

Abstract
The Piwi-piRNA pathway is important for germ cell maintenance, genome integrity, DNA methylation and retrotransposon control and thus may be involved in cancer development. In the present study, we comprehensively analyzed prognostic roles of 3,116 common SNPs in PIWI-piRNA pathway genes in melanoma disease-specific survival. A published genome-wide association study (GWAS) by The University of Texas M.D. Anderson Cancer Center was used to identify associated SNPs, which were later validated by another GWAS from the Harvard Nurses’ Health Study and Health Professionals Follow-up Study. After multiple testing correction, we found that there were 27 common SNPs in two genes (PIWIL4 and DCP1A) with false discovery rate < 0.2 in the discovery dataset. Three tagSNPs (i.e., rs7933369 and rs508485 in PIWIL4; rs11551405 in DCP1A) were replicated. The rs11551405 A allele, located at the 3’ UTR microRNA binding site of DCP1A, was associated with an increased risk of melanoma disease-specific death in both discovery dataset [adjusted Hazards ratio (HR) = 1.66, 95% confidence interval (CI) = 1.21–2.27, P =1.50×10−3] and validation dataset (HR = 1.55, 95% CI = 1.03–2.34, P = 0.038), compared with the C allele, and their meta-analysis showed an HR of 1.62 (95% CI,1.26–2.08, P =1.55×10−4). Using RNA-seq data from the 1000 Genomes Project, we found that DCP1A mRNA expression levels increased significantly with the A allele number of rs11551405. Additional large, prospective studies are needed to validate these findings.
Keywords: cutaneous melanoma, PIWI-piRNA pathway, disease specific survival, single nucleotide polymorphisms, Cox regression
INTRODUCTION
In recent years, it has become increasingly apparent that non-coding RNAs, especially small regulatory RNAs, play crucial roles in cancer development.1 To date, three major classes of small regulatory RNAs have been identified, including microRNAs (miRNAs), short-interfering RNAs and PIWI-interacting RNAs (piRNA). piRNA is a novel class of small, endogenous, regulatory, non-coding RNA molecules expressed in animal cells.2 piRNAs form RNA-protein complexes through binding to PIWI proteins (a subset of Argonaute family proteins). Consequently, these complexes regulate both epigenetic and post-transcriptional gene silencing of transposons and other genetic elements to maintain genome integrity in germline cells2, and an abnormal PIWI-piRNA pathway increases repeats of retrotransposons, component parts of telomeres.3 Initially, most studies showed that piRNAs mainly existed in germline cells,2 but later, some studies reported a widespread presence of piRNAs in multiple tissues of fruit fly, mouse and rhesus macaque samples,4 and piRNAs were found to accumulate at the onset of meiosis or during spermatogenesis.5 Afterwards, piRNAs were also revealed to influence cell proliferation, viability, cell invasion and trans-well motility.1
In humans, independent studies have accumulated evidence that aberrantly expressed piRNAs may play a role in the biogenesis of different types of cancers, including cancers of the stomach,6 colon,7 lung,7 liver,8 mesothelium,7 breast9 and ovaries10. The peripheral blood levels of piRNAs were also suggested to be valuable biomarkers for detecting circulating gastric cancer cells with a favorable area under curve.11 The functions of PIWI in the germline cells have been extensively investigated; for instance, studies have indicated that altered human PIWI proteins (HIWI and HILI) are aberrantly expressed in a variety of cancers12–15 and involved in cell growth, adhesion,15, 16 apoptosis17 and cancer invasion.13 There are additional clinical reports suggesting a potential use for PIWI expression in evaluating cancer clinical outcome, such as cancers of the pancreas,18 colon,19 esophagus,20 liver,21 and stomach22 as well as gliomas23 and sarcoma.24
Multiple genetic alterations, either germline or somatic, are believed to be involved in cutaneous melanoma (CM) development and progression, and the PIWI-piRNA pathway is suggested to have been involved in tumorigenesis. Thus, we hypothesize that genetic variants (single-nucleotide polymorphisms, SNPs) of the PIWI-piRNA pathway genes are associated with CM survival. To test this hypothesis, we used available genotyping data of PIWI-piRNA pathway genes from a previously published genome-wide association study (GWAS) of CM,25 followed by validation in another GWAS dataset from Harvard University.26 We also tried to provide biological evidence in support for positive associations by performing online gene function prediction27 and gene expression Quantitative Trait Loci (e-QTL) analyses.
MATERIALS AND METHODS
Study populations
MD Anderson discovery dataset
As described previously,25 all patients in the discover GWAS were accrued for a hospital-based case-control study of CM at The University of Texas MD Anderson Cancer Center (MDACC). Characteristic details of the subjects have also been previously described.28, 29 Briefly, all patients with CM stages I/II (primary tumors without evidence of regional or distant metastasis), stage III (locoregional disease, including in transit, satellite, and/or regional lymph node metastasis), and stage IV (distant metastasis) were classified according to the 7th edition of the American Joint Committee on Cancer (AJCC) staging system.30 Follow-up was conducted according to standardized guidelines.31 Stage of the disease and length of the follow-up were determined from the date of diagnosis. Among the 1,804 patients, 943 patients were excluded because of no questionnaire data. Three additional patients were excluded due to loss to the follow-up after diagnosis. Hence, the final analysis included 858 patients who had complete information about both questionnaire and clinical prognostic variables. All individuals provided a written informed consent under an Institutional Review Board-approved protocol.
Harvard validation dataset
The Harvard dataset consisted of two studies: Nurses’ Health Study (NHS) and Health Professionals Follow-up Study (HPFS). Sampling, genotyping and quality control procedures have been described previously.26 In short, eligible cases in both NHS (317 CM cases) and HPFS (177 CM cases) cohorts were participants with histopathologically confirmed invasive melanoma, diagnosed at any time after baseline up to the 2008 follow-up cycle for both cohorts. These 494 cases with survival data were included in final analysis, and all were US non-Hispanic Caucasians.
SNP selection and genotyping
Based on the KEGG (http://www.genome.jp/kegg/) databases for the piRNAs/PIWI pathway, there were 23 core genes (TDRKH, TDRD5, MAEL, XRN1, DCP1A, DDX4, TDRD6, ASZ1, PIWIL2, TDRD7, TDRD1, PIWIL4, DDX6, PIWIL1, RNF17, TDRD9, TNRC6A, TNRC6C, PLD6, TDRD12, PIWIL3, TNRC6B and MOV10L1) that are located on autosomes.
Genotyping and quality control (QC) of MDACC genome-wide scan dataset have been previously described.25 Briefly, genomic DNA extracted from the whole blood was genotyped with the Illumina HumanOmni-Quad_v1_0_B array, and genotypes were called by using the BeadStudio algorithm, at John Hopkins University Center for Inherited Disease Research (CIDR). Genome-wide imputation was also performed using the MACH software based on the 1000 Genome project, phase I V2 CEPH (Utah residents with ancestry from northern and western Europe) or CEU data. The typed or imputed common SNPs (with a minor allele frequency ≥ 0.05, a genotyping successful rate ≥ 95%, and a Hardy-Weinberg equilibrium P value ≥ 0. 001, and from imputation for those SNPs with r2 ≥ 0.8) within these genes were selected. As a result, 3116 SNPs in 23 PIWI-piRNA pathway genes were extracted from the MDACC GWAS dataset and used for the analyses, of which there were only 105 independent SNPs after performing the LD pruning using SECA with the criterion of r2 < 0.1.32 The Hardy-Weinberg equilibrium P value for those discussed SNPs in the present study were detailed in Supporting Information Table S1.
Genotyping in the Harvard dataset was performed using the Illumina HumanHap550 array, HumanHap610 array and Affymetrix 6.0 array.26 Imputation was performed based on genotyped SNPs and haplotype information from phase II HapMap CEU data using the program MACH.33 Only SNPs with imputation quality r2 > 0.95 were included, and a total of 1,579,307 SNPs passed through the filter. Finally, we extracted interested SNPs from Harvard dataset for validation.
Statistical methods
Disease-specific survival (DSS) was the primary endpoint of the present study, which was calculated from the date of diagnosis to the date of death from melanoma or the date of the last follow-up, whichever came first. Using data from the MDACC dataset, associations between SNPs and DSS, presented as hazards ratios (HRs) in an additive model, were obtained by both univariate and multivariate Cox proportional hazards regression models performed with the GenABEL package of R software34 with adjustment for age, sex, Breslow thickness, tumor stage, tumor cell mitotic rate and ulceration of tumor. A false discovery rate (FDR) cut-off of 0.2 was applied to limit the probability of false positive findings arising from multiple comparisons.35 Kaplan-Meier survival curves and log-rank tests were also used to evaluate effects of SNPs on DSS. Using linkage disequilibrium (LD) information from the latest 1000 Genomes Project for CEU populations,36 we selected tagSNPs based on r2>0.8 and LD analysis. Next, the identified tagSNPs were further validated in the Harvard dataset, and pooled HRs and 95% CIs were obtained from the meta-analysis using a conservative random-effects model, and the inter-study heterogeneity was assessed with Cochrane's Q test.
A Fine-Gray37 competing-risks regression model was further used for univariate and multivariate regression analyses, which results in sub-distribution HR from a proportional hazards model. It assesses the SNPs of interest and cumulative incidence of melanoma-specific death, where deaths due to other causes were modeled as a competing event rather than a censoring event as in a Cox model.
Finally, SNP rs11551405, which was significantly associated with risk of melanoma death in both MDACC and Harvard datasets, was predicted to regulate protein translation by affecting microRNA (miRNA) binding sites activity by SNPinfo.27 The e-QTL analyses were also used to test for trends in associations between rs11551405 genotypes and corresponding gene expression levels obtained from RNA-seq data both from all populations and European descendants, which are part of the GEUVADIS RNA sequencing project for the 1000 Genomes Project samples.38 The GEUVADIS RNA sequencing project for the 1000 Genomes Project samples have combined transcriptome and genome sequencing data generated by mRNA and miRNA sequencing on 465 lymphoblastoid cell lines (LCLs) derived from five populations of the 1000 Genomes Project: the CEPH (CEU), Finns (FIN), British (GBR), Toscani (TSI) and Yoruba (YRI). Of these samples, 423 were part of the 1000 Genomes Project Phase 1 dataset39 with low-coverage whole genome and high-coverage exome sequencing data, and the remaining 42 are part of the later phases of the 1000 Genomes Project with Omni 2.5M SNP array data available at time of this study; these genotypes were imputed from the array data using Phase 1 as the reference.40 All other analyses were performed using SAS software (Version 9.3; SAS institute, Cary, NC). All reported P values were two-sided, and P<0.05 was considered statistically significant. The flow chart of this study is illustrated in Supporting Information Fig S1.
RESULTS
Patient characteristics
The final analysis included 858 patients from MDACC and 494 patients from Harvard University (Table 1). All patients with CM were non-Hispanic white, with complete information regarding follow-up and GWAS data. In the MDACC dataset, patients had an age range between 17 and 94 years (with a mean age of 52.4 ±14.4 years) at diagnosis, with more men (496, 57.8%) than women (362, 42.2%). There were more patients with stages I/II melanoma (709, 82.6%) than with stages III/IV melanoma (149, 17.4%). Pathological information was shown in Supporting Information Table S2. The 858 MDACC patients had a median follow-up time (MFT) of 81 months (95% CI = 82.9–88.7; interquartile range: 67.2–103.0), during which 95 (11.07%) died of CM; while the 494 Harvard patients had a mean age 60.1±10.6 years at diagnosis and a relatively longer MFT (179 months, 95% CI = 177–203; interquartile range: 140.0–287.0), during which 57 (11.5%) patients died of CM.
Table 1.
Characteristics of the study populations at the time of analysis
| MDACC | Harvard | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | Patients | Death (%) | MFT | HR (95%CI)* | P* | Patients | Death (%) | MFT | HR (95%CI)* | P* |
| Total | 858 | 95 (11.1) | 81.0 | 494 | 57 (11.5) | 179 | ||||
| Age (years old) | ||||||||||
| ≤50 | 371 | 31 (8.4) | 85.8 | 1.00 | 77 | 3 (3.9) | 340 | 1.00 | ||
| >50 | 487 | 64 (13.1) | 78.1 | 1.69 (1.10 – 2.59) | 0.017 | 417 | 54 (13.0) | 161 | 4.19 (1.31 – 13.46) | 0.016 |
| Sex | ||||||||||
| Male | 496 | 69 (13.9) | 77.8 | 1.00 | 177 | 22 (12.4) | 191 | 1.00 | ||
| Female | 362 | 26 (7.2) | 85.9 | 0.48 (0.31 – 0.76) | 0.002 | 317 | 35 (11.0) | 152 | 1.20 (0.70 –2.04) | 0.508 |
MDACC, MD Anderson Cancer Center, MFT, median follow-up time (months); HR, hazards ratio; 95%CI, 95% confidence interval;
univariate analysis.
Discovery in the MADCC dataset
To assess associations of 3116 SNPs in 23 PIWI-piRNA pathway genes (Supporting Information Table S3) with DSS, we performed both univariate and multivariate Cox hazards regression analyses with adjustment for age, sex, tumor stage, Breslow thickness, ulceration of tumor and tumor cell mitotic rate. Specifically, 257 SNPs were individually significantly associated with DSS at P < 0.05 in an additive genetic model (Supporting Information Fig S2). When FDR was used to control the probability of false positive associations arising from multiple comparisons, 27 SNPs were still considered noteworthy, including 19 SNPs of DCP1A and 8 SNPs of PIWIL4 (Supporting Information Fig S2 and Supporting Information Table S4). The regional association plots for DCP1A and PIWIL4 in the additive genetic model are presented in Supporting Information Fig S3. Next, with r2>0.8 among SNPs in the same gene as the cut-off value, DCP1A rs11551405 C>A and PIWIL4 rs7933369 G>A, rs508485 T>C were chosen as the representative tagSNPs (Supporting Information Fig S4). We found that the rs11551405 A allele showed a strong association with a shorter DSS [A vs C: adjusted HR = 1.66, 95% confident interval (CI) = 1.21–2.27, P = 1.50×10−3 in an additive model]. Besides, rs7933369 A allele carriers exhibited a significantly increased HR of early melanoma-specific death (adjusted HR = 1.97, 95% CI = 1.45–2.67, P = 1.43×10−5), compared with G allele carriers. Furthermore, the rs508485 C allele was associated with a statistically significantly favorable DSS (adjusted HR = 0.57, 95% CI = 0.42–0.77, P = 0.0002), compared with the T allele (Table 2).
Table 2.
Associations between DSS of CM patients and selected SNPs in the PIWI-piRNA pathway in the MDACC dataset.
| Genotype | No. of patients | Death (%) | Univariate analysis | Multivariate analysis* | ||
|---|---|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | |||
| PIWIL4 | ||||||
| rs7933369 | ||||||
| GG | 250 | 22 (8.8) | 1.00 | 1.00 | ||
| AG | 427 | 40 (9.4) | 1.07 (0.64–1.80) | 0.797 | 1.44 (0.83–2.51) | 0.193 |
| AA | 181 | 33 (18.2) | 2.20 (1.28–3.78) | 0.004 | 3.66 (2.04–6.56) | 1.31×10−5 |
| Trend | 1.53 (1.15–2.05) | 0.004 | 1.97 (1.45–2.67) | 1.43×10−5 | ||
| PIWIL4 | ||||||
| rs508485 | ||||||
| TT | 224 | 34 (15.2) | 1.00 | 1.00 | ||
| CT | 430 | 46 (10.7) | 0.68 (0.44–1.07) | 0.092 | 0.59 (0.38–0.93) | 0.024 |
| CC | 204 | 15 (7.4) | 0.46 (0.25–0.84) | 0.012 | 0.32 (0.17–0.60) | 0.0004 |
| Trend | 0.68 (0.51–0.91) | 0.009 | 0.57 (0.42–0.77) | 0.0002 | ||
| DCP1A | ||||||
| rs11551405 | ||||||
| CC | 530 | 47 (8.9) | 1.00 | 1.00 | ||
| AC | 287 | 39 (13.6) | 1.60 (1.05–2.44) | 0.030 | 2.02 (1.30–3.15) | 1.80×10−3 |
| AA | 41 | 9 (22.0) | 2.97 (1.45–6.07) | 0.003 | 2.23 (1.03–4.81) | 0.041 |
| Trend | 1.67 (1.22–2.29) | 0.001 | 1.66 (1.21–2.27) | 1.50×10−3 | ||
SNP, single nucleotide polymorphisms; CM, cutaneous melanoma; HR, hazards ratio; DSS, disease-specific survival; HR, hazards ratio; 95%CI, 95% confidence interval;
Adjusted by age, sex, tumor stage, Breslow thickness, ulceration of tumor, tumor cell mitotic rate in the Cox models, in additive model.
Validation in the Harvard dataset
As shown in Table 2 and Supporting Information Fig S2, the initial Cox regression analyses indicated that three SNPs (DCP1A rs11551405 C>A and PIWIL4 rs7933369 G>A and rs508485 T>C) were important predictors for DSS of CM patients. We further validated the effects on risk of DSS in the Harvard dataset. As shown in Table 3, per-unit increase of the rs11551405 A allele (the trend measure) was associated with an increased risk of melanoma-specific death (HR =1.55, 95% CI = 1.03–2.34, P = 0.038). However, associations with PIWIL4 rs7933369 A and, rs508485 C failed to reach significance (P = 0.767 and 0.806, respectively).
Table 3.
Meta-analysis of three tagSNPs of the PIWI-piRNA pathway for the effects on CM DSS in an additive model
| SNP | Gene | MDACC cohort (Discovery) |
Harvard cohort (Replication) |
Joint analysis | Heterogeneit y test |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HR (95% CI) + | P+ | HR (95% CI) ※ | P※ | HR (95% CI) + | P+ | HR (95% CI) | P* | Phet | |||
| DSS | |||||||||||
| rs11551405 | DCP1A | 1.64 (1.21–2.23) | 0.003 | 1.66 (1.21–2.27) | 1.50×10−3 | 1.55 (1.03–2.34) | 0.038 | 1.62 (1.26–2.08) | 1.55×10−4 | 0.792 | |
| rs7933369 | PIWIL4 | 1.57 (1.18–2.09) | 0.002 | 1.97 (1.45–2.67) | 1.43×10−5 | 0.95 (0.66–1.36) | 0.767 | 1.37 (0.67–2.82) | 0.393 | 0.002 | |
| rs508485 | PIWIL4 | 0.66 (0.50–0.89) | 0.006 | 0.57 (0.42–0.77) | 0.0002 | 1.05 (0.73–1.50) | 0.806 | 0.77 (0.42–1.39) | 0.386 | 0.011 | |
| Competing risk model | |||||||||||
| rs11551405 | DCP1A | 1.63 (1.20–2.21) | 0.002 | 1.61 (1.15–2.26) | 0.006 | 1.53 (1.03–2.25) | 0.033 | 1.58 (1.22–2.03) | 3.47×10−4 | 0.847 | |
| rs7933369 | PIWIL4 | 1.58 (1.17–2.14) | 0.003 | 1.99 (1.45–2.74) | 2.50×10−5 | 0.93 (0.65–1.35) | 0.714 | 1.37 (0.65–2.88) | 0.406 | 0.002 | |
| rs508485 | PIWIL4 | 0.66 (0.49–0.88) | 0.005 | 0.56 (0.41–0.76) | 0.0002 | 1.06 (0.74–1.52) | 0.742 | 0.77 (0.41–1.43) | 0.408 | 0.008 | |
HR, hazards ratio; 95%CI, 95% confidence interval; DSS, disease specific survival.
Adjusted by age, sex, tumor stage, Breslow thickness, ulceration of tumor, tumor cell mitotic rate in the Cox models;
If Phet>0.05, meta-analysis was performedwith fixed effect model; if Phet ≤ 0.05, meta-analysis was performed using random effects model.
Adjusted by age, sex.
Meta-analysis
When we combined the results from the MDACC and the Harvard datasets together, there was no heterogeneity for rs11551405 among the two datasets (Pheterogeneous = 0.792), and this SNP was associated with a significantly increased risk of melanoma-specific death by 1.62 fold (95% CI = 1.26–2.08, P = 1.55×10−4, Supporting Information Fig S5). As shown in Fig. 1 with the Kaplan-Meier curves, patients with an increased number of the DCP1A rs11551405 A allele had a poorer DSS (log-rank P = 0.003 in the MDACC dataset, and log-rank P = 0.059 in the Harvard dataset), compared with those with C allele. The results for other two SNPs remained statistically non-significant.
Figure 1.
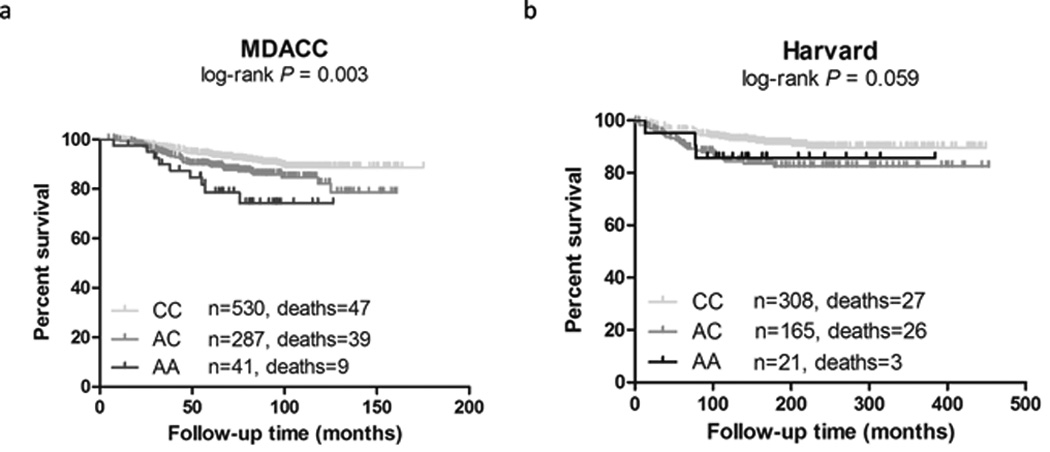
Kaplan-Meier survival curve plots of CM DSS with different rs11551405 genotypes in (a) the MDACC dataset and (b) the Harvard dataset.
Competing-risks regression model
In the Fine and Gray competing-risks regression model, cumulative incidence of an event of interest (i.e., melanoma-specific mortality) was calculated in the presence of competing risks (death from other causes). During the follow-up, 38 and 117 patients died as a result of other causes in MDACC and Harvard datasets, respectively. Table 3 lists univariate and multivariate competing-risks regression models. In multivariate competing-risks regression models, rs11551405 was a statistically significant predictor of melanoma-specific death, after accounting for other-cause mortality in both datasets (with a sub-distribution HR of 1.61 in the MDACC and 1.53 in Harvard study populations).
Expression Quantitative Trait Loci (e-QTL) analyses
We further evaluated correlations between rs11551405 genotypes and mRNA expression levels of DCP1A in normal cells, a possible functional basis for the observed associations, by using the gene expression data of the publically available RNA-seq data both from all populations (457 individuals) and European descendants (370 individuals, from the 1000 Genomes Project), whose genotyping data were available for DCP1A rs11551405. Consistent with the observed associations, the rs11551405 A allele were associated with significantly higher mRNA expression levels of DCP1A in all populations (P = 0.039) and European descendants (P = 0.043) in an additive genetic model (Fig. 2).
Figure 2.
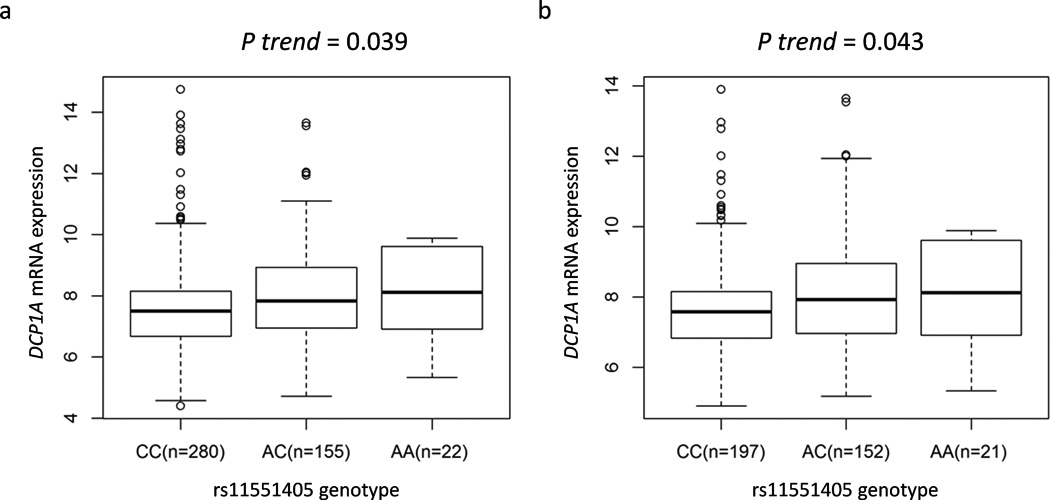
Association between expression quantitative trait loci (eQTL) and DCP1A rs11551405 genotypes. The eQTL analyses were performed in additive models. We used RNA-seq data both from (a) all populations (457 individuals) and (b) European descendants (370 individuals), which are part of the 1000 Genomes Project.
DISCUSSION
To our knowledge, this is the first study to evaluate associations between genetic variants in PIWI-piRNA pathway genes and CM DSS. We found that DCP1A rs11551405 was likely to modulate DSS of CM patients, possibly through a mechanism of modulating gene expression.
Regulation of mRNA translation and degradation is critical in control of cell growth and survival. Processing bodies (P-bodies) are dynamic foci with untranslated mRNA and can serve as sites of mRNA degradation.41 The full name of DCP1A is mRNA-Decapping Enzyme 1a (Dcp1a), and DCP1A is a component of the decapping enzyme and has been shown to reside in P-bodies in mammals.42 In animal experiments, Dcp1a is also suggested to be involved in modulation of, or signal transduction from, P-bodies.43 Meanwhile, the turnover of mRNA is a critical step in the regulation of gene expression, and an important step in mRNA decay is removal of the 5’ cap.43 Following the poly(A) tail shortening, the 5-methylguanosine cap is removed through the action of Dcp1a and Dcp2.42 Subsequent to decapping, Xrn1 degrades mRNA in a 5-3 fashion.44 Meanwhile, Dcp1a and other proteins involved in mRNA degradation or translation repression are key factors in the messenger ribonucleoprotein granule assembly.42 Dcp1a has also been reported to have roles in cellular signaling. Dcp1a contains an N-terminal EVH1 (enabled vasodilator-stimulated protein homology 1) domain, and the EVH1 domain of Dcp1a has a role in transforming growth factor-β signaling through a SMAD4 interaction.45 In addition, Dcp1a is found to induce translational arrest through protein kinase R (PKR) activation that requires the EVH1 Domain.46 Interestingly, in a recent investigation of the status of Dcp1a and P-bodies during stages of the cell cycle, Dcp1a was found to be hyper-phosphorylated during mitosis.42
In the present study, we revealed some significant associations between genetic variants in DCP1A and CM DSS. Specifically, The DCP1A rs11551405 C>A SNP showed a prognostic role in both MDACC and Harvard datasets. This variant was computationally predicted to be located in the binding sites of hsa-miR-1322, hsa-miR-144, hsa-miR-203, hsa-miR-299-3p, hsa-miR-302c, hsa-miR-302d, hsa-miR-338-3p, hsa-miR-372, hsa-miR-373, hsa-miR-520a-3p, hsa-miR-520b, hsa-miR-520c-3p, hsa-miR-520d-3p, hsa-miR-520e, hsa-miR-520f, hsa-miR-522, hsa-miR-573 and hsa-miR-584.27 Among the above-mentioned miRNAs, miR-203 was reported to be associated with melanoma survival.47 Therefore, rs11551405 may regulate protein translation by affecting miRNA-binding site activity.27 Genetic variants in the predicted miRNA binding sites are suggested to be deleterious, and they are likely to be candidates for causal variants of human disease48 or associated with disease survival.49 At the same time, in the published expression data of the 1000 Genomes Project,38 we found that the DCP1A mRNA expression levels changed in a linear manner with an increasing number of the rs11551405A allele in an additive genetic model. We also performed FDR multiple comparison correction tests to assess the possibility of false positive associations with adjustment for some statistically significant and clinically important variables that could confound genetic effects on DSS.
In MDACC and Harvard datasets, none of the top five components showed significant association with melanoma-specific survival, so they were not controlled in the survival analyses. Another caveat regarding the present study is that the adjustment in the Harvard dataset only contained age and sex, which were inconsistent with the MDACC dataset, due to limited information available to us. But in the MDACC dataset, we found that the results of baseline model (adjusted for age and sex) and the multivariate model (adjusted for age, sex, tumor stage, Breslow thickness, ulceration of tumor and tumor cell mitotic rate) were similar, suggesting that our results did not dramatically change in different adjustment models. In addition, the identified SNP just has moderate effect on melanoma prognosis, which limited its application to personal prognostic management. Considering this, if could be validated by other independent studies, this SNP needs to be combined with identified genetic factors and clinical variables for personal prognostic assessment.
In summary, we performed a comprehensive assessment of genetic variants in genes involved in the PIWI-piRNA pathway, and we identified that DCP1A rs11551405 may have a prognostic effect on survival of CM patients. However, our findings need to be validated in other independent, larger patient populations.
Supplementary Material
Novelty and Impact.
The PIWI-piRNA pathway is suggested to be involved in tumorigenesis. In the present study, the authors identified one tagSNP, rs11551405 C>A in the PIWI-piRNA pathway gene DCP1A, was associated with an increased risk of melanoma disease-specific death in both discovery and validation datasets. The discovery offers a translational potential for using genetic variants in the PIWI-piRNA pathway gene DCP1A as biomarkers for developing improved prognostic assessment and personalized management of cutaneous melanoma patients.
Acknowledgments
Financial Support
This work was partially supported by the National Institutes of Health/National Cancer Institute Grants (R01CA100264 and P50CA093459); the Marit Peterson Fund for Melanoma Research; the Start-Up funds from Duke University and support from the Duke Cancer Institute as part of National Institutes of Health/National Cancer Institute Cancer Center Support Grant (P30CA014236); and the Harvard cohort grants (UM1CA186107, P01CA87969, R01CA49449, and UM1CA167552). Additional funding was provided by philanthropic contributions to The University of Texas MD Anderson Cancer Center Moon Shots Program, the Miriam and Jim Mulva Melanoma Research Fund and the Marit Peterson Fund for Melanoma Research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We thank the individuals who participated in this project. We thank the John Hopkins University Center for Inherited Disease Research for conducting high-throughput genotyping for this study. We would like to thank the participants and staff of the Nurses’ Health Study (NHS) and Health Professionals Follow-up Study (HPFS) for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. We also thank Dr. Yuanyuan Lin from the Department of Epidemiology at Indiana University Fairbanks School of Public Health for her help on LD pruning analysis. The authors assume full responsibility for analyses and interpretation of these data.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 2.Siomi MC, Sato K, Pezic D, et al. PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Bio. 2011;12:246–258. doi: 10.1038/nrm3089. [DOI] [PubMed] [Google Scholar]
- 3.Savitsky M, Kwon D, Georgiev P, et al. Telomere elongation is under the control of the RNAi-based mechanism in the Drosophila germline. Gene Dev. 2006;20:345–354. doi: 10.1101/gad.370206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yan Z, Hu HY, Jiang X, et al. Widespread expression of piRNA-like molecules in somatic tissues. Nucleic Acids Res. 2011;39:6596–6607. doi: 10.1093/nar/gkr298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toth KF, Pezic D, Stuwe E, et al. The piRNA Pathway Guards the Germline Genome Against Transposable Elements. Adv Exp Med Biol. 2016;886:51–77. doi: 10.1007/978-94-017-7417-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng J, Deng H, Xiao B, et al. piR-823, a novel non-coding small RNA, demonstrates in vitro and in vivo tumor suppressive activity in human gastric cancer cells. Cancer Lett. 2012;315:12–17. doi: 10.1016/j.canlet.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 7.Cheng J, Guo J-M, Xiao B-X, et al. piRNA, the new non-coding RNA, is aberrantly expressed in human cancer cells. Clin Chim Acta. 2011;412:1621–1625. doi: 10.1016/j.cca.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 8.Xie Y, Yang Y, Ji D, et al. Hiwi downregulation, mediated by shRNA, reduces the proliferation and migration of human hepatocellular carcinoma cells. Mol Med Rep. 2015;11:1455–1461. doi: 10.3892/mmr.2014.2847. [DOI] [PubMed] [Google Scholar]
- 9.Krishnan P, Ghosh S, Graham K, et al. Piwi-interacting RNAs and PIWI genes as novel prognostic markers for breast cancer. Oncotarget. 2016 May 10; doi: 10.18632/oncotarget.9272. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim SL, Ricciardelli C, Oehler MK, et al. Overexpression of piRNA pathway genes in epithelial ovarian cancer. PLoS One. 2014;9:e99687. doi: 10.1371/journal.pone.0099687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui L, Lou Y, Zhang X, et al. Detection of circulating tumor cells in peripheral blood from patients with gastric cancer using piRNAs as markers. Clin Biochem. 2011;44:1050–1057. doi: 10.1016/j.clinbiochem.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 12.Qiao D, Zeeman A-M, Deng W, et al. Molecular characterization of hiwi, a human member of the piwi gene family whose overexpression is correlated to seminomas. Oncogene. 2002;21:3988–3999. doi: 10.1038/sj.onc.1205505. [DOI] [PubMed] [Google Scholar]
- 13.Liu W-K, Jiang X-Y, Zhang Z-X. Expression of PSCA, PIWIL1 and TBX2 and its correlation with HPV16 infection in formalin-fixed, paraffin-embedded cervical squamous cell carcinoma specimens. Arch Virol. 2010;155:657–663. doi: 10.1007/s00705-010-0635-y. [DOI] [PubMed] [Google Scholar]
- 14.Liu X, Sun Y, Guo J, et al. Expression of hiwi gene in human gastric cancer was associated with proliferation of cancer cells. Int J Cancer. 2006;118:1922–1929. doi: 10.1002/ijc.21575. [DOI] [PubMed] [Google Scholar]
- 15.Lee JH, Schütte D, Wulf G, et al. Stem-cell protein Piwil2 is widely expressed in tumors and inhibits apoptosis through activation of Stat3/Bcl-XL pathway. Hum Mol Genet. 2006;15:201–211. doi: 10.1093/hmg/ddi430. [DOI] [PubMed] [Google Scholar]
- 16.Lee JH, Jung C, Javadian-Elyaderani P, et al. Pathways of proliferation and antiapoptosis driven in breast cancer stem cells by stem cell protein piwil2. Can Res. 2010;70:4569–4579. doi: 10.1158/0008-5472.CAN-09-2670. [DOI] [PubMed] [Google Scholar]
- 17.Lu Y, Zhang K, Li C, et al. Piwil2 suppresses p53 by inducing phosphorylation of signal transducer and activator of transcription 3 in tumor cells. PLoS One. 2012;7:e30999. doi: 10.1371/journal.pone.0030999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grochola L, Greither T, Taubert H, et al. The stem cell-associated Hiwi gene in human adenocarcinoma of the pancreas: expression and risk of tumour-related death. Brit J Cancer. 2008;99:1083–1088. doi: 10.1038/sj.bjc.6604653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu C, Qu L, Dong B, et al. Combined phenotype of 4 markers improves prognostic value of patients with colon cancer. Am J Med Sci. 2012;343:295–302. doi: 10.1097/MAJ.0b013e31822cb4cd. [DOI] [PubMed] [Google Scholar]
- 20.He W, Wang Z, Wang Q, et al. Expression of HIWI in human esophageal squamous cell carcinoma is significantly associated with poorer prognosis. BMC cancer. 2009;9:426. doi: 10.1186/1471-2407-9-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao YM, Zhou JM, Wang LR, et al. HIWI is associated with prognosis in patients with hepatocellular carcinoma after curative resection. Cancer. 2012;118:2708–2717. doi: 10.1002/cncr.26524. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Liu Y, Shen X, et al. The PIWI protein acts as a predictive marker for human gastric cancer. Int J Clin Exp Pathol. 2012;5:315–325. [PMC free article] [PubMed] [Google Scholar]
- 23.Sun G, Wang Y, Sun L, et al. Clinical significance of Hiwi gene expression in gliomas. Brain Res. 2011;1373:183–188. doi: 10.1016/j.brainres.2010.11.097. [DOI] [PubMed] [Google Scholar]
- 24.Taubert H, Greither T, Kaushal D, et al. Expression of the stem cell self-renewal gene Hiwi and risk of tumour-related death in patients with soft-tissue sarcoma. Oncogene. 2007;26:1098–1100. doi: 10.1038/sj.onc.1209880. [DOI] [PubMed] [Google Scholar]
- 25.Amos CI, Wang LE, Lee JE, et al. Genome-wide association study identifies novel loci predisposing to cutaneous melanoma. Hum Mol Genet. 2011;20:5012–5023. doi: 10.1093/hmg/ddr415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song F, Qureshi AA, Zhang J, et al. Exonuclease 1 (EXO1) gene variation and melanoma risk. DNA Repair. 2012;11:304–309. doi: 10.1016/j.dnarep.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu Z, Taylor JA. SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res. 2009;37:W600–W605. doi: 10.1093/nar/gkp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, Liu H, Liu Z, et al. Functional Variants in Notch Pathway Genes NCOR2, NCSTN and MAML2 Predict Survival of Patients with Cutaneous Melanoma. Cancer Epidemiol Biomarkers Prev. 2015;24:1101–1110. doi: 10.1158/1055-9965.EPI-14-1380-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin J, Liu H, Liu Z, et al. Genetic variants in Fanconi Anemia Pathway Genes BRCA2 and FANCA Predict Melanoma Survival. J Invest Dermatol. 2015;135:542–550. doi: 10.1038/jid.2014.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gershenwald JE, Ross MI. Sentinel-lymph-node biopsy for cutaneous melanoma. New Engl J Med. 2011;364:1738–1745. doi: 10.1056/NEJMct1002967. [DOI] [PubMed] [Google Scholar]
- 32.Nyholt DR. SECA: SNP effect concordance analysis using genome-wide association summary results. Bioinformatics. 2014;30:2086–2088. doi: 10.1093/bioinformatics/btu171. [DOI] [PubMed] [Google Scholar]
- 33.Biernacka JM, Tang R, Li J, et al. Assessment of genotype imputation methods. BMC Proc. 2009;3(Suppl 7):S5. doi: 10.1186/1753-6561-3-s7-s5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aulchenko YS, Ripke S, Isaacs A, et al. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–1296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- 35.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Statist Soc Ser B. 1995;57:289–300. [Google Scholar]
- 36.Genomes Project C, Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94:496–509. [Google Scholar]
- 38.Montgomery SB, Sammeth M, Gutierrez-Arcelus M, et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abecasis GR, Auton A, Brooks LD, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.AC't Hoen P, Friedländer MR, Almlöf J, et al. Reproducibility of high-throughput mRNA and small RNA sequencing across laboratories. Nat Biotechnol. 2013;31:1015–1022. doi: 10.1038/nbt.2702. [DOI] [PubMed] [Google Scholar]
- 41.Aizer A, Brody Y, Ler LW, et al. The dynamics of mammalian P body transport, assembly, and disassembly in vivo. Mol Biol Cell. 2008;19:4154–4166. doi: 10.1091/mbc.E08-05-0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aizer A, Kafri P, Kalo A, et al. The P body protein Dcp1a is hyper-phosphorylated during mitosis. PLoS One. 2013;8:e49783. doi: 10.1371/journal.pone.0049783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chiang P-Y, Shen Y-F, Su Y-L, et al. Phosphorylation of mRNA decapping protein Dcp1a by the ERK signaling pathway during early differentiation of 3T3-L1 preadipocytes. PloS One. 2013;8:e61697. doi: 10.1371/journal.pone.0061697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arribere JA, Doudna JA, Gilbert WV. Reconsidering movement of eukaryotic mRNAs between polysomes and P bodies. Mol Cell. 2011;44:745–758. doi: 10.1016/j.molcel.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Callebaut I. An EVH1/WH1 domain as a key actor in TGFβ signalling. FEBS Lett. 2002;519:178–180. doi: 10.1016/s0014-5793(02)02751-5. [DOI] [PubMed] [Google Scholar]
- 46.Dougherty JD, Reineke LC, Lloyd RE. mRNA decapping enzyme 1a (Dcp1a)-induced translational arrest through protein kinase R (PKR) activation requires the N-terminal enabled vasodilator-stimulated protein homology 1 (EVH1) domain. J Biol Chem. 2014;289:3936–3949. doi: 10.1074/jbc.M113.518191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noguchi S, Mori T, Hoshino Y, et al. MicroRNAs as tumour suppressors in canine and human melanoma cells and as a prognostic factor in canine melanomas. Vet Comp Oncol. 2013;11:113–123. doi: 10.1002/vco.306. [DOI] [PubMed] [Google Scholar]
- 48.Chen K, Rajewsky N. Natural selection on human microRNA binding sites inferred from SNP data. Nat Genet. 2006;38:1452–1456. doi: 10.1038/ng1910. [DOI] [PubMed] [Google Scholar]
- 49.Christensen BC, Moyer BJ, Avissar M, et al. A let-7 microRNA-binding site polymorphism in the KRAS 3′ UTR is associated with reduced survival in oral cancers. Carcinogenesis. 2009;30:1003–1007. doi: 10.1093/carcin/bgp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.