

Abstract
Background
Neuropathic pain (NPP) arises from a lesion or dysfunction of the somatosensory nervous system. Recent studies have demonstrated multiple microRNAs (miRNAs) play key roles in NPP development. This study aimed to investigate the effects of miR-128 on microglial cells.
Material/Methods
We established a compressive spinal cord injury (SCI) model and collected the spinal cord segment-derived conditioned medium (CM). We then measured the expression of miR-128 in the murine microglial cell line BV2 treated with CM-SCI or CM obtained from control (CM-NC). Furthermore, lentivirus production of miR-128 and scrambled control were transfected into BV2 cells, which were first treated with CM-SCI or CM-NC. Moreover, the effects of miR-128 on cell viability, M1/M2 microglial gene expression, inflammatory cytokines concentration, and the protein expression of P38 and phosphorylated P38 (P-P38) were investigated.
Results
The expression of miR-128 was downregulated in murine microglial BV2 cells treated with CM-SCI. Overexpression of miR-128 markedly promoted the viability of murine microglial cells. In addition, miR-128 overexpression significantly decreased the expression levels of microglial M1 phenotypic markers CD86 and CD32, and increased the expression levels of M2 phenotypic markers Arg1 and CD206. Furthermore, miR-128 overexpression obviously decreased the concentration of TNF-α, IL-1β, and IL-6. We found that miR-128 overexpression significantly downregulated the expression levels of P38 andP-P38.
Conclusions
Our findings indicate that down-regulation of miR-128 in murine microglial cells may contribute to the development of NPP following SCI via activation of P38. MiR-128 may be a potential intervention target for NPP.
MeSH Keywords: Gene Expression, MicroRNAs, Neuralgia, p38 Mitogen-Activated Protein Kinases
Background
Neuropathic pain (NPP) arises from a lesion or dysfunction of the somatosensory nervous system, adversely impacting quality of life [1,2]. A considerable proportion of patients with NPP have been observed to have an increased risk of social defect symptoms, including anxiety, depression, and suicidal ideation. In a significant number of patients, the treatment of NPP is inadequate and remains a great challenge [4–6]. A better understanding of the molecular mechanisms underlying NPP may lead to the development of more efficient therapeutics for patients.
MicroRNAs (miRNAs), small non-coding RNAs, have attracted considerable attention recently due to their key role in a variety of neurological diseases [7,8]. miRNAs can regulate the expression of multiple genes by binding to 3′-untranslated region (3′-UTR) of target mRNAs [9]. Recent studies have showed that miRNAs function as critical mediators in the regulation of neuroinflammation and NPP [10–12]. Chen et al. showed that intrathecal miR-96 could alleviates NPP via inhibiting the expression of Nav1.3 in rats following chronic construction injury [13]. Favereaux et al. demonstrated that miR-103 was downregulated in neuropathic animals and implied that miR-103 might be a possible therapeutic target because intrathecal applications of miR-103 relieved neuropathic pain successfully [14]. It is also reported that miR-195 may be a potential target for therapy of NPP because increased miR-195 can aggravate NPP via inhibiting autophagy after peripheral nerve injury [15]. Aberrant miRNA expression may play key roles in NPP development; therefore, exploring more miRNAs involved in NPP will provide an avenue for elucidation the molecular mechanism or identification of potential therapeutic targets. However, little is known about the key miRNAs associated with NPP to data.
In the present study we established a compressive spinal cord injury (SCI) model and collected the spinal cord segment-derived conditioned medium (CM). We then measured the expression of miR-128 in the murine microglial cell line BV2 treated with CM-SCI or CM obtained from control (CM-NC). Furthermore, lentivirus production of miR-128 and scrambled control were transfected into BV2 cells treated with CM-SCI or CM-NC to detect the effect of miR-128 overexpression. Our study aimed to investigate the biological functions of miR-128 on microglial cells, as well as to elucidate the regulatory mechanism underlying NPP.
Material and Methods
Animals
All the experiments were approved and reviewed by the Animal Care Committee of the Fourth Military Medical University. Male Sprague-Dawley rats (weighing 220–250 g) were obtained from the Experimental Animal Center of the Fourth Military Medical University and housed at a temperature 22–25°C under a 12/12 h light/dark cycle and had free access to food and water.
Construction of compressive SCI model
The compressive SCI model was established as previously described. Briefly, the rats were anesthetized by intraperitoneal injection of 1% sodium pentobarbital (50 mg/kg). The dorsal midline (about 30–40 mm) of rats was incised and the spinal cord was exposed at the position of the T8 vertebra using a bilateral laminectomy without damage to the dura. The vertebral column was then fixed and a metal rod was placed onto the spinal cord to make the compression injury. The tip of the rod was covered with a plastic plate, which was perpendicular to the longitudinal axis of spinal cord, and attached the dura surface. A metal tube was used to hold the rod and was then moved down vertically at a speed of 0.5 mm/min via connection with the stereotaxic apparatus to produce compression onto the spinal cord. After 5 min, the plastic plate was reached to the bottom of the vertebral canal and the rod was remained here for 1 min, followed by a slow withdraw within 1 min. Over the vertebral column, the wound was closed by suturing muscles and skin. The rats were kept in cages and the urinary bladder was manual evacuated twice daily.
Collection of spinal cord segment-derived CM
The central lesion of each spinal cord was distinguished macroscopically at the Th8–Th9 level. From the rostral (Th2–Th6), central lesion (Th7–Th11), and caudal (Th12–L3) segments to the site of injury, approximately 1.0 cm of samples were taken out and then cut into three 0.3-cm – thick sections per segment. These samples were then seeded on a 12-well culture plate containing DMEM without FBS. After 24-h incubation, CM obtained from SCI (CM-SCI) were collected (rostral, lesion, and caudal segments). The same procedure was performed for obtaining CM from control (CM-NC). Samples were subjected to trypsin digestion, desalted using C18 ziptips (Millipore), and dried under vacuum. Finally, samples were resuspended in water/5% acetonitrile/0.1% formic acid before use.
Cell culture
The murine microglial cell line BV2 was cultured on Poly-L-Ornithine-coated glass coverslips containing Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% fetal bovine serum (FBS) for 24 h and then treated with CM-SCI or CM-NC.
Lentivirus production and transduction
The flanking sequence of miRNA-128 and scrambled control were synthesized from GenePharma (Shanghai, China). To generate miRNA-128 and control lentivirus plasmid, the sequences of miRNA-128 and scrambled control were cloned into pMD18-T vector (Takara, Kyoto, Japan) and then subcloned into lentiviral vector pCDH-CMV-MCS-EF1-copGFP (System Bioscience, USA). The sequences of these recombinant plasmids were confirmed by gene sequencing. To generate lentivirus, the plasmids encoding miRNA-128 or scrambled control were cotransfected into 293T cells combined with the plasmids pHelper 1.0 and pHelper 2.0 (Genechem, Shanghai, China) using Lipofectamine 2000 (Invitrogen, CA) according to the manufacturer’s recommended protocol. To perform lentiviral transduction, the BV2 cells treated with CM-SCI or CM-NC were plated at 40–50% confluence and incubation was continued overnight.
MTT assay
Cells in logarithmic growth phase were adjusted to 5×104/mL and then cultured in a 96-well plate at 37°C in an atmosphere with 5% CO2. After transfection for 24, 48, and 72 h, 20 μL of fresh medium with 0.5 mg/mL MTT was added into each well to incubate for another 4 h before termination. Then, 200 μL dimethyl sulfoxide (DMSO) was added into each well. Optical density (OD) values for each well at 492 nm were measured with an Infinite M200 device (Tecan, Mannedorf, Switzerland). Each sample was tested 3 times.
Enzyme-linked immunosorbent assay (ELISA)
After transfection of 24 h, the media were removed from each culture well for quantitation of tumor-necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-6. Concentrations of these secreted cytokines were then measured using commercially available mouse colorimetric sandwich ELISA plates (R&D Systems, Minneapolis, MN) following the recommend protocols of the manufacturer.
Quantitative real-time PCR (qRT-PCR)
Total RNA were extracted from treated cells using Trizol reagent (Invitrogen) in accordance with the manufacturer’s instructions. Then, 2 μg total RNA was reverse-transcribed to cDNA using reverse transcriptase (iScript™ cDNA Synthesis Kit; Bio-Rad Laboratories). The SYBR green-based qRT-PCR was then performed to detect the relative expression level of miR-128. Expression of U6 and β-actin was used as an endogenous control for miR-128. The primers used in our study were: miR-128, sense: 5′-ACACTCCAGCTGGGTCACAGTGAACCGGTC-3′, anti-sense: 5′-TGGTGTCGTGGAGTCG-3′; U6, sense: 5′-CTCGCT TCGGCAGCACA-3′, anti-sense: 5′-AACGCTTCACGAATTTGCGT-3′. The relative expression of targets was calculated using the method of comparative threshold (Ct) cycle (2−ΔΔCt), as previously described. All the qRT-PCR reactions were performed in triplicate.
Western blot analysis
Cells were lysed using radioimmunoprecipitation assay (RIPA) buffer (Roche, Basel, Switzerland). After protein concentrations were tested with Bradford (Bio-Rad, Madrid, Spain), the same concentration of protein samples was separated with 10–12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto polyvinylidene difluoride (PVDF) membranes. After blocking with PBST (0.1% triton in 19 PBS), the membrane was probed with anti-P38 antibody (Rabbit, SC7149, 1:200, Santa Cruz Co., CA), and anti-P-p38antibody (Rabbit, SC-101759, 1: 400) overnight at 4°C. The membranes were then probed with the secondary antibody, Abexcel (anti-rabbit, Abcam, 1:1000), for 2 h at room temperature. The expression of GAPDH was used as a loading control. The protein bands were developed by enhanced chemiluminescence and analyzed using use of ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Statistical analysis
Statistical analyses were performed with SPSS version 19.0 (SPSS Inc, Chicago, IL). All experimental data are presented as the mean ±SD. The normal distribution assessed with the one-sample K-S test, after which the P-values were calculated using one-way ANOVA or t-test. A statistically significant difference was defined at P<0.05.
Results
The expression of miR-128 was downregulated by CM-SCI
To determine whether miR-128 plays a role in NNP, BV2 cells were treated with CM-SCI or CM-NC, and then the mRNA level expression of miR-128 was monitored by RT-PCR analysis. As shown in Figure 1, the expression of miR-128 in BV2 cells treated with CM-SCI was significantly lower than that in BV2 cells or BV2 cells treated with CM-NC (P<0.05).
Figure 1.
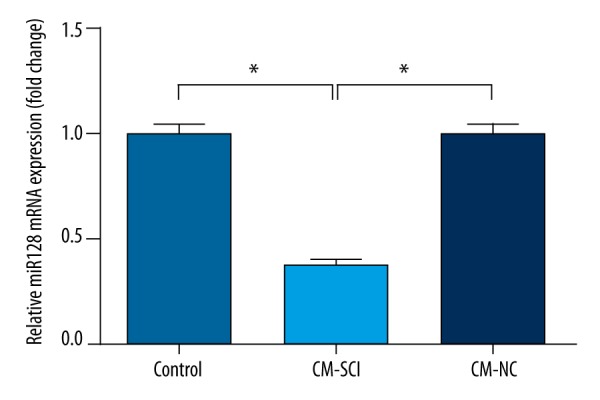
RT-PCR analysis showed the expression of miR-128 in BV2 cells treated with CM-SCI or CM-NC. The results are presented as the mean ±SD. * P<0.05.
Next, to further confirm the relationship between CM-SCI treatment and the expression of miR-128, BV2 cells were first treated with CM-SCI or CM-NC, and transfected with miRNA-128 or control vector, then the expression level of miR-128 was measured by RT-PCR analysis again (Figure 2). CM-NC The results showed that the expression of miR-128 was significantly downregulated by CM-SCI, and transfection with miR-128 vector can weaken its down-regulative effect (P<0.05). In addition, the expression of miR-128 in the CM-NC+vector group was significantly higher than that in the CM-SCI+vector group, and transfection with miR-128 significantly enhanced this up-regulative effect (P<0.05). These results indicate that miR-128 was successfully overexpressed.
Figure 2.
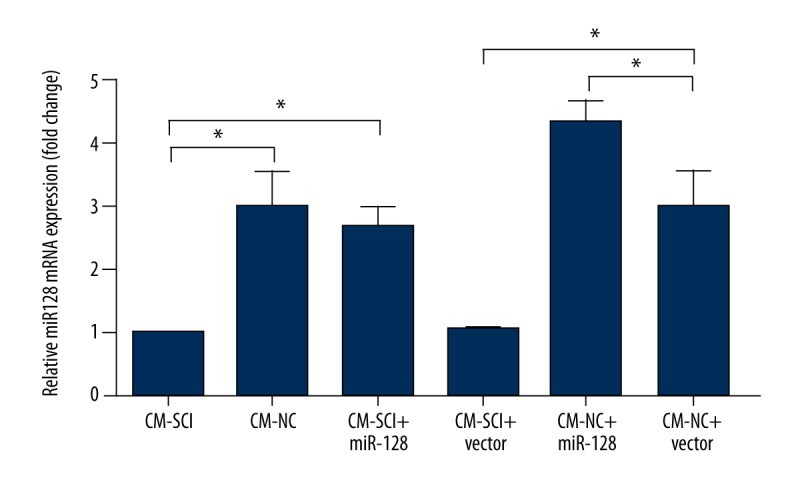
RT-PCR analysis showed the expression of miR-128 in BV2 cells first transfected with miRNA-128 or control vector, and then treated with CM-SCI or CM-NC. The results are presented as the mean ±SD. * P<0.05.
Overexpression of miR-128 promoted cell viability
Cell viability was also evaluated by MTT assay after miR-128 overexpression. As shown in Figure 3, we found that the cell viability was significantly increased by miR-128 overexpression (P<0.05) in a time-dependent manner. These data indicate that overexpression of miR-128 can significantly promote cell viability.
Figure 3.
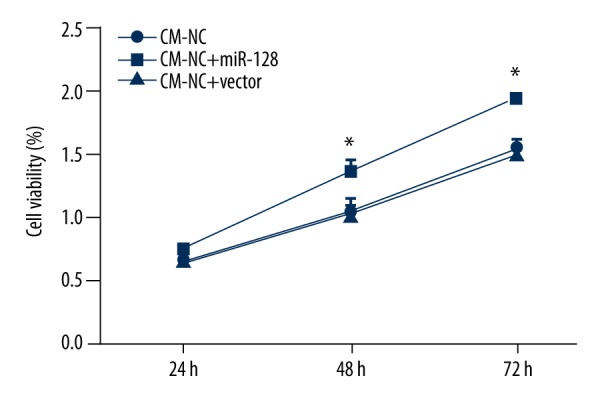
MTT assay showed the cell viability of BV2 cells after transduction of miRNA-128 or control vector in an experimental period of 72 h. The results are presented as the mean ±SD. * P<0.05.
Overexpression of miR-128 altered M1/M2 microglial gene expression
To detect whether miR-128 overexpression affected microglial M1 and M2 phenotypes, the expression levels of microglial phenotypic markers CD86, CD32, Arg1, and CD206 were determined. Figure 4 shows that the expression levels of microglial M1 phenotypic markers, including CD86 and CD32, were significantly decreased in the CM-SCI+miR-128 group in comparison with that in the CM-SCI group or the CM-SCI+vector group (P<0.05), while the expression levels of microglial M2 phenotypic markers, such as CD86 and CD32, were markedly increased (P<0.05).
Figure 4.
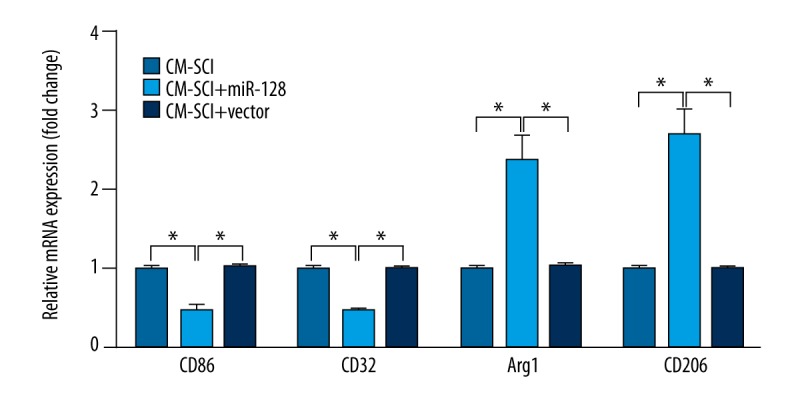
RT-PCR analysis showed the expressions of microglial phenotypic markers CD86, CD32, Arg1, and CD206 after transduction of miRNA-128 or control vector. The results are presented as the mean ±SD. * P<0.05.
Overexpression of miR-128 decreased the concentration of TNF-α, IL-1β and IL-6
We further detected the concentration of inflammatory cytokines TNF-α, IL-1β and IL-6 to verify whether miR-128 function is a critical mediator in the regulation of neuroinflammation. The results of ELISA in Figure 5 show that the concentrations of TNF-α, IL-1β, and IL-6 were all significantly decreased after miR-128 overexpression (P<0.05).
Figure 5.
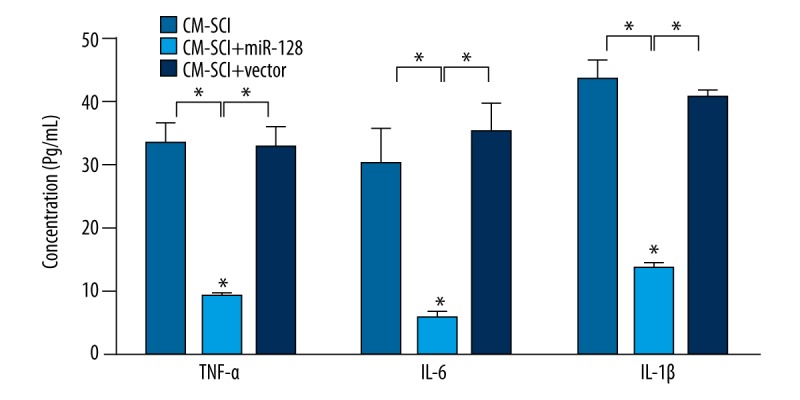
ELISA showed the concentration of TNF-α, IL-1β and IL-6 after transduction of miRNA-128 or control vector. The results are presented as mean ±SD. * P<0.05.
Overexpression of miR-128 downregulated the expression of P38 and phosphorylated P38 (P-P38)
To further investigate the possible mechanism of miR-128 in the development of NPP, the expression levels of P38 and P-P38 were explored (Figure 6). The results showed that the expression levels of P38 and P-P38 proteins were significantly decreased in the CM-SCI+miR-128 group compared with the CM-SCI group or the CM-SCI+vector group (P<0.05)
Figure 6.

Western bolt analysis of the expression levels of P38 and P-P38 after transduction of miRNA-128 or control vector. The results are presented as mean ±SD. GAPDH was used as internal control. * P<0.05.
Discussion
MiR-128 is a neuronal enriched miRNA, whose expression is correlated with central nervous system development [16,17]. MiR-128 is also reported to be dysregulated in neurodegenerative disease [18]. The present study is the first to show the crucial roles of miR-128 in the regulation of NPP following SCI. We found that miR-128 was downregulated in murine microglial cells following SCI. In addition, overexpression of miR-128 promoted the viability of murine microglial cells, facilitated the activation of microglial M2 phenotype, and decreased the concentration of TNF-α, IL-1β and IL-6. We found that miR-128 overexpression significantly downregulated the expression levels of P38 and P-P38, suggesting the regulatory roles of P38 activation in miR-128-mediated NPP development. Collectively, these findings suggest that miR-128 may be a key regulator of NPP development.
Emerging evidence has confirmed the critical role of microglia in the pathogenesis of NPP [19]. It has been confirmed that there are 2 functional subtypes of macrophages/microglia in the injured spinal cord: pro-inflammatory (M1 phenotype) and anti-inflammatory (M2 phenotype) macrophages/microglia. M2 activation is transient in the injured spinal cord following SCI [20], suggesting that M2 activation facilitates tissue repair via inflammatory responses following SCI. Jetten et al. demonstrated that M2 macrophages have higher angiogenic potential than M1 macrophages [21]. In our study, miR-128 was downregulated in murine microglial cells following SCI, and overexpression of miR-128 promoted the viability of murine microglial cells and increased the expression levels of M2 phenotypic markers Arg1 and CD206. Although the roles of miR-128 in microglia are unclear, we speculate that down-regulation of miR-128 may promote NPP development following SCI via suppressing the activation of microglial M2 phenotype. In addition, we found that miR-128 overexpression significantly decreased the concentration of inflammatory cytokines such as TNF-α, IL-1β, and IL-6. Inflammatory cytokines are key mediators of NPP. TNF-α is an NPP-related cytokine and plays a role in the peripheral mediation of NPP [22]. Silencing of TNF-α in dorsal root ganglia can reduce neuronal cell death and relieve the pathogenesis of NPP [23]. Intraneural injection of TNF-α and IL-1β into rat sciatic nerves can effectively induce signs of NPP [24]. The increased IL-6 in dorsal root ganglial neurons and spinal cord is also shown to result in the development of NPP following motor fiber injury [25]. Previous studies have demonstrated the strong correlation between NPP and neuroinflammation [26,27]. Taken together, these results suggest that miR-128 may be involved in NPP development via regulating the expression of inflammatory cytokines.
Furthermore, activation of p38 mitogen-activated protein kinase (MAPK) in spinal microglia has been shown to be the key mechanism involved in the pathogenesis of NPP following spinal cord injury [28,29]. It has been reported that brain-derived neurotrophic factor (BDNF) secreted from spinal microglia leads to the development of NPP, possibly via activation of the p38-MAPK system [30,31]. In addition, TNF-α can trigger mechanical allodynia after spinal nerve ligation (SNL) through activation of p38-MAPK [32]. Activation of p38 in microglia can contribute to the increased concentration of pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6 [33]. To further explore the potential regulatory mechanism of miR-128, we investigated the expression of P38 and P-P38. We found that miR-128 overexpression significantly downregulated the expression levels of P38 and P-P38. In addition to the results that miR-128 overexpression significantly decreased the concentration of inflammatory cytokines such as TNF-α, IL-1β, and IL-6, it is intriguing to speculate that activation of P38 in murine microglial cells may be the key mechanism mediating the roles of miR-128 in the development of NPP following SCI.
Conclusions
Our findings indicate that down-regulation of miR-128 in murine microglial cells may contribute to the development of NPP following SCI via activation of P38. MiR-128 may be a potential intervention target for NPP. However, the precise role of miR-128 in the regulation of NPP needs to be verified.
Footnotes
Disclosure of conflicts of interest
There are no conflicts of interest.
Source of support: Departmental sources
References
- 1.Baron R, Binder A, Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010;9:807–19. doi: 10.1016/S1474-4422(10)70143-5. [DOI] [PubMed] [Google Scholar]
- 2.Costigan M, Scholz J, Woolf CJ. Neuropathic pain: A maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X, Feng SW, Wang F, Xu S. Modeled behavior of neuropathic pain with social defect in rats: A preliminary methodology evaluation. Med Sci Monit Basic Res. 2014;20:164–69. doi: 10.12659/MSMBR.892615. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4.Dworkin RH, O’connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: Evidence-based recommendations. Pain. 2007;132:237–51. doi: 10.1016/j.pain.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 5.Baron R. Mechanisms of disease: Neuropathic pain – a clinical perspective. Nat Clin Pract Neurol. 2006;2:95–106. doi: 10.1038/ncpneuro0113. [DOI] [PubMed] [Google Scholar]
- 6.Croft P, Blyth FM, van der Windt D. Chronic pain epidemiology: From aetiology to public health. Oxford University Press; 2010. [Google Scholar]
- 7.Wang W, Kwon EJ, Tsai L-H. MicroRNAs in learning, memory, and neurological diseases. Learn Mem. 2012;19:359–68. doi: 10.1101/lm.026492.112. [DOI] [PubMed] [Google Scholar]
- 8.Rao P, Benito E, Fischer A. MicroRNAs as biomarkers for CNS disease. Front Mol Neurosci. 2013;6:39. doi: 10.3389/fnmol.2013.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 10.Von Schack D, Agostino MJ, Murray BS, et al. Dynamic changes in the microRNA expression profile reveal multiple regulatory mechanisms in the spinal nerve ligation model of neuropathic pain. PLoS One. 2011;6:e17670. doi: 10.1371/journal.pone.0017670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andersen HH, Duroux M, Gazerani P. MicroRNAs as modulators and biomarkers of inflammatory and neuropathic pain conditions. Neurobiol Dis. 2014;71:159–68. doi: 10.1016/j.nbd.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Imai S, Saeki M, Yanase M, et al. Change in microRNAs associated with neuronal adaptive responses in the nucleus accumbens under neuropathic pain. J Neurosci. 2011;31:15294–99. doi: 10.1523/JNEUROSCI.0921-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen H-P, Zhou W, Kang L-M, et al. Intrathecal miR-96 inhibits Nav1. 3 expression and alleviates neuropathic pain in rat following chronic construction injury. Neurochem Res. 2014;39:76–83. doi: 10.1007/s11064-013-1192-z. [DOI] [PubMed] [Google Scholar]
- 14.Alexandre F, Olivier T, Rabia BB, et al. Bidirectional integrative regulation of Cav1.2 calcium channel by microRNA miR-103: Role in pain. EMBO J. 2011;30:3830–41. doi: 10.1038/emboj.2011.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi G, Shi J, Liu K, et al. Increased miR-195 aggravates neuropathic pain by inhibiting autophagy following peripheral nerve injury. Glia. 2013;61:504–12. doi: 10.1002/glia.22451. [DOI] [PubMed] [Google Scholar]
- 16.Krichevsky AM, King KS, Donahue CP, et al. A microRNA array reveals extensive regulation of microRNAs during brain development. RNA. 2003;9:1274–81. doi: 10.1261/rna.5980303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krichevsky AM, Sonntag KC, Isacson O, Kosik KS. Specific microRNAs modulate embryonic stem cell-derived neurogenesis. Stem Cells. 2006;24:857–64. doi: 10.1634/stemcells.2005-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saba R, Goodman CD, Huzarewich RL, et al. A miRNA signature of prion induced neurodegeneration. PLoS One. 2008;3:e3652. doi: 10.1371/journal.pone.0003652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trang T, Beggs S, Salter MW. Purinoceptors in microglia and neuropathic pain. Pflügers Archiv. 2006;452:645–52. doi: 10.1007/s00424-006-0074-5. [DOI] [PubMed] [Google Scholar]
- 20.Kigerl KA, Gensel JC, Ankeny DP, et al. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–44. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jetten N, Verbruggen S, Gijbels MJ, et al. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis. 2014;17:109–18. doi: 10.1007/s10456-013-9381-6. [DOI] [PubMed] [Google Scholar]
- 22.Leung L, Cahill CM. Review TNF-α and neuropathic pain – a review. J Neuroinflammation. 2010;7:27. doi: 10.1186/1742-2094-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ogawa N, Terashima T, Kawai H, et al. Gene therapy for neuropathic pain by silencing of TNF-α expression with lentiviral vectors targeting the dorsal root ganglion in mice.(S10. 004) Neurology. 2015;84 doi: 10.1371/journal.pone.0092073. S10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zelenka M, Schäfers M, Sommer C. Intraneural injection of interleukin-1β and tumor necrosis factor-alpha into rat sciatic nerve at physiological doses induces signs of neuropathic pain. Pain. 2005;116:257–63. doi: 10.1016/j.pain.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 25.Wei X-H, Na X-D, Liao G-J, et al. The up-regulation of IL-6 in DRG and spinal dorsal horn contributes to neuropathic pain following L5 ventral root transection. Exp Neurol. 2013;241:159–68. doi: 10.1016/j.expneurol.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 26.Kiguchi N, Kobayashi Y, Kishioka S. Chemokines and cytokines in neuroinflammation leading to neuropathic pain. Curr Opin Pharmacol. 2012;12:55–61. doi: 10.1016/j.coph.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 27.Clark AK, Old EA, Malcangio M. Neuropathic pain and cytokines: Current perspectives. J Pain Res. 2013;6:803–14. doi: 10.2147/JPR.S53660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crown ED, Ye Z, Johnson KM, et al. Increases in the activated forms of ERK 1/2, p38 MAPK, and CREB are correlated with the expression of at-level mechanical allodynia following spinal cord injury. Exp Neurol. 2006;199:397–407. doi: 10.1016/j.expneurol.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 29.Crown ED, Gwak YS, Ye Z, et al. Activation of p38 MAP kinase is involved in central neuropathic pain following spinal cord injury. Exp Neurol. 2008;213:257–67. doi: 10.1016/j.expneurol.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ulmann L, Hatcher JP, Hughes JP, et al. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci. 2008;28:11263–68. doi: 10.1523/JNEUROSCI.2308-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gong QJ, Li YY, Xin WJ, et al. ATP induces long-term potentiation of C-fiber-evoked field potentials in spinal dorsal horn: The roles of P2X4 receptors and p38 MAPK in microglia. Glia. 2009;57:583–91. doi: 10.1002/glia.20786. [DOI] [PubMed] [Google Scholar]
- 32.Schäfers M, Svensson CI, Sommer C, Sorkin LS. Tumor necrosis factor-α induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci. 2003;23:2517–21. doi: 10.1523/JNEUROSCI.23-07-02517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taves S, Berta T, Liu D-L, et al. Spinal inhibition of p38 MAP kinase reduces inflammatory and neuropathic pain in male but not female mice: Sex-dependent microglial signaling in the spinal cord. Brain Behav Immun. 2016;55:70–81. doi: 10.1016/j.bbi.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]