

Abstract
Schistosomiasis is a neglected tropical disease that affects millions of people worldwide. Thioredoxin glutathione reductase of Schistosoma mansoni (SmTGR) is a validated drug target that plays a crucial role in the redox homeostasis of the parasite. We report the discovery of new chemical scaffolds against S. mansoni using a combi-QSAR approach followed by virtual screening of a commercial database and confirmation of top ranking compounds by in vitro experimental evaluation with automated imaging of schistosomula and adult worms. We constructed 2D and 3D quantitative structure–activity relationship (QSAR) models using a series of oxadiazoles-2-oxides reported in the literature as SmTGR inhibitors and combined the best models in a consensus QSAR model. This model was used for a virtual screening of Hit2Lead set of ChemBridge database and allowed the identification of ten new potential SmTGR inhibitors. Further experimental testing on both shistosomula and adult worms showed that 4-nitro-3,5-bis(1-nitro-1H-pyrazol-4-yl)-1H-pyrazole (LabMol-17) and 3-nitro-4-{[(4-nitro-1,2,5-oxadiazol-3-yl)oxy]methyl}-1,2,5-oxadiazole (LabMol-19), two compounds representing new chemical scaffolds, have high activity in both systems. These compounds will be the subjects for additional testing and, if necessary, modification to serve as new schistosomicidal agents.
Graphical abstract
1. Introduction
Schistosomiasis is one of the major neglected tropical diseases (NTDs) that affects millions of people worldwide.1 Recent estimates suggest that at least 261 million people required preventive treatment for this disease in 2013. This parasitosis is reported in 78 countries located in sub-Saharan Africa, the Middle East, the Caribbean, and South America, resulting in 20,000 to 200,000 deaths annually.2 The disease is caused by flatworms of the genus Schistosoma (S. mansoni, S. japonicum, S. haematobium, S. intercalatum, and S. mekongi).3, 4 In the absence of a vaccine, praziquantel (PZQ) has been the drug of choice recommended by the World Health Organization for the treatment and control of all the major Schistosoma species in mass drug administration programs for almost three decades.5 However, the disseminated and repeated use of this drug in endemic areas, because of the high incidence of reinfection, brings concerns about the development of parasitic resistance.6, 7 This problem is further emphasized by the known lack of efficacy of PZQ against juvenile worms,8 which is a potential cause of treatment failure in endemic areas. For these reasons, the development of new schistosomicidal drugs is urgently required.6–10
The complete genome sequencing of S. mansoni has brought the possibility of exploring a great variety of biological targets in the search for new drugs against this parasite.11, 12 Thioredoxin glutathione reductase of S. mansoni (SmTGR, E.C. 1.8.1.9) plays a crucial role in the redox homeostasis of the parasite.13 SmTGR is a multifunctional enzyme that acts in the detoxification of reactive oxygen species (ROS) present in the blood vessels of the mammalian host, thus allowing parasite survival. While mammalian cells use several enzymes of the glutathione and thioredoxin systems, S. mansoni ROS detoxification relies only on SmTGR. 13, 14 Moreover, it has been validated as a potential drug target as demonstrated in studies silencing SmTGR expression using RNA interference.13 Validation studies have also been performed in S. japonicum, confirming the importance of TGR in parasite survival.15, 16
Advances in computational hardware and software over the last few decades have enabled the development of new strategies for computer-aided/assisted drug design (CADD), which has the advantages of reducing the time and costs in the identification of new drug candidates.17–23 Quantitative structure-activity relationship (QSAR) has been widely-used as a lead optimization tools as well as for pharmacokinetics property optimization and in virtual screening campaigns.24, 25 Our group has been working on developing and applying many CADD strategies aiming at discovering new drug candidates for neglected tropical diseases. In this context, different QSAR methods have been applied for identification of new hits for these diseases.26–28 Another important technological advance impacting drug discovery was the introduction of automated microscopes along with powerful image analysis software enabling high-throughput phenotypic assays of cells and small organisms, a technique known as high-content screening (HCS), imaging (HCI) or analysis (HCA).29 Whole-organism antihelminthic screens employing the HCS approach have already proved useful for the larval stage of S. mansoni.30
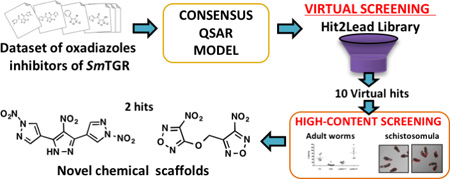
The goal of this study was the identification of novel anti-schistosomal agents, preferably representing new chemical scaffolds. To achieve this goal, we used combi-QSAR approach, uniting 2D and 3D QSAR models, followed by virtual screening of Hit2Lead chemical library and experimental evaluation of the potential SmTGR inhibitors against schistosomula and adult lifecycle stages of S. mansoni. For the latter test, novel medium-throughput assay methodology using 96-well microplate and HCI technology is implemented.
2. Methods
All critical steps of the study are summarized in the workflow presented in Figure 1. Our workflow followed the best practices of QSAR modeling and computer-assisted molecular design.21, 31 The main steps of the workflow are the following: (i) data curation; (ii) models building; (iii) virtual screening; and (iv) experimental validation of suggested/designed hits.
Figure 1.

General workflow of our study.
Dataset
The QSAR studies were performed using a series of 35 oxadiazoles-2-oxides reported in the literature as inhibitors of SmTGR. Theirs in vitro enzymatic potency values (measured by IC50) were obtained by the same experimental protocol.32, 33 The molecules were drawn manually and resulting chemical structures as well as associated activity values were verified following our standard curation protocols.34, 35 Then the IC50 values were converted to negative logarithmic units, pIC50 (−log IC50) with approximate range of three orders of magnitude. The dataset was manually divided into training and external test sets, ensuring a representative coverage across the entire range of pIC50 values. Curated chemical structures and corresponding IC50 and pIC50 values are listed in Table 1.
Table 1.
Chemical structures and corresponding IC50 and pIC50 experimental values of SmTGR inhibitors.
| Cpd | Structure | R | X | IC50 (µM) | pIC50 |
|---|---|---|---|---|---|
| 1 | 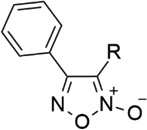 |
CN | - | 6.30 | 5.20 |
| 2 | CH3 | - | 50.1 | 4.30 | |
| 3 | CH2OH | - | 11.2 | 4.95 | |
| 4a | CHO | - | 0.11 | 6.95 | |
| 5 | COOH | - | 0.63 | 6.20 | |
| 6a | CONH2 | - | 17.7 | 4.75 | |
| 7 | 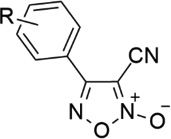 |
3-NO2 | - | 2.23 | 5.65 |
| 8 | 3-CF3 | - | 2.51 | 5.60 | |
| 9 | 3-Br, 4-F | - | 2.81 | 5.55 | |
| 10* | 3-Br | - | 2.81 | 5.55 | |
| 11 | 3-Cl | - | 3.54 | 5.45 | |
| 12 | 4-Br | - | 3.54 | 5.45 | |
| 13* | 4-Cl | - | 4.07 | 5.39 | |
| 14 | 4-CF3 | - | 7.07 | 5.15 | |
| 15 | 3-OH | - | 7.07 | 5.15 | |
| 16* | 4-F | - | 7.94 | 5.10 | |
| 17 | 2-OMe | - | 7.94 | 5.10 | |
| 18 | 3,4,5-OMe | - | 8.91 | 5.05 | |
| 19* | 3-OMe | - | 8.91 | 5.05 | |
| 20 | 4-OMe | - | 10.0 | 5.00 | |
| 21 | 4-Me | - | 11.2 | 4.95 | |
| 22 | 4-Ph | - | 15.8 | 4.80 | |
| 23 | 4-OH | - | 18.1 | 4.74 | |
| 24 | 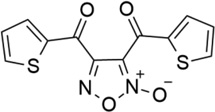 |
- | - | 0.04 | 7.39 |
| 25* | 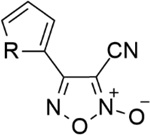 |
O | - | 2.81 | 5.55 |
| 26 | S | - | 3.54 | 5.45 | |
| 27 | 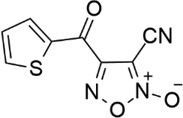 |
- | - | 0.063 | 7.20 |
| 28 | 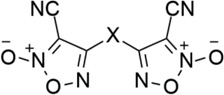 |
- | 1,3-Ph | 3.54 | 5.45 |
| 29 | - | 1,4-Ph | 1.00 | 6.00 | |
| 30 | - | 5-F-1,3-Ph | 0.47 | 6.32 | |
| 31 | - | 2,5-tiophene | 0.40 | 6.39 | |
| 32* | - | 2,4-tiophene | 0.35 | 6.45 | |
| 33 | 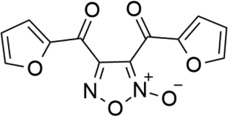 |
- | - | 0.038 | 7.42 |
| 34 | 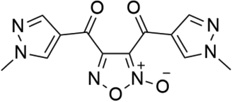 |
- | - | 1.28 | 5.89 |
| 35* | 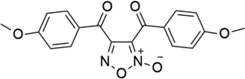 |
- | - | 0.10 | 6.98 |
Cpd=compound
Test set compounds
outliers.
The 3D structures of compounds were generated using OMEGA software v.2.5.1.4.36, 37 OMEGA generates various initial conformations for each compound based on a database of pre-calculated fragments and the structures are optimized by MMFF94 force field. All QSAR models were generated and analyzed using SYBYL-X software v.1.2.38
HQSAR
The HQSAR module available at SYBYL-X software v.1.2 38 was used for generation of HQSAR models. The models were derived using the standard fragment size (4–7 atoms) and various combinations of fragment distinctions. Only the models with q2LOO > 0.5 were chosen for external validation.
HQSAR models were obtained using combinations of different fragment distinctions (A, atoms; B, bonds; C, connectivity; H, hydrogen; DA, donor and acceptors of hydrogen bonds), fragment size (2–5, 3–6, 4–7, 5–8, 6–9, 7–10 atoms), and hologram length (53–401).
Atomic charges assignment
Two different charge assignment methods were used, the empirical method Gasteiger-Hückel,39, 40 available at SYBYL-X v.1.2 platform 38 and the semi-empirical AM1-BCC charges 41, 42 available at QUACPAC software v.1.6.3.143
Molecular alignment
The shape-based alignment and alignment based on a morphological similarity function were evaluated. The former was performed using ROCS software v.3.2.1.4.44, 45 First, several conformers were calculated for each molecule in OMEGA v.2.5.1.4,36, 37 which generated various initial conformations obtained from a database of pre-calculated fragments. Then, the conformers were superimposed, using ROCS, with the most potent SmTGR inhibitor (33). The conformers were classified according to the TanimotoCombo score function.45 The alignment based on morphological similarity function was done in Surflex-Sim, available at SYBYL-X v.1.2.38 Two most potent SmTGR inhibitors, compounds 24 and 33, were chosen for template definition. Remaining compounds were superimposed with this template. The maximal number of poses generated per molecule was 20. The best pose of each compound was chosen based on the calculated similarity to the template.
CoMFA
The aligned training set molecules were placed in a 3D lattice box, with a grid spacing of 0.5, 1.0, 1.5 and 2.0 Å in the x, y, and z directions. CoMFA steric and electrostatic fields were calculated at each grid point with the Tripos force field using a carbon atom probe with sp3 hybridization (Csp3) and charge +1.0. The energy cutoff was set to 30 kcal/mol. The standard deviation coefficients (SDC) were used for region focusing, with values ranging from 0.3 to 1.5.
CoMSIA
CoMSIA models were generated using the same molecular alignments used for CoMFA. The aligned compounds were placed in a 3D lattice box, with a grid spacing of 2.0 Å. In addition to the steric and electrostatic fields, hydrophobic, hydrogen bond donor and acceptor descriptors were included in CoMSIA studies. A probe carbon atom with radius of 1.0 Å and charge +1.0 was used for obtaining the similarity indices. A Gaussian function was used to describe the energetic terms in function of the distance between the probe atom and the aligned molecules with an attenuation factor of 0.3.
QSAR models building and validation
Partial least squares regression (PLS)46–48 was used for development of statistical models for all 3 descriptors sets. The internal validation of the QSAR models was performed using the full cross-validation r2 (q2), leave-one-out (LOO), and leave-many-out (LMO) methods. The latter was used to evaluate the stability of the best models and was performed using five groups and 25 runs. The predictive ability of the models was assessed by Q2ext estimated on external set compounds that were not used for model building or selection. Four consensus models were generated using different combinations of the best individual HQSAR, CoMFA, and CoMSIA models. Three consensus models were obtained by pairwise combinations of individual models (HQSAR+CoMFA, HQSAR+CoMSIA, and CoMFA+CoMSIA) and one model was obtained by combination of three models (HQSAR+CoMFA+CoMSIA). The combination did not occur during models building. The models were built and used separately for predictions. The predicted activity of each compound by a consensus model was the result of the arithmetic mean of individual models predictions. The external validation of these models was done using the same metrics as for individual models.
Virtual screening of new potential Sm TGR inhibitors
The virtual screening of potential SmTGR inhibitors was performed based on similarity and common substructure search in the Hit2Lead chemical library from ChemBridge database.49 First, the most potent inhibitor of the dataset (33) was used as a template for the similarity search. The molecular access system (MACCS) structural key fingerprints50–52 were calculated for compound 33 and molecules from Hit2Lead chemical library. Subsequently, Tanimoto coefficient was calculated between compound 33 and compounds from database. Compounds with Tanimoto coefficient (Tc ≥ 0.6) were selected. Additionally, the substructure search was applied to find compounds containing the common substructure, i.e., the oxadiazole ring. All compounds were prepared using the same protocol and software used in QSAR dataset preparation, i.e., 3D structure and conformer generation, partial atomic charges calculation and molecular alignment. The chosen method of alignment and partial charges calculation was the same of the best individual CoMFA and CoMSIA models. In the next step, the best consensus model was used to predict the biological activity of the potential SmTGR inhibitors. The most promising compounds, with highest predicted pIC50, were selected for biological evaluation. Furthermore, five highly-predictive in-house models, described elsewhere,53, 54 were used to predict some ADME properties of the compounds, such as logP, Caco-2 cell permeability, blood-brain barrier penetration (BBBP), hERG inhibition, CYP3A4 inhibition, and water solubility.
Biological evaluation on S. mansoni
Compounds and Media
Ten selected compounds were purchased from ChemBridge (San Diego-CA, USA) and given the identifiers LabMol-13 to LabMol-22. Compounds were resuspended in 100% DMSO and used immediately in the assays. DMEM and M169 media were purchased from Vitrocell (Campinas, SP, Brazil). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Larval Schistosoma mansoni (schistosomula) in vitro assay
Schistosomula were produced by the mechanical method adapted from both Mansour et al. (2010) 55 and Marxer et al. (2012).56 The cercariae (S. mansoni, BH strain) were vortexed at maximum speed for 5 minutes for tail shedding and cercariae transformation into schistosomula. The schistosomula were resuspended in Medium 169, plated in 384 well plates (120 per well) and maintained in an incubator with 5% CO2 overnight before compound addition. Schistosomula were divided into three groups: negative control (0.625% DMSO), positive control (10 µM of PZQ or Oltipraz (OLT)), and treated (LabMol compounds at a concentration range of 0.3125-20 µM). The effect of the compounds on schistosomula motility and phenotypes was assessed at 48h after compound addition using an automated analysis method described below.
Automated scoring of schistosomula motility and phenotype
The automated image-based method for scoring schistosomula motility and phenotype was performed as described previously.57 Bright-field images were collected using an ImageXpressMicro HCS microscope (IXM; Molecular Devices, Wokingham, UK). For motility analysis 5 × 6 sec interval time-lapse images were collected using a 4x objective. For detailed morphology a 10x objective was used to collect 4 adjacent images fields from within a well, which were considered together to maximize larval numbers for phenotype analysis. Analysis of both the larval phenotype and motility was then carried out in Pipeline Pilot 9 as described by Paveley et al. (2012).57 Phenotype analysis of individual parasites was carried out by a two class Laplacian-modified Bayesian categorization model analysis of 80 image descriptors which constituted shape, size, image intensity, and texture statistics and compared to a training set of data comprising 20,000 parasites. Motility analysis of individual parasites was also done by the average object displacement from the origin point in subsequent 4x image across the time-frame series. Both the Bayesian phenotype and motility scores are subsequently adjusted to the control wells (DMSO treated) on each plate.57
Adult Schistosoma mansoni ex vivo assays
Three- to six-days-old Swiss mice were individually infected percutaneously with 150 ± 10 S. mansoni cercariae (BH strain). The animals were placed into cylindrical vials under incandescent light with a thin water layer containing the cercariae for a period of 30 min. At 42–49 days after infection (i.e., the time required for S. mansoni to reach maturity), the animals were euthanized, and the worms were perfused (with 0.85% sodium chloride and 0.75% sodium citrate solution) from mesenteric and portal hepatic veins. Worms were rinsed and individually transferred into 96 well plates with complete DMEM media (i.e., DMEM plus 10% fetal calf serum, 2mM L-glutamine, 100 µM/ml penicillin, 100 µg/ml streptomycin). Male and female worms were distributed in three groups of six individuals each: negative control (0.02% DMSO), positive control (10 µM PZQ) and treatment (10 µM LabMol compounds). The plates were maintained at 37 °C in a humidified atmosphere of 5% CO2 throughout the entire experiment. The effect of the compounds on adult worm motility was assessed either immediately or 24, 48, or 72h after compound addition using the automated analysis method (see below).
Automated measurement of adult worm mobility
Our strategy was based on sequential pairwise comparison of 100 time-lapse images captured every 250–300 ms using an automated bright-field microscope with a 2x objective lens (ImageXpress Micro XLS, Molecular Devices, CA). Subsequent quantitative image analysis used a custom-developed pipeline for detecting changes in parasite motility using the open-source CellProfiler software version 2.1.2.58 The pipeline along with its validation will be thoroughly described in a subsequent publication and the pipeline itself will be made freely available. Briefly, at each cycle of the pipeline, an image captured at a given instant (tn) is compared with the image captured at the preceding instant (tn-1) and so on until all images are processed. Two different motility measurements were calculated. First, a precursor metric, “AdjustedRandIndex” is calculated by comparing worm objects identified on images captured at times tn and tn-1 with CellProfiler’s Overlap module. This measure ranges from 0 to 1, with 1 meaning two objects are perfectly aligned (no movement). Hence, we created an “Overlap” mobility score, which is directly proportional to the amount of movement, by subtracting 1-”AdjustedRandIndex”. Another motility measure, “DiffWorms”, is the mean pixel intensity of the image calculated from the absolute difference of the parasite images in tn-1 and tn. The higher the DiffWorms score higher is the parasite mobility measured. Both measures are iteratively taken for the 99 image pairs and scores per well are calculated by averaging over all measurements.
Statistical Analysis of biological evaluation
All statistical analysis and graphs were performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, La Jolla, CA, USA, www.graphpad.com).
Ethics statement
Animals were maintained and experiments carried out in accordance with the Institutional Ethics Committee for Laboratory Animal Use at the Oswaldo Cruz Foundation (CEUA/FIOCRUZ, Brazil; license number, LW-78/12) or using the NC3Rs and ARRIVE guidelines under the United Kingdom Animal’s Scientific Procedures Act 1986 with approval from the London School of Hygiene and Tropical Medicine Ethics committee.
Molecular Docking
The structure of SmTGR was obtained from Protein Data Bank59(PDB ID: 2×8H, resolution of 1.9 Å).60 The structure was imported to Maestro v. 10.0 61 and prepared using Protein Preparation Wizard workflow as follows: hydrogen atoms were added according to Epik v. 2.7 62, 63 (pH 7.4 ± 0.5) and minimized using the OPLS-2005 force field.64 Then the structures of compounds to be docked were drawn on MarvinSketch65 and a maximal of 2,000 conformations was generated using OMEGA v.2.5.1.4.36 Subsequently, the conformers had their most favorable ionization state calculated at pH 7.4, using ‘fixpka’ function available on QUACPAC v.1.6.3.1.43 Additionally, AM1-BCC charges were added using QUACPAC v.1.6.3.1.43 Prior to docking studies, two different grids were defined: one for the thioredoxin domain (Trx) and another for glutaredoxin domain (Grx). The Trx grid was built with dimensions of 30.73 Å × 19.18 Å × 19.61 Å (x, y and z) and volume of 11,562 Å3. The Grx grid had dimensions of 19.89 Å × 14.89 Å × 13.61 Å (x, y and z) and volume of 4,029 Å3. Finally, molecular docking of selected compounds was performed in FRED, available in OEDocking suite v. 3.0.166, using the high resolution precision and ChemGauss4 scoring function. In addition, a covalent docking was performed on the package Prime v.3.8.67, 68 Prior to covalent docking, the ligands were prepared on LigPrep69 using OPLS-2015 force field64 and Epik v. 2.7 62, 63 (pH 7.4 ± 0.5).
3. Results and Discussion
QSAR models
Hologram length and fragment size and distinction can affect the quality of HQSAR models.46, 70 In this work, various combinations of these parameters were tested. The final HQSAR models were obtained using combinations of different fragment distinctions (A, atoms; B, bonds; C, connectivity; H, hydrogen; DA, donor and acceptors of hydrogen bonds), fragment size (2–5, 3–6, 4–7, 5–8, 6–9, 7–10 atoms) and hologram length (53–401).
Three best HQSAR models are presented in Table S1 (Supporting Information). They have similar statistical characteristics but model 2 showed a slight superiority when evaluated the external set. The best HQSAR model was obtained using the fragment distinction A/DA (Table 2). The predicted activity (pIC50) for the test set compounds using this model (Table S2, Supporting Information) indicated that only two compounds had their predicted values greater than the standard deviation of residuals, indicating a good predictive capacity of the model. The plot of the experimental versus predicted biological activity of the best HQSAR model is displayed in Supporting Information (Figure S1A).
Table 2.
Statistical characteristics for the best individual QSAR models obtained.
| Model | q2LOO | q2LMO | r2 | N | Q2ext |
|---|---|---|---|---|---|
| HQSAR | 0.61 | 0.57 | 0.85 | 4 | 0.94 |
| CoMFA-model I | 0.71 | 0.66 | 0.99 | 6 | 0.90 |
| CoMFA-model II | 0.72 | 0.67 | 0.99 | 6 | 0.82 |
| CoMSIA-model I | 0.51 | 0.48 | 0.99 | 6 | 0.95 |
| CoMSIA-model II | 0.60 | 0.55 | 0.99 | 6 | 0.96 |
q2LOO, leave-one-out cross-validated determination coefficient; q2LMO, leave-many-out cross-validated determination correlation coefficient; r2, non-cross-validated determination coefficient; N, optimal number of latent variables in PLS analysis; Q2ext, determination coefficient for external set.
Besides predicting the biological activity of untested compounds, HQSAR models also give information regarding the relationships between the structural fragments and the biological activity, which can be visualized through the contribution maps. These maps indicate the individual contribution of each atom or fragment for the activity by color-coded schemes. Colors around red spectrum (orange, red orange, and red) indicate negative contribution, while colors around green (yellow, green blue, and green) indicate positive contribution to biological activity. The contribution maps for the most (33) and the less (2) potent compounds of our data set are presented in Figure 2.
Figure 2.

HQSAR contribution maps for the most (A, 33) and less (B, 2) potent SmTGR inhibitors. The 1,2,5-oxadiazole ring is highlighted in blue, which is the maximal common substructure.
The contribution map for the most potent inhibitor of the dataset (33, Figure 2) suggests that the oxygen atom (O11) from carbonyl group has positive contribution for the biological activity. Moreover, the furan ring is important for the biological activity since the O17 has a positive contribution for activity. The carbon atoms C10, C13, and C14 also have positive contribution to the biological activity. Despite the fragments in green and yellow, the contribution map for the less potent inhibitor (2) suggests that the hydrogen atom attached to the carbon atom C12 negatively contribute to biological activity. The absence of the two carbonyl groups and furan rings in compound 2 suggests that these groups may play a critical role in SmTGR inhibition, because the activity decreased three logarithmic units in comparison to compound 33.
Previous study carried out by Gasco and co-workers 71 indicated that the oxadiazoles are capable of releasing nitric oxide in solution containing thiols by nucleophilic attack in C3 and C4 carbon atoms. Because SmTGR has a selenocysteine (Sec) residue in the C-terminal end, the oxadiazoles may undergo nucleophilic attack mainly by this residue due to its superior reactivity in comparison to cysteine. The resulting nitric oxide release may be the reason for the antiparasitic activity described for these compounds.32 The presence of the carbonyl group in the most potent inhibitors of the dataset suggests an important role of this group, which behaves as a linker that favors the nucleophilic attack in C3 and C4 atoms.
In CoMFA and CoMSIA studies, the calculation of electrostatic descriptors depends on the assignment of partial atomic charges of the compounds. Therefore, the charge assignment method is critical to the success and may affect the quality of the developed models.26 Furthermore, the contour maps may have some differences depending on the method used for partial atomic charges assignment.72, 73 Two different charge assignment methods were tested in this study, the empirical method Gasteiger-Hückel,39, 40 and the semi-empirical AM1-BCC charges.41, 42
Another crucial aspect in 3D-QSAR studies is the structural alignment, which is used to represent the probable bioactive conformation of the compounds. The quality of the 3D-QSAR models can be directly affected by the structural alignment.74 Therefore, two ligand-based alignment hypotheses were tested, the shape-based alignment using ROCS software v.3.2.1.444, 45 and the morphological similarity alignment, implemented in Surflex-Sim, available at SYBYL-X v.1.2 platform.38 The data set was aligned using the two schemes, displayed in Figure 3. In alignment 1, the best conformer of each molecule, after superposition, was classified according to TanimotoCombo score function. TanimotoCombo is a combination of the functions ShapeTanimoto, which compares the molecules according to the best molecular volume superposition, and ColorTanimoto which is related to the appropriate superposition of groups with certain properties like hydrogen bond donors and acceptors, hydrophobic, cations, anions, and rings.45 In alignment 2, no previous conformers were generated since Surflex-Sim has fast techniques to generate poses. Additionally, a morphological similarity function is used to align the molecules. This function is defined as a Gaussian function of the distances of two molecules to observation points of a grid.75
Figure 3.

Molecular alignment used in 3D-QSAR studies. (A) Shape-based alignment performed in ROCS; (B) Surflex-Sim alignment which uses a morphological similarity function.
In 3D-QSAR, similarly to HQSAR, compounds 4 and 6 were identified as outliers, the same identified in HQSAR modeling. The outlier detection was performed using a CoMFA model generated with all compounds of the dataset and default grid spacing of 2.0 Å. The compounds 4 and 6 were identified as outliers, since their calculated residues were near or higher than two times the standard deviation of residues.
CoMFA and CoMSIA models were investigated by PLS analysis, using the full cross-validated r2 (q2) leave-one-out (LOO) method. The leave-many-out (LMO) method was used to evaluate the stability of the best models. To evaluate the 3D-QSAR models predictive power, we used Q2ext. The full results of the best CoMFA and CoMSIA models are displayed in Supporting Information (Tables S3 and S4, respectively).
Two best CoMFA models presented good internal consistence and high external predictivity (Table 2). The models generated using the AM1-BCC charges and ROCS alignment (Models I and II) showed superior performance in comparison with those generated using Gasteiger-Hückel charges and Surflex-Sim alignment (Table 2 and Table S3, Supporting Information).
The best CoMSIA models were obtained using the steric, electrostatic, hydrophobic, and H-bond acceptor fields (Table S4, Supporting Information). The two best CoMSIA models were obtained using ROCS alignment, one using Gasteiger-Hückel charges (Model I, Table 2) and the other using AM1-BCC charges (Model II, Table 2). These two models have similar results for internal validation and external predictivity. The difference between the two best CoMSIA models can be better visualized in the prediction of the activity of test set compounds (Table S2, Supporting Information). Observed versus predicted activities for both training and test sets are displayed in Figure S1B and S1C (Supporting Information).
The best CoMFA and CoMSIA models were used to generate contour maps. These maps indicate regions where certain types of interactions are favorable or unfavorable for biological activity.76 The interpretation of contour maps is useful to guide the design of new potent inhibitors of SmTGR. CoMFA and CoMSIA contour maps were generated using the STDEV*COEFF field type and the function Contour by actual. Figure 4 shows contour maps obtained from (A) CoMFA steric (green/yellow) and (B) electrostatic (red/blue) fields; (C–F) CoMSIA steric (green/yellow), electrostatic (red/blue), hydrophobic (yellow/gray) and hydrogen bond acceptor (purple/magenta) fields with the most potent compound of the data set (33).
Figure 4.

CoMFA steric and electrostatic (A–B) and CoMSIA steric, electrostatic, hydrophobic, and H-bond acceptor (C–F) contour maps surrounding the most potent inhibitor (33). Steric fields: green contours indicate region where bulky groups are favorable for biological activity; electrostatic fields: red contours indicate regions where electronegative groups are favorable to biological activity and blue contours indicate regions where electronegative groups are unfavorable; Hydrophobic fields: yellow contours indicate regions where hydrophobic groups are favorable and gray contours indicate regions where these groups are unfavorable; H-bond acceptor fields: purple contours indicate regions where hydrogen bond acceptors are favorable for biological activity and magenta contours indicate regions where hydrogen bond acceptors are unfavorable.
The steric contour map of the best CoMFA model (Figure 4A) shows green contours surrounding the furan ring of the compound (33), indicating that bulky substituents could be favorable to biological activity. Figure 4B shows red regions near the oxygen atoms O12 and O11 of the carbonyl groups, where substitution for electronegative groups can favor the biological activity. Furthermore, this map shows blue regions surrounding the carbon atoms of the furan rings, indicating that electronegative substituents at the furan rings are unfavorable. This map indicates that the carbonyl group represented by C7 and O12 atoms is important for biological activity. For the CoMFA and CoMSIA models, steric and electrostatic contribution maps were similar. The steric contour map of the best CoMSIA (Figure 4C) model also indicates that bulky groups in the region near the furan ring are favorable to biological activity. The electrostatic map (Figure 4D) indicates that electronegative groups in the region of the oxygen atoms of the carbonyl groups (O12 and O11) are favorable. Furthermore, electropositive groups in the region near the carbon atom C7 are favorable. In addition, the electrostatic CoMSIA map indicates that electronegative groups near the oxygen atom O17 of the furan ring are favorable to biological activity. The hydrophobic contour map (Figure 4E) shows two gray contours near the oxygen atoms of the carbonyl groups (O12 and O11), indicating that hydrophobic groups are unfavorable in this region. Figure 4F shows a purple region near the oxygen atoms of the carbonyl groups (O12 and O11), suggesting that hydrogen bond acceptors groups in this region are favorable, whereas in regions near the two furan rings there are magenta contours, indicating that substitution for hydrogen bond acceptors is unfavorable to biological activity.
Four different consensus models were obtained using different combinations of the best 2D- and 3D-QSAR models (Table 3). The external validation of consensus models was performed using the same test set and metrics used in individual models. Model 4 (Table 3) was selected as the best consensus model because it had good performance and unites the characteristics of the three QSAR approaches explored.
Table 3.
Statistical characteristics for the consensus QSAR models.
| Model | Models | RMSEP | Q2ext |
|---|---|---|---|
| 1 | HQSAR + CoMFA | 0.16 | 0.96 |
| 2 | HQSAR + CoMSIA | 0.12 | 0.98 |
| 3 | CoMFA + CoMSIA | 0.18 | 0.95 |
| 4 | HQSAR + CoMFA + CoMSIA | 0.14 | 0.97 |
The best consensus model is highlighted in bold font. RMSEP, root mean square error of prediction; Q2ext, determination coefficient for external set.
Virtual screening
The best consensus model was used to activity prediction after a virtual screening of new potential SmTGR inhibitors. Firstly, a similarity search on the Hit2Lead library of ChemBridge database49 identified 80 compounds with TC≥0.6 with compound 33. Additionally, 377 compounds containing the oxadiazole ring (common substructure) were identified. Duplicates including compounds already used to derive the QSAR models were excluded. Then, model 4 was used to predict the SmTGR inhibitory activity of remaining 194 compounds. The consensus prediction of the biological activity was calculated using the arithmetic mean of the predictions from individual HQSAR, CoMFA, and CoMSIA models (Table S5, Supporting Information).
The use of similarity search in virtual screening is an example of computational approach for identification of new chemical scaffolds, as described elsewhere.77, 78 In the review by Heikamp & Bajorath78, the use of molecular fingerprints is highlighted as an important way to search novel active compounds. Other examples of approaches for scaffold hopping are shape matching, 3D pharmacophore searching and fragment replacement.77 In a study performed by Gardiner and coworkers, the use of 2D fingerprint similarity has proven to be a simple and efficient way to perform the search of new chemical scaffolds. In their study, they evaluated the effectiveness of six different types of 2D fingerprints for scaffold hopping in three different databases.79 Several successful examples of drug design by scaffold hopping approaches are also described elsewhere.80
Poor pharmacokinetic properties are important causes of costly late-stage failures in drug development.81 Our laboratory has been working to overcome or reduce these failures using in silico tools for early prediction and optimization of ADME properties, such as Caco-2 cell permeability, blood-brain barrier penetration (BBBP), hERG inhibition, CYP3A4 inhibition and water solubility. Five in-house highly-predictive models were developed using large datasets of diverse compounds to cover the chemical space for the prediction of new compounds and are described elsewhere.53, 54 Table 4 shows the structure, consensus predicted potency against SmTGR (IC50 in µM), and some predicted ADME properties of the ten new potential SmTGR inhibitors.
Table 4.
Chemical structures, predicted potency against SmTGR and some calculated ADME properties of the lead compound (33) and the virtual hits.
| Cpd ID | Structure | IC50 preda (µM) |
clogPb | Caco2 | BBB | Water Solubility (logS) |
CYP3A4 | hERG |
|---|---|---|---|---|---|---|---|---|
| 33* | 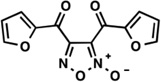 |
0.04 | 0.96 | Moderate -poor |
BBB+ | −1.91 | Non- Inhibitor |
Non- blocker |
| LabMol-13 | 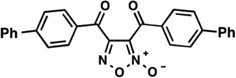 |
0.23 | 5.10 | Moderate -poor |
BBB+ | −2.20 | Non- Inhibitor |
Non- blocker |
| LabMol-14 | 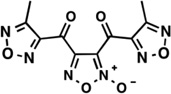 |
0.03 | −0.85 | Moderate -poor |
BBB+ | −1.86 | Non- Inhibitor |
Non- blocker |
| LabMol-15 | 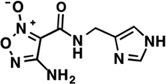 |
1.23 | −1.41 | Moderate -poor |
BBB+ | −2.47 | Non- Inhibitor |
Weak blocker |
| LabMol-16 | 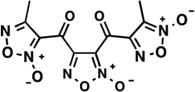 |
0.06 | −2.37 | Moderate -poor |
BBB+ | −2.07 | Non- Inhibitor |
Non- blocker |
| LabMol-17 | 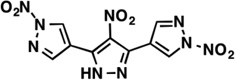 |
2.40 | −0.20 | Moderate -poor |
BBB+ | −1.75 | Non- Inhibitor |
Non- blocker |
| LabMol-18 | 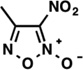 |
1.54 | −0.58 | Moderate -poor |
BBB+ | −2.94 | Non- Inhibitor |
Non- blocker |
| LabMol-19 | 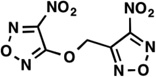 |
0.51 | −0.37 | Moderate -poor |
BBB+ | −2.37 | Non- Inhibitor |
Non- blocker |
| LabMol-20 | 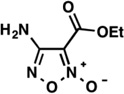 |
1.00 | −0.93 | Moderate -poor |
BBB+ | −1.98 | Non- Inhibitor |
Non- blocker |
| LabMol-21 | 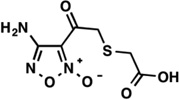 |
1.12 | −1.11 | Moderate -poor |
BBB+ | −2.17 | Non- Inhibitor |
Non- blocker |
| LabMol-22 | 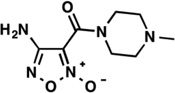 |
0.89 | −1.72 | Moderate -poor |
BBB+ | −2.47 | Non- Inhibitor |
Weak blocker |
IC50 prediction by the best consensus model.
Calculated octanol/water partition coefficient using RDKit 2.4.0.
As we can see from Table 4, ten compounds selected as virtual hits presented high predicted potency against SmTGR, using the consensus QSAR model. Moreover, the selected hits were predicted to present favorable ADME properties and did not show any potential of being hERG blocker or CYP3A4 inhibitors. These compounds were selected for subsequent in vitro biological evaluation against schistosomula and adult S. mansoni worms.
Biological evaluation on S. mansoni
The whole-organism screening (phenotypic screening) is an indispensable method to identify new anti-schistosomal compounds. In our study, despite the prediction of compounds as potential SmTGR inhibitors, it was necessary to evaluate their activity in a phenotypic screening. A promising anti-schistosomal compound, to reach the target, must be able to cross many biological membranes and resist degradation by enzymes of the parasite. Thus, a hit identified in early phenotypic screening has more biological relevance than one identified in an enzyme inhibition assay. Two out of ten tested compounds (LabMol-17 and LabMol-19) were confirmed experimentally as new hits against Schistosoma mansoni.
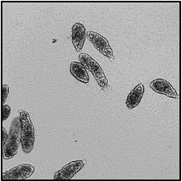
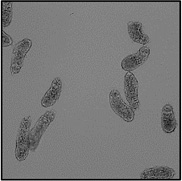
Motility and phenotypic scores of LabMol-17 and LabMol-19 at 10 µM along with the negative (DMSO 0.625%) and positive controls (PZQ or OLT 10 µM) are shown in Table 5. The phenotypic and mobility scores calculated by the Bayesian model for both hits indicate their effects on schistosomula are equivalent or even more pronounced than produced by the reference drugs PZQ and OLT. EC50 values could be calculated from dose-response curves using either motility score (LabMol-19 EC50 1.00 ± 0.11 µM) or phenotype score as the response (LabMol-17 EC50 4.76 ± 1.15 µM). The motility EC50 value determined for LabMol-19 was comparable to PZQ (EC50: 2.2 µM).82
Table 5.
Motility and phenotype adjusted index values for S. mansoni schistosomula exposed for 48h to hit compounds or standard drugs at 10 µM.
| Compound | Motility adjusted index (mean ± SD) |
Phenotype adjusted index (mean ± SD) |
Motility image | Phenotype image |
|---|---|---|---|---|
| Control (DMSO 0.625%) |  |
 |
||
| LabMol-17 | −0.95 ± 0.01 | −0.43 ± 0.11 |  |
 |
| LabMol-19 | −0.95 ± 0.01 | −0.63 ± 0.02 | 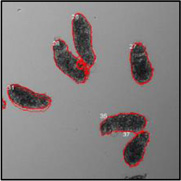 |
 |
| PZQ | −0.48 ± 0.04 | −0.17 ± 0.02 | 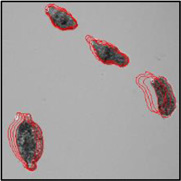 |
 |
| OLT | −0.90 ± 0.04 | −0.34 ± 0.07 |  |
 |
Motility images represent the position of each parasite over 5 time points (11s interval).
Furthermore, the Bayesian model was able to classify the phenotype induced by both hit compounds as OLT-like indicating that the phenotype induced by these compounds was closest to OLT in comparison to the other modeled schistosomacides (PZQ, dihydroartemisinin, methylclonazepam, Ro15-5458, and oxamniquine). This result is consistent with the fact that both LabMol-17 and LabMol-19 are potential inhibitors of SmTGR, an enzyme involved in ROS detoxification in the S. mansoni, similar to the OLT mechanism of action which is also thought to interfere with the parasite’s redox defense system.83
Compounds active on schistosomula were then tested on S. mansoni adult worms in vitro. Chemicals were assayed by a new methodology that utilizes HCS technology to automatically score changes on parasite motility. Besides avoiding human bias, this quantitative method is more robust and sensitive to subtle changes in parasite movement than the standard assay using manual microscopic visualization.84 Figures 5 and 6 shows percent motilities of male and female worms, respectively, measured after exposure to drugs at 10 µM for up to 72h incubation time.
Figure 5.
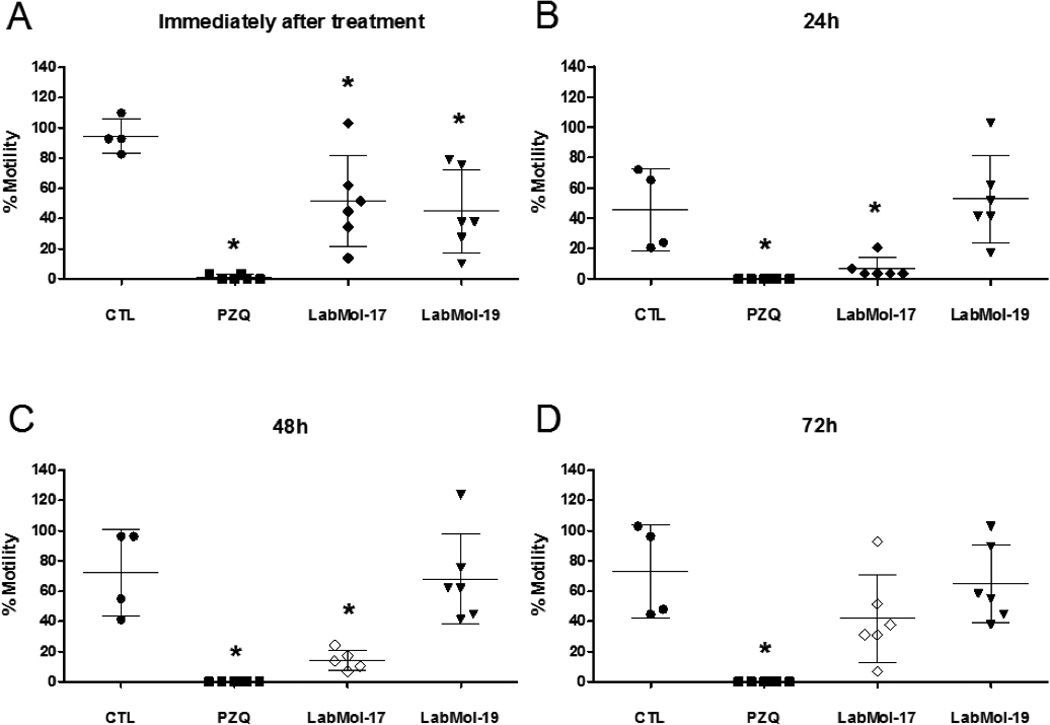
The effect of compounds on male adult worm motility as analyzed by HCS analysis for 0–72 h. PZQ, LabMol-17, and LabMol-19 were screened at 10 µM and DMSO at an equivalent % concentration. The percentage motility values were determined immediately (A); 24 h (B); 48 h (C); or at 72 h (D) by comparison to the average motility of the worms before compound addition. CTL - DMSO 0.02%, PZQ - Praziquantel. Statistical significance (* = p<0.05) was calculated by One-way ANOVA analysis followed by Tuckey´s post-hoc test.
Figure 6.
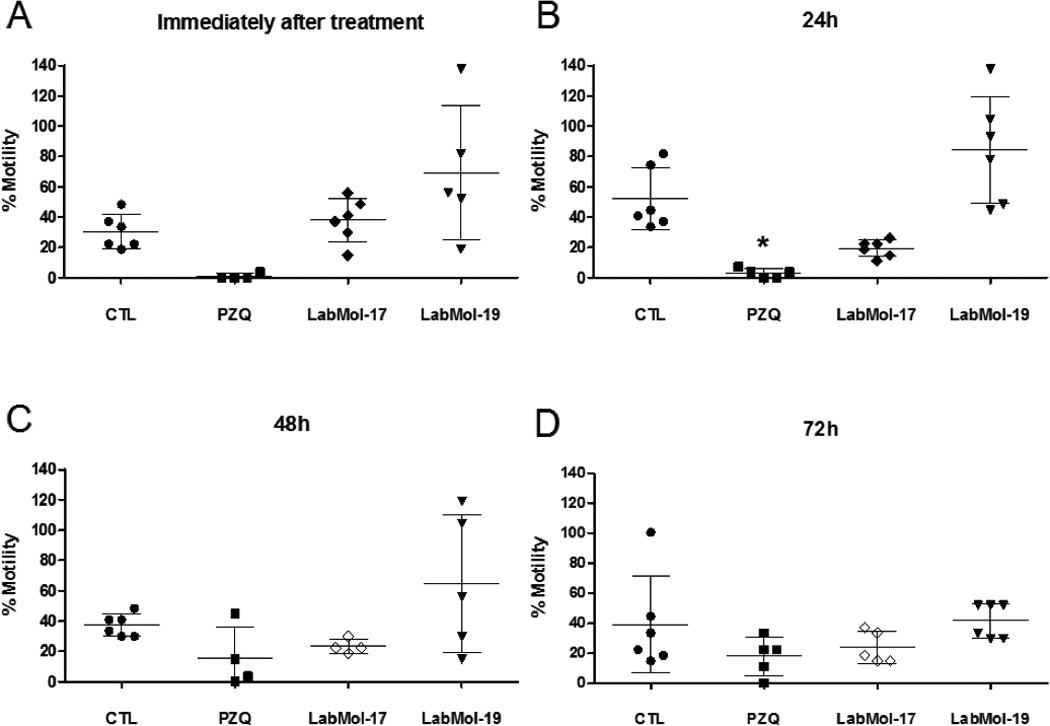
The effect of compound on female adult worm motility as analyzed by HCS analysis for 0 −72 h. PZQ, LabMol-17, and LabMol-19 were screened at 10 µM and DMSO at an equivalent % concentration. The percentage motility values were determined immediately (A); 24 h (B); 48 h (C); or at 72 h (D) by comparison to the average motility of the worms before compound addition. CTL - DMSO 0.02%, PZQ - Praziquantel. Statistical significance (* = p<0.05) was calculated by One-way ANOVA analysis followed by Tuckey´s post-hoc test.
Like PZQ, both LabMol-17 and LabMol-19 were more active on male worms. LabMol-17 was the most active hit, reducing male worm motility up to five times compared to untreated control worms. Significant reduction (p<0.05) of male worm motility was observed immediately after addition of the drugs to the microplate well and in the case of LabMol-17 peaked after 48h incubation. Effects on female worms were less pronounced. A discernible, although not statistically significant, trend could still be observed for LabMol-17 after 24 h and 48 h of drug exposure. Interestingly, although not statistically significant for the whole treatment group, LabMol-19 induced augmented motility in at least half of the female worms in the group on exposure times up to 48h. Further experiments will elucidate if this different behavior on drug sensitivity between male and female worms may be due to different expression patterns of SmTGR or other reasons.
Both experimentally validated hits, LabMol-17 and LabMol-19, are dissimilar from the most potent compound (33) in the training set (Tc of 0.60 and 0.63, respectively, see Table S5, Supporting Information). Moreover, the most active hit, LabMol-17, is very dissimilar from the current schistosomicidal drug, PZQ (Tc = 0.07) and oltipraz (Tc = 0.08), as well as from other known antischistosomal drugs (Figure 7).
Figure 7.
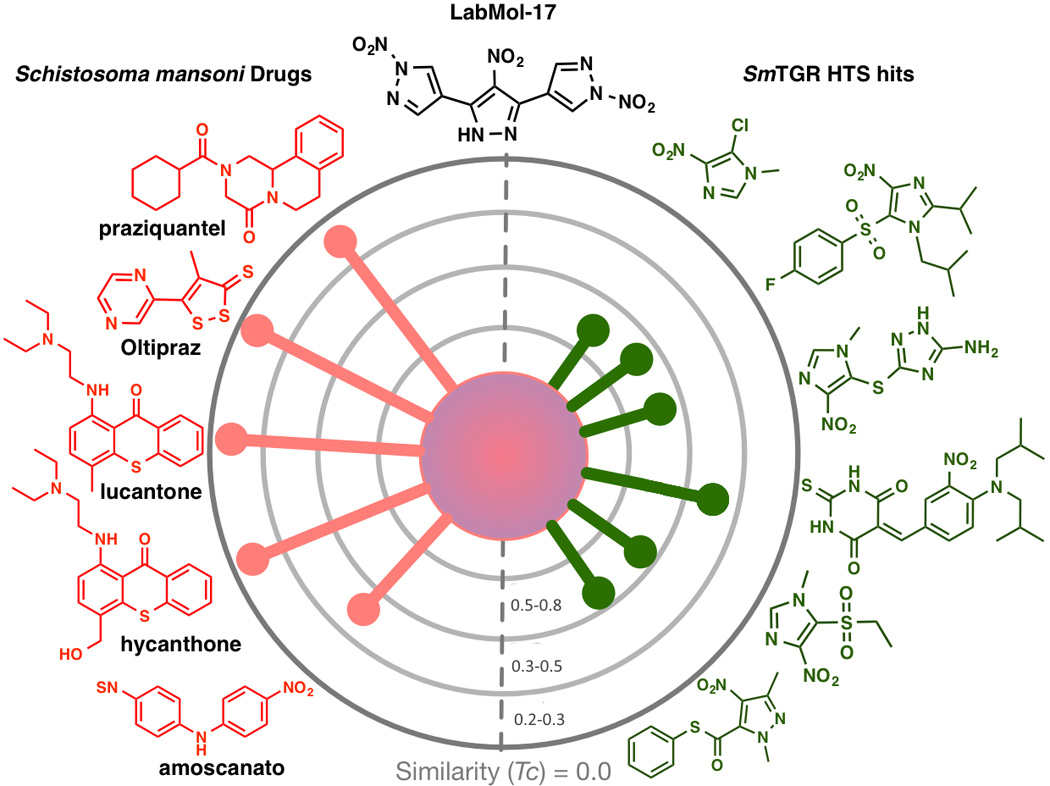
Radial plots for similarity of LabMol-17 with known schistosomicidal drugs and known SmTGR hits. The central node represents the target compound surrounded by known schistosomicidal drugs (left-hand side) and known SmTGR hits from a HTS screen retrieved from PubChem BioAssay (AID: 485364) (right-hand side). The similarity was calculated using Tanimoto coefficient (Tc) using MACCS structural key fingerprints.
Moreover, we have also analyzed the similarity between LabMol-17 and LabMol-19 and known SmTGR hits discovered in a HTS screening (PubChem BioAssay, AID: 485364). As we can see from Figure 7, LabMol-17 represents a new chemical scaffold very dissimilar (Tc = 0.20 – 0.47) from known drugs and active compounds against S. mansoni. The situation with LabMol-19 is pretty much the same (Tc = 0.24 – 0.38). High activity against schistosomula and adult worms of LabMol-17 and LabMol-19 made them promising potential SmTGR inhibitors representing novel not yet explored chemical scaffolds highly dissimilar with known anti-schistosomal drugs.
Molecular docking
The two hits identified in this study were docked in two important catalytic domains of SmTGR: (i) Trx domain where reducing equivalents are transferred from reduced FAD to the Cys154-Cys159 pair and (ii) the Grx domain, which contains the Cys28-Cys31 pair and receives reducing equivalents from the highly mobile C-terminal end. The C-terminal end, which has cysteine and a selenocysteine residues (Cys596-Sec597 pair) was not used for docking for two reasons: (i) The C-terminal end is missing in the structure from PDB (ID: 2X8H); and (ii) The PDB structure containing the C-terminal end (ID: 2X8C) has low quality in terms of resolution (3.1 Å).
Two different docking methods were used: a conventional docking for evaluation of molecular interactions and a covalent docking. Figure 8 shows the interactions, after conventional docking, between the hits and SmTGR. It can be observed that both compounds establish important interactions in both domains of SmTGR. The ChemGauss4 score of LabMol-17 and LabMol-19, in Grx domain (Figure 8A and 8C), was −3.292 and −3.802 kcal/mol, respectively. However, the scores were better in Trx domain, −9.569 and −8.209 kcal/mol, respectively. It suggests a possible higher affinity of these hits to the Trx domain. Furthermore, the binding modes of these ligands, near to cysteine residues, suggest the possibility of nucleophilic attacks from these residues and formation of covalent bonds.
Figure 8.

Interactions between the two hits and SmTGR after conventional docking. The yellow dashed lines show important interactions of LabMol-17 with residues of Grx domain (A) and Trx domain (B). The interactions of LabMol-19 with Grx domain (C) and Trx domain (D) are also highlighted. Both compounds have established hydrogen bonds in both domains and the proximity of cysteines (Cys28 and Cys159) indicate the possibility of ligands to undergo nucleophilic attack by these residues.
The covalent docking was performed because SmTGR has, in both domains, a pair of cysteine residues which have important role in the catalytic process. Cysteine residues are nucleophiles and, as demonstrated in previous studies, some SmTGR inhibitors including the oxadizole-2-oxides, can undergo nucleophilic attack by cysteines and bind covalently in these residues. As result, these compounds release nitric oxide, an anti-parasitic molecule.32 Figure 9 shows the interactions between the two hits and SmTGR after covalent docking. In addition to covalent bond, the hydrogen bonds between the compounds and residues seem to be important to the inhibition mechanism, helping the ligands to be placed in favorable positions to undergo nucleophilic attack and form a covalent bond with cysteine. These results suggest that these compounds have potential to be irreversible inhibitors of SmTGR.
Figure 9.

Interactions between the two hits and SmTGR after covalent docking. The yellow dashed lines show important interactions of LabMol-17 with residues of Grx domain (A) and Trx domain (B). The interactions of LabMol-19 with Grx domain (C) and Trx domain (D) are also highlighted. Both compounds have established hydrogen bonds in both domains and covalent bonds were formed with cysteines (Cys28 and Cys159). These results suggest an irreversible mechanism of inhibition of SmTGR.
One of the future directions of our study is the addition of LabMol-17 and LabMol-19, after their testing in enzymatic assay against SmTGR, into training set of next generation of models. This will provide new structural information and will increase AD of QSAR models. Although the introduction of new scaffolds will not affect HQSAR because of fragment-based nature of the approach, it will strongly influence the superposition of compounds in CoMFA and CoMSIA. This limitation was discussed in the original publication85 and was resolved in the follow-up publication.86 However, when the dataset consists of various scaffolds, we would recommend to use fragment-based approaches like HQSAR70, SiRMS87, etc.
4. Conclusions
We succeed to develop robust and externally predictive consensus model merging 2D (HQSAR) and 3D (CoMFA and CoMSIA) QSAR models for SmTGR inhibition. We applied this model for virtual screening of Hit2Lead library of ChemBridge database and identified ten new potential SmTGR inhibitors. The ten virtual hits were tested against S. mansoni on both schistosomula and adult worms in vitro. Two of them, LabMol-17 and LabMol-19, showed high activity, represent new chemical scaffolds and are highly dissimilar (Tc = 0.20 – 0.47) with known anti-schistosomal drugs. As a future direction, we will perform structural modification of discovered scaffolds (compounds) to improve their properties and develop on their base new schistosomicidal agents.
Supplementary Material
Acknowledgments
The authors would like to thank Brazilian funding agencies, CNPq, CAPES, FAPERJ, and FAPEG for financial support and fellowships. AEC and LK were supported by the National Institutes of Health (R01 GM095672). EM thanks the financial support from NIH (GM 096967 and GM66940), CNPq (grant 400760/2014-2), and UNC for Junior Faculty Development Award. We are grateful for Molecular Devices for providing the HCS equipment for this study as well as OpenEye Scientific Software Inc. and ChemAxon for providing us with academic licenses for their software. Authors also thank the Bioassays and Drug Screening Platform (FIOCRUZ RPT11-I subunit) for technological support and the Malacology Laboratory (Dr. Silvana C. Thiengo) from IOC/FIOCRUZ for providing S. mansoni cercariae.
Footnotes
Associated content
Supporting Information. Statistical characteristics for the three best HQSAR models obtained using different combinations of fragment distinction, fragment size, and hologram length (Table S1); Experimental and predicted potencies (pIC50) for the test set compounds using the best HQSAR, CoMFA, and CoMSIA models (Table S2); Best CoMFA models obtained using different charge-assigning and alignment methods (Table S3); Best CoMSIA models generated using different charge-assigning and alignment methods (Table S4); Chemical structures and predicted biological activities (pIC50 and IC50) using the best consensus QSAR model for the lead compound (33) and the identified hits (Table S5); Experimental versus predicted biological activity of the best HQSAR (A); CoMFA (B); and CoMSIA (C) models (Figure S1). This material is available free of charge via the Internet at http://pubs.acs.org.
Authors Contributions
All authors contributed in the writing of the manuscript. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
REFERENCES
- 1.Colley DG, Bustinduy AL, Secor WE, King CH. Human Schistosomiasis. Lancet. 2014;383:2253–2264. doi: 10.1016/S0140-6736(13)61949-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. [(accessed Jul 14, 2015)];Schistosomiasis. http://www.who.int/mediacentre/factsheets/fs115/en/
- 3.Gryseels B. Schistosomiasis. Infect. Dis. Clin. North Am. 2012;26:383–397. doi: 10.1016/j.idc.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Gryseels B, Polman K, Clerinx J, Kestens L. Human Schistosomiasis. Lancet. 2006;368:1106–1118. doi: 10.1016/S0140-6736(06)69440-3. [DOI] [PubMed] [Google Scholar]
- 5.World Health Organization. Progress Report 2001–2011 and Strategic Plan 2012–2020. Geneva, Switzerland: 2012. [Google Scholar]
- 6.Hagan P, Appleton CC, Coles GC, Kusel JR, Tchuem-Tchuenté L-A. Schistosomiasis Control: Keep Taking the Tablets. Trends Parasitol. 2004;20:92–97. doi: 10.1016/j.pt.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 7.Loukas A, Bethony JM. New Drugs for an Ancient Parasite. Nat. Med. 2008;14:365–367. doi: 10.1038/nm0408-365. [DOI] [PubMed] [Google Scholar]
- 8.Wang W, Wang L, Liang Y-S. Susceptibility or Resistance of Praziquantel in Human Schistosomiasis: A Review. Parasitol. Res. 2012;111:1871–1877. doi: 10.1007/s00436-012-3151-z. [DOI] [PubMed] [Google Scholar]
- 9.Caffrey CR. Chemotherapy of Schistosomiasis: Present and Future. Curr. Opin. Chem. Biol. 2007;11:433–439. doi: 10.1016/j.cbpa.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 10.Neves BJ, Andrade CH, Cravo PVL. Natural Products as Leads in Schistosome Drug Discovery. Molecules. 2015;20:1872–1903. doi: 10.3390/molecules20021872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berriman M, Haas BJ, LoVerde PT, Wilson RA, Dillon GP, Cerqueira GC, Mashiyama ST, Al-Lazikani B, Andrade LF, Ashton PD, Aslett Ma, Bartholomeu DC, Blandin G, Caffrey CR, Coghlan A, Coulson R, Day Ta, Delcher A, DeMarco R, Djikeng A, Eyre T, Gamble Ja, Ghedin E, Gu Y, Hertz-Fowler C, Hirai H, Hirai Y, Houston R, Ivens A, Johnston Da, Lacerda D, Macedo CD, McVeigh P, Ning Z, Oliveira G, Overington JP, Parkhill J, Pertea M, Pierce RJ, Protasio AV, Quail Ma, Rajandream MA, Rogers J, Sajid M, Salzberg SL, Stanke M, Tivey AR, White O, Williams DL, Wortman J, Wu W, Zamanian M, Zerlotini A, Fraser-Liggett CM, Barrell BG, El-Sayed NM. The Genome of the Blood Fluke Schistosoma Mansoni. Nature. 2009;460:352–358. doi: 10.1038/nature08160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Protasio AV, Tsai IJ, Babbage A, Nichol S, Hunt M, Aslett Ma, De Silva N, Velarde GS, Anderson TJC, Clark RC, Davidson C, Dillon GP, Holroyd NE, LoVerde PT, Lloyd C, McQuillan J, Oliveira G, Otto TD, Parker-Manuel SJ, Quail Ma, Wilson RA, Zerlotini A, Dunne DW, Berriman M. A Systematically Improved High Quality Genome and Transcriptome of the Human Blood Fluke Schistosoma Mansoni. PLoS Negl. Trop. Dis. 2012;6:e1455. doi: 10.1371/journal.pntd.0001455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuntz AN, Davioud-Charvet E, Sayed Aa, Califf LL, Dessolin J, Arnér ESJ, Williams DL. Thioredoxin Glutathione Reductase from Schistosoma Mansoni: An Essential Parasite Enzyme and a Key Drug Target. PLoS Med. 2007;4:e206. doi: 10.1371/journal.pmed.0040206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Angelucci F, Miele AE, Boumis G, Dimastrogiovanni D, Brunori M, Bellelli A. Glutathione Reductase and Thioredoxin Reductase at the Crossroad: The Structure of Schistosoma Mansoni Thioredoxin Glutathione Reductase. Proteins. 2008;72:936–945. doi: 10.1002/prot.21986. [DOI] [PubMed] [Google Scholar]
- 15.Song L, Li J, Xie S, Qian C, Wang J, Zhang W, Yin X, Hua Z, Yu C. Thioredoxin Glutathione Reductase as a Novel Drug Target: Evidence from Schistosoma Japonicum. PLoS One. 2012;7:e31456. doi: 10.1371/journal.pone.0031456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han Y, Fu Z, Hong Y, Zhang M, Han H, Lu K, Yang J, Li X, Lin J. Inhibitory Effects and Analysis of RNA Interference on Thioredoxin Glutathione Reductase Expression in Schistosoma Japonicum . J. Parasitol. 2014;100:463–469. doi: 10.1645/13-397.1. [DOI] [PubMed] [Google Scholar]
- 17.Iskar M, Zeller G, Zhao X-M, van Noort V, Bork P. Drug Discovery in the Age of Systems Biology: The Rise of Computational Approaches for Data Integration. Curr. Opin. Biotechnol. 2012;23:609–616. doi: 10.1016/j.copbio.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 18.Bajorath J. Chemoinformatics: Recent Advances at the Interfaces between Computer and Chemical Information Sciences, Chemistry, and Drug Discovery. Bioorg. Med. Chem. 2012;20:5316. doi: 10.1016/j.bmc.2012.08.051. [DOI] [PubMed] [Google Scholar]
- 19.Duffy BC, Zhu L, Decornez H, Kitchen DB. Early Phase Drug Discovery: Cheminformatics and Computational Techniques in Identifying Lead Series. Bioorg. Med. Chem. 2012;20:5324–5342. doi: 10.1016/j.bmc.2012.04.062. [DOI] [PubMed] [Google Scholar]
- 20.Jorgensen WL. The Many Roles of Computation in Drug Discovery. Science. 2004;303:1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 21.Cherkasov A, Muratov EN, Fourches D, Varnek A, Baskin II, Cronin M, Dearden J, Gramatica P, Martin YC, Todeschini R, Consonni V, Kuz’min VE, Cramer R, Benigni R, Yang C, Rathman J, Terfloth L, Gasteiger J, Richard A, Tropsha A. QSAR Modeling: Where Have You Been? Where Are You Going To? J. Med. Chem. 2014;57:4977–5010. doi: 10.1021/jm4004285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ha H, Debnath B, Odde S, Bensman T, Ho H, Beringer PM, Neamati N. Discovery of Novel CXCR2 Inhibitors Using Ligand-Based Pharmacophore Models. J. Chem. Inf. Model. 2015;55:1720–1738. doi: 10.1021/acs.jcim.5b00181. [DOI] [PubMed] [Google Scholar]
- 23.Bisignano P, Burford NT, Shang Y, Marlow B, Livingston KE, Fenton AM, Rockwell K, Budenholzer L, Traynor JR, Gerritz SW, Alt A, Filizola M. Ligand-Based Discovery of a New Scaffold for Allosteric Modulation of the µ-Opioid Receptor. J. Chem. Inf. Model. 2015;55:1836–1843. doi: 10.1021/acs.jcim.5b00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jorgensen WL. Challenges for Academic Drug Discovery. Angew. Chem. Int. Ed. Engl. 2012;51:11680–11684. doi: 10.1002/anie.201204625. [DOI] [PubMed] [Google Scholar]
- 25.Andrade CH, Pasqualoto KFM, Ferreira EI, Hopfinger AJ. 3D-Pharmacophore Mapping of Thymidine-Based Inhibitors of TMPK as Potential Antituberculosis Agents. J. Comput. Aided. Mol. Des. 2010;24:157–172. doi: 10.1007/s10822-010-9323-y. [DOI] [PubMed] [Google Scholar]
- 26.Melo-Filho CC, Braga RC, Andrade CH. 3D-QSAR Approaches in Drug Design: Perspectives to Generate Reliable CoMFA Models. Curr. Comput. Aided. Drug Des. 2014;10:148–159. doi: 10.2174/1573409910666140410111043. [DOI] [PubMed] [Google Scholar]
- 27.Neves BJ, Bueno RV, Braga RC, Andrade CH. Discovery of New Potential Hits of Plasmodium Falciparum Enoyl-ACP Reductase through Ligand- and Structure-Based Drug Design Approaches. Bioorg. Med. Chem. Lett. 2013;23:2436–2441. doi: 10.1016/j.bmcl.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 28.Andrade CH, Pasqualoto KFM, Ferreira EI, Hopfinger AJ. 4D–QSAR: Perspectives in Drug Design. Molecules. 2010;15:3281–3294. doi: 10.3390/molecules15053281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zanella F, Lorens JB, Link W. High Content Screening: Seeing Is Believing. Trends Biotechnol. 2010;28:237–245. doi: 10.1016/j.tibtech.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Paveley Ra, Bickle QD. Automated Imaging and Other Developments in Whole-Organism Anthelmintic Screening. Parasite Immunol. 2013;35:302–313. doi: 10.1111/pim.12037. [DOI] [PubMed] [Google Scholar]
- 31.Tropsha A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010;29:476–488. doi: 10.1002/minf.201000061. [DOI] [PubMed] [Google Scholar]
- 32.Rai G, Sayed Aa, Lea Wa, Luecke HF, Chakrapani H, Prast-Nielsen S, Jadhav A, Leister W, Shen M, Inglese J, Austin CP, Keefer L, Arnér ESJ, Simeonov A, Maloney DJ, Williams DL, Thomas CJ. Structure Mechanism Insights and the Role of Nitric Oxide Donation Guide the Development of Oxadiazole-2-Oxides as Therapeutic Agents against Schistosomiasis. J. Med. Chem. 2009;52:6474–6483. doi: 10.1021/jm901021k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sayed AA, Simeonov A, Thomas CJ, Inglese J, Austin CP, Williams DL. Identification of Oxadiazoles as New Drug Leads for the Control of Schistosomiasis. Nat. Med. 2008;14:407–412. doi: 10.1038/nm1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fourches D, Muratov E, Tropsha A. Trust, but Verify: On the Importance of Chemical Structure Curation in Cheminformatics and QSAR Modeling Research. J. Chem. Inf. Model. 2010;50:1189–1204. doi: 10.1021/ci100176x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fourches D, Muratov E, Tropsha A. Curation of Chemogenomics Data. Nat. Chem. Biol. 2015;11:535–535. doi: 10.1038/nchembio.1881. [DOI] [PubMed] [Google Scholar]
- 36.OMEGA 2.5.1.4: OpenEye Scientific Software. Santa Fe, NM: http://www.eyesopen.com. [Google Scholar]
- 37.Hawkins PCD, Skillman aG, Warren GL, Ellingson Ba, Stahl MT. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.SYBYL-X 1.2. Tripos International, 1699 South Hanley Rd., St. Louis, Missouri, 63144, USA: [Google Scholar]
- 39.Gasteiger J, Marsili M. A New Model for Calculating Atomic Charges in Molecules. Tetrahedron Lett. 1978;19:3181–3184. [Google Scholar]
- 40.Purcell WP, Singer JA. A Brief Review and Table of Semiempirical Parameters Used in the Hückel Molecular Orbital Method. J. Chem. Eng. Data. 1967;12:235–246. [Google Scholar]
- 41.Jakalian A, Bush BL, Jack DB, Bayly CI. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: I. Method. J. Comput. Chem. 1999;21:132–146. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 42.Jakalian A, Jack DB, Bayly Fast, Efficient Generation of High-Quality Atomic Charges. Parameterization and Validation. J. Comput. Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 43.QUACPAC 1.6.3.1: OpenEye Scientific Software. Santa Fe, NM: Http://ww.eyesopen.com. [Google Scholar]
- 44.ROCS 3.2.1.4: OpenEye Scientific Software. Santa Fe, NM: Http://www.eyesopen.com. [Google Scholar]
- 45.Hawkins PCD, Skillman aG, Nicholls A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2007;50:74–82. doi: 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- 46.Seel M, Turner DB, Willett P. Effect of Parameter Variations on the Effectiveness of HQSAR Analyses. Mol. Inform. 1999;18:245–252. [Google Scholar]
- 47.Lindberg W, Persson J-A, Wold S. Partial Least-Squares Method for Spectrofluorimetric Analysis of Mixtures of Humic Acid and Lignin Sulfonate. Anal. Chem. 1983;55:643–648. [Google Scholar]
- 48.Wold S, Sjöström M, Eriksson L. PLS-Regression: A Basic Tool of Chemometrics. Chemom. Intell. Lab. Syst. 2001;58:109–130. [Google Scholar]
- 49. [(accessed Apr 14, 2015)];ChemBridge Online Chemical Store. http://www.hit2lead.com/
- 50.Dill JD, Hounshell WD, Marson S, Peacock S, Wipke WT. 182nd National Meeting of the American Chemical Society. New York: 1981. Search and Retrieval Using an Automated Molecular Access System; pp. 23–28. [Google Scholar]
- 51.Anderson S. Graphical Representation of Molecules and Substructure-Search Queries in MACCS. J. Mol. Graph. 1984;2:83–90. [Google Scholar]
- 52.Durant JL, Leland BA, Henry DR, Nourse JG. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002;42:1273–1280. doi: 10.1021/ci010132r. [DOI] [PubMed] [Google Scholar]
- 53.Braga RC, Alves VM, Silva AC, Liao LM, Andrade CH. Virtual Screening Strategies in Medicinal Chemistry: The State of the Art and Current Challenges. Curr. Top. Med. Chem. 2014;14:1899–1912. doi: 10.2174/1568026614666140929120749. [DOI] [PubMed] [Google Scholar]
- 54.Braga R, Alves V, Silva M, Muratov E, Fourches D, Tropsha A, Andrade C. Tuning hERG Out: Antitarget QSAR Models for Drug Development. Curr. Top. Med. Chem. 2014;14:1399–1415. doi: 10.2174/1568026614666140506124442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mansour NR, Bickle QD. Comparison of Microscopy and Alamar Blue Reduction in a Larval Based Assay for Schistosome Drug Screening. PLoS Negl. Trop. Dis. 2010;4:e795. doi: 10.1371/journal.pntd.0000795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marxer M, Ingram K, Keiser J. Development of an in Vitro Drug Screening Assay Using Schistosoma Haematobium Schistosomula. Parasit. Vectors. 2012;5:165. doi: 10.1186/1756-3305-5-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paveley Ra, Mansour NR, Hallyburton I, Bleicher LS, Benn AE, Mikic I, Guidi A, Gilbert IH, Hopkins AL, Bickle QD. Whole Organism High-Content Screening by Label-Free, Image-Based Bayesian Classification for Parasitic Diseases. PLoS Negl. Trop. Dis. 2012;6:1–11. doi: 10.1371/journal.pntd.0001762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, Carpenter AE. Improved Structure, Function and Compatibility for Cellprofiler: Modular High-Throughput Image Analysis Software. Bioinformatics. 2011;27:1179–1180. doi: 10.1093/bioinformatics/btr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Angelucci F, Dimastrogiovanni D, Boumis G, Brunori M, Miele AE, Saccoccia F, Bellelli A. Mapping the Catalytic Cycle of Schistosoma Mansoni Thioredoxin Glutathione Reductase by X-Ray Crystallography. J. Biol. Chem. 2010;285:32557–32567. doi: 10.1074/jbc.M110.141960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maestro, Version 10.0. New York, NY: Schrödinger, LLC; 2014. [Google Scholar]
- 62.Epik, Version 3.0. New York, NY: Schrödinger, LLC; 2014. [Google Scholar]
- 63.Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. Epik: A Software Program for pK( a ) Prediction and Protonation State Generation for Drug-like Molecules. J. Comput. Aided. Mol. Des. 2007;21:681–691. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- 64.Banks JL, Beard HS, Cao Y, Cho AE, Damm W, Farid R, Felts AK, Halgren TA, Mainz DT, Maple JR, Murphy R, Philipp DM, Repasky MP, Zhang LY, Berne BJ, Friesner RA, Gallicchio E, Levy RM. Integrated Modeling Program, Applied Chemical Theory (IMPACT) Journal of Computational Chemistry. 2005:1752–1780. doi: 10.1002/jcc.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.MarvinSketch 15.10.26. 2015 ChemAxon ( http://www.chemaxon.com)
- 66.OEDocking 3.2.0.2: OpenEye Scientific Software. Santa Fe, NM: http://www.eyesopen.com. [Google Scholar]
- 67.Jacobson MP, Friesner RA, Xiang Z, Honig B. On the Role of the Crystal Environment in Determining Protein Side-Chain Conformations. J. Mol. Biol. 2002;320:597–608. doi: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 68.Jacobson MP, Pincus DL, Rapp CS, Day TJF, Honig B, Shaw DE, Friesner RA. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins Struct. Funct. Genet. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 69.LigPrep, Version 3.2. New York, NY: Schrödinger, LLC; 2014. [Google Scholar]
- 70.Lowis DR. HQSAR: A New, Highly Predictive QSAR Technique. Tripos Tech. notes. 1997;1:1–17. [Google Scholar]
- 71.Gasco A, Fruttero R, Sorba G, Di Stilo A, Calvino R. NO Donors: Focus on Furoxan Derivatives. Pure Appl. Chem. 2004;76:973–981. [Google Scholar]
- 72.Kroemer RT, Hecht P, Guessregen S, Liedl KR. Improving the Predictive Quality of CoMFA Models. In: Kubinyi H, Folkers G, Martin YC, editors. 3D QSAR in Drug Design Recent Advances. Dordrecht: Kluwer Academic Publishers; 1998. pp. 41–56. [Google Scholar]
- 73.Tsai K-C, Chen Y-C, Hsiao N-W, Wang C-L, Lin C-L, Lee Y-C, Li M, Wang B. A Comparison of Different Electrostatic Potentials on Prediction Accuracy in CoMFA and CoMSIA Studies. Eur. J. Med. Chem. 2010;45:1544–1551. doi: 10.1016/j.ejmech.2009.12.063. [DOI] [PubMed] [Google Scholar]
- 74.Doweyko AM. 3D-QSAR Illusions. J. Comput. Aided. Mol. Des. 2004;18:587–596. doi: 10.1007/s10822-004-4068-0. [DOI] [PubMed] [Google Scholar]
- 75.Jain AN. Ligand-Based Structural Hypotheses for Virtual Screening. J. Med. Chem. 2004;47:947–961. doi: 10.1021/jm030520f. [DOI] [PubMed] [Google Scholar]
- 76.Kubinyi H. QSAR and 3D QSAR in Drug Design Part 1: Methodology. Drug Discov. Today. 1997;2:457–467. [Google Scholar]
- 77.Böhm HJ, Flohr A, Stahl M. Scaffold Hopping. Drug Discov. Today Technol. 2004;1:217–224. doi: 10.1016/j.ddtec.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 78.Heikamp K, Bajorath J. Fingerprint Design and Engineering Strategies: Rationalizing and Improving Similarity Search Performance. Future Med. Chem. 2012;4:1945–1959. doi: 10.4155/fmc.12.126. [DOI] [PubMed] [Google Scholar]
- 79.Gardiner EJ, Holliday JD, O’Dowd C, Willett P. Effectiveness of 2D Fingerprints for Scaffold Hopping. Future Med. Chem. 2011;3:405–414. doi: 10.4155/fmc.11.4. [DOI] [PubMed] [Google Scholar]
- 80.Sun H, Tawa G, Wallqvist A. Classification of Scaffold-Hopping Approaches. Drug Discov. Today. 2012;17:310–324. doi: 10.1016/j.drudis.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van de Waterbeemd H, Gifford E. ADMET in Silico Modelling: Towards Prediction Paradise? Nat. Rev. Drug Discov. 2003;2:192–204. doi: 10.1038/nrd1032. [DOI] [PubMed] [Google Scholar]
- 82.Ingram-Sieber K, Cowan N, Panic G, Vargas M, Mansour NR, Bickle QD, Wells TNC, Spangenberg T, Keiser J. Orally Active Antischistosomal Early Leads Identified from the Open Access Malaria Box. PLoS Negl. Trop. Dis. 2014;8:e2610. doi: 10.1371/journal.pntd.0002610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nare B, Smith JM, Prichard RK. Oltipraz-Induced Decrease in the Activity of Cytosolic Glutathione S-Transferase in Schistosoma Mansoni. Int. J. Parasitol. 1991;21:919–925. doi: 10.1016/0020-7519(91)90167-6. [DOI] [PubMed] [Google Scholar]
- 84.Ramirez B, Bickle Q, Yousif F, Fakorede F, Mouries M-A, Nwaka S. Schistosomes: Challenges in Compound Screening. Expert Opin. Drug Discov. 2007;2:S53–S61. doi: 10.1517/17460441.2.S1.S53. [DOI] [PubMed] [Google Scholar]
- 85.Cramer RD, Patterson DE, Bunce JD. Comparative Molecular Field Analysis (CoMFA). 1. Effect of Shape on Binding of Steroids to Carrier Proteins. J. Am. Chem. Soc. 1988;110:5959–5967. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- 86.Cramer RD. Topomer CoMFA: A Design Methodology for Rapid Lead Optimization. J. Med. Chem. 2003;46:374–388. doi: 10.1021/jm020194o. [DOI] [PubMed] [Google Scholar]
- 87.Kuz’min VE, Artemenko AG, Polischuk PG, Muratov EN, Hromov AI, Liahovskiy AV, Andronati SA, Makan SY. Hierarchic System of QSAR Models (1D-4D) on the Base of Simplex Representation of Molecular Structure. J. Mol. Model. 2005;11:457–467. doi: 10.1007/s00894-005-0237-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.