SM, Nascimento I, Schaer R, Barreto LMS, Meyer R. Evaluation of the humoral and cellular immune response to different antigens of Corynebacterium pseudotuberculosis in Canindé goats and their potential protection against caseous lymphadenitis. Vet Immunol Immunopathol. 2008;126:131–41. doi: 10.1016/j.vetimm.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 32.Pinto AC, de Sá PHCG, Ramos RTJ, Barbosa S, Barbosa HPM, Carneiro AR, Silva WM, Rocha FS, Santana MP, Castro TLP, Miyoshi A, Schneider MPC, Silva A, Azevedo V. Differential transcriptional profile of Corynebacterium pseudotuberculosis in response to abiotic stress. BMC Genomics. 2014;15:14. doi: 10.1186/1471-2164-15-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spier SJ, Toth B, Edman J, Quave A, Habasha F, Garrick M, Byrne BA. Survival of Corynebacterium pseudotuberculosis biovar equi in soil. Veterinary Record. 2012. doi: 10.1136/vr.100543. [DOI] [PubMed]