

Abstract
Allosteric enzymes regulate a wide range of catalytic transformations, including biosynthetic mechanisms of important human pathogens, upon binding of substrate molecules to an orthosteric (or active) site and effector ligands at distant (allosteric) sites. We find that enzymatic activity can be impaired by small molecules that bind along the allosteric pathway connecting the orthosteric and allosteric sites, without competing with endogenous ligands. Noncompetitive allosteric inhibitors disrupted allostery in the imidazole glycerol phosphate synthase (IGPS) enzyme from Thermotoga maritima as evidenced by nuclear magnetic resonance, microsecond time-scale molecular dynamics simulations, isothermal titration calorimetry, and kinetic assays. The findings are particularly relevant for the development of allosteric antibiotics, herbicides, and antifungal compounds because IGPS is absent in mammals but provides an entry point to fundamental biosynthetic pathways in plants, fungi, and bacteria.
Graphical abstract
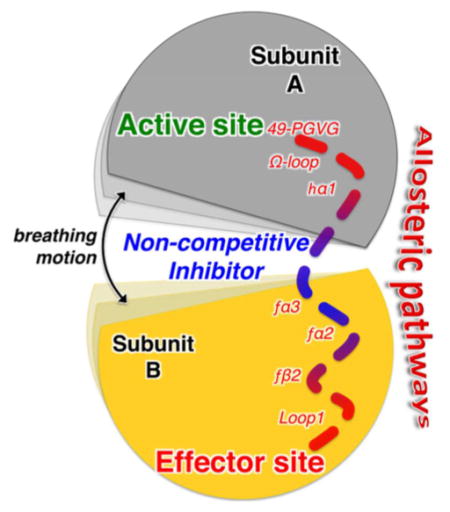
Allosteric enzymes are ubiquitous biological catalysts that control and regulate fundamental chemical processes in cells. The transformation of the primary ligand (substrate) occurs at the orthosteric (or active) site and is regulated by binding of a second ligand (i.e., effector) at a distant and topographically distinct (i.e., allosteric) site (Figure 1). In positive allosteric modulation, effector binding stabilizes an active protein conformation, ultimately enhancing the enzymatic activity by either increasing the affinity of the substrate for the orthosteric site (in K-type enzymes) or improving its catalytic conversion rate (in V-type enzymes). The molecular details of allostery, however, remain to be fully resolved,1 despite the acknowledged validity of historical (symmetric2 and sequential3) phenomenological models4 and the recognition of structure-based and ensemble views of allostery.5,6 Changes in dynamics and disorder critical to inhibition of enzymatic activation have been found to be associated with the allosteric communication between the orthosteric and allosteric sites,6–8 which is expected to propagate through conserved allosteric pathways.9
Figure 1.

Basal (low or negligible) catalytic activity of the apoenzyme observed in the presence of substrate (green) but no endogenous effector (red). Positive allosteric modulation by effector binding >10 Å from the active site increases enzymatic activity. The two sites communicate through an allosteric pathway (red dotted line). Enzymatic activity is inhibited upon binding of competitive exogenous ligands at the active or effector sites. Noncompetitive ligands disrupt allosteric communication upon binding at critical sites along the allosteric pathway.
Advanced experimental techniques such as nuclear magnetic resonance (NMR) combined with computer simulations10 can provide fundamental insights for structural, dynamical, and energetic characterization of allosteric enzymes,11–17 especially in the absence of available crystallographic data. The combination of molecular dynamics (MD) simulations and NMR experiments has improved our understanding of the connection among allostery, ligand binding, and protein flexibility, allowing characterization of specific allosteric pathways at the molecular level and correlation between structural dynamics and small molecule binding.18
A detailed understanding of protein allostery can permit the development of allosteric drugs.4,7,19–21 However, a better mechanistic understanding of allosteric inhibitor binding effects, particularly in enzymes where allostery occurs primarily through changes in dynamics, would enhance the outcome of those therapeutic efforts. Here, we show that characterization of allosteric pathways in a model enzyme allows the discovery of small molecules that interfere with allosteric signaling, ultimately impairing enzymatic activity without directly competing for the endogenous ligand sites.
Traditional enzyme inhibitor discovery has been based on the optimization of lead molecules that bind to an active site (Figure 1) and compete with endogenous agonists to alter (generally inhibit) the natural physiological reaction. Allosteric systems offer a second target site for ligands, the effector binding site, where an exogenous ligand can bind and modulate the natural function. Thus, endogenous allosteric modulators can be substituted with exogenous ligands designed to bind at the effector site and alter the allosteric signaling mechanism at its origin (Figure 1).22,23 However, discovering effective exogenous allosteric ligands is hampered by the fact that allosteric sites often remain unidentified and “orphan” of their endogenous ligands. Nevertheless, allosteric ligands can offer pharmacological advantages over traditional orthosteric agonists by exerting their effects only when endogenous agonists are present, providing exceptional spatial and temporal selectivity, and offering saturability of their effects and intrinsic safety in overdosage.22,24 Once the allosteric sites are occupied, no further effect can be produced, even with excessive doses. Here, we focus on targeting specific protein domains that are essential for allosteric communication to screen allosteric molecules that do not compete with the natural effectors and still suppress catalytic activity. The identification of such allosteric inhibitors requires detailed molecular insights into the allosteric mechanism and, in particular, characterization of the allosteric pathways with atomistic resolution.
We combine NMR experiments, computational modeling, isothermal titration calorimetry (ITC), and kinetic assays to find clear evidence of small heterocyclic organic molecules that function as noncompetitive allosteric inhibitors of the enzyme imidazole glycerol phosphate synthase (IGPS) from Thermotoga maritima,13,25 a V-type allosteric enzyme essential for purine and histidine biosynthesis in plants and important human pathogens.26 This work is motivated by the absence of X-ray structures establishing the binding modes of small molecules (effectors and/or inhibitors) bound to the flexible V-type enzyme. IGPS is a key metabolic enzyme of the amidotransferase family that links amino acid and nucleotide biosynthesis in bacteria, plants, and fungi but is absent in mammals. Because of its metabolic role, IGPS is also a potential target for herbicides and antifungal agents.27,28 IGPS from T. maritima is a heterodimeric enzyme consisting of two noncovalently associated subunits, HisH (23 kDa, glutaminase domain) and HisF (28 kDa, cyclase domain), depicted in Figure 2. In fungi and plants, the two IGPS subunits reside on a single polypeptide.29 IGPS catalyzes glutamine (Gln) hydrolysis at the active site of HisH, where there is a conserved catalytic triad (C84, H178, and E180) that produces ammonia (NH3) and glutamate. The generated NH3 travels from the HisH active site to the HisF effector site passing, more than 25 Å away, through the (β/α)8 barrel (see Figure 2) and reacts to break down the N′-[(5′-phosphoribulosyl)formimino]-5-ami-noimidazole-4-carboxamide ribonucleotide (PRFAR) effector into two products [IGP and AICAR (Figure 2)], which are entry points to the histidine and purine biosynthetic pathways, respectively.25,26,30 The glutaminase half-reaction of IGPS exhibits V-type allosteric modulation, with the Gln binding affinity being minimally altered by the presence of PRFAR, while a 4900-fold increase in reaction rate is observed for the PRFAR-bound complex over the basal glutaminase activity of IGPS.26,30
Figure 2.

Glutamine hydrolysis, catalyzed by IGPS at the HisH active site, initiated after binding of the effector (PRFAR) to HisF and allosteric communication. (a) Tertiary structures of the HisH and HisF domains of IGPS, showing the active site (green), the allosteric site (red), and a schematic representation of the internal ammonia (blue) channel (gray tube). The allosteric pathway (red dashed line) involves secondary structures (labeled in red) on the right side (side R) of the complex. (b) Schematic representations of glutamine hydrolysis and effector decomposition at the active and effector sites. (c–f) The allosteric communication involves structural changes induced by binding of PRFAR (red) to the apo complex (blue), affecting the IGPS breathing motion targeted by small molecule inhibitors that bind at the HisF–HisH interface.
In the absence of X-ray structures, the study of these challenging systems must rely on state-of-the-art NMR and computational methods. The allosteric pathways of IGPS have been described by a combined NMR and MD study of the apo and PRFAR-bound IGPS complexes.13,31–33 Essential changes in conformational fluctuations were found upon PRFAR binding at HisF, which propagate along the right side (side R) of the IGPS complex (Figure 2) and promote an active conformation of the HisH glutaminase site. In particular, when PRFAR binds at the bottom of the HisF barrel, it alters hydrophobic contacts at the fβ2 strand of the (β/α)8 barrel (involving residues f V48, f L50, and f I52), affecting the motion of the fα2 helix and the flexible loop 1. The f and h prefixes indicate residues or secondary structure elements of HisF and HisH, respectively. These changes at the bottom of side R propagate toward HisH, leading to stabilization of salt bridges between charged side chains of the fα2 and fα3 helices of HisF and the hα1 helix of HisH. Changes in ionic interactions at the heterodimer interface affect the “breathing motion” of the complex, i.e., the relative motion of HisH hinged to HisF at the f R249–hW123 joint (Figure 2).
While the two subunits are anchored together by a salt bridge (f D98–hK181), the changes induced by PRFAR binding are enough to alter the opening/closing motion of the IGPS complex, reducing the correlations in motions at the bottom of the HisH domain. Such poorly correlated motions allow more flexibility of the oxyanion strand, i.e., the hP49-hG50-hV51-hG52 (49-PGVG) amino acid sequence whose H-bond to the Ω-loop (hV51–hP10) breaks rather easily, facilitating rotation of the strand and stabilizing a charged tetrahedral intermediate (oxyanion hole) in the active site,13,34 a critical intermediate in glutaminase catalysis.35 These data suggest that the difficulty of achieving PRFAR-bound X-ray structures is likely due to the enhanced flexibility of the enzyme when PRFAR binds and demonstrated that a combination of MD and NMR studies can provide a detailed understanding of allostery in this challenging system.
MATERIALS AND METHODS
Computational Modeling
Molecular Docking
Molecular docking calculations were performed with the Maestro suite (Schrödinger, LLC, New York, NY), using the extra-precision procedure implemented in the Glide software,36 including an exhaustive screening of ligand binding over the entire Maybridge library containing a collection of 83766 heterocyclic organic molecules. Of the ~400000 pharmacophores in the world drug index, ~87% are expressed by the Maybridge Screening collection. The docking score is determined by the Glide score (i.e., the sum of the Coulomb, binding, and van der Waals energetic contributions) and the state penalty for a given protonation or tautomeric state of the ligand. The ligands were prepared using Maestro’s Ligprep utility, and Epik software37 was used to generate protonation states at the target pH (7.0 ± 2.0).38 The protein structure of IGPS was obtained from the Protein Data Bank (PDB entry 1GPW), the crystal structure of the enzyme complex from T. maritima. The position of PRFAR in the HisF active site was determined after superimposing the phosphate groups in the T. maritima IGPS crystal and the PRFAR-bound complex in the yeast enzyme (PDB entry 1OX5). The standard-precision (SP) calculation was first done, and the resulting top 2% molecules and their poses based on the SP glide score were sent to extra-precision (XP) screening. The resulting molecular structures and interactions within their nearby environment were carefully examined, and eliminations were made when undesired functional groups and structures were observed. The top three candidates from the interface XP screening were selected for further analysis and MD simulations.
Molecular Dynamics Simulations
The structural models for all IGPS complexes were prepared using the coordinates of the docked ligands obtained from Glide screening. MD simulations of all IGPS complexes were performed with NAMD239 using the AMBER-ff99SB40 force field for the protein and the generalized Amber force field41 for ligands and PRFAR. The system pre-equilibration and production run procedures followed methods previously reported.13 Data analysis and figure rendering were performed using VMD.42
Anton MD Simulations
All microsecond-time simulations were performed using Anton,43 a special-purpose computer for long molecular dynamics simulations, starting from well-equilibrated structures after at least 0.1 μs of the NAMD simulations. The simulated system and the force field used in Anton are the same as in NAMD simulations. Three independent Anton simulations were performed using the reversible multiple-time step algorithm44 to integrate the equations of motion with a time step of 2 fs for short-range nonbonded and bonded forces and 6 fs for the long-range nonbonded forces, for a total simulation time of ~7 μs. Long-range electrostatic interactions were calculated using the k-space Gaussian split Ewald method45 with a 64 × 64 × 64 grid and a cutoff of 11 Å for the Lennard-Jones and short-range electrostatic interactions. The simulations were performed at constant temperature (303 K) and pressure (1 atm) using the multigrator integrator as implemented in Anton.46 All bond lengths involving hydrogen atoms were held fixed using the SHAKE algorithm.47
Experimental Procedures
Sample Preparation
Labeled samples of HisH- and HisF-IGPS were expressed and purified as previously described.13,34,48 Escherichia coli BL21(DE3) cells supplemented with 15NH4Cl were grown in deuterated M9 minimal medium to an OD600 of 0.9–1.0. Following induction with 1.0 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), cell pellets for HisH and HisF were harvested, resuspended, and co-lysed by ultrasonication. Purification of the IGPS complex was facilitated through a C-terminal histidine tag (HisH) for Ni/NTA affinity chromatography. The purity and uniformity of IGPS samples were confirmed through sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and dynamic light scattering, respectively.
NMR Chemical Shift Perturbation of Apo IGPS by 3
NMR spectroscopic data were collected at 30 °C on a Varian INOVA 600 MHz spectrometer. NMR chemical shift perturbations were determined through titrations of 3 into 15N-labeled samples of HisF- and HisH-IGPS and were conducted in the following manner. Concentrated stocks (~15 mM) of 3 were prepared by dissolving solid aliquots in 20 μL of dimethyl sulfoxide (DMSO) (to aid solubility) and diluting with 380 μL of NMR buffer (10 mM HEPES, 10 mM KCl, 0.5 mM EDTA, and 1.0 mM DTT). The final pH of the stock solution was adjusted to 7.3. For [15N]HisF-IGPS, spectra were recorded in the presence of 0, 0.2, 0.755, 1.34, 1.69, 2.05, 2.41, 2.79, and 3.17 mM 3 (0.74 mM IGPS). For [15N]HisH-IGPS, spectra were recorded at 0, 2.05, and 3.17 mM 3 (0.52 mM IGPS). Spectra were processed with NMRPipe49 and analyzed with Sparky.50
NMR Chemical Shift Perturbation of Binary PRFAR-Bound IGPS by 3
NMR experiments were also conducted on PRFAR-bound HisF-IGPS under similar conditions. PRFAR was synthesized as previously described26,34,48 and added to a concentration of 0.96 mM to a sample of HisF-IGPS (0.68 mM IGPS), and NMR confirmed saturation. The binary PRFAR complex was then titrated with a 16 mM stock solution of 3 to concentrations of 1.1, 4.5, 6.0, 7.2, and 9.4 mM. Spectra were processed and analyzed as described above.
Isothermal Titration Calorimetry
IGPS was expressed in LB medium without isotopic enrichment and purified as described for the NMR titration studies. IGPS samples used in ITC experiments were dialyzed twice in 4.0 L of 50 mM HEPES (pH 7.6) and 10 mM KCl. 7-Benzyl-8-[(1-{[(2-hydroxyethyl)amino]methyl}propyl)amino]-1,3-dimethyl-2,3,6,7-tetrahydro-1H-2,6-purinedione (3) (product no. RJC03502) was purchased from Ryan Scientific. 3 was dissolved in DMSO and then diluted with ITC buffer to a final concentration of 12.4 mM and 10% DMSO; 10% DMSO was added to each IGPS sample immediately prior to beginning the experiment for a final IGPS concentration of 377.0 μM. Stocks of 3 and IGPS were adjusted to a pH of 7.60 as needed. ITC experiments were performed on a MicroCal iTC200 instrument at 303 K with 14 injections of 2.5 μL of 12.4 mM 3 into 377.0 μM IGPS.
Kinetic Glutaminase Assays
Kinetic assays monitoring the glutaminase activity of IGPS in the presence and absence of 3 were adapted from the procedure of Klem and Davission.26 Stock solutions of IGPS (0.3 mM), PRFAR (5 mM), 3 (12 mM), and glutamine (165 mM) were prepared in a 50 mM Tris buffer (pH 8.0). Samples containing IGPS, PRFAR, and 3 in the same buffer were incubated at 30 °C with varying concentrations of glutamine (0–12 mM) for 15–20 min. Following incubation, samples were boiled for 3 min and immediately frozen at –80 °C to quench the reaction. Identical reactions were performed with and without the inhibitor 3. Quantification of glutamate production required stock solutions of glutamate dehydrogenase (10 mg/mL) and 3-acetylpyridine adenine dinucleotide (APAD, 70 mM) in an identical 50 mM Tris buffer (pH 8.0). Aliquots of previously quenched reactions were thawed, and 0.5 mM APAD and 100 μg of glutamate dehydrogenase were added. The reaction mixtures were incubated at 37 °C for an additional 65 min, and the concentration of APADH, formed during the conversion of glutamate to 2-oxoglutarate, was determined by measuring the sample absorbance at 363 nm (ε363 = 8900 M−1 cm−1). Plots of k (inverse seconds) versus glutamine concentration (Figure 5) were analyzed using nonlinear regression with noncompetitive inhibition modeling in GraphPad Prism 6 (GraphPad Software).
Figure 5.

Suppression of PRFAR-enhanced glutaminase activity upon binding of 3. (a) Inhibition of glutaminase activity of the PRFAR–IGPS binary complex (blue) by 3 (red) as measured by kinetic assays. Nonlinear least-squares fitting of a noncompetitive binding model yields a Ki of 0.9 ± 0.07 mM. (b) Microsecond MD simulations of inhibitor binding. Binding of 3 to the PRFAR-bound IGPS binary complex is monitored along a representative 2.5 μs MD trajectory, showing two main poses (1, 2, and a transient pose 2′) characterized by specific H-bonds (highlighted with dotted lines and colored circles) and hydrophobic contacts between the benzyl functional group of 3 and HisF (ring down) or alternatively HisH (ring up).
RESULTS AND DISCUSSION
Identification of Allosteric Inhibitors
IGPS serves as a particularly interesting model for investigating the potential for disrupting allosteric communication with noncompetitive inhibitors because of its importance in amino acid and nucleotide biosynthesis. Here, we explore the binding of small organic molecules at the HisH–HisF interface to block allosteric information transfer between PRFAR and Gln binding sites (Figure 2). Our strategy is inspired by the allosteric mechanism suggested by the earlier computational and NMR work of us and others, involving interactions at the HisH–HisF interface that affect the breathing motion of the IGPS complex.13,31,33,34,48 The interfacial inhibitor concept has been successfully applied for desynchronizing other macro-molecular machines and for arresting allosteric enzymes.51 Here, we explore the interfacial inhibitor concept at the molecular level, as correlated to the allosteric signal inhibition, providing an original approach for the identification of noncompetitive inhibitors. We performed molecular docking targeting the HisF–HisH interface, previously determined by NMR and MD simulations to exhibit conformational flexibility, with small heterocyclic organic molecules, from the Maybridge screening collection of 83766 heterocyclic small molecules.38 Computational docking was performed by defining a docking area embracing the domain interface.
Figure 3 shows the docking scores and binding sites of the most promising compounds. Upon careful structure examination, we found that the top three potential inhibitors, N1-[3,5-di(trifluoromethyl)phenyl]-2-({[5-hydroxy-4-(hydroxymethyl)-6-methyl-3-pyridyl]methyl}thio)acetamide (1), 7-{3-[bis(2-hydroxyethyl)amino]-2-hydroxypropyl}-1,3-dimethyl-3,7-dihydro-1H-purine-2,6-dione (2), and 7-benzyl-8-[(1-{[(2-hydroxyethyl)amino]methyl}propyl)amino]-1,3-dimethyl-2,3,6,7-tetrahydro-1H-2,6-purinedione (3), had specific interactions with the IGPS residues directly involved in the allosteric mechanism. The three ligands were found to dock at the same region of the HisH–HisF interface, in the proximity of the Ω-loop of HisH and the fα3–fβ3 turn (Figure 3). The initial poses of 2 and 3 involved H-bonding interactions with critical IGPS elements, such as f D98, which is part of the f D98–hK181 anchor that regulates the relative interdomain breathing motion and is important for IGPS catalysis, as demonstrated by a decrease in glutaminase activity after mutation to alanine (D98A).32 The three potential inhibitors also make H-bonds with hE180, a constituent of the catalytic triad. More importantly, all ligands interact with the hN12 residue and bind close to the HisH Ω-loop, a crucial structural element for the allosteric signal transduction in IGPS.13 These fundamental interactions are important because prior community network analysis of correlated motions in IGPS has shown that the communication flow between HisH and HisF in the PRFAR-bound complex involves the Ω-loop, with hN15, hN12, and hP10 in HisH and f D98 in HisF being the protein network elements most altered by effector binding.13
Figure 3.

Docking of allosteric inhibitors 1–3 and their effects on IGPS motions. (a) Inhibitors (black) dock at the HisF–HisH interface, affecting the motion of HisH relative to HisF (breathing motion). The breathing motion is measured by the angle ϕ defined by the α-carbon atoms (Cα) of f F120, hW123, and hG52. (b–d) Docking poses of allosteric inhibitors 1–3. (e–g) Evolution of ϕ during the MD simulation of the apo complex (blue), the binary PRFAR-bound complex (red), and the ternary complex with inhibitors 1–3 (black). (h, i, and l) The average root-mean-square fluctuations (RMSF) of Cα atoms, relative to average configurations, are reported for the apo complex (blue), the binary PRFAR-bound complex (red), and the ternary complex with inhibitors 1–3 (black). (m) Inhibitors 1–3 were selected according to the docking scores of the top 400 molecules screened by Glide from the 83766 heterocyclic small molecules of the Maybridge library. (n–p) The three inhibitors affect IGPS motions differently. The Cα atoms of the amino acid residues showing the highest differences in the RMSF average values (ΔRMSF > 1.1 Å) between the ternary and binary complexes (i.e., the RMSF of those residues that have been largely affected by the presence of potential inhibitors) are mapped onto the structures of the ternary IGPS complexes.
The docking results for 1–3 at the IGPS interface suggest that binding of these small molecules might affect the interdomain motions and enzyme dynamics necessary for activity. Submicrosecond dynamics of the PRFAR-bound complex show that effector binding enhances the overall interdomain motion, increasing the correlations at side R, which remains more rigidly closed than side L during breathing. An allosteric inhibitor of IGPS should thus disrupt the PRFAR-induced signal by inhibiting the breathing motion and releasing the side R motions. We have evaluated the inhibition effect of binding exogenous ligands 1–3 by comparing the protein motions of the PRFAR-bound binary complex with those of the ternary (PRFAR and inhibitor) complexes.
Figure 3 compares the HisH–HisF breathing motion amplitude during MD simulations of apo and PRFAR-bound IGPS to those in the 1–, 2–, and 3–IGPS ternary complexes. The apo complex exhibits slow (a single opening motion in 0.1 μs) and ample interdomain motions, with a breathing angle (ϕ) between 12° and 30°. The PRFAR-bound complex shows more recurrent (multiple opening/closing events in 0.1 μs) and smaller breathing oscillations (17° < ϕ < 25°), while all ternary complexes exhibit distinct interdomain motions. The presence of the interfacial ligands stabilizes a “static” open structure, with fluctuations of the breathing angle of 5–6° with respect to the average angles, which are similar for 1– and 2–IGPS complexes (~26–27°) and slightly smaller (~23°) for the 3–IGPS complex. Notably, binding of interfacial ligands has a significant influence on the motions large distances from the HisF–HisH interface. The ionic interactions between residues of the fα2 and fα3 helices (and the relative displacement of the two secondary structure elements) and the hydrophobic contacts at the fβ2 strand of the (β/α)8 barrel are significantly altered in the ternary complexes when compared to those in the PRFAR-bound binary IGPS complex (Figures S1 and S2 and Table S1). Interestingly, these changes in motion are correlated with a change in the accessibility of ammonia to the HisF barrel. Residues f R5, f E46, f K99, and f E167 create salt bridges that serve as an ammonia gate for the α/β barrel,25 which easily opens (in ~0.03 μs) upon PRFAR binding but remains closed in the ternary (PRFAR and inhibitor) complexes (like in apo IGPS) along the MD trajectories (Figure S3). Therefore, the barrier for NH3 tunneling toward HisF must be higher in the ternary complexes.
The protein motions of 1–, 2–, and 3–IGPS ternary complexes showed similar features among the various ternary complexes, which in some cases significantly differ from those of the PRFAR-bound binary complex. Figure 3 shows, for each complex, the averaged root-mean-square (RMS) deviations of α-carbon atoms, relative to the average configurations along the MD trajectories, which represent the averaged RMS fluctuations (RMSF), i.e., the mobility of each residue. PRFAR binding induces significant changes in the RMSF of amino acid residues belonging to loop 1 as well as the fα2 and fα3 helices, which unsurprisingly are components of the IGPS allosteric pathways.13 The RMSF indicate that exogenous ligand binding enhances the mobility of IGPS amino acid residues with respect to those of both apo and PRFAR-bound complexes to a different extent with each ligand. In particular, the RMSF of the 1–IGPS complex is not considerably different from those of the apo or binary complexes, while both 2– and 3–IGPS complexes show much larger RMSF values, with the 2–IGPS complex being the largest. Highlighting the residues with the largest RMSF differences (Figure 3) between the ternary and binary complexes (ΔRMSF > 1.1 Å) shows how only a few residues are largely affected by binding of 1, while the effect of 2 and 3 is spread over different regions of IGPS. In particular, the effect of 2 involves almost the entire side L of HisF and side R of HisH, comprising some secondary structure elements involved in allostery, i.e., loop 1, fα3, hα1, and the Ω-loop. Notably, binding of 3 affects mostly amino acid residues that belong to HisF side R and are involved in allosteric communication, suggesting that 3 is a good candidate for inhibition of the PRFAR allosteric signal. Therefore, in the following sections, we focus on the characterization of the binding of 3, its ability to reduce IGPS enzymatic activity, and the mechanism of allosteric communication disruption by means of NMR experiments and MD simulations extended to the microsecond time scale.43
Inhibitor Binding and Allosteric Effects
Binding of 3 to apo IGPS was monitored by isothermal titration calorimetry (ITC), which revealed an exothermic process with a dissociation constant Kd of 1.5 ± 0.3 mM (see Figure 4). Favorable enthalpic (ΔH = −566 ± 54 cal/mol) and entropic (−TΔS = −378 cal/mol) contributions lead to a free energy change of −0.94 kcal/mol. These results suggest a loose binding mode, in agreement with docking and MD simulations, showing binding in a shallow and rather water-exposed interface pocket. Titration of 3 into apo IGPS, containing 15N-labeled HisF, caused substantial chemical shift perturbations (Δδ) in several resonances at increased inhibitor concentrations (Figure 4). A total of 31 HisF residues were found to have significant Δδ values (≥0.08 ppm) determined from a trimmed mean of all resonance positions (Figures S4 and S5). The four amino acid residues that undergo the largest chemical shift changes (Δδ > 0.14) upon binding of 3 are in loop 1 and at the HisH–HisF interface. Notably, f D98 displays the most substantial chemical shift perturbation (Δδ > 0.15) of any interfacial residue. Interestingly, the other three residues most affected by 3 binding (f V12, f K13, and f V18) belong to the flexible loop 1 with displacements correlated to the breathing motion,13,31 as influenced by the interfacial ligand.
Figure 4.

Characterization of inhibitor binding by chemical shifts changes in apo- and effector-bound IGPS, induced upon inhibitor binding. (a) Raw (top) and integrated (bottom) calorimetric isotherm collected upon binding of 3 (12.4 mM) to apo IGPS (0.377 mM). (b and c) Correlation peaks (of selected residues mapped onto the structure of the 3–IGPS complex) from 1H–15N HSQC NMR experiments with 15N-labeled HisF-IGPS. Titration of 3 into apo IGPS (blue) at 3.17 mM (orange) causes distinct shifts and/or exchange broadening in several resonances. Residues with Δδ values of >0.1 (green spheres at Cα) are mapped onto the structure of the 3–IGPS complex, highlighting the four most affected resonances (orange spheres) with Δδ values of >0.14. (d and e) Analogous correlation peaks are shown for the binary IGPS complex, with a PRFAR concentration of 0.96 mM. Titration of 3 into binary PRFAR-bound IGPS (red) to a concentration of 9.4 mM (orange) causes distinct shifts in several resonances. Residues with Δδ values of >0.1 (blue spheres at Cα) are mapped onto the ternary IGPS structure (3 and PRFAR) (e), highlighting the six most affected resonances (black spheres) with Δδ values of >0.14.
PRFAR binding causes widespread chemical shift perturbations throughout HisF, with several residues affected by as much as 0.5 ppm, a magnitude consistent with previously reported NMR titrations.34,48 Subsequent addition of saturating concentrations of 3 results in the chemical shift changes of 27 residues beyond the significance cutoff of 0.08 ppm, with an average perturbation of 0.128 ppm (Figures S4, S7, and S8). The magnitudes and identities of the shifted residues vary when compared to those of the apo enzyme, with a large cluster of perturbations near the PRFAR binding site and a smaller group of shifted residues at the dimer interface (Figure S8). Therefore, the chemical shift perturbation studies suggest that 3 binds to the apo complex and the binary PRFAR–IGPS complex and interacts somewhat differently with the binary complex, perhaps because of a more preferable enzyme conformation induced by initial effector binding. A more favorable arrangement of the binary structure would result in several key resonances showing less substantial shifts when interacting with 3, as they have already been perturbed by PRFAR.
Figure 4 shows the residues with larger changes in chemical shifts (Δδ > 0.14), induced upon binding of 3 to the binary IGPS complex. In addition to the loop 1 residues affected by binding of 3 to the apo structure (fV12, fK13, and fV18), two significant changes in chemical shifts are observed for residues fD85 and fS90. Interestingly, these polar residues belong to the fα3 helix, one of the secondary structure elements, along with loop 1, involved in the IGPS allosteric pathway. While not emerging as the most affected residue, f D98 and other interface residues are still involved in chemical shift changes upon binding of 3 to the binary complex (Figure S8). However, PRFAR perturbs these regions significantly, perhaps masking the full extent of variations induced by the binding of 3. In summary, NMR experiments consistently indicate binding of 3 at the HisH–HisF interface, with the most significant changes in chemical shifts in HisF involving side R, in agreement with MD simulations and supporting 3 as a potential inhibitor of the allosteric signal in IGPS.
We examined the inhibitory properties of 3 with kinetic assays monitoring the glutaminase activity of IGPS. Figure 5 shows that addition of 3 to a PRFAR-bound binary complex of IGPS inhibits glutaminase catalysis substantially. The kinetic profile supports 3 as a noncompetitive inhibitor with respect to glutamine. Gln and 3 bind with similar affinities (millimolar), which is 3 orders of magnitude weaker than PRFAR (micromolar) binding.34
The following section provides a detailed description of the mechanism of allosteric signal disruption, due to binding of 3, inducing inhibition of glutaminase activity.
Mechanism of Allosteric Signal Disruption
Microsecond MD simulations provide fundamental insights into the effect of binding of 3 to the PRFAR-bound ternary complex and the inhibition of allosteric communication between HisF and HisH in the IGPS heterodimer. Figure 5 shows representative poses of 3 during a 2.5 μs MD simulation, during which time 3 remained bound to the HisH–HisF interface. In the first pose (pose 1), the terminal hydroxyethyl group of 3 is H-bonded to the carboxylic side chain of hE180, with the 7-benzyl substituent group of the purinedione aromatic ring pointing toward the hydrophobic part of the HisF barrel (ring down conformation), in the proximity of the fα3–fβ3 turn. These polar and hydrophobic contacts stabilize the ligand between the two protein domains for more than 1.3 μs, after which the 3–hE180 H-bond exchanges with a new 3–f D98 H-bond. The change in the H-bond acceptor residue from HisH to HisF moves the inhibitor to a slightly more solvent-exposed pose (pose 2), in which the benzyl ring is pointing toward the hydrophobic area of the 49-PGVG sequence in HisH (ring up conformation), keeping the ligand sandwiched between HisH and HisF. Thermal fluctuations allow transient changes in the H-bonding interactions of the hydroxyethyl group of 3, which can form H-bonds with hP10 (an important residue in the IGPS protein network), without significantly changing the position of the ligand relative to the interface (pose 2′). Thus, microsecond time-scale MD simulations indicate that exchange of polar and hydrophobic interactions between 3 and the two domains holds the inhibitor at the interface, stabilizing a rigid open structure with limited breathing motion (Figure S9), as observed in shorter (nanosecond) time-scale simulations. In summary, microsecond MD simulations support the binding mode of 3 in the proximity of secondary structure elements that are crucial to the allosteric communication of IGPS, including the Ω-loop and the oxyanion strand in HisH, and the fα3 helix in HisF, which is consistent with the docking analysis.
Figure 6 describes the 3–IGPS contacts that affect allosteric communication in IGPS and inhibit glutaminase activity. Submicrosecond MD simulations of the allosteric signal transduction promoted by PRFAR binding showed how changes in hydrophobic contacts at the bottom of the HisF barrel propagate to HisF side R, changing ionic interactions among the solvent-exposed helices fα2, fα3, and hα1, with changes in the relative positions of helices fα2 and fα3 due to formation of specific salt bridges (Figure 6).13 Strong interactions of 3 with the IGPS fα3–fβ3 turn (by H-bonding with f D98 and establishing hydrophobic contacts with the benzyl ring) perturb thermal fluctuations of the fα3 residues, promoting ionic contacts with fα2 side chains that differ from those stabilized by PRFAR binding. An important aspect is the fact that the presence of the inhibitor promotes formation of the f R59–f D85 salt bridge, an ionic interaction that brings the fα2 and fα3 helices closer together, never observed in apo IGPS or binary complexes. Notably, NMR measurements indicate that fα3, including the f D85 residue, is a structural element significantly disturbed by binding of 3 to the binary PRFAR-bound complex.
Figure 6.

Mechanism of IGPS allosteric signal disruption by inhibitor binding. Changes in ionic interactions among the fα2, fα3, and hα1 helices (bottom row) in the apo IGPS complex (blue, center column) induced by PRFAR binding (red, right column) and subsequently altered by inhibitor binding (black, left column). Disruption of the allosteric signal launched by PRFAR manifested as hampered oxyanion strand flip, which is initiated on the submicrosecond time scale in the binary complex while absent in the 3–IGPS ternary complex (middle row). The flip of the 49-PGVG strand is crucial for glutaminase activity because it allows stabilization of a four-center intermediate (FCI) in the oxyanion hole (top row).
The allosteric effect of binding of PRFAR to HisF is transduced by rupture of the hP10–hV51 H-bonding interaction between the Ω-loop (hP10) and the 49-PGVG sequence (hV51) in HisH, which is an essential event for freeing the oxyanion strand.13 In fact, if the 49-PGVG strand can easily flip along the backbone, the NH amide group of hV51 can likely be found in a catalytically active conformation13 that stabilizes a charged four-center tetrahedral intermediate in the “oxyanion hole” during Gln hydrolysis (Figure 6). Our microsecond MD simulations of the ternary 3–IGPS complex clearly indicate how the hP10–hV51 H-bond is quickly broken and transiently substituted with the hP10–3 H-bonding interaction, because of the presence of the interfacial inhibitor near the Ω-loop and the 49-PGVG strand (Figure 5). Despite the rupture of the hP10–hV51 H-bond as in the PRFAR-bound complex, however, the oxyanion flip does not occur in the ternary complex during the 2.5 μs MD trajectory (Figure 6), although it is rapidly initiated in the binary PRFAR complex (<50 ns). The origin of the hindrance for oxyanion flipping can be traced back to the effect of the interfacial inhibitor on the breathing motion and the direct interactions with the oxyanion strand. These results suggest that breaking of the hP10–hV51 H-bond is necessary but not sufficient for flipping the oxyanion. Thus, inhibitor binding at the HisF–HisH interface uncouples the changes in motion induced by PRFAR binding at the bottom of the HisF barrel and the HisH motions that lead to a catalytically active conformation in the glutaminase active site. This likely leads to a reduction in the degree of conformational sampling of the fully active enzyme that is normally induced by PRFAR binding, thereby causing a decrease in glutaminase activity.
CONCLUSIONS
Disruption and characterization of the allosteric signal in IGPS have been achieved by docking of a small organic molecule that interferes with the IGPS allosteric pathways and inhibits the catalytic activity. These findings are significant for the development of allosteric ligands in general and potentially for IGPS because it is involved in fundamental biosynthetic pathways of important human pathogens. Characterization of secondary structure elements that are essential for IGPS allostery by MD simulations and NMR methods has revealed a noncompetitive allosteric inhibitor of IGPS for the first time, i.e., a potential druglike molecule that suppresses catalytic activity without competing with the substrate or the effector for their binding sites. Molecular docking and microsecond MD simulations in conjunction with ITC, NMR techniques, and enzyme kinetics support certain binding modes of the ligands and disruption of allosteric mechanisms, while further work would be required to unambiguously establish both of these fundamental aspects. We observed that the largest changes in chemical shifts due to binding of 3 to the binary PRFAR–IGPS complex at important sites of allosteric communication are consistent with MD simulations. Kinetic assays show that IGPS glutaminase activity is substantially suppressed upon binding of the inhibitor to the HisH–HisF interface. Microsecond MD simulations provide a detailed characterization of the inhibition mechanism at the molecular level. The allosteric inhibitor is able to uncouple motions induced by the effector ligand with essential motions in the distant active site, favoring an inactive conformation of this V-type enzyme. Moreover, our findings pave the way for the development of conceptually new molecular inhibitors, involving noncompetitive ligands that target specific protein structural elements found by NMR experiments and comprehensive computational modeling rather than resulting from a serendipitous search.
Supplementary Material
Acknowledgments
Funding
Funding for this work was supported by National Institutes of Health Grant GM106121 (1R01GM10621-01A1) to J.P.L. and V.S.B., National Institutes of Health Training Grant T32GM008283 to G.M., École Normale Supérieure de Lyon Fonds Recherche Grant 900/S81/BS81-FR14 to I.R., and National Institutes of Health Grant RC2GM093307 to the National Resource for Biomedical Supercomputing and the Pittsburgh Supercomputing Center.
We acknowledge supercomputer time from the National Energy Research Scientific Computing Center (NERSC), High Performance Computing facilities at Yale University (New Haven, CT). Anton computer time was provided by the National Resource for Biomedical Supercomputing and the Pittsburgh Supercomputing Center (PSC). I.R. thanks Dr. Markus Dittrich and other staff at NRBSC and PSC for the support while using the Anton machine.
Footnotes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.6b00859.
•Supplementary analysis of computational and NMR results (PDF)
References
- 1.Fenton AW. Allostery: an illustrated definition for the ‘second secret of life’. Trends Biochem Sci. 2008;33:420–425. doi: 10.1016/j.tibs.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monod J, Wyman J, Changeux JP. On the Nature of Allosteric Transitions: a Plausible Model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 3.Koshland DE, Nemethy G, Filmer D. Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits. Biochemistry. 1966;5:365–368. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 4.Changeux JP. 50 years of allosteric interactions: the twists and turns of the models. Nat Rev Mol Cell Biol. 2013;14:819–829. doi: 10.1038/nrm3695. [DOI] [PubMed] [Google Scholar]
- 5.Laskowski RA, Gerick F, Thornton JM. The structural basis of allosteric regulation in proteins. FEBS Lett. 2009;583:1692–1698. doi: 10.1016/j.febslet.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 6.Motlagh HN, Wrabl JO, Li J, Hilser VJ. The ensemble nature of allostery. Nature. 2014;508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nussinov R, Tsai CJ. Allostery in Disease and in Drug Discovery. Cell. 2013;153:293–305. doi: 10.1016/j.cell.2013.03.034. [DOI] [PubMed] [Google Scholar]
- 8.Kar G, Keskin O, Gursoy A, Nussinov R. Allostery and population shift in drug discovery. Curr Opin Pharmacol. 2010;10:715–722. doi: 10.1016/j.coph.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Süel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 10.Cui Q, Karplus M. Allostery and cooperativity revisited. Protein Sci. 2008;17:1295–1307. doi: 10.1110/ps.03259908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manley G, Rivalta I, Loria JP. Solution NMR and Computational Methods for Understanding Protein Allostery. J Phys Chem B. 2013;117:3063–3073. doi: 10.1021/jp312576v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.VanWart AT, Eargle J, Luthey-Schulten Z, Amaro RE. Exploring Residue Component Contributions to Dynamical Network Models of Allostery. J Chem Theory Comput. 2012;8:2949–2961. doi: 10.1021/ct300377a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rivalta I, Sultan MM, Lee NS, Manley GA, Loria JP, Batista VS. Allosteric pathways in imidazole glycerol phosphate synthase. Proc Natl Acad Sci U S A. 2012;109:E1428–1436. doi: 10.1073/pnas.1120536109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feher VA, Durrant JD, Van Wart AT, Amaro RE. Computational approaches to mapping allosteric pathways. Curr Opin Struct Biol. 2014;25:98–103. doi: 10.1016/j.sbi.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mino-Galaz GA. Allosteric Communication Pathways and Thermal Rectification in PDZ-2 Protein: A Computational Study. J Phys Chem B. 2015;119:6179–6189. doi: 10.1021/acs.jpcb.5b02228. [DOI] [PubMed] [Google Scholar]
- 16.Perica T, Kondo Y, Tiwari SP, McLaughlin SH, Kemplen KR, Zhang XW, Steward A, Reuter N, Clarke J, Teichmann SA. Evolution of oligomeric state through allosteric pathways that mimic ligand binding. Science. 2014;346:1254346. doi: 10.1126/science.1254346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ricci CG, Silveira RL, Rivalta I, Batista VS, Skaf MS. Allosteric Pathways in the PPARγ-RXRα nuclear receptor complex. Sci Rep. 2016;6:19940. doi: 10.1038/srep19940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lisi GP, Manley GA, Hendrickson H, Rivalta I, Batista VS, Loria JP. Dissecting Dynamic Allosteric Pathways Using Chemically Related Small-Molecule Activators. Structure. 2016;24:1155–1166. doi: 10.1016/j.str.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gohara DW, Di Cera E. Allostery in trypsin-like proteases suggests new therapeutic strategies. Trends Biotechnol. 2011;29:577–585. doi: 10.1016/j.tibtech.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discovery. 2009;8:733–750. doi: 10.1038/nrd2927. [DOI] [PubMed] [Google Scholar]
- 21.Changeux JP. The concept of allosteric modulation: an overview. Drug Discovery Today: Technol. 2013;10:e223–228. doi: 10.1016/j.ddtec.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 22.van der Westhuizen ET, Valant C, Sexton PM, Christopoulos A. Endogenous Allosteric Modulators of G Protein-Coupled Receptors. J Pharmacol Exp Ther. 2015;353:246–260. doi: 10.1124/jpet.114.221606. [DOI] [PubMed] [Google Scholar]
- 23.Wootten D, Christopoulos A, Sexton PM. Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discovery. 2013;12:630–644. doi: 10.1038/nrd4052. [DOI] [PubMed] [Google Scholar]
- 24.Christopoulos A. Allosteric binding sites on cell-surface receptors: novel targets for drug discovery. Nat Rev Drug Discovery. 2002;1:198–210. doi: 10.1038/nrd746. [DOI] [PubMed] [Google Scholar]
- 25.Douangamath A, Walker M, Beismann-Driemeyer S, Vega-Fernandez MC, Sterner R, Wilmanns M. Structural evidence for ammonia tunneling across the (beta/alpha)(8) barrel of the imidazole glycerol phosphate synthase bienzyme complex. Structure. 2002;10:185–193. doi: 10.1016/s0969-2126(02)00702-5. [DOI] [PubMed] [Google Scholar]
- 26.Klem TJ, Davisson VJ. Imidazole glycerol phosphate synthase: the glutamine amidotransferase in histidine biosynthesis. Biochemistry. 1993;32:5177–5186. doi: 10.1021/bi00070a029. [DOI] [PubMed] [Google Scholar]
- 27.Gomez MJ, Neyfakh AA. Genes Involved in Intrinsic Antibiotic Resistance of Acinetobacter baylyi. Antimicrob Agents Chemother. 2006;50:3562–3567. doi: 10.1128/AAC.00579-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Breitbach K, Köhler J, Steinmetz I. Induction of protective immunity against Burkholderia pseudomallei using attenuated mutants with defects in the intracellular life cycle. Trans R Soc Trop Med Hyg. 2008;102:S89–S94. doi: 10.1016/S0035-9203(08)70022-1. [DOI] [PubMed] [Google Scholar]
- 29.Chittur SV, Chen Y, Davisson VJ. Expression and purification of imidazole glycerol phosphate synthase from Saccharomyces cerevisiae. Protein Expression Purif. 2000;18:366–377. doi: 10.1006/prep.2000.1207. [DOI] [PubMed] [Google Scholar]
- 30.Klem TJ, Chen Y, Davisson VJ. Subunit interactions and glutamine utilization by Escherichia coli imidazole glycerol phosphate synthase. J Bacteriol. 2001;183:989–996. doi: 10.1128/JB.182.3.989-996.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amaro RE, Sethi A, Myers RS, Davisson VJ, Luthey-Schulten ZA. A network of conserved interactions regulates the allosteric signal in a glutamine amidotransferase. Biochemistry. 2007;46:2156–2173. doi: 10.1021/bi061708e. [DOI] [PubMed] [Google Scholar]
- 32.Myers RS, Amaro RE, Luthey-Schulten ZA, Davisson VJ. Reaction Coupling through Interdomain Contacts in Imidazole Glycerol Phosphate Synthase. Biochemistry. 2005;44:11974–11985. doi: 10.1021/bi050706b. [DOI] [PubMed] [Google Scholar]
- 33.Amaro RE, Myers RS, Davisson VJ, Luthey-Schulten ZA. Structural elements in IGP synthase exclude water to optimize ammonia transfer. Biophys J. 2005;89:475–487. doi: 10.1529/biophysj.104.058651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lipchock JM, Loria JP. Nanometer propagation of millisecond motions in V-type allostery. Structure. 2010;18:1596–1607. doi: 10.1016/j.str.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.List F, Bocola M, Haeger MC, Sterner R. Constitutively Active Glutaminase Variants Provide Insights into the Activation Mechanism of Anthranilate Synthase. Biochemistry. 2012;51:2812–2818. doi: 10.1021/bi201618v. [DOI] [PubMed] [Google Scholar]
- 36.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 37.Greenwood JR, Calkins D, Sullivan AP, Shelley JC. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J Comput-Aided Mol Des. 2010;24:591–604. doi: 10.1007/s10822-010-9349-1. [DOI] [PubMed] [Google Scholar]
- 38.Sharma H, Landau MJ, Sullivan TJ, Kumar VP, Dahlgren MK, Jorgensen WL, Anderson KS. Virtual screening reveals allosteric inhibitors of the Toxoplasma gondii thymidylate synthase-dihydrofolate reductase. Bioorg Med Chem Lett. 2014;24:1232–1235. doi: 10.1016/j.bmcl.2013.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins: Struct, Funct, Genet. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 42.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 43.Shaw DE, Deneroff MM, Dror RO, Kuskin JS, Larson RH, Salmon JK, Young C, Batson B, Bowers KJ, Chao JC, Eastwood MP, Gagliardo J, Grossman JP, Ho CR, Ierardi DJ, Kolossvary I, Klepeis JL, Layman T, Mcleavey C, Moraes MA, Mueller R, Priest EC, Shan YB, Spengler J, Theobald M, Towles B, Wang SC. Anton, a special-purpose machine for molecular dynamics simulation. Commun ACM. 2008;51:91–97. [Google Scholar]
- 44.Tuckerman M, Berne BJ, Martyna GJ. Reversible multiple time scale molecular dynamics. J Chem Phys. 1992;97:1990–2001. [Google Scholar]
- 45.Shan YB, Klepeis JL, Eastwood MP, Dror RO, Shaw DE. Gaussian split Ewald: A fast Ewald mesh method for molecular simulation. J Chem Phys. 2005;122:054101. doi: 10.1063/1.1839571. [DOI] [PubMed] [Google Scholar]
- 46.Lippert RA, Predescu C, Ierardi DJ, Mackenzie KM, Eastwood MP, Dror RO, Shaw DE. Accurate and efficient integration for molecular dynamics simulations at constant temperature and pressure. J Chem Phys. 2013;139:164106. doi: 10.1063/1.4825247. [DOI] [PubMed] [Google Scholar]
- 47.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 48.Lipchock J, Loria JP. Millisecond dynamics in the allosteric enzyme imidazole glycerol phosphate synthase (IGPS) from Thermotoga maritima. J Biomol NMR. 2009;45:73–84. doi: 10.1007/s10858-009-9337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRpipe - a Multidimensional Spectral Processing System Based on Unix Pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 50.Goddard TD, Kneller DG. SPARKY. University of California; San Francisco: 2008. [Google Scholar]
- 51.Pommier Y, Marchand C. Interfacial inhibitors: targeting macromolecular complexes. Nat Rev Drug Discovery. 2012;11:233. doi: 10.1038/nrd3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.