

Abstract
Cross‐presentation by MHC class I molecules allows the detection of exogenous antigens by CD8+ T lymphocytes. This process is crucial to initiate cytotoxic immune responses against many pathogens (i.e., Toxoplasma gondii) and tumors. To achieve efficient cross‐presentation, dendritic cells (DCs) have specialized endocytic pathways; however, the molecular effectors involved are poorly understood. In this work, we identify the small GTPase Rab22a as a key regulator of MHC‐I trafficking and antigen cross‐presentation by DCs. Our results demonstrate that Rab22a is recruited to DC endosomes and phagosomes, as well as to the vacuole containing T. gondii parasites. The silencing of Rab22a expression did not affect the uptake of exogenous antigens or parasite invasion, but it drastically reduced the intracellular pool and the recycling of MHC‐I molecules. The knockdown of Rab22a also hampered the cross‐presentation of soluble, particulate and T. gondii‐associated antigens, but not the endogenous MHC‐I antigen presentation through the classical secretory pathway. Our findings provide compelling evidence that Rab22a plays a central role in the MHC‐I endocytic trafficking, which is crucial for efficient cross‐presentation by DCs.
Keywords: cross‐presentation, dendritic cells, MHC‐I molecules, small GTPase Rab22a, Toxoplasma gondii
Subject Categories: Immunology, Membrane & Intracellular Transport
Introduction
Dendritic cells (DCs) are the most potent antigen‐presenting cells capable of initiating adaptive immune responses. These cells process and present antigens in the context of class I or class II molecules of the major histocompatibility complex (MHC) to trigger CD8+ or CD4+ T‐cell activation, respectively. Antigens presented by MHC‐I can be either endogenous or exogenous. The former antigen presentation scenario is referred to as the “classical pathway” and the latter as “cross‐presentation”. Cross‐presentation is pivotal to establish CD8+ cytotoxic immune responses against tumors and several pathogens, including the intracellular parasite Toxoplasma gondii 1. Compared to other professional phagocytes, DCs have developed highly specialized mechanisms of internalization to achieve an efficient cross‐presentation 2. Indeed, the acidification of the phagosomal content of DCs has proved to be a very slow process due to the production of reactive oxygen species by the NADPH oxidase NOX2 3 and to an incomplete activation of the V‐ATPase 4. The combination of a high pH and low levels of lysosomal proteases limits the proteolytic activity in DC phagosomes 5. This limited phagosomal proteolysis may leave intact proteins or large polypeptides for export to the cytosol, most likely via a Sec61‐dependent mechanism 6, for further degradation by the proteasome. Then, the processed peptides are translocated by TAP1/2 transporters into the lumen of the ER or back into phagosomes, since these organelles recruit components from the ER–Golgi intermediate compartment by the action of the SNARE Sec22b 7. The translocated peptides are trimmed by ER aminopeptidases 8 and their analogues, such as IRAP 9, prior to loading on MHC‐I molecules by the peptide loading complex 10. Finally, the MHC‐I/peptide complexes are transported to the cell surface in order to trigger CD8+ T‐cell activation.
Although some major advances in the understanding of MHC‐I intracellular trafficking during cross‐presentation have recently been made, in particular those regarding the importance of Rab11a and endosomal recycling compartments (ERC) 11, the molecular effectors of the endocytic network that regulate MHC‐I presentation remain poorly understood. Even less understood are the interactions that the parasitophorous vacuole (PV) of T. gondii establishes with the endocytic and exocytic pathways to intercept MHC‐I molecules for efficient CD8+ T‐cell activation during DC infection 12. In this study, we focused on the small GTPase Rab22a, which is a molecule that modulates central aspects of MHC‐I and CD1a endocytic recycling 13, 14. Rab proteins mediate most intracellular membrane trafficking events by cycling between an active GTP‐bound state and an inactive GDP‐bound state. Working as molecular switches, different Rabs associate with specific membrane compartments giving a unique surface identity, which is required for the recruitment of molecules involved in targeting specificity 15. Rab22a has been described in early and late endosomes, but not in lysosomes in CHO cells 16. Rab22a also controls the transport of the transferrin receptor (TfR) from sorting to recycling endosomes 17, and it is associated with tubular recycling intermediates containing MHC‐I molecules and transferrin 13. In the latter work, the authors have also suggested that the activation of Rab22a is required for tubule formation, while the inactive state of Rab22a is necessary for the final fusion of these tubules derived from recycling compartments with the plasma membrane 13. Moreover, Rab22a participates in the phagosome/vacuole physiology during infection and the intracellular trafficking of several pathogens, such as Anaplasma phagocytophilum 18, Neisseria meningitidis 19, Borrelia burgdorferi 20 and Mycobacterium tuberculosis 21. Nevertheless, although this Rab‐GTPase plays important roles in the endocytic pathway by modulating endosomal and phagosomal functions, its role in DCs remains unknown.
In this work, we demonstrate that Rab22a is widely distributed in the endocytic network of DCs and that it is early recruited to endosomes and phagosomes, as well as to the T. gondii PV, a parasite‐modified compartment known to be distinct from a phagosome in infected DCs. We demonstrate that Rab22a controls MHC‐I intracellular distribution, recycling and trafficking to DC phagosomes. The silencing of Rab22a expression in DCs impacts on the ability of these cells to achieve an efficient cross‐presentation of soluble and particulate exogenous antigens, as well as T. gondii‐derived soluble antigens. We conclude that Rab22a plays an essential role in MHC‐I endocytic trafficking during antigen cross‐presentation by DCs.
Results and Discussion
Early recruitment of Rab22a to DC endosomes and phagosomes
It is known that Rab22a regulates the intracellular transport of MHC‐I and TfR in several cell lines 13, 14, 17. Thus, it was interesting to assess the localization of this protein in the endocytic pathway of DCs. By immunofluorescence (IF), we analyzed the colocalization of endogenous Rab22a with three endocytic markers (EEA1 for early endosomes, Lamp1 for late endosomes/lysosomes, and TfR for recycling compartments) in mouse bone marrow‐derived dendritic cells (BMDCs). Rab22a was present in the three compartments; however, in contrast to previous reports, the Pearson correlation coefficient (Pcc) determined for Lamp1 was markedly high (0.8412), suggesting that in DCs, Rab22a was present not only in early endosomes (Pcc, 0.4317 for EEA1) but also in late endocytic compartments (Fig 1A, upper and middle panels). Rab22a was also present in recycling compartments as indicated by a high localization with the TfR (Pcc, 0.6409; Fig 1A, lower panel). The presence of Rab22a in the recycling center was confirmed by co‐staining with Rab11a, a GTPase that is specific for this compartment (Fig EV1A). In summary, our results indicate that Rab22a has a ubiquitous distribution in the endocytic and recycling compartments of DCs.
Figure 1. Cellular distribution of Rab22a and its recruitment to BMDC endosomes and phagosomes.
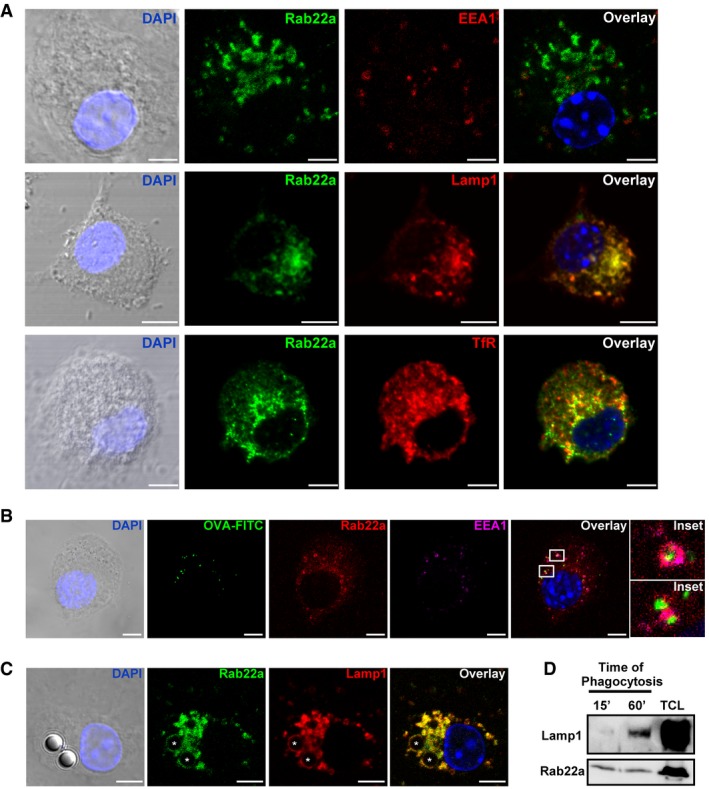
- Confocal microscopy analysis showing the localization of endogenous Rab22a (green) and different endocytic markers (red): early endosomal marker EEA1, late endosome/lysosomal marker Lamp1, and the recycling compartment marker TfR in BMDCs at steady state. The means ± SEM of the Pearson correlation coefficients (Pcc) from Rab22a and EEA1 (0.4317 ± 0.05101), Lamp1 (0.8412 ± 0.03562), and TfR (0.6409 ± 0.02999) were estimated from 15 images analyzed for each marker. Scale bars: 5 μm.
- Colocalization of endogenous Rab22a (red) and EEA1 (magenta) around endosomes containing fluorescent soluble OVA (OVA‐FITC, green) after 30 min of internalization by BMDCs. More than 60% of the endosomes were positive for Rab22a and EEA1. The indicated boxes are shown at higher magnification in the insets. Scale bars: 5 μm.
- IF detection of endogenous Rab22a (green) and Lamp1 (red) after 1 h of phagocytosis of 3‐μm latex beads (LB) in BMDCs. Around 50% of the phagosomes were double positive for Rab22a and Lamp1. Asterisks indicate the LB. Scale bars: 5 μm.
- BMDCs were incubated with 3‐μm magnetic beads for 15 min at 37°C and chased for 0 or 45 min. The panel shows immunoblotting of purified phagosomes, and the total cell lysates (TCL) analyzed for Lamp1 and Rab22a. A total protein amount of 10 μg and 50 μg was loaded for purified phagosomes and TCL, respectively. The blot is representative of three independent experiments.
Figure EV1. Subcellular localization of Rab22a in BMDC and JAWS‐II DC.
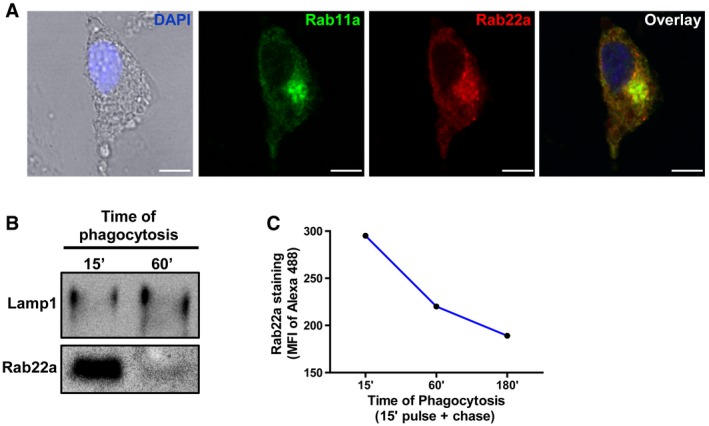
- Confocal microscopy analysis showing endogenous Rab22a (red) co‐localizing with endogenous Rab11a (green) at the recycling center in BMDCs at steady state. The nuclear marker DAPI (blue) and DIC images are shown on the left panel. Scale bars: 5 μm.
- JAWS‐II DC was incubated with 3‐μm magnetic beads for 15 min at 37°C and chased for 0 and 45 min. Immunoblotting of purified phagosomes was analyzed for Lamp1 and Rab22a. Ten micrograms of protein was loaded on each lane for purified phagosomes.
- JAWS‐II DC was incubated with 3‐μm LB for 15 min at 37°C and chased for 0, 45, or 165 min. Rab22a staining on isolated phagosomes was measured by FACS at the indicated time periods. Data are representative of three independent experiments.
Source data are available online for this figure.
The recruitment of Rab22a to compartments containing soluble and particulate exogenous antigens was then explored. We observed that many early endosomes, as labeled with the EEA1 marker, were positive for Rab22a and internalized soluble fluorescent ovalbumin (OVA) after 30 min of uptake (Fig 1B). When BMDCs were fed during 1 h with 3‐μm latex beads (LB), a clear recruitment of Rab22a and Lamp1 was observed around LB phagosomes (Fig 1C). To confirm these observations, the presence of Rab22a in purified BMDC phagosomes was assessed by Western blot. As expected, an increase in Lamp1 over time was observed, whereas Rab22a was recruited as soon as 15 min after internalization and remained in the phagosomes for up to 60 min (Fig 1D). Similar results were obtained with the JAWS‐II DC line, where Rab22a was detected in purified phagosomes at 15 min after internalization and decreased at 60 min (Fig EV1B). These observations were confirmed through the quantification of Rab22a levels in isolated phagosomes by flow cytometry (Fig EV1C). Altogether, these experiments demonstrate that Rab22a is distributed widely in the endocytic pathway of DCs and that it is early and efficiently recruited to endosomes and phagosomes containing internalized material.
The localization of Rab22a in other cell types is mainly restricted to early, late, and recycling endosomes, but it is excluded from other compartments with higher proteolytic capacity, such as lysosomes 16. Intriguingly, the location of Rab22a in DCs seems to be slightly different. Indeed, this GTPase was found to be more abundant in recycling endosomes and late endosomes/lysosomes. It can be hypothesized that this difference in Rab22a location, with a wider distribution along the endocytic pathway of DCs, is important for the interception of exogenous antigens after their internalization.
The intracellular trafficking of MHC‐I molecules is a Rab22a‐dependent process
To assess the role of Rab22a in the endocytic pathway of DCs, we generated stable knockdown JAWS‐II DCs (Rab22a KD cells) by means of lentivirus‐delivered short hairpin RNAs (shRNA). As shown in the immunoblot of Fig 2A, the expression of Rab22a was lowered to 40% as compared to DCs transduced with the scramble shRNA used as control (Scramble cells). This level of KD efficiency was confirmed by RNA extraction and qPCR (Fig 2B). Despite this modest KD efficacy, we observed a drastic effect on the intracellular distribution of MHC‐I molecules, as shown by IF detection and confocal microscopy. In particular, we noticed the disappearance of the perinuclear MHC‐I pool corresponding to the recycling center that markedly colocalized with Rab22a in Scramble DCs (Fig 2C). In contrast, the plasma membrane‐associated labeling did not seem to be affected (Fig 2C). This alteration in MHC‐I distribution was then assessed by flow cytometry. MHC‐I molecules exposed on the plasma membrane were labeled with a FITC‐conjugated anti‐H‐2Kb‐specific antibody in non‐permeabilized cells, while the internal pool was labeled after cell fixation and permeabilization with saponin. As depicted in Figs 2D and EV2A, the amount of MHC‐I expressed on the cell surface did not exhibit significant differences between KD and control cells. In contrast, the total amount of MHC‐I (cell surface + intracellular pool) was strongly decreased in the Rab22a KD DCs. To confirm these results, we performed this set of experiments also with primary BMDCs. We silenced the expression of Rab22a and we observed, by confocal microscopy (Fig EV2B) and by flow cytometry (Fig EV2C and D), a clear disappearance of the MHC‐I intracellular pool, while the amount of MHC‐I present at the cell surface remained unchanged, as compared to Scramble BMDCs. Therefore, it can be assumed that the reduction in the expression of Rab22a causes an important decrease in the intracellular pool of MHC‐I molecules in both, JAWS‐II DCs and BMDCs.
Figure 2. Rab22a controls the intracellular trafficking of MHC‐I molecules in DCs.
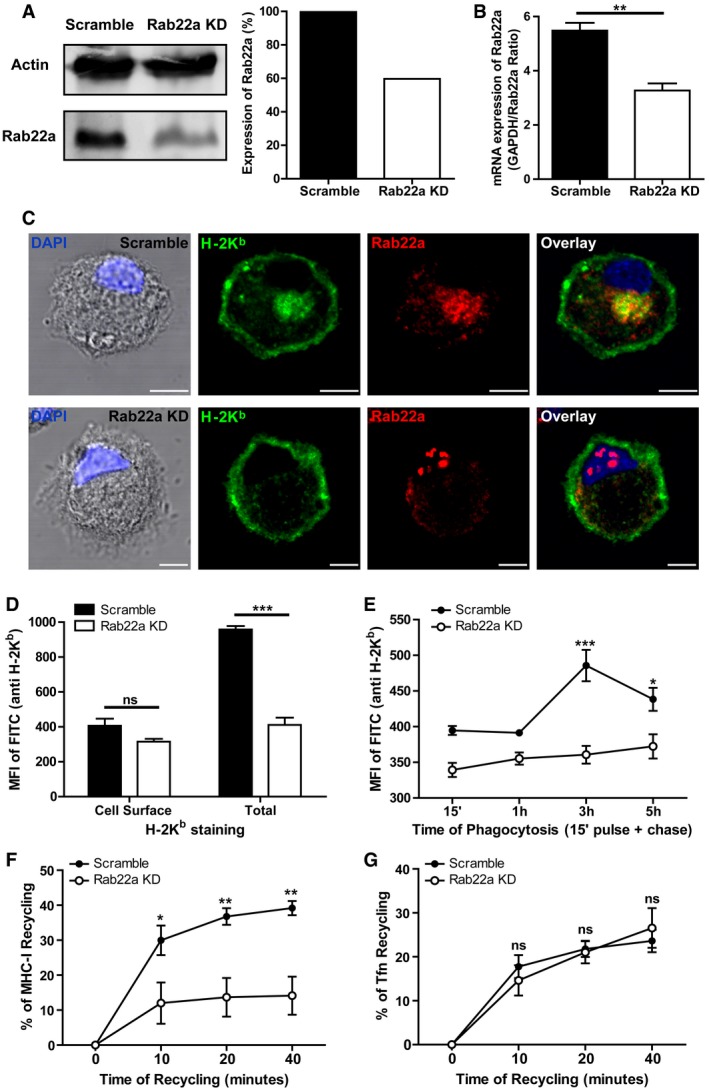
- Immunoblotting and densitometry quantification of Rab22a in JAWS‐II DCs infected with lentiviruses encoding a random sequence (Scramble) and a shRNA specific for silencing Rab22a (Rab22a KD). Data are representative of at least three independent experiments.
- RNA extraction and qPCR quantification of Rab22a in the same cells analyzed in (A). Data show mean ± SEM of triplicate values and are representative of two independent experiments. **P = 0.0047.
- IF labeling and confocal microscopy analysis showing the distribution of MHC‐I molecules (H‐2Kb, green) and Rab22a (red) in Scramble and Rab22a KD JAWS‐II DCs. Nuclei stained with DAPI and DIC images are shown on the left. Overlay is shown in the right panels. Scale bars: 5 μm. Data are representative of at least 30 images analyzed for each experimental condition from three independent experiments.
- FACS analysis of MHC‐I labeled in intact (cell surface) and permeabilized (total) Scramble and Rab22a KD JAWS‐II cells. Data represent mean ± SEM of triplicate values and are representative of three independent experiments. P = 0.1082 (ns) and ***P = 0.0003.
- MHC‐I staining on isolated phagosomes was measured by FACS at the indicated time periods after 3‐μm LB internalization by Scramble and Rab22a KD JAWS‐II DCs. Data represent mean ± SEM of three independent experiments. ***P < 0.001 at 3 h and *P < 0.05 at 5 h between both DC types.
- MHC‐I molecules recycling ability was measured by FACS at the indicated time periods by Scramble and Rab22a KD JAWS‐II DCs. Data represent mean ± SEM of three independent experiments. *P < 0.05 at 10 min and **P < 0.01 at 20 and 40 min.
- The transferrin (Tfn) recycling ability was measured by FACS at the indicated time periods by Scramble and Rab22a KD JAWS‐II DCs. Data represent mean ± SEM of three independent experiments. P > 0.05 (ns) at 10, 20, and 40 min.
Figure EV2. Rab22a expression controls the intracellular pool of MHC‐I molecules in DCs.
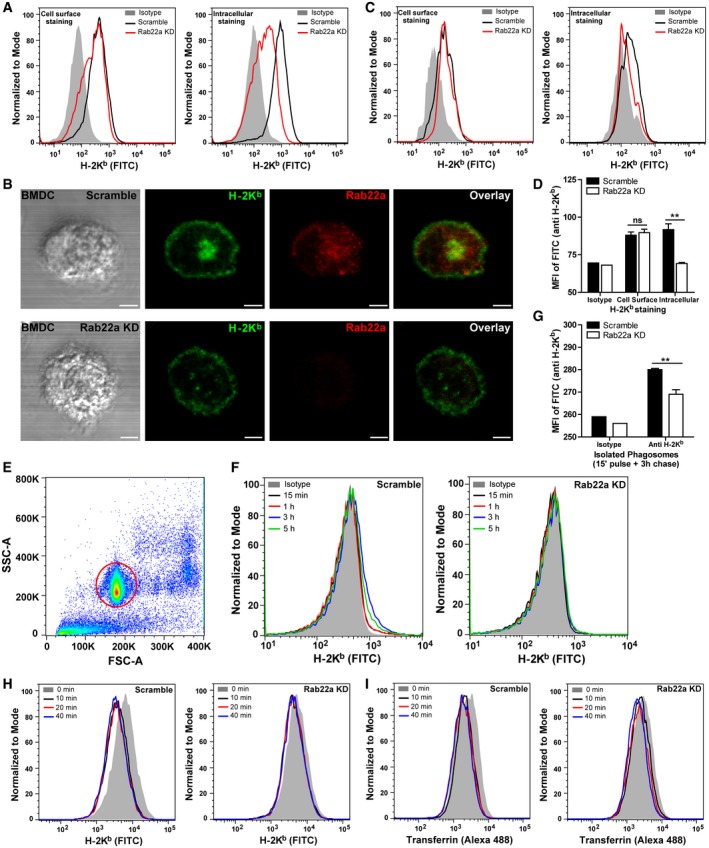
- Representative FACS profile of intact (left panel) and saponin‐permeabilized (right panel) Scramble and Rab22a KD JAWS‐II DCs after H‐2Kb staining.
- IF labeling and confocal microscopy analysis showing the distribution of MHC‐I molecules (H‐2Kb, green) and Rab22a (red) in Scramble and Rab22a KD BMDCs at steady state. DIC images are shown in the left panels. Overlay is shown in the right panels. Scale bars: 5 μm.
- Representative FACS profiles of intact (left panel) and saponin‐permeabilized (right panel) Scramble and Rab22a KD BMDCs after H‐2Kb staining.
- FACS analysis of MHC‐I labeled in intact (cell surface) and permeabilized (intracellular) Scramble and Rab22a KD BMDCs. Data represent mean ± SEM of triplicate values. P = 0.6283 (ns) and **P = 0.0046. The two‐tailed Student's unpaired t‐test was performed.
- Representative dot plot obtained from flow cytometry analysis after the isolation of 3‐μm LB phagosomes. The population corresponding to individual phagosomes (red circle) is easily distinguished from the cellular debris generated after sample processing. More than ten thousand phagosomes from each condition were analyzed in each experiment.
- Representative FACS profiles of H‐2Kb staining on isolated phagosomes at the indicated time periods after 3‐μm LB internalization by Scramble and Rab22a KD JAWS‐II DCs.
- MHC‐I staining on isolated phagosomes was measured by FACS at 3 h post‐internalization of 3‐μm LB by Scramble and Rab22a KD BMDCs. Data represent mean ± SEM of triplicate values. **P = 0.0070. The two‐tailed Student's unpaired t‐test was performed.
- Representative FACS profiles of MHC‐I molecules (H‐2Kb FITC) recycling at the indicated time periods by Scramble and Rab22a KD JAWS‐II DCs.
- Representative FACS profiles of TfR (Alexa 488‐transferrin) recycling at the indicated time periods by Scramble and Rab22a KD JAWS‐II DCs.
The remarkable changes observed in the intracellular pool of MHC‐I molecules prompted us to investigate whether Rab22a was controlling the acquisition of MHC‐I molecules to DC phagosomes, a process already described for Rab11a KD DCs 11. To address this issue, we isolated LB‐containing phagosomes from Scramble and Rab22a KD JAWS‐II DCs at 15 min, 1, 3, and 5 h post‐internalization and we analyzed the presence of MHC‐I by flow cytometry (Fig EV2E). A striking impairment in the phagosomal recruitment of MHC‐I molecules was observed in Rab22a KD DCs, as compared to Scramble DCs (Figs 2E and EV2F). Interestingly, results showed the existence of two different kinetics for both cell types: while in Scramble DCs, phagosomal MHC‐I labeling decreased after 3 h post‐internalization, in Rab22a KD DCs, the staining was lower but sustained over time (Fig 2E), indicating that Rab22a may also be playing an important role during MHC‐I molecules export from the phagosome to the cell surface. To confirm the defect of MHC‐I phagosomal acquisition after the silencing of Rab22a, we isolated phagosomes at 3 h post‐internalization from primary transduced BMDCs. Again, the flow cytometric analysis demonstrated a significant reduction in MHC‐I molecules in Rab22a KD BMDC phagosomes, as compared to Scramble cells (Fig EV2G). Our results demonstrate that Rab22a has a crucial role in the regulation of the MHC‐I phagosomal recruitment by DCs.
Although previous studies carried out in other cell types have demonstrated that Rab22a controls the recycling of MHC‐I molecules, data regarding the role of this GTPase in the fast recycling of TfR are controversial 13, 17. Therefore, these aspects of the endocytic trafficking were assessed herein. We evaluated the MHC‐I and TfR recycling capacities of Scramble and Rab22a KD JAWS‐II DCs by flow cytometry. Thus, the decrease in the mean fluorescent intensities (MFI) of FITC‐conjugated H‐2Kb, and Alexa 488‐conjugated transferrin were measured in order to assess the efficiency of molecular recycling to the plasma membrane after binding, internalization, and cell surface acid stripping. Interestingly, in Rab22a KD DCs, we observed a significant recycling capacity inhibition for MHC‐I (Figs 2F and EV2H), but not for TfR (Figs 2G and EV2I), as compared to Scramble DCs.
Taken together, these results support the hypothesis that Rab22a is crucial to stabilize the intracellular pool of MHC‐I molecules at the ERC, where it colocalizes with Rab11a and TfR. Moreover, we observed that Rab22a is necessary for the normal recruitment of MHC‐I molecules to DC phagosomes. In this sense, it is worth mentioning that the acquisition kinetics of MHC‐I to Scramble DC phagosomes is in accordance with the phagosomal recruitment and subsequent disappearance of Rab22a. The role of Rab22a in MHC‐I molecules recycling has been already described by others 13, 14; however, the modulatory capacity of this small GTPase has never been addressed in immune cells. In our study, a severe defect of MHC‐I molecules recycling by Rab22a KD DCs was observed. Interestingly, the fast recycling, as evaluated by the use of fluorescent‐labeled transferrin, is not altered in these cells.
Rab22a is required for antigen cross‐presentation by DC
Since Rab22a impacted on the intracellular trafficking of MHC‐I molecules to DC phagosomes, we analyzed the effect of Rab22a silencing on antigen cross‐presentation. Scramble and Rab22a KD JAWS‐II DCs were fed with soluble OVA or OVA‐coated beads during 5 h. Then, DCs were extensively washed, fixed, and incubated with the β‐galactosidase‐inducible B3Z T‐cell hybrid, which is specific for the OVA peptide (SIINFEKL) in association with H‐2Kb MHC‐I molecules. A strong inhibition of the cross‐presentation capacity for soluble OVA and OVA‐coated LB in Rab22a KD DCs was observed, as compared to Scramble DCs (Fig 3A and B, respectively). The synthetic peptide SIINFEKL, which does not need further processing in intracellular compartments, was presented equally well in both DC types (Fig 3C), demonstrating that the impairment of cross‐presentation in Rab22a KD DCs was due to a defect in endosomal and phagosomal uptake and/or antigen processing.
Figure 3. Rab22a controls cross‐presentation by DCs without affecting antigen internalization, phagosomal degradation, or endogenous MHC‐I presentation.
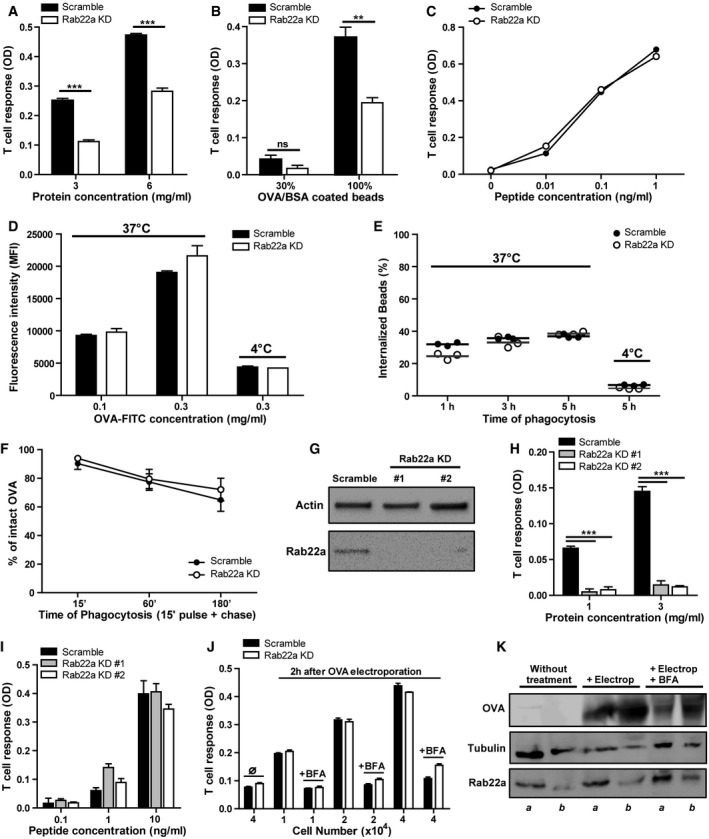
-
A–CThe cross‐presentation ability after incubation with (A) soluble OVA, (B) OVA/BSA‐coated beads, and (C) the SIINFEKL control peptide at the indicated concentrations by Scramble and Rab22a KD JAWS‐II DCs was evaluated with the B3Z hybridoma. Data represent mean ± SEM of triplicate values and are representative of three independent experiments. (A) ***P = 0.0001 and (B) P = 0.1432 (ns); **P = 0.0044. The two‐tailed Student's unpaired t‐test was performed.
-
D, EEvaluation of endocytosis and phagocytosis in Scramble and Rab22a KD JAWS‐II DCs. (D) The endocytosis of fluorescent OVA after 1 h of internalization and (E) the phagocytosis of 3‐μm fluorescent LB at different times of internalization were assessed by FACS analysis. The antigen internalization was conducted at 37°C for effective uptake and at 4°C as negative control. In (D), data represent mean ± SEM of triplicate values and are representative of three independent experiments.
-
FThe kinetics of OVA degradation, as percentage of proteases inhibitors, in isolated phagosomes at the indicated time periods post‐internalization from Scramble and Rab22a KD JAWS‐II DCs was assessed by FACS analysis. Data represent mean ± SEM of three independent experiments.
-
GImmunoblotting of Rab22a and Actin in BMDCs infected with lentiviruses encoding a random sequence (Scramble) and two shRNA specific for silencing Rab22a (Rab22a KD #1 and #2).
-
H, IThe cross‐presentation capacity after the incubation with (H) soluble and (I) the SIINFEKL control peptide at the indicated concentrations by Scramble, Rab22a KD #1, and Rab22a KD #2 BMDCs was evaluated as described before for JAWS‐II DCs. Data represent mean ± SEM of triplicate values and are representative of two independent experiments. ***P = 0.0001. The two‐tailed Student's unpaired t‐test was performed.
-
JSoluble OVA was electroporated into the cytosol of Scramble and Rab22a KD JAWS‐II DCs, and T‐cell activation was determined 2 h later with the B3Z hybridoma. To control endogenous MHC‐I antigen presentation specificity, DCs were also treated with brefeldin A (BFA). The use of this drug markedly reduced CD8+ T‐cell response to similar levels obtained by DCs without any antigen (∅). Data represent mean ± SEM of triplicate values and are representative of three independent experiments.
-
KImmunoblotting showing the amount of OVA incorporated by Scramble (a) and Rab22a KD (b) JAWS‐II DCs after electroporation and BFA treatment.
Source data are available online for this figure.
Whether the reduced cross‐presentation ability of Rab22a KD JAWS‐II DCs could be due to a defect in antigen uptake was then evaluated. To test this hypothesis, we evaluated the endocytosis of FITC‐coupled soluble OVA by FACS. No significant differences between both cell types were observed regarding such endocytic capacity (Figs 3D and EV3A). In order to assess whether these findings were due to a defect on phagocytosis, DCs were incubated with 3‐μm fluorescent LB coated with OVA. After different internalization time periods, LB associated with the cell surface were labeled with an anti‐OVA antibody. With this experimental approach, we could differentiate between surface‐associated and fully internalized beads. When only completely phagocytosed particles were considered, no significant differences in phagocytosis between both DC types were observed (Fig 3E). Since Rab22a has previously been found to be involved in the modulation of phagosome maturation 21, we investigated if Rab22a is also playing a role in this function in DCs. To this end, we analyzed the phagosomal degradation of OVA (the same antigen used to assess cross‐presentation) by FACS, as described previously 7. OVA‐coated LB were internalized during 15 min, 1 h, and 3 h by Scramble and Rab22a KD JAWS‐II DCs. Then, the isolated phagosomes were stained for OVA and the amount of intact and degraded antigen was determined. As depicted in Figs 3F and EV3B, Rab22a KD DCs exhibited a similar phagosomal degradation capacity, as compared to Scramble DCs. Indeed, we observed that antigen phagosomal degradation was slow and unaggressive, a specialization of DC phagosomes that has been extensively documented 3, 4. These results indicate that neither antigen uptake nor antigen processing is affected by the silencing of Rab22a in DCs.
Figure EV3. Rab22a silencing does not affect antigen uptake, phagosomal degradation, or BMDC maturation.
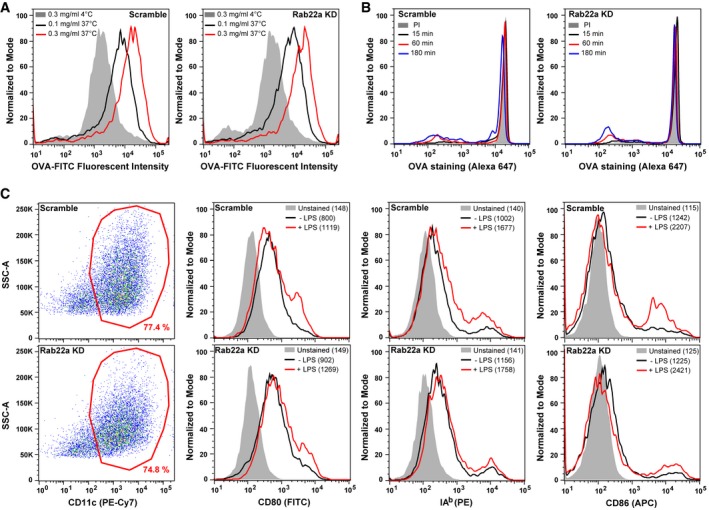
- Scramble and Rab22a KD JAWS‐II DCs were incubated with soluble FITC‐OVA for 60 min. DCs were fixed and analyzed by FACS. Histograms represent the fluorescent intensities of two different concentrations of soluble OVA after internalization at 37°C or the highest concentration at 4°C. Data are representative of three independent experiments.
- Representative FACS profiles showing the OVA staining on isolated phagosomes of 15 min, 1 h, and 3 h post‐internalization of 3‐μm LB from Scramble and Rab22a KD JAWS‐II DCs. The PI condition represents 3‐h phagosomes treated with proteases inhibitors. Data are representative of three independent experiments.
- Scramble and Rab22a KD BMDCs ± LPS‐treated were labeled for CD11c, CD80, IAb, and CD86, and were analyzed by FACS. The gatings on CD11c‐positive cells (red) are depicted in the left panels. The MFI values are specified between parentheses in the FACS profiles. Data are representative of two independent experiments.
To confirm the role of Rab22a in antigen cross‐presentation in primary DCs, we transduced BMDCs transiently with two different shRNAs to achieve Rab22a silencing (Rab22a KD #1 and #2) and with the control shRNA (Scramble). In this case, and differently from our stable Rab22a KD DC line, we obtained an almost complete depletion of the protein for both BMDCs lentiviral treatments (Fig 3G). Accordingly, the impact on soluble OVA cross‐presentation was much more drastic in these cells, as compared to Scramble BMDCs (Fig 3H). As before, Rab22a KD BMDCs did not exhibit any defect in the control peptide presentation (Fig 3I). To assess whether Rab22a silencing induced an alteration on BMDC maturation, we labeled LPS‐treated and non‐treated transduced BMDCs, and we analyzed by FACS the cell surface expression of different activation/maturation markers: CD80, CD86, and MHC‐II molecules. No significant differences in the expression of these markers or in the percentages of CD11c‐positive cells were observed between Scramble and Rab22a KD BMDCs (Fig EV3C).
The constitutive trafficking of newly synthetized MHC‐I molecules to the plasma membrane depends on the normal functioning of the Golgi apparatus. Besides, the trans‐Golgi network establishes frequent interactions with endosomal recycling vesicles. Hence, we studied if the morphology of the Golgi apparatus was somehow altered in the Rab22a KD DCs. To this end, we performed IF staining and confocal microscopy to evaluate the shape of the cis‐ and trans‐Golgi networks by means of the GM130 and TGN46 markers, respectively. No obvious modifications of the two different Golgi markers tested were observed (Fig EV4A and B), suggesting a normal functioning of the secretory pathway for both DC types. To confirm this hypothesis, we tested the endogenous MHC‐I antigen presentation capacity of Rab22a KD DCs (“classical pathway”). This type of MHC‐I antigen presentation uses the classical secretory pathway, which can be interrupted by brefeldin A (BFA), an inhibitor of Golgi‐mediated secretion. Briefly, Scramble and Rab22a KD JAWS‐II DCs were pre‐treated or not with BFA during 30 min. After incubation, soluble OVA was electroporated into the cytosol of DCs 22 and cells were incubated for 2 h with or without BFA, fixed, and confronted with CD8+ T cells. As shown in Fig 3J, no significant differences in T‐cell activation were observed between Scramble and Rab22a KD DCs at any condition tested. By Western blot, we also controlled the amount of antigen incorporated by both DC types after the electroporation treatment (Fig 3K). These experiments corroborate the notion that the implication of Rab22a in MHC‐I trafficking only impacts the cross‐presentation pathway.
Figure EV4. The morphology of the Golgi apparatus is not altered by the KD of Rab22a in DCs.
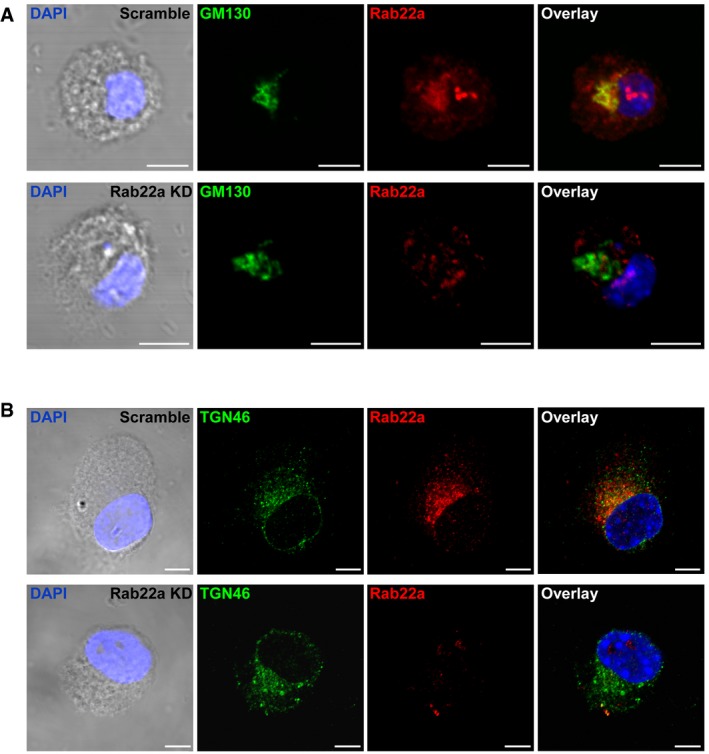
-
A, BIF labeling and confocal microscopy analysis showing the distribution of endogenous Rab22a (red) and (A) the cis‐Golgi marker GM130 (green) or (B) the trans‐Golgi marker TGN46 (green) in Scramble and Rab22a KD JAWS‐II DCs at steady state. The nuclear marker DAPI (blue) and DIC images are shown in the left panels. Overlays are shown in the right panels. Scale bars: 5 μm. Data are representative of at least 30 images analyzed from two independent experiments.
Consistent with the effect on MHC‐I trafficking, our results indicate that Rab22a is critical for optimal cross‐presentation of soluble OVA and OVA‐coated LB in DCs. Rab22a does not participate in classical MHC‐I antigen presentation, and its impact is not related to a defect in neither fluid phase endocytosis, phagocytosis, nor antigen phagosomal degradation. Interestingly, in our Rab22a KD DC line, and depending on the antigen concentration, the cross‐presentation inhibition was 40–60%, a percentage comparable to the percentage of Rab22a silencing in those cells. Stronger inhibitions of cross‐presentation were obtained in Rab22a KD BMDCs, which also exhibit stronger Rab22a silencing. Rab3b/3c‐positive recycling vesicles have already been reported to be involved in antigen cross‐presentation 23. However, the first study that directly showed a crucial role for the ERC during this immunological process is more recent 11. In the latter work, the authors have shown that Rab11a exhibits a very similar role to Rab22a in our study, in terms of MHC‐I intracellular distribution pattern and antigen cross‐presentation. Our results also suggest that the fast recycling pathway could be used to compensate other endocytic functions, such as OVA‐mediated endocytosis, a process not affected in DCs by the silencing of Rab22a. Surprisingly, the phagosomal antigen degradation is not enhanced in Rab22a KD DCs, since it was previously described that Rab22a controls the maturation of Mycobacterium tuberculosis‐containing phagosomes 21. The discrepancies found between the latter work and our results could be due to differences in the models used for both studies, including the cell types (macrophages vs. DCs) and the nature of phagosomes (M. tuberculosis vs. inert LB).
Cross‐presentation of Toxoplasma gondii‐derived antigens is compromised by Rab22a silencing
To study the function of Rab22a in a different context of exogenous antigen presentation, we analyzed DCs infected by the T. gondii, an obligate intracellular parasite that actively shapes and replicates in a specialized parasitophorous vacuole (PV). While the PV is clearly different from a phagolysosome 24, it interacts with, and potentially sequesters, host organelles, such as the ER 25, the Golgi apparatus 26, and lysosomes 27. The PV also shares an important feature of cross‐presenting compartments such as the SNARE Sec22b‐dependent ER recruitment 7, 28, which is decisive for efficient activation of OVA‐specific CD8+ T lymphocytes. During T. gondii infection, overexpression studies suggested that Rab14, Rab30, and Rab43 are recruited to the parasite PV and that they may ensure sphingolipids scavenging from the Golgi 26.
To investigate whether Rab22a plays a role in the PV‐endocytic compartment communication, we first evaluated if this GTPase was recruited to vacuoles containing YFP‐expressing TgRH parasites after 8‐h infection. As shown in Fig EV5A, BMDCs infected by T. gondii exhibited an impressive recruitment of endogenous Rab22a and most of the intracellular location of this GTPase was redistributed toward the PVs, suggesting that it is involved in the trafficking in and out of the vacuole. Since host ER recruitment and cross‐presentation of T. gondii‐derived antigens are completely dependent on active infection 28, we evaluated whether the Rab22a recruitment to the PV is also conditioned by the infective status of the parasite. To address this issue, the IF assays included a staining for GRA6, a protein secreted by the parasite only upon active infection. By this approach, we could discriminate between infective PVs (GRA6‐positive PV) from phagocytosed parasites (GRA6‐negative PV). Contrary to what we observed during active infection, GRA6‐negative PVs showed a faint Rab22a staining and the intracellular distribution of this protein was not visibly altered (Fig EV5B). This finding is similar to previous results obtained with LB phagosomes (Fig 1C). In summary, < 30% of the GRA6‐negative PVs were Rab22a‐positive, while the percentage of Rab22a/GRA6‐double‐positive PVs was near 80%.
Figure EV5. Rab22a recruitment to the parasitophorous vacuole of Toxoplasma gondii in BMDCs.
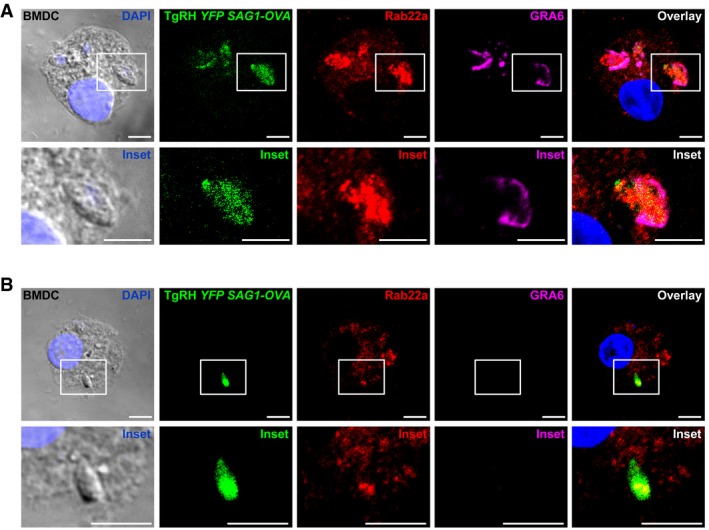
-
A, BBMDCs were infected with OVA‐YFP‐expressing T. gondii (TgRH YFP SAG1‐OVA) for 8 h and confocal images detecting the parasite (green), endogenous Rab22a (red), and GRA6 (magenta) were taken. White boxes are shown at higher magnification in the insets. The nuclear marker DAPI (blue) and DIC images and are shown in the left panels. Overlays are shown in the right panels. Scale bars: 5 μm. Data are representative of more than 50 PVs analyzed from two independent experiments.
Next, we studied if GRA6 was equally detected in the PV of Scramble and Rab22a KD JAWS‐II DCs. Indeed, GRA6 was present in both DC types, but a clear Rab22a labeling around PVs was observed only in Scramble DCs (Fig 4A). Hence, defects on Rab22a could influence the presentation of T. gondii‐derived antigens. To test this possibility, we measured presentation of the OVA‐derived SIINFEKL peptide by H‐2Kb on the surface of DCs infected for 8 h with TgRH parasites that express and secrete SAG1‐OVA into the vacuole (TgRH YFP SAG1‐OVA parasites) 29. A dramatic inhibition of OVA cross‐presentation in Rab22a KD DCs was observed, as compared to control cells (Fig 4B). Importantly, FACS analysis showed that both DC types displayed a similar degree of infection regardless of the multiplicity of infection (MOI) employed (Fig 4C), indicating that the defect in B3Z CD8+ T lymphocyte activation was not due to a differential infection rate of Rab22a KD cells. Additionally, we verified that the total amount of parasite‐secreted SAG1‐OVA antigen present in DCs infected at two different MOI and two different times post‐infection was similar between Rab22a KD and Scramble DCs (Fig 4D).
Figure 4. Rab22a is recruited to the parasitophorous vacuole of Toxoplasma gondii and is necessary for cross‐presentation of a parasite‐derived soluble antigen.
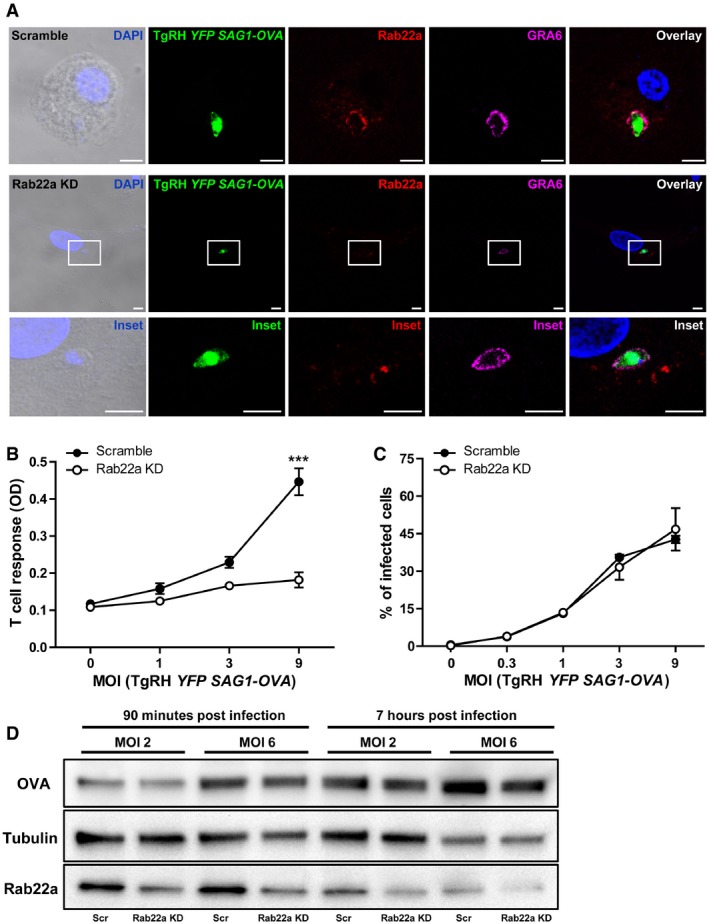
- Scramble and Rab22a KD JAWS‐II DCs were infected with OVA‐YFP‐expressing T. gondii (TgRH YFP SAG1‐OVA) for 8 h and confocal images detecting the parasite (green), endogenous Rab22a (red), and GRA6 (magenta) were taken. Top panels: Scramble cells; bottom panels: Rab22a KD cells. White boxes are shown at higher magnification in the insets. The nuclear marker DAPI (blue) and DIC images are shown in the left panels. Overlays are shown in the right panels. Scale bars: 5 μm. Images are representative of at least 30 analyzed from three independent experiments.
- The cross‐presentation of OVA secreted by T. gondii (TgRH YFP SAG1‐OVA) after 8 h of infection at the indicated MOI was evaluated by B3Z T‐cell activation. Data represent mean ± SEM of triplicate values and are representative of three independent experiments. ***P < 0.001. A two‐way ANOVA and the Bonferroni post‐test were performed.
- The efficiency of infection (8 h) of TgRH YFP SAG1‐OVA in Scramble and Rab22a KD JAWS‐II DCs was measured by FACS analysis at the indicated MOI. Data represent mean ± SEM of duplicate values and are representative of three independent experiments.
- Immunoblotting showing the total amount of OVA in Scramble and Rab22a KD JAWS‐II DCs after 90 min and 7 h of infection with TgRH YFP SAG1‐OVA parasite at MOI 2 and 6. Fifty micrograms of total cell lysates was loaded onto each lane. Data are representative of two independent experiments.
Source data are available online for this figure.
These observations indicate that Rab22a plays a major role in another cross‐presentation context: the case of soluble T. gondii‐derived antigens secreted into the PV. The T. gondii PV is different from a phagosome as it is actively shaped by parasite secretory proteins during invasion and replication, and it does not fuse with lysosomes 24. Some of these secretory proteins (e.g., GRA6) are natural MHC‐I antigens, whose processing is regulated by their membrane association properties 30. Beyond the soluble OVA model antigen used in this report, it will now be key to study the implication of Rab22a in cross‐presentation of endogenous T. gondii antigens. Intriguingly, we also report a strong recruitment of endogenous Rab22a toward the T. gondii PV exclusively upon active infection, similarly of what accounts for host ER recruitment 28. This phenomenon would suggest that, besides playing a role in antigen presentation, the Rab22a‐dependent recruitment of ERC components could regulate host–parasite interactions. Remarkably, very few host Rab GTPases have been described to be recruited by the parasite PV. In one recent study, in which overexpression of Rab14, Rab30, or Rab43 was induced in HeLa cells, the three GTPases were present in the vacuole and they were important for sphingolipids scavenging from the Golgi 26. Whether Rab22a may support or restrict parasite growth is an interesting issue to be further addressed. In line with the importance of Rab22a during pathogen invasion, a recent work demonstrates that the spirochete Borrelia burgdorferi is phagocytosed and compacted by macrophages in a mechanism coordinated by the action of Rab22a and Rab5, showing that these endosomal GTPases are essential for the processing and elimination of this bacterium 20. Furthermore, the Gram‐negative bacteria Neisseria meningitidis subverts host cell polarization and the intracellular trafficking to cross the epithelial barrier by association with ERC, as the phagosome‐containing this pathogen is positive for several Rab proteins typical of recycling vesicles, such as Rab22a 19.
The establishment of protective CD8+ T‐cell responses through the cross‐presentation by DCs involves adequate MHC‐I endocytic trafficking and antigenic peptide loading, a process that is only starting to be explored. Taking into account this information, and considering that CD8+ T‐cell responses elicited by the cross‐presentation of tumor antigens are fundamental for the design of novel immunotherapies, we consider that Rab22a could play a central role in future approaches based on the stimulation of DCs and for new vaccination strategies against intracellular parasite infections.
Materials and Methods
Mice and cells
C57BL/6 mice from 6 to 10 weeks of age were used to obtain bone marrow stem cells from the femur and tibia. The mice were maintained in specific pathogen‐free conditions (SPF), housed in temperature‐controlled rooms (22–25°C), and received water and food ad libitum. All animal procedures were performed according to the bioethics rules of the “Comité Institucional para el Cuidado y Uso de Animales de Laboratorio (CICUAL), Facultad de Ciencias Médicas, Universidad Nacional de Cuyo”. BMDCs were produced after 7–14 days of bone marrow cells culture in GM‐CSF‐containing medium (IMDM), as previously described 31. JAWS‐II DCs were also maintained in culture by using GM‐CSF‐containing medium. The J558 GM‐CSF‐producing cell line and the BMDC line JAWS‐II were kindly provided by S. Amigorena (INSERM U932, Institute Curie, France). T. gondii parasites were grown and maintained by infecting monolayers of the HFF cell line in DMEM complete medium.
Material and buffers
Ovalbumin, lyophilized powder (Worthington Biochemical Corporation). 3‐μm latex beads and 3‐μm blue latex beads (Polysciences Inc.). 3‐μm magnetic latex beads Dynabeads M‐280 (Invitrogen). OVA peptide 257–264, SIINFEKL (Polypeptide Group). Transferrin from human serum conjugated to Alexa 488 and OVA conjugated to Alexa 488 (Molecular Probes). IMDM (Laboratorio Microvet SRL). Poly‐L‐lysine, saponin, sucrose, protease inhibitor cocktail, brefeldin A, lipopolysaccharide (LPS) from Salmonella typhimurium and qPCR primers (Sigma‐Aldrich). Ammonium persulfate (Bio Basic Inc.). BSA (Santa Cruz). Tricine, Tris Base, and TEMED (Calbiochem). Glycine (Bio‐Rad). Acrylamide (Promega). Imidazole and NP‐40 (ICN Biomedicals Inc.).
Antibodies
The following antibodies were used: purified rabbit polyclonal anti‐OVA, mouse monoclonal anti‐α‐tubulin and mouse monoclonal anti‐β‐actin (Sigma‐Aldrich), mouse monoclonal anti‐Rab22a (Santa Cruz), mouse monoclonal anti‐TGN46 (Abcam), purified mouse anti‐GM130, purified rat anti‐Lamp1, FITC mouse anti‐H‐2Kb, FITC hamster anti‐CD80, PE mouse anti I‐Ab, PE‐Cy7 hamster anti‐CD11c and APC rat anti‐CD86 (BD Pharmingen), mouse monoclonal anti‐TfR H68.4 (ATCC), rabbit polyclonal anti‐Rab22a, anti‐Rab11a and anti‐EEA1 (Aviva Systems Biology), purified rabbit anti‐HY10, GRA6 (custom made, Biotem 30). Anti‐species conjugated to Alexa 488, 568, or 647 (Molecular Probes) or peroxidase (Jackson Laboratories) were used as secondary antibodies.
Lentiviral shRNA knockdown of Rab22a
Generation and titer of lentivirus
Bacterial glycerol stocks of plasmids encoding lentiviruses expressing shRNAs were purchased from Sigma‐Aldrich. Plasmids were first purified with the QiaPrep miniprep kit (QIAGEN) and were transfected into HEK293T cells with a three‐plasmid system to produce lentivirus with a very high titer of ~107 CFU/ml 32, 33. Rab22a #1 shRNA (target sequence GTACCGGGACGCCACCTCATGCTCTTTACTCGAGTAAAGAGCATGAGGTGGCGTCTTTTTTG), Rab22a #2 shRNA (target sequence GTACCGGTGTCAGAGTCGTATCAGTAAGCTCGAGCTTACTGATACGACTCTGACATTTTTTG), and the control hairpin (a scramble sequence against GFP) were produced.
Dendritic cell infection
Dendritic cells infection was performed as described before 34. Briefly, JAWS‐II cells were plated on a 96‐well plate (round bottom) at a concentration of 105 cells per well with 200 μl of GM‐CSF‐containing medium (IMDM). After 48 h, the medium was carefully removed without disturbing the cells and 20 μl of virus (Rab22a #1 and Scramble) was added and the pellet was mixed by pipetting 3–5 times. After this, 30 μl of polyB in GM‐CSF medium was added (8 μg/ml final concentration) and the plate was centrifuged at 800 g for 90 min at 37°C. After centrifugation, all media were removed and replaced by 200 μl/well of fresh GM‐CSF medium. Plates were incubated for 48 h, and cells were selected with 20 μg/ml of puromycin. Cells were expanded until the establishment of stable cell lines. Transient BMDC infection was performed by using Rab22a #1, Rab22a #2, and Scramble lentiviruses. When not specified, only Rab22a #1 lentivirus was used to silence Rab22a expression in BMDCs. The protocol of infection was the same as for JAWS‐II DCs, but puromycin selection was done with 5 μg/ml since BMDCs are more sensible to this antibiotic. Cells were collected for experiments 72 h after selection.
Immunofluorescence assays
Bone marrow‐derived dendritic cells or JAWS‐II DCs were placed on poly‐l‐lysine‐coated glass coverslips at room temperature during 30 min. After washing with PBS, complete medium was added and the cells were incubated for 30 min at 37°C in an atmosphere of 5% CO2. Phagocytosis was performed by adding to the cells 3‐μm latex beads (dilution 1:500) for 1 h, endocytosis was performed by adding 100 μg/ml of OVA‐FITC during 30 min, and T. gondii infection was done at MOI 3 during 8 h, and all antigens were incubated at 37°C. After extensive washing with cold PBS, cells were fixed with 2% PFA during 10 min at 4°C and quenched by adding 0.2 M glycine. Cells were permeabilized in PBS/0.05% saponin/0.2% BSA for 20 min at room temperature, washed, and incubated first with primary antibodies over night at 4°C and then with secondary antibodies 45 min at 4°C. After washing, the coverslips were mounted with Vectashield (with DAPI from Vector Laboratories). Image acquisition was performed either on an Olympus FV‐1000 (IHEM microscopy platform) or a Zeiss LSM 710 (CPTP microscopy platform) confocal microscope with a 63×/1.4 NA oil immersion objective. One z‐stack plane is shown from the acquired images.
Phagosome purification
Bone marrow‐derived dendritic cells and JAWS‐II cells were incubated with 3‐μm magnetic beads for 20 min at 18°C and 15 min at 37°C in incomplete IMDM medium (pulse). After washing once with 1 ml of serum and twice with cold 2% PBS/BSA, cells were incubated at 37°C for the indicated times with complete medium. Cells were then disrupted with a syringe (22G needle) in homogenization buffer (PBS 8% sucrose, 3 mM imidazole, 1 mM DTT, and 1× protease inhibitor cocktail), as described previously 35. Magnetic phagosomes were removed from the post‐nuclear supernatant using a magnet and washed three times in cold PBS. Phagosomes were then lysed in lysis buffer (50 mM Tris–HCl pH 7.4, 0.5% NP‐40, and 1× protease inhibitor cocktail) during 30 min at 4°C, and cellular debris were excluded from the solution by centrifugation 5 min at 17,000 g.
Immunoblotting
Purified phagosomes (10 μg) or total cell lysates from BMDCs and JAWS‐II DCs (50 μg) were subjected to SDS–PAGE on 10% gel. After transferring, the membranes were blocked in 10% Milk/PBS during 1 h and incubated with primary antibodies and peroxidase‐conjugated secondary antibodies. Bound antibodies were revealed using the kit Chemiluminescent Peroxidase Substrate‐3 (Sigma‐Aldrich), according to the manufacturers' instructions. The intensity of the bands was quantified by densitometry using Quantity One 4.6.6 software (Bio‐Rad) and was expressed as arbitrary units.
qPCR
Cell homogenization and RNA extraction were performed by using the TRIzol Reagent protocol (Invitrogen). After determining RNA concentration, cDNA was produced with the iScript cDNA Synthesis Kit (Bio‐Rad) and PCR amplification was performed by using 5× HOT FIREPol EvaGreen qPCR Mix (Soli BioDyne) during 40 cycles. GAPDH/Rab22a normalization was done by midpoint slope determination.
FACS analysis
Scramble and Rab22a KD DCs were either directly labeled with anti‐H‐2Kb coupled to FITC during 30 min at 4°C or fixed with 2% PFA during 10 min at 4°C and permeabilized in PBS/0.05% saponin/0.2% BSA for 20 min at room temperature prior to labeling.
For phagosomal isolation, DCs were pulsed for 20 min at 18°C and 15 min at 37°C with 3‐μm LB and washed once with cold 2% PBS/BSA and twice in 1 ml of serum. Cells were then chased for the indicated time points with GM‐CSF complete medium at 37°C and washed once with cold 2% PBS/BSA to stop phagocytosis. To assess Rab22a and MHC‐I phagosomal recruitment, cells were disrupted with a syringe (22G needle) in homogenization buffer (PBS 8% sucrose, 3 mM imidazole, 1 mM DTT, and 1× protease inhibitor cocktail) as it was previously described for the purification of magnetic phagosomes. Then, samples were centrifuged at 150 g during 5 min at 4°C and supernatants were placed on a 96‐well plate (round bottom). Phagosomes (beads) were washed with cold 2% PBS/BSA by centrifugation at 1,050 g during 5 min at 4°C and fixed with 1% PBS/PFA during 10 min on ice. After two washes with 0.2 M glycine and one wash with 2% PBS/BSA, samples labeled ON at 4°C with anti‐Rab22a (Aviva Systems Biology) or anti‐H‐2Kb FITC‐coupled antibodies. For Rab22a staining, a secondary antibody incubation was performed with an anti‐rabbit coupled to Alexa 488 during 45 min at 4°C. To evaluate OVA degradation on isolated phagosomes, pulse and chase steps were performed as detailed above. After the chase times, DCs were washed with cold 2% PBS/BSA, placed on a 96‐well plate (round bottom), and lysed ON at 4°C in lysis buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 0.5% NP‐40, 1 mM DTT, and 1× protease inhibitor cocktail). Next day, samples were washed once with lysis buffer (without DTT and protease inhibitors) and twice with cold 2% PBS/BSA by centrifugation at 1,050 g during 5 min at 4°C. Finally, phagosomes were stained with anti‐OVA for 45 min at 4°C, washed three times, and incubated with an anti‐rabbit coupled to Alexa 647 during 45 min at 4°C. Preparations were analyzed after gating on a particular FSC/SSC region corresponding to single beads in solution, as shown in Fig EV2E.
For BMDC phenotyping, a group of cells were first treated with 10 μg/ml of LPS during 20 h. Then, cells were collected and surface stained on ice during 45 min. After washing with cold PBS, the MFI of CD80, MHC‐II (IAb), and CD86 were analyzed on CD11c‐positive cells.
For antigen uptake experiments, phagocytic and the endocytic capacities of Scramble and Rab22a KD DCs were assessed using 3‐μm blue latex beads and OVA‐FITC respectively, as detailed in Figs 3 and EV3.
These assays were performed by using the FACSARIA‐III (BD Biosciences) from the Flow Cytometry Facility of the “Facultad de Ciencias Médicas, Universidad Nacional de Cuyo” (Argentina) and the MACSQuant10 (Milteny Biotec) at the CPTP Flow Cytometry Platform (France).
Recycling experiments
JAWS‐II DCs were surface labeled either with anti‐H‐2Kb FITC‐coupled or transferrin Alexa 488‐coupled for 30 min at 4°C (binding). After extensive washing with 0.5% PBS/BSA, cells were incubated during 30 min at 37°C in complete medium to allow internalization. DCs were then spun down and resuspended in buffer stripping solution (0.5 M NaCl and 0.5% acetic acid, pH 3) for 10 min on ice. Then, cells were washed twice with cold PBS and twice with complete medium (pulse), resuspended again in complete medium and incubated at 37°C for 0, 10, 20, and 40 min (chase) to allow recycling of MHC‐I and TfR to the cell surface. After each time period, DCs were resuspended again in buffer stripping solution for 10 min on ice, washed, and fixed with 1% PFA. The MFI of FITC and Alexa 488 were analyzed by FACS, and the percentage of recycled MHC‐I and TfR was measured as described by others 36, using the equation (T0 – Tx)/T0 × 100. T0 represents the MFI of cells at 0 min, and Tx is the MFI of cells at 10, 20, and 40 min.
Antigen cross‐presentation assays
JAWS‐II DCs were incubated during 5 h with 3 or 6 mg/ml of soluble OVA, or with 3‐μm LB coated with different ratios of OVA and BSA proteins (OVA 10 mg/ml alone; OVA 3 mg/ml‐BSA, 7 mg/ml; and BSA 10 mg/ml alone), or during 1 h with different concentrations of the SIINFEKL peptide at 37°C. For transduced BMDCs, we performed incubations with 1 or 3 mg/ml of soluble OVA and the control peptide. Then, DCs were washed with 0.5% of PBS/BSA, fixed with 0.008% glutaraldehyde during 3 min at 4°C, and quenched with 0.2 M glycine. After one final wash with PBS, B3Z hybrid T cells were added during 16 h at 37°C. T‐cell activation was measured detecting β‐galactosidase activity by optical density (absorbance at 595–655 nm) using CPRG as substrate for the reaction. For T. gondii cross‐presentation experiments, the remaining intracellular TgRH YFP SAG1‐OVA parasites were released from the fibroblasts by forcing them through a 23‐G needle and added to Scramble and Rab22a KD DCs at various MOI. After 8 h of infection, DCs were washed, fixed, and confronted with B3Z T cells, as described above. Alternatively, 8 h after parasite infection, the percentage of YFP (+) JAWS‐II cells at different MOI were analyzed by FACS. To evaluate the amount of SAG1‐OVA secreted by T. gondii in Scramble and Rab22a DCs, cells were infected for 90 min and 7 h at MOI 2 and 6. Immunoblotting for OVA, Tubulin, and Rab22a detection was performed by charging 105 lysed cells in every condition.
Endogenous MHC‐I antigen presentation
JAWS‐II DCs were pre‐treated or not with 7.5 μg/ml of BFA during 30 min at 37°C. After this time, cells were extensively washed with PBS, suspended in 1 mM HEPES/PBS pH 7.4 + OVA at 3 mg/ml, and electroporated by using a Bio‐Rad Gene Pulser II Porator (0.45 kV and 250 μF of voltage and capacitance, respectively) to introduce the antigen into the cytosol. After electroporation, DCs were immediately put on ice and washed once with cold PBS. Then, cells were suspended in complete medium and incubated for 2 h at 37°C in the presence or absence of BFA. Finally, DCs were washed, fixed with glutaraldehyde, and confronted with B3Z CD8+ T cells as described before for cross‐presentation experiments. T‐cell activation was measured detecting β‐galactosidase activity by optical density (absorbance at 595–655 nm). Immunoblotting for OVA, Tubulin, and Rab22a detection was performed by charging 105 lysed cells in every condition.
Statistical analysis
The two‐tailed Student's unpaired t‐test, and a two‐way ANOVA and the Bonferroni post‐test were performed at the indicated Figures. The Pearson correlation coefficients were determined by using the ImageJ software. The GraphPad Prism 5 software was used for all analysis.
Author contributions
IC, NB, and LSM conceived and designed the study. IC performed most of the experiments and wrote the manuscript. CC and NAG performed the experiments. NB and LSM contributed to manuscript editing.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
This work was supported by grants from the “Agencia Nacional de Promoción Científica y Tecnológica” (PICT 2012‐0089 to I.C. and PICT 2013‐1433 to L.S.M.), the program of Scientific Cooperation between the “Ministerio de Ciencia, Tecnología e Innovación Productiva de la República Argentina” and ECOS‐Sud from France (action A13S01 to L.S.M and N.B.), the EMBO Short Term Fellowship (ASTF 361‐2013 to I.C.), the “Agence Nationale de la Recherche” (ANR‐11‐BSV3‐01002 to N.B.), the “Institut National de la Santé et de la Recherche Médicale” (Avenir Grant to N.B.), the Human Frontier Science Program Organization (CDA00047/2011 to N.B.), the Marie Curie International Reintegration Grant to N.B., the PIA Parafrap Consortium (ANR‐11‐LABX0024 to N.B.), and PIA Aninfimip equipment (ANR‐11‐EQPX‐0003 to NB). We would like to thank S. Amigorena and M. Ostrowski for helpful discussions.
EMBO Reports (2016) 17: 1753–1765
Contributor Information
Ignacio Cebrian, Email: icebrian@mendoza-conicet.gob.ar.
Luis S Mayorga, Email: lmayorga@fcm.uncu.edu.ar.
References
- 1. Blanchard N, Gonzalez F, Schaeffer M, Joncker NT, Cheng T, Shastri AJ, Robey EA, Shastri N (2008) Immunodominant, protective response to the parasite Toxoplasma gondii requires antigen processing in the endoplasmic reticulum. Nat Immunol 9: 937–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Joffre OP, Segura E, Savina A, Amigorena S (2012) Cross‐presentation by dendritic cells. Nat Rev Immunol 12: 557–569 [DOI] [PubMed] [Google Scholar]
- 3. Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon‐Dumenil AM, Seabra MC, Raposo G, Amigorena S (2006) NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126: 205–218 [DOI] [PubMed] [Google Scholar]
- 4. Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I (2003) Activation of lysosomal function during dendritic cell maturation. Science 299: 1400–1403 [DOI] [PubMed] [Google Scholar]
- 5. Savina A, Amigorena S (2007) Phagocytosis and antigen presentation in dendritic cells. Immunol Rev 219: 143–156 [DOI] [PubMed] [Google Scholar]
- 6. Zehner M, Marschall AL, Bos E, Schloetel JG, Kreer C, Fehrenschild D, Limmer A, Ossendorp F, Lang T, Koster AJ et al (2015) The translocon protein Sec61 mediates antigen transport from endosomes in the cytosol for cross‐presentation to CD8(+) T cells. Immunity 42: 850–863 [DOI] [PubMed] [Google Scholar]
- 7. Cebrian I, Visentin G, Blanchard N, Jouve M, Bobard A, Moita C, Enninga J, Moita LF, Amigorena S, Savina A (2011) Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell 147: 1355–1368 [DOI] [PubMed] [Google Scholar]
- 8. Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N (2002) ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature 419: 480–483 [DOI] [PubMed] [Google Scholar]
- 9. Saveanu L, Carroll O, Weimershaus M, Guermonprez P, Firat E, Lindo V, Greer F, Davoust J, Kratzer R, Keller SR et al (2009) IRAP identifies an endosomal compartment required for MHC class I cross‐presentation. Science 325: 213–217 [DOI] [PubMed] [Google Scholar]
- 10. Wearsch PA, Cresswell P (2007) Selective loading of high‐affinity peptides onto major histocompatibility complex class I molecules by the tapasin‐ERp57 heterodimer. Nat Immunol 8: 873–881 [DOI] [PubMed] [Google Scholar]
- 11. Nair‐Gupta P, Baccarini A, Tung N, Seyffer F, Florey O, Huang Y, Banerjee M, Overholtzer M, Roche PA, Tampe R et al (2014) TLR signals induce phagosomal MHC‐I delivery from the endosomal recycling compartment to allow cross‐presentation. Cell 158: 506–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blanchard N, Shastri N (2010) Topological journey of parasite‐derived antigens for presentation by MHC class I molecules. Trends Immunol 31: 414–421 [DOI] [PubMed] [Google Scholar]
- 13. Weigert R, Yeung AC, Li J, Donaldson JG (2004) Rab22a regulates the recycling of membrane proteins internalized independently of clathrin. Mol Biol Cell 15: 3758–3770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barral DC, Cavallari M, McCormick PJ, Garg S, Magee AI, Bonifacino JS, De LG, Brenner MB (2008) CD1a and MHC class I follow a similar endocytic recycling pathway. Traffic 9: 1446–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grosshans BL, Ortiz D, Novick P (2006) Rabs and their effectors: achieving specificity in membrane traffic. Proc Natl Acad Sci USA 103: 11821–11827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mesa R, Salomon C, Roggero M, Stahl PD, Mayorga LS (2001) Rab22a affects the morphology and function of the endocytic pathway. J Cell Sci 114: 4041–4049 [DOI] [PubMed] [Google Scholar]
- 17. Magadan JG, Barbieri MA, Mesa R, Stahl PD, Mayorga LS (2006) Rab22a regulates the sorting of transferrin to recycling endosomes. Mol Cell Biol 26: 2595–2614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang B, Hubber A, McDonough JA, Roy CR, Scidmore MA, Carlyon JA (2010) The Anaplasma phagocytophilum‐occupied vacuole selectively recruits Rab‐GTPases that are predominantly associated with recycling endosomes. Cell Microbiol 12: 1292–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barrile R, Kasendra M, Rossi‐Paccani S, Merola M, Pizza M, Baldari C, Soriani M, Arico B (2015) Neisseria meningitidis subverts the polarized organization and intracellular trafficking of host cells to cross the epithelial barrier. Cell Microbiol 17: 1365–1375 [DOI] [PubMed] [Google Scholar]
- 20. Naj X, Linder S (2015) ER‐coordinated activities of Rab22a and Rab5a drive phagosomal compaction and intracellular processing of Borrelia burgdorferi by macrophages. Cell Rep 12: 1816–1830 [DOI] [PubMed] [Google Scholar]
- 21. Roberts EA, Chua J, Kyei GB, Deretic V (2006) Higher order Rab programming in phagolysosome biogenesis. J Cell Biol 174: 923–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gil‐Torregrosa BC, Lennon‐Dumenil AM, Kessler B, Guermonprez P, Ploegh HL, Fruci D, van Endert P, Amigorena S (2004) Control of cross‐presentation during dendritic cell maturation. Eur J Immunol 34: 398–407 [DOI] [PubMed] [Google Scholar]
- 23. Zou L, Zhou J, Zhang J, Li J, Liu N, Chai L, Li N, Liu T, Li L, Xie Z et al (2009) The GTPase Rab3b/3c‐positive recycling vesicles are involved in cross‐presentation in dendritic cells. Proc Natl Acad Sci USA 106: 15801–15806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Joiner KA, Fuhrman SA, Miettinen HM, Kasper LH, Mellman I (1990) Toxoplasma gondii: fusion competence of parasitophorous vacuoles in Fc receptor‐transfected fibroblasts. Science 249: 641–646 [DOI] [PubMed] [Google Scholar]
- 25. Magno RC, Straker LC, de Souza W, Attias M (2005) Interrelations between the parasitophorous vacuole of Toxoplasma gondii and host cell organelles. Microsc Microanal 11: 166–174 [DOI] [PubMed] [Google Scholar]
- 26. Romano JD, Sonda S, Bergbower E, Smith ME, Coppens I (2013) Toxoplasma gondii salvages sphingolipids from the host Golgi through the rerouting of selected Rab vesicles to the parasitophorous vacuole. Mol Biol Cell 24: 1974–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Coppens I, Dunn JD, Romano JD, Pypaert M, Zhang H, Boothroyd JC, Joiner KA (2006) Toxoplasma gondii sequesters lysosomes from mammalian hosts in the vacuolar space. Cell 125: 261–274 [DOI] [PubMed] [Google Scholar]
- 28. Goldszmid RS, Coppens I, Lev A, Caspar P, Mellman I, Sher A (2009) Host ER‐parasitophorous vacuole interaction provides a route of entry for antigen cross‐presentation in Toxoplasma gondii‐infected dendritic cells. J Exp Med 206: 399–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gubbels MJ, Striepen B, Shastri N, Turkoz M, Robey EA (2005) Class I major histocompatibility complex presentation of antigens that escape from the parasitophorous vacuole of Toxoplasma gondii . Infect Immun 73: 703–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lopez J, Bittame A, Massera C, Vasseur V, Effantin G, Valat A, Buaillon C, Allart S, Fox BA, Rommereim LM et al (2015) Intravacuolar membranes regulate CD8 T cell recognition of membrane‐bound Toxoplasma gondii protective antigen. Cell Rep 13: 2273–2286 [DOI] [PubMed] [Google Scholar]
- 31. Winzler C, Rovere P, Rescigno M, Granucci F, Penna G, Adorini L, Zimmermann VS, Davoust J, Ricciardi‐Castagnoli P (1997) Maturation stages of mouse dendritic cells in growth factor‐dependent long‐term cultures. J Exp Med 185: 317–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272: 263–267 [DOI] [PubMed] [Google Scholar]
- 33. Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK et al (2006) A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high‐content screen. Cell 124: 1283–1298 [DOI] [PubMed] [Google Scholar]
- 34. Savina A, Peres A, Cebrian I, Carmo N, Moita C, Hacohen N, Moita LF, Amigorena S (2009) The small GTPase Rac2 controls phagosomal alkalinization and antigen crosspresentation selectively in CD8(+) dendritic cells. Immunity 30: 544–555 [DOI] [PubMed] [Google Scholar]
- 35. Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, van Endert P, Amigorena S (2003) ER‐phagosome fusion defines an MHC class I cross‐presentation compartment in dendritic cells. Nature 425: 397–402 [DOI] [PubMed] [Google Scholar]
- 36. Osborne DG, Piotrowski JT, Dick CJ, Zhang JS, Billadeau DD (2015) SNX17 affects T cell activation by regulating TCR and integrin recycling. J Immunol 194: 4555–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4