

Abstract
TANK‐binding kinase 1 (TBK1) activation is a central event in type I interferon production in anti‐virus innate immunity. However, the regulatory mechanism underlying TBK1 activation remains unclear. Here we report that Raf kinase inhibitory protein (RKIP) is essential for TBK1 activation and type I interferon production triggered by viral infection. Upon viral infection, RKIP is phosphorylated at serine 109 (S109) by TBK1. Phosphorylation of RKIP enhances its interaction with TBK1 and in turn promotes TBK1 autophosphorylation. Mutation of RKIP S109 to alanine abrogates the interaction between RKIP and TBK1, and the anti‐viral function of RKIP. RKIP deficiency inhibits intracellular double‐stranded RNA‐ or DNA‐induced type I interferon production. Consistently, RKIP deficiency renders the mice more susceptible to vesicular stomatitis virus (VSV) and herpes simplex virus (HSV) infections. This study reveals a previously unrecognized positive feedback loop between RKIP and TBK1 that is essential for type I interferon production in anti‐viral innate immunity.
Keywords: anti‐viral immunity, RKIP, TBK1, type I interferon
Subject Categories: Immunology
Introduction
The innate immune system serves as the first‐line sentinel of the host defense against invading pathogens by recognizing various conserved molecular motifs termed the pathogen‐associated molecular patterns (PAMPs) (Ausubel, 2005; Vance et al, 2009). PAMPs are detected by several classes of host pattern recognition receptors (PRRs) (Lee & Kim, 2007), including Toll‐like receptors (TLRs) (Werling et al, 2009), RIG‐I‐like receptors (RLRs) (Loo & Gale, 2011), NOD‐like receptors (NODs) (Chen et al, 2009), C‐type lectins (Geijtenbeek & Gringhuis, 2009), and double‐strand DNA (dsDNA) receptors (Paludan & Bowie, 2013). Engagement of the PRRs response triggers the activation of downstream signaling events and thus induces proinflammatory cytokine and type I interferon production to mediate innate immunity (Takeuchi & Akira, 2010). Upon viral infection, most of the PRRs turn on the activity of TBK1 through phosphorylation, which subsequently activates a transcription factor interferon regulatory factor 3 (IRF3), to induce type I interferon production (Barbalat et al, 2011; Gack, 2014).
TBK1 is composed of an N‐terminal kinase domain (KD), and three regulatory elements such as a ubiquitin‐like domain (ULD), a dimerization domain (DD), and a small protein interaction module at the C‐terminus, named as adaptor‐binding (AB) motif (Hacker & Karin, 2006; Ikeda et al, 2007). The three domains, KD, ULD, and DD, join together to form a tri‐modular subunit and further assemble to a compact dimer. Upon stimulation, TBK1 is recruited to signaling complexes via its AB motif. Local clustering of TBK1 molecules allows inter‐dimer KD interactions and leads to trans‐autophosphorylation (Helgason et al, 2013). Furthermore, conformational changes in TBK1 have been indicated to control its activation (Helgason et al, 2013; Shu et al, 2013). However, how TBK1 activation is finely regulated remains elusive.
RKIP is the prototypical member of the PEBP family (Bernier & Jolles, 1984; Serre et al, 2001). It was originally identified as a Raf1‐binding protein that inhibits MEK phosphorylation and activation (Yeung et al, 1999). Ever since then, emerging results have demonstrated RKIP as a fundamental modulator of many signal cascades, including MAPK (Yeung et al, 1999; Trakul et al, 2005), G‐protein‐coupled receptor (GPCR) (Lorenz et al, 2003), NF‐κB (Yeung et al, 2001), GSK3β (Al‐Mulla et al, 2011), and microRNA (miR) let‐7 (Dangi‐Garimella et al, 2009) signal transduction cascades. RKIP plays important roles in many processes, including cell differentiation, migration, apoptosis, and cell cycle (Zeng et al, 2008; Al‐Mulla et al, 2013). It was reported that PEBP‐1(RKIP) also serves as a protective role against the host immunological responses in nematodes and Drosophila (Morgan et al, 2006; Reumer et al, 2009). Recently, we had demonstrated that RKIP contributes to colitis via mediating intestinal epithelial cell apoptosis (Lin et al, 2016). However, in mammalian, its role in the regulation of innate or adaptive immunity remains unknown.
In this study, we report that RKIP serves as a positive regulator of type I interferon production in innate immunity by promoting TBK1 activation. TBK1 phosphorylates RKIP at S109, which is essential for the interaction of RKIP with TBK1, and triggers a positive feedback control of TBK1 activation. Our finding identifies the TBK1–RKIP loop as an “impeller” for TBK1 activation during anti‐viral innate immunity.
Results
RKIP positively regulates RIG‐I indution of type I interferon and pro‐inflammatory cytokines
We first explored the correlation between RKIP expression level and viral infection based on public available gene array data (GEO accession GSE51808). RKIP mRNA is significantly upregulated in the peripheral blood samples of the patient from the dengue hemorrhagic fever or dengue fever group comparing to those of the healthy control or convalescent group (Fig EV1A), indicating that RKIP may be involved in the anti‐viral response. We next used primary peritoneal macrophage from wild‐type and RKIP‐deficient mice to assess the effects of RKIP deficiency on viral infection‐induced proinflammatory cytokine and type I interferon production. As shown in Fig 1A and B, RKIP deficiency significantly inhibits vesicular stomatitis virus (VSV) or Sendai virus (SeV) infection‐induced IFN‐β production and modestly decreases VSV‐induced or SeV infection‐induced TNF‐α and IL‐6 production both in mRNA and protein level in macrophages. We then detected poly(I:C) intracellular transfection infection‐induced IFN‐β production in wild‐type or RKIP‐deficient macrophages. As shown in Fig 1C, poly(I:C) transfection triggered much less Ifnb1 mRNA expression and IFN‐β secretion in RKIP‐deficient macrophages. We also examined the role of RKIP in anti‐DNA viral and Listeria monocytogenes infection. Consistently, RKIP deficiency significantly inhibits herpes simplex virus (HSV), Listeria monocytogenes (LM)‐induced Ifnb1 mRNA expression (Fig EV1B) or cGAMP [cyclic guanosine monophosphate–adenosine monophosphate; an endogenous second messenger triggered by DNA virus infection (Sun et al, 2013)]‐induced Ifnb1 mRNA expression and IFN‐β production (Fig EV1C). In RAW264.7 cells, RKIP knockdown inhibits VSV‐induced IFN‐β production (Fig EV1D). Conversely, RKIP overexpressed stable cell line infected by VSV (Fig EV1E) or HSV (Fig EV1F) produces much more IFN‐β, and proinflammatory cytokines than the control transfectants. Furthermore, replication of VSV expressing GFP in RAW 264.7 cells was determined by fluorescence‐activated cell sorting (FACS) analysis and 50% tissue culture infective dose (TCID50) assay. While RKIP overexpression inhibits VSV replication (Fig 1D), knockout of RKIP renders the cells susceptible to viral infection (Fig 1E), indicating that RKIP plays an important role in host defense against viral infection by positively regulating type I interferon production. Immunoblot assay confirmed the efficiency of RKIP knockout, knockdown, and overexpression in macrophage (Fig EV1G).
Figure EV1. RKIP is required for virus infection‐induced IFN‐β and proinflammatory cytokine production in macrophages.
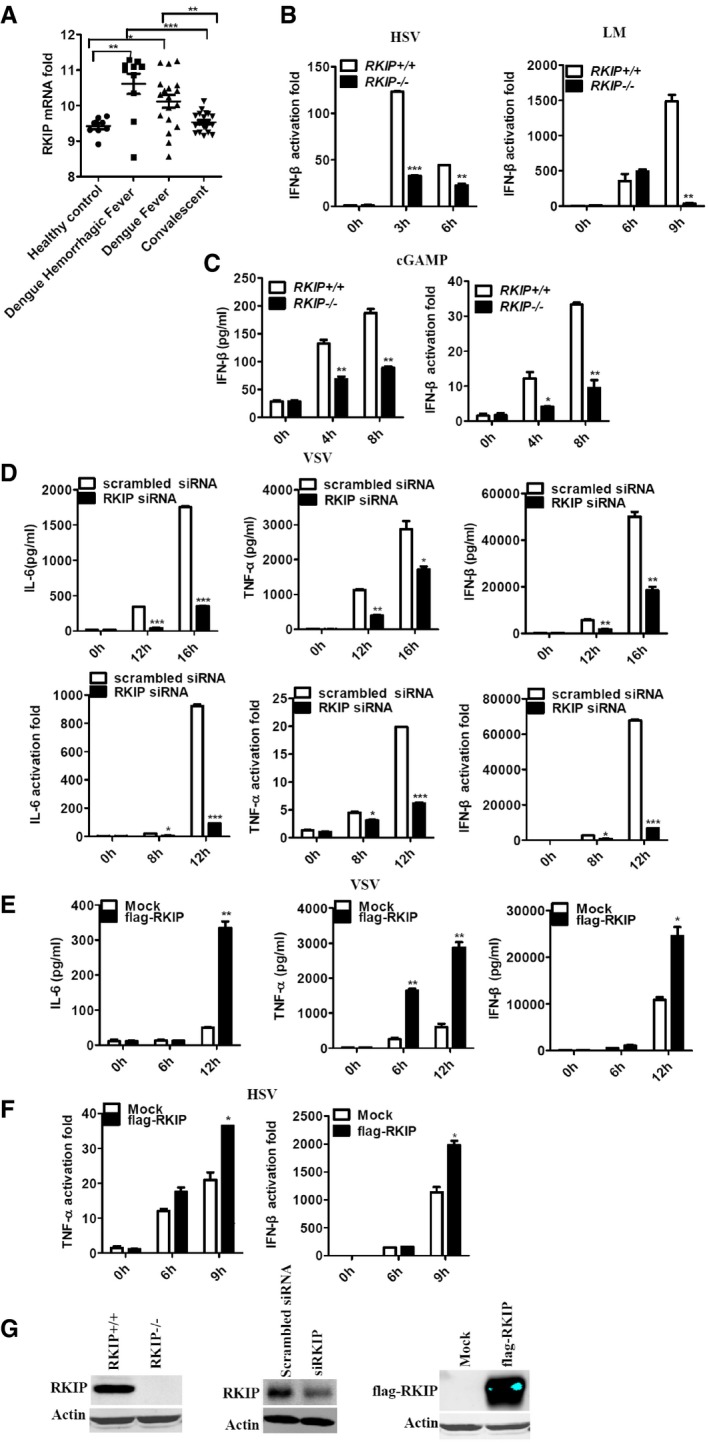
- Gene array analysis of RKIP mRNA (GEO profile GDS5093) expression in peripheral blood samples from healthy donors and patients with dengue virus infection. Each symbol represents an individual subject; horizontal lines indicate the mean.
- Real‐time PCR analysis of Ifnb1 mRNA in peritoneal macrophage isolated from RKIP +/+ or RKIP −/− mice infected with HSV (MOI 5) or Listeria monocytogenes (MOI 10).
- cGAMP (100 nM) was delivered to BMDMs for the indicated times, and then, IFN‐β RNA and secreted protein were measured by real‐time PCR and ELISA, respectively.
- ELISA of IL‐6, TNF‐α, and IFN‐β protein and real‐time PCR analysis of Il6, Tnf, and Ifnb1 mRNA in RAW264.7 cells treated with RKIP‐specific or scrambled siRNA, followed by infection with VSV (MOI 0.1).
- ELISA of IL‐6, TNF‐α, and IFN‐β protein in RAW264.7 cells stably overexpressing RKIP, followed by treatment with VSV (MOI 0.1).
- Real‐time PCR analysis of Tnf and Ifnb1 mRNA in RAW264.7 cells stably overexpressing RKIP and then followed by infection with HSV (MOI 5).
- Immunoblot analysis of the expression of RKIP protein in primary macrophages with RKIP knockout or knockdown or RKIP stably overexpressing RAW264.7 transfectants.
Figure 1. RKIP is required for viral infection and poly(I:C)‐induced IFN‐β and proinflammatory cytokine production in macrophages.

-
A–CEnzyme‐linked immunosorbent assay (ELISA) of IL‐6, TNF‐α, and IFN‐β protein and real‐time PCR analysis of Il6, Tnf, and IIfnb1 mRNA in peritoneal macrophages isolated from wild‐type and RKIP‐deficient mice treated with VSV (MOI 0.1) (A) and SEV (MOI 10) (B) or transfected with poly(I:C) (1 μg/ml) (C).
-
DFluorescence‐activated cell sorting (FACS) and TCID50 (50% tissue culture infective dose) assessing the proliferation level of VSV. RAW264.7 cells line stably overexpressing flag‐RKIP and infected with VSV‐eGFP at MOI of 1 for the indicated time. The fusion protein fluorophore was analyzed by FACS. Then, fresh medium with the VSV‐eGFP infection was serially diluted on the monolayer of HEK293 cells for 3–7 days and TCID50 was measured.
-
EFACS analysis of wild‐type and RKIP‐deficient peritoneal macrophages infected with VSV‐eGFP at MOI of 1. The fresh medium with the VSV‐eGFP infection was collected after 9 h for TCID50 assessing.
RKIP is critical for anti‐viral immunity in vivo
To unveil the physiological function of RKIP for host defense against viral infection, we treated wild‐type (RKIP +/+) and RKIP knockout (RKIP −/−) mice with intraperitoneal injection of VSV. We noted that mice deficient in RKIP have lower concentrations of type I interferon and IL‐6 in serum than wild‐type mice (Fig EV2A). At the same time, a significantly stronger VSV replication and impaired IFN‐β production are recorded in the livers from RKIP −/− mice (Fig EV2B). Consequently, inflammatory responses in internal and external hepatic biliary ducts are much more severe in RKIP −/− mice (Fig EV2C). Moreover, VSV infection results in rapid death of RKIP −/− mice, and no mice survival beyond 24 h post‐infection, while RKIP +/+ mice exhibit a higher resistance to VSV infection, as half of the mice survived longer than 48 h (Fig EV2D). Western blot data confirmed RKIP deficiency in liver in the RKIP knockout mouse (Fig EV2E). Furthermore, RKIP −/− mice are more vulnerable to DNA virus such as HSV. As shown in Fig EV2F, after infection with HSV, RKIP −/− mice produce less IFN‐β in serum compared with RKIP +/+ mice. In similarly, RKIP −/− peritoneal macrophages show lower expression of IIfnb1 mRNA than do their wild‐type counterparts (Fig EV2G). Consistent with this, viral titers are much higher in the brain from RKIP −/− mice than the wild‐type counterparts after HSV infection (Fig EV2H).
Figure EV2. RKIP is required for anti‐viral innate immunity in vivo .

- ELISA of IFN‐β and IL‐6 in serum obtained from wild‐type (RKIP +/+) and RKIP −/− mice 12 h after intraperitoneal injection with VSV (1 × 107 pfu/g) (n = 5 per group).
- Real‐time PCR analysis of VSV expression and Ifnb1 mRNA in the liver from wild‐type and RKIP‐deficient mice treated as in (A).
- Hematoxylin and eosin staining of liver sections from mice in (A). Scale bar, 100 μm.
- Survival of ˜8‐week‐old RKIP +/+ and RKIP −/− mice given intravenous tail injection of VSV (1 × 108 pfu/g, i.p.) (n = 7 per group; Wilcoxon test).
- Immunoblot assay of RKIP expression in lung, liver, and spleen from wild‐type and RKIP‐deficient mice.
- ELISA of IFN‐β in serum from RKIP +/+ and RKIP −/− mice 6 h after intravenous injection of HSV (1 × 106 pfu/mouse) (n = 5 per group).
- Real‐time PCR analysis of Ifnb1 gene expression in peritoneal macrophages 24 h after intravenous injection of HSV (1 × 106 pfu/mouse) (n = 5 per group).
- Viral titers in brains obtained from RKIP +/+ and RKIP −/− mice 3 days after intravenous injection of HSV (1 × 106 pfu/mouse) (n = 5 per group).
To further evaluate the anti‐viral function of RKIP in hematopoietic cells, we generated RKIP bone marrow chimeras. The bone marrow cells from wild‐type or RKIP‐deficient mice were transferred into lethally irradiated wild‐type mice. Eight weeks after bone marrow reconstitution, mice were challenged with VSV. RKIP‐deficient chimeras produce lower levels of IFN‐β in serum than wild‐type chimeras in response to infection of VSV (Fig 2A). VSV titer in organs from RKIP‐deficient chimera mice is significantly enhanced compared to that in organs from wild‐type chimeras (Fig 2B). Increased VSV replication and decreased type I interferon production were found in the liver from RKIP‐deficient chimeras (Fig 2C). More infiltration of inflammatory cells is observed in the lungs from RKIP‐deficient chimeras after infection with VSV (Fig 2D). Analysis of liver parenchyma reveals a richer portal inflammatory infiltrate in RKIP‐deficient chimeras, which consists of mixed eosinophil and monocyte cells (Fig 2D). Ifnb1 mRNA expression induced by VSV infection in the peritoneal macrophages from RKIP‐deficient chimeras is much lower than that in macrophage from wild‐type chimeras (Fig 2E). Consistently, RKIP‐deficient chimeras are more susceptible to lethal VSV infection compared to wild‐type chimeras in overall survival assays (Fig 2F). Thus, RKIP plays an important role in mediating type I interferon expression in anti‐viral innate immunity response in vivo.
Figure 2. RKIP is required for anti‐virus innate immunity in vivo .

- Wild‐type (RKIP +/+) and RKIP‐deficient (RKIP −/−) chimera mice were intraperitoneally injected with VSV (1 × 107 pfu/g); 12 h later ELISA of IFN‐β and IL‐6 in serum was performed (n = 6 per group).
- Determination of VSV loads in organs by TCID50 assay 12 h after RKIP +/+ and RKIP −/− chimera mice (n = 6 per group) were intraperitoneally injected with VSV (1 × 107 pfu/g).
- Real‐time PCR analysis of Ifnb1 mRNA and VSV expression in liver from RKIP +/+ and RKIP −/− chimera mice 12 h after intraperitoneal injection with VSV (1 × 107 pfu/g) (n = 6 per group).
- Real‐time PCR analysis of Ifnb1 mRNA expression in peritoneal macrophages from RKIP +/+ and RKIP −/− chimera mice 12 h after intraperitoneal injection with VSV (1 × 107 pfu/g) (n = 6 per group).
- Survival curve for 8‐week‐old wild‐type and RKIP‐deficient chimera mice challenged with VSV (1 × 108 pfu/g, i.v.) (n = 10 per group; Wilcoxon test).
RKIP selectively regulates the activation of TBK1 and P38
VSV infection induces type I interferon production by triggering double‐strand RNA sensor RIG‐I and downstream kinases such as TAK1 and TBK1, which mediate activation of NF‐κB, MAPKs, and IRFs pathways (Honda et al, 2006). We further examined the effect of RKIP deficiency on VSV infection‐ or poly(I:C) transfection‐induced activation of NF‐κB, MAPKs, and IRFs pathways. RKIP deficiency does not affect the capacity of VSV‐ or intracellular poly(I:C)‐induced activation of ERK1/2, JNK1/2, and NF‐κB pathways in primary peritoneal macrophage (Fig 3A and B). However, it remarkably inhibits VSV‐ or intracellular poly(I:C)‐induced TBK1, IRF3, and P38 phosphorylation (Fig 3A and B). While RKIP knockdown inhibits VSV‐induced TBK1, IRF3, and P38 phosphorylation in peritoneal macrophages (Fig 3C) and RAW264.7 cells (Fig EV3A), RKIP overexpressing increases VSV‐ or intracellular poly(I:C)‐induced TBK1, IRF3, and P38 phosphorylation in RAW264.7 macrophages (Fig 3D and E). Similarly, RKIP deficiency remarkably inhibits HSV‐ or LM‐induced TBK1, IRF3, and P38 phosphorylation in mouse peritoneal macrophages (Fig EV3B and C). Consistent with our findings in mouse cells, knockdown of RKIP in human peripheral blood mononuclear cells (PBMCs) markedly decreases the activation of TBK1 and downstream IRF3 phosphorylation upon VSV infection (Fig EV3D), suggesting a cross‐species function of RKIP on anti‐viral defense. The Ifnb1 promoter contains DNA‐binding sites for several transcription factors: NF‐κB and IRF3‐IRF7. RKIP can promote RIG‐I CARD (a mutant containing N‐terminal CARD domains of RIG‐I acts as a constitutive active RIG‐I mutant) or TBK1 overexpression‐induced ISRE‐luc and IFN‐β‐luc reporter gene expression. However, RKIP does not affect the capability of IKKi or IRF3‐5D, a constitutive active IRF3 mutant (Lin et al, 1998), to induced ISRE‐luc and IFN‐β‐luc reporter gene expression (Fig 3F). Taken together, these data indicate that RKIP increases type I interferon production by positively regulating TBK1 activation in anti‐viral innate immune responses.
Figure 3. RKIP is required for VSV‐ and poly(I:C)‐induced TBK1, IRF3, and p38 phosphorylation.
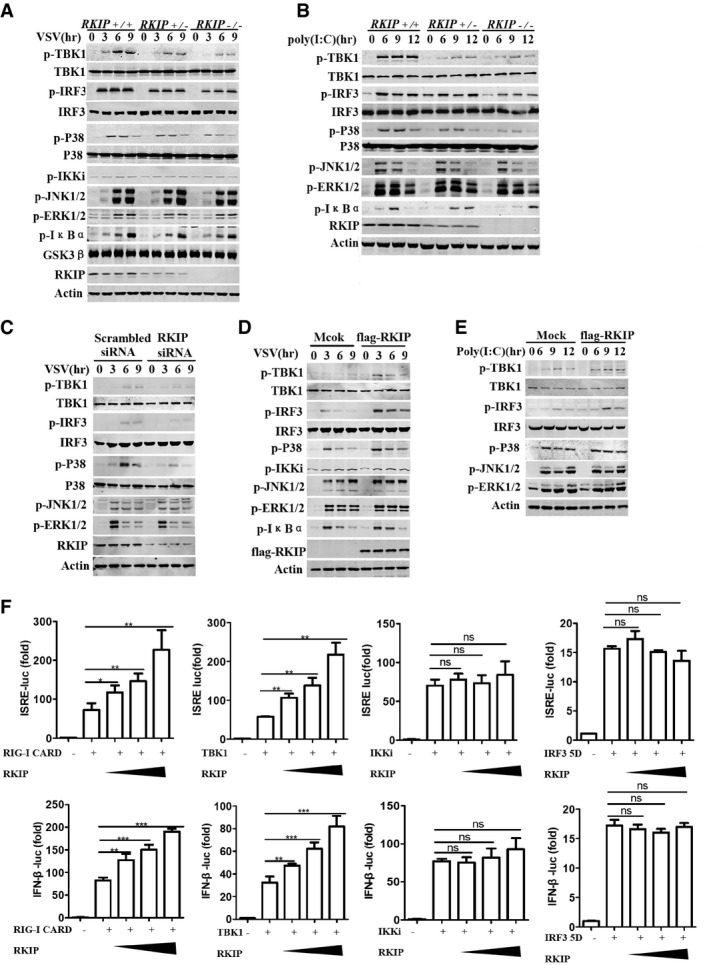
- Immunoblot analysis of p‐TBK1, p‐IKKi, p‐IRF3, p‐P38, p‐JNK1/2, p‐ERK1/2, and p‐IκBα in lysates of RKIP +/+ , RKIP +/−, and RKIP −/− peritoneal macrophages stimulated with VSV (MOI 0.1) for the indicated time. Actin was used as a loading control.
- Immunoblot assay of RKIP +/+ , RKIP +/−, and RKIP −/− peritoneal macrophages treated with intracellular poly(I:C) (1 μg/ml). Actin was used as a loading control.
- Immunoblot analysis of phosphorylated p‐TBK1, p‐IKKi, p‐IRF3, p‐P38, p‐JNK1/2, and p‐ERK1/2 in peritoneal macrophages transfected with RKIP‐specific or scrambled siRNA, followed by infection with VSV (MOI 0.1). Actin was used as a loading control.
- Immunoblot analysis of p‐TBK1, p‐IKKi, p‐IRF3, p‐P38, p‐JNK1/2, p‐ERK1/2, and p‐IκBα in lysates of VSV‐treated RAW264.7 cells stably overexpressing RKIP. Actin was used as a loading control.
- Immunoblot analysis in lysates of RAW264.7 cells stably overexpressing RKIP and treated with intracellular poly(I:C) (1 μg/ml). Actin was used as a loading control.
- Luciferase activity in 293T cells transfected with plasmid encoding a luciferase reporter for IFN‐β (IFN‐β‐luc) or ISRE (ISRE‐luc), and an expression vector for RKIP (0, 50, 100, 200 ng each), together with plasmids encoding RIG‐I CARD, TBK1, IKKi, and IRF3 5D.
Figure EV3. RKIP is required for virus‐induced TBK1, IRF3, and p38 phosphorylation.
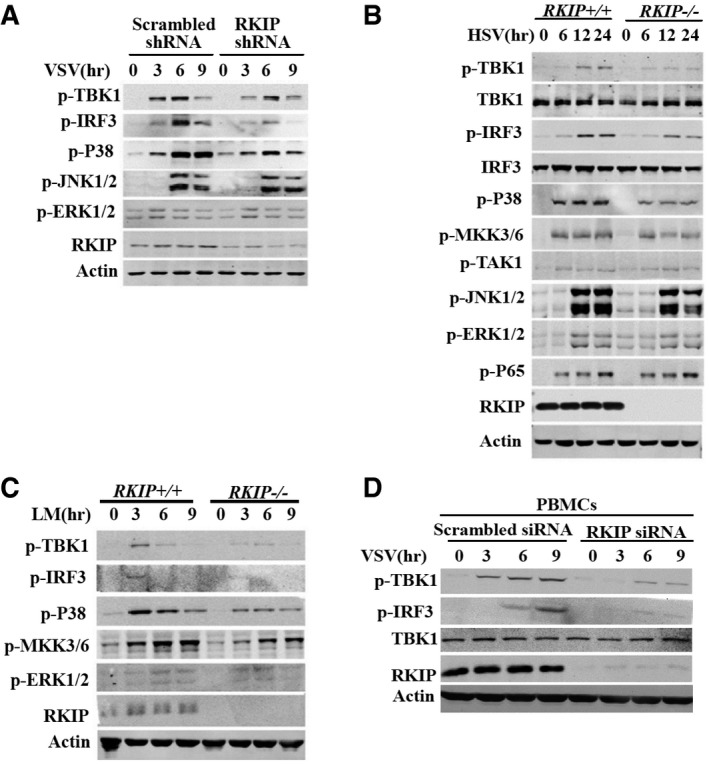
-
AImmunoblot analysis of p‐TBK1, p‐IRF3, p‐P38, p‐JNK1/2, p‐ERK1/2 in RAW264.7 cells stably transfected with RKIP‐specific or scrambled shRNA, followed by infection with VSV (MOI 0.1).
-
B, CImmunoblot analysis of p‐TBK1, p‐IRF3, p‐P38, p‐MKK3/6, p‐JNK1/2, p‐ERK1/2, p‐P65, and p‐TAK1 in lysates of wild‐type and RKIP‐deficient peritoneal macrophages stimulated with HSV (MOI 5) (B) or Listeria monocytogenes (MOI 10) (C).
-
DImmunoblot analysis of p‐TBK1 and p‐IRF3 in PBMCs treated with scrambled or RKIP‐specific siRNA for 48 h followed by stimulation with VSV (MOI 0.1) for the indicated times.
RKIP mediates autophosphorylation of TBK1
It is reported that glycogen synthase kinase 3β (GSK3β) regulates anti‐viral response via activation of TBK1 in a kinase‐independent manner (Lei et al, 2010). RKIP has been demonstrated to stabilize the expression GSK3β in HEK293 and HEK499 cells (Al‐Mulla et al, 2011). However, we did not observe any difference in the expression of GSK3β between the RKIP‐deficienct and wild‐type macrophages (Fig 3A), which excludes the possibility that RKIP affects TBK1 activation via GSK3β. We further analyzed whether RKIP affects the association between TBK1 and GSK3β. As shown in the Fig EV4A, RKIP has no effect on the interaction TBK1 with GSK3β in the macrophages treated with or without VSV infection. To investigate the underlying mechanism of the positive regulation of TBK1 pathway by RKIP, we immunoprecipitated RKIP from lysates of VSV‐infected RAW264.7 macrophages stably overexpressing flag‐RKIP. RKIP is physically associated with TBK1, but not IKKi, RIG‐I, MAVS, and STING in RAW264.7 cells (Fig 4A). VSV infection promotes the interaction between TBK1 and RKIP in a time‐dependent manner, as the interaction between these proteins peaks at 3 h after VSV infection, and then declines to the base line in resting cells (Fig 4A). It has been reported that the ULD of TBK1 regulates its kinase activity, playing an important role in signal transduction and mediating interactions of TBK1 with other molecules in the IFN pathway (Welchman et al, 2005; Ikeda et al, 2007). Domain mapping experiments demonstrate that the ubiquitin‐like domain (ULD) (aa 311‐388) is required for TBK1 to interact with RKIP (Fig 4B). However, we could not detect the interaction between RKIP and ULD truncation (Fig EV4B), indicating that ULD domain is necessary but not sufficient for the interaction of TBK1 with RKIP. As shown in Fig 4C, RKIP markedly enhances TBK1 phosphorylation, stronger than GSK3β, the positive control, which has been reported to associate with TBK1 and promote TBK1 autophosphorylation at Ser172 (Lei et al, 2010), when co‐overexpressed in 293T cells. Consistent with the data from RKIP‐deficient macrophage, overexpression of RKIP has no effect on the protein level of GSK3β (Fig 4C). We performed experiments to determine whether RKIP promoting TBK1 activation via GSK3β. Knockdown of GSK3β with GSK3β siRNA does not impair TBK1 activation induced by RKIP overexpression (Fig EV4C). In contrast, depletion of endogenous RKIP not only suppresses TBK1 activation but also abolishes GSK3β‐induced phosphorylation of TBK1 (Fig EV4D). These findings suggest that RKIP regulates TBK1 activation in a GSK3β‐independent way, whereas GSK3β promotes TBK1 activation in an RKIP‐dependent manner. To determine whether RKIP promotes TBK1 autophosphorylation of serine 172, we conducted in vitro kinase assay using the purified recombinant GST‐his‐RKIP and flag‐TBK1 protein (Fig EV4E). As shown in Fig 4D, RKIP directly enhances the autophosphorylation at serine 172 in vitro. We also examined whether RKIP regulates TBK1 dimerization or oligomerization. In transient transfection and coimmunoprecipitation experiments, we found RKIP has no effect on TBK1 self‐association (Fig EV4F). Taken together, these data suggest that RKIP promotes TBK1 autophosphorylation at Ser172, which is critical for TBK1‐mediated IRF3 activation and IFN‐β production.
Figure EV4. RKIP promotes TBK1 autophosphorylation.
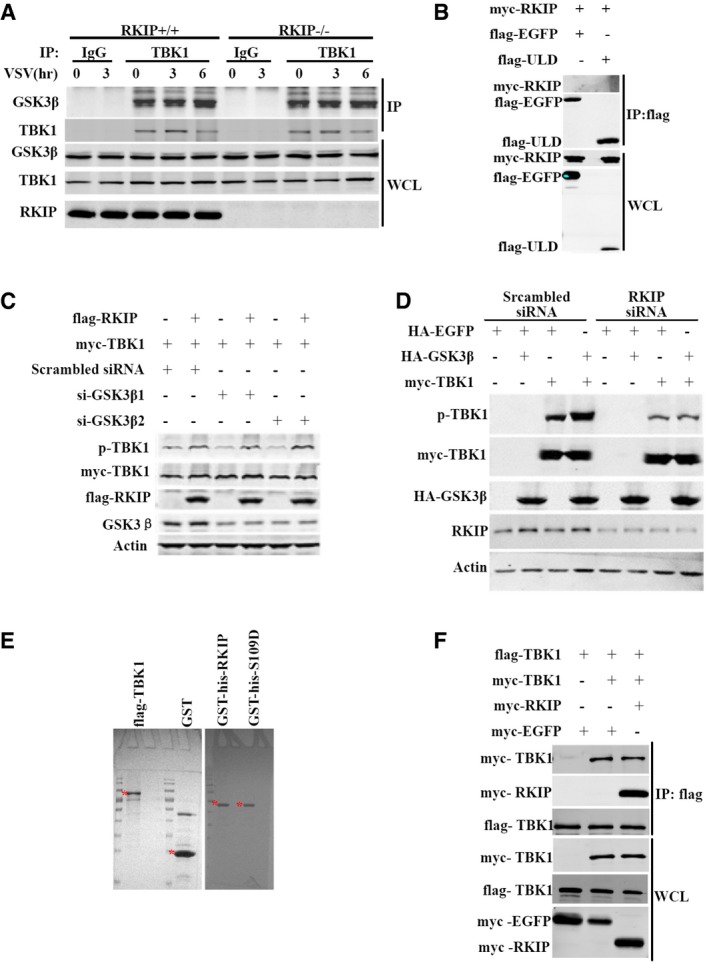
- Coimmunoprecipitation analysis with anti‐TBK1 antibody in RKIP +/+ and RKIP −/− peritoneal macrophages infected with VSV (MOI 0.1), followed by immunoblotting with the indicated antibody.
- Co‐immunoprecipitation and immunoblot analysis of 293T cells transfected with flag‐ULD alone or with myc‐RKIP. ULD, ubiquitin‐like domain.
- RKIP promoting TBK1 phosphorylation is GSK3β independent. Immunoblot of extracts of 293T cells transfected with plasmid for myc‐TBK1, flag‐RKIP, together with GSK3β‐specific or scrambled siRNA.
- GSK3β promotes TBK1 activation through a RKIP‐dependent mechanism. Immunoblot of extracts of 293T cells transfected with plasmid for myc‐TBK1, HA‐GSK3β, together with RKIP‐specific or scrambled siRNA.
- Purified flag‐TBK1, GST, GST‐his‐RKIP, or GST‐his‐S109D proteins were detected with Coomassie brilliant blue. * indicates targeted band.
- RKIP does not affect TBK1 self‐association. Coimmunoprecipitation and immunoblot analysis of 293T cells transfected with indicated combination of plasmid for flag‐TBK1, myc‐TBK1, myc‐RKIP, followed by immunoblot analysis with anti‐myc and anti‐flag antibodies.
Figure 4. RKIP interacts with TBK1 and promotes TBK1 autophosphorylation.
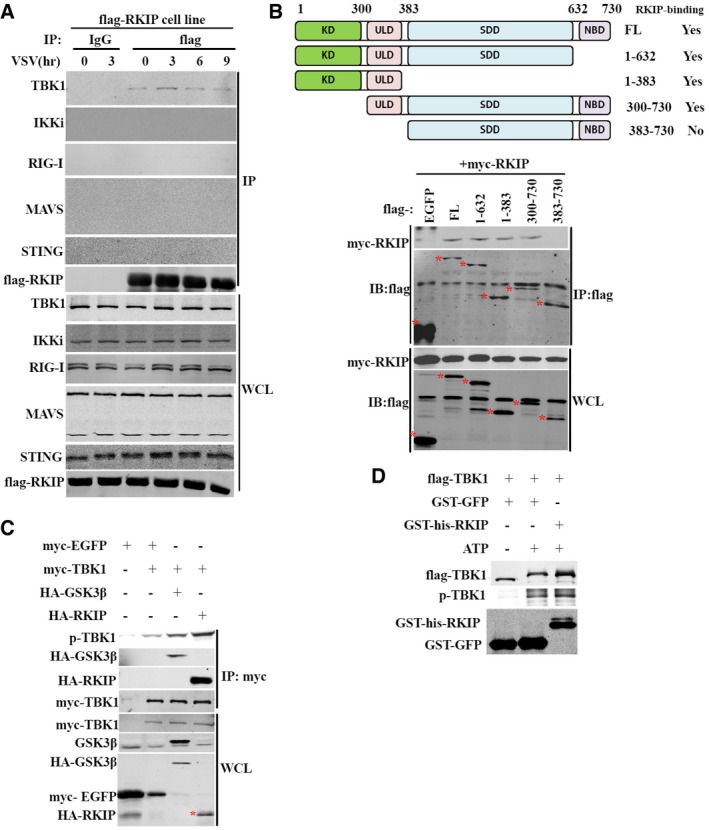
- Immunoblot of extracts of RAW264.7 cells stably overexpressing flag‐RKIP and infected with VSV (MOI 0.1) for the indicated times followed by immunoprecipitation (IP). WCL, whole‐cell lysates.
- Coimmunoprecipitation and immunoblot analysis of 293T cells transfected with vector for myc‐RKIP alone or with the deletion mutants of TBK1. Numbers in parentheses indicate amino acids included in construct. FL, full length; KD, kinase domain; ULD, ubiquitin‐like domain; SDD, α‐helical scaffold domain; NBD, C‐terminal domain. * indicates targeted band.
- Immunoblot of 293T cells transfected with plasmid for myc‐TBK1 together with plasmid for HA‐GSK3β or HA‐RKIP, followed by immunoprecipitation. * indicates targeted band.
- In vitro kinase assay was carried out in the reaction mixture containing 2 mM ATP, 100 μM GST‐RKIP or 100 μM GST‐GFP, and 5 μM flag‐TBK1 at 30°C for 30 min and then followed by immunoblotting with the indicated antibody.
S109 phosphorylation is essential for RKIP anti‐virus function
RKIP has been reported as an inhibitor of Raf/MEK/ERK1/2 and NF‐κB pathway (Yeung et al, 2001). Phosphorylation of RKIP at serine 153 by protein kinase C (PKC) switches RKIP from an inhibitor of Raf/MEK/ERK signaling pathway to an enhancer of GPCR pathway (Corbit et al, 2003; Lorenz et al, 2003), indicating that the post‐translational modification of RKIP is important for RKIP to execute multiple physiological functions. We purified flag‐tagged RKIP from VSV‐treated and untreated RAW264.7 cells using affinity purification processes, followed by mass spectrometry to examine whether viral infection induces post‐translation modification of RKIP. VSV infection does not affect the methylation or acetylation of RKIP (data not shown). Phosphorylation at serine 109 (S109) is observed only in RKIP purified from VSV‐treated cells (Fig EV5A) while that at serine 52 (S52) is observed in RKIP from both VSV‐treated or untreated cells (data not shown). To evaluate the roles of S52 and S109 phosphorylation in RKIP anti‐virus function, each serine was individually mutated to alanine (S52A or S109A) to mimic non‐phosphorylation, to aspartic or glutamic acid (S52D or S109D) to mimic constitutive phosphorylation. RAW264.7 cells stably expressed these mutants were collected and exposed to VSV infection. As shown in Figs 5A and EV5B, the flag‐S109A mutant fails to promote virus‐triggered production of type I IFN and proinflammatory cytokines, whereas flag‐RKIP and flag‐S109D mutants enhance the production of type I IFN and pro‐inflammatory cytokines. We next evaluated whether both IRF3 activation and p38 activation are affected by phosphorylation of S109. Phosphorylation levels of TBK1, IRF3, and p38 are elevated after VSV or HSV infection in RAW264.7 macrophages overexpressing flag‐RKIP or flag‐S109D, comparing to that in control cells. In contract, flag‐S109A fails to enhance the phosphorylation of TBK1, IRF3, and p38 triggered by viral infection (Figs 5B and EV5C). Overexpression of flag‐RKIP or mutant flag‐S109D, but not flag‐S109A, renders RAW264.7 cells resistant to viral infection (Fig 5C). However, S52A and S52D mutants show comparable effects to wild‐type RKIP on anti‐viral innate immune response (data not shown). Rescue with RKIP‐deficient macrophage was performed to confirm the role of serine 109 phosphorylation in regulating the production of type I interferon. As shown in the Fig 5D, RKIP‐deficient macrophage showed the impaired phosphorylation of P38, TBK1, and IRF3 induced by VSV, which was rescued to comparable level to wild‐type macrophage upon overexpression of flag‐RKIP or flag‐S109D but not flag‐S109A. Measurement of type I interferon and TNF‐α production using ELISA revealed similar results (Fig 5E). Taken together, the phosphorylation at S109 is critical for RKIP to promote IFN‐β production and virus clearance.
Figure EV5. RKIP promotes anti‐viral immune response in a S109 phosphorylation‐dependent manner.
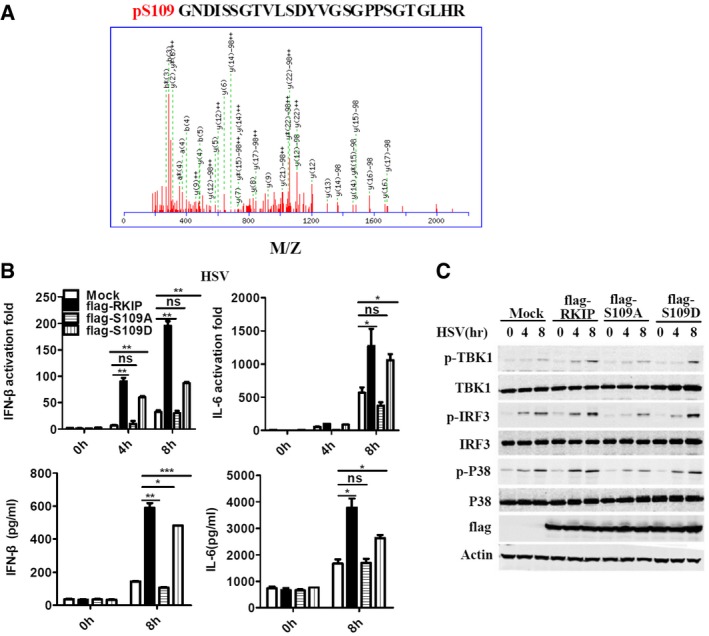
- Tandem MS of phosphorylation sites S109 of RKIP.
- ELISA of IL‐6 and IFN‐β, and real‐time PCR analysis of Il6 and Ifnb1 mRNA in RAW264.7 cells stably overexpressing flag‐RKIP, flag‐S109A, or flag‐S109D, followed by infection with HSV (MOI 5).
- Immunoblot analysis of p‐TBK1, p‐IRF3, p‐P38 of cells in (B).
Figure 5. RKIP promotes anti‐viral immune response in a S109 phosphorylation‐dependent manner.
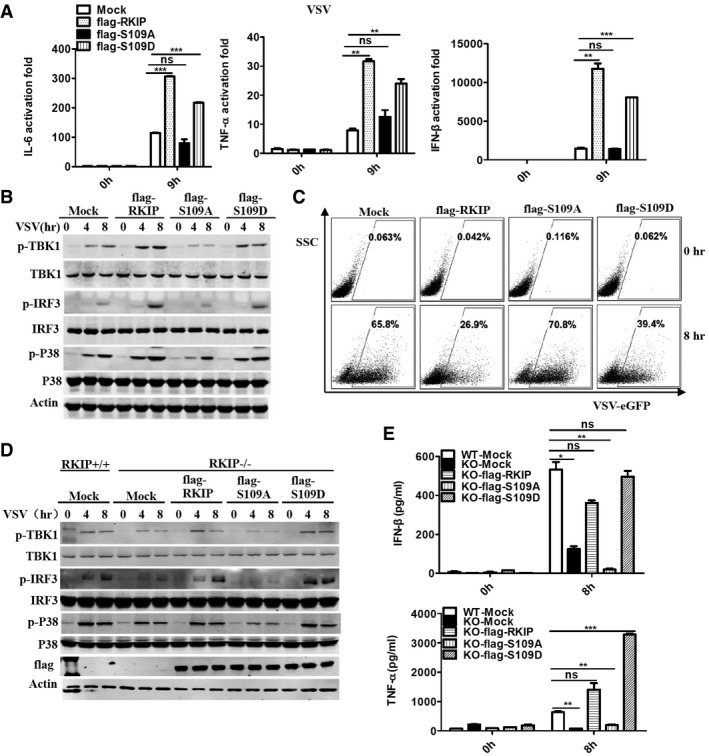
- Real‐time PCR analysis of Il6, Tnf, and Ifnb1 mRNA in RAW264.7 cells stably overexpressing flag‐RKIP, flag‐S109A, and flag‐S109D, followed by infection with VSV (MOI 0.1).
- Immunoblot analysis of p‐TBK1, p‐IRF3, and p‐P38 of cells as in (A).
- FACS analysis of RAW264.7 cells stably overexpressing flag‐RKIP, flag‐S109A, and flag‐S109D infected with VSV‐eGFP at MOI of 1.
- Immunoblot analysis of phosphorylated TBK1, IRF3, and P38 in lysates of RKIP +/+ or RKIP −/− bone marrow‐derived macrophages (BMDM) transfected with flag‐RKIP, flag‐S109A, and flag‐S109D, and then stimulated with VSV (MOI 0.1).
- ELISA of IFN‐β and TNF‐α of cells in (D).
TBK1 phosphorylates RKIP at S109
To facilitate phosphorylation detection of RKIP at S109, we generated pSer109‐RKIP‐specific antibody (p‐RKIP). This antibody specifically recognizes wild‐type RKIP but not the S109A mutants overexpressed in 293T cells (Appendix Fig S1A), and calf intestine phosphatase (CIP) treatment diminishes the phosphorylation of RKIP S109 in VSV‐treated BMDM cells (Appendix Fig S1B). We examined the phosphorylation of RKIP S109 in BMDM cells infected with VSV or HSV, the phosphorylation increases to highest level at 3 h after infection, and then restores to the rest condition (Fig 6A). RKIP physically interacts with TBK1, TGF‐activated kinase 1 (TAK1), and Raf1 in RAW264.7 macrophage (data not shown). To illuminate how RKIP is phosphorylated, we used TBK1‐, TAK1‐, or Raf1‐specific siRNA to investigate their roles in RKIP phosphorylation in BMDM. The phosphorylation of RKIP at S109 is only reduced by TBK1 knockdown (Fig 6B and Appendix Fig S1C). Simultaneously, the phosphorylation of RKIP at S109 is abolished in the TBK1 knockout mouse embryonic fibroblasts (MEFs) with or without VSV treatment (Fig 6C). We further performed in vitro kinase assay to examine whether TBK1 is the kinase responsible for S109 phosphorylation. As shown in Fig 6D, TBK1 directly phosphorylates RKIP at S109 in vitro. A shift of RKIP bands is also observed in the TBK1‐treated group, indicating that TBK1 is responsible for the viral infection triggered RKIP phosphorylation.
Figure 6. TBK1 phosphorylates RKIP at serine 109.
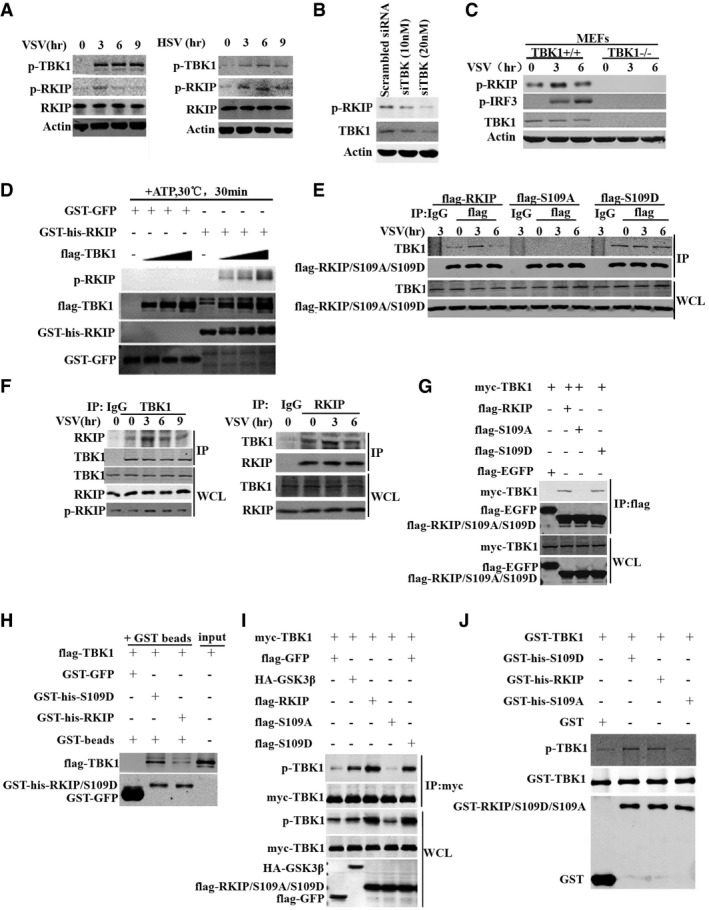
- BMDM cells were infected with VSV (MOI 0.1) or HSV (MOI 5) for the indicated times. Cell lysates were analyzed by immunoblotting with the indicated antibodies.
- BMDM cells were transfected with control or TBK1 siRNA. 72 h after transfection, cells were analyzed by immunoblotting with antibodies for phospho‐RKIP or TBK1.
- Immunoblot analysis of RKIP phosphorylation in control or TBK1 −/− mouse embryonic fibroblasts after VSV (MOI 0.1) infection for the indicated times.
- Recombinant flag‐tagged TBK1 was immunoprecipitated with anti‐flag M2 beads in flag‐TBK1‐transfected 293T cells. Followed this, in vitro kinase assay was carried out in the reaction mixture containing 2 mM ATP, 100 μM GST‐RKIP, or 100 μM GST‐GFP together with 5 μM flag‐TBK1 at 30°C for 30 min. Assay mixtures were immunoblotted with the indicated antibodies for phospho‐RKIP, flag, GST, or his.
- Immunoblot analysis of extracts of RAW264.7 cells stably overexpressing flag‐RKIP, flag‐S109A, and flag‐S109D, and infected with VSV (MOI 0.1) for the indicated times, followed by immunoprecipitation with anti‐flag beads.
- BMDM cells were infected with VSV (MOI 0.1) for the indicated times. The cell lysates were immunoprecipitated with anti‐TBK1 (left) or anti‐RKIP (right). The immunoprecipitates were analyzed by immunoblot with anti‐RKIP or anti‐TBK1 antibody. The levels of the endogenous RKIP, TBK1, and p‐RKIP were detected by immunoblot analysis.
- Co‐immunoprecipitation and immunoblot analysis of 293T cells transfected with myc‐TBK1 alone or with flag‐RKIP, flag‐S109A, or flag‐S109D.
- GST pull‐down experiment was performed with 1 μg fusion protein GST‐GFP, GST‐his‐RKIP, or GST‐his‐S109D mixed with 20 μl pre‐cleared agarose beads in 800 μl reaction medium, followed by adding 1 μg flag‐TBK1 and incubating at 4°C for 3 h with gentle rotation. Pull‐down (lane 1–3) and input samples (lane 4) were separated by SDS–PAGE followed by immunoblotting with anti‐GST or anti‐flag antibody.
- Immunoprecipitation and immunoblot analysis of 293T cells transfected with indicated combinations of vector for myc‐TBK1, flag‐RKIP, flag‐S109A, flag‐S109D, HA‐GSK3β or flag‐GFP.
- In vitro assay was carried out in the reaction mixture containing 2 mM ATP, 50 μM GST‐TBK1 together with 100 μM GST‐his‐RKIP, 100 μM GST‐his‐S109D, 100 μM GST‐his‐S109A, or 100 μM GST, respectively, at 30°C for 30 min, followed by immunoblotting with anti‐p‐TBK1 or anti‐GST antibody.
RKIP widely exists in a variety of different mammalian species such as monkey, rat, chicken, and human (Appendix Fig S1D). The crystal structure of RKIP in complex with a phosphotyrosine (PDB ID: 2QYQ) reveals two noteworthy features (Appendix Fig S2A). First, S109 is buried in a hydrophobic pocket formed by residues Pro43, Val46, Tyr120, Tyr169, and Ala171. The hydroxyl group of S109 forms a short hydrogen bond (2.7 Å) with the hydroxyl group of tyrosine 169, stabilizing the interactions. Second, the phosphotyrosine binding pocket is closed to S109, with a short distance of 5 Å. These structural features suggest a phosphorylation‐induced molecular switch function of RKIP: RKIP first undergoes an unfolding process to expose S109 for phosphorylation. As the phosphate moiety is negative charged and bulky, phosphorylated S109 no longer folds back to the original hydrophobic pocket. Instead, it tends to shift to the phosphate binding site nearby. Such a structural transition further causes the rearrangement of the tertiary structure of RKIP and switches the functional state of RKIP (Appendix Fig S2B).
To verify the molecular switch function of RKIP, we hypothesized whether the phosphorylation at S109 of RKIP influences its interaction with TBK1. Coimmunoprecipitation experiments showed that RKIP and S109D but not S109A associate with TBK1 in RAW264.7 cell line. The association between RKIP and TBK1 is strengthened by VSV treatment at 3 h and then restored to basal level in RKIP transfectants (Fig 6E). In BMDM cells, the VSV‐induced association of RKIP with TBK1 correlates with RKIP S109 phosphorylation (Fig 6F). Cytosolic RNA/DNA sensing pathways facilitated by either MAVS or STING binding to cytosolic sensors RLG‐I or cGAS, leads to activation of TBK1 (Rathinam & Fitzgerald, 2011; Jensen & Thomsen, 2012). Thus, we next examined whether TBK1–RKIP interaction depends on MAVS or STING recruitment. As shown in the Appendix Fig S2C, the interaction between TBK1 and RKIP is observed in the MAVS −/− or STING −/− BMDMs as in the wild‐type BMDMs, indicating MAVS or STING recruitment is not required for the physical TBK1–RKIP interaction. As expected, this interaction is enhanced by VSV or HSV infection in the wild‐type BMDMs but not in the MAVS −/− or STING −/− BMDMs. Consistently, TBK1 is coimmunoprecipitated with flag‐RKIP and flag‐S109D but not with flag‐S109A in 293T cells (Fig 6G). GST pull‐down assay showed that RKIP directly interacts with TBK1, and mutation of S109D in RKIP strongly increases the interaction between RKIP and TBK1, indicating that phosphorylation of S109 facilitates the interaction between RKIP and TBK1 (Fig 6H). Meanwhile, the overexpression of flag‐RKIP and flag‐S109D but not flag‐S109A causes an increased phosphorylation of TBK1 (Fig 6I). In vitro kinase activity assay showed that recombinant protein GST‐his‐RKIP and GST‐his‐S109D but not GST‐his‐S109A promotes GST‐TBK1 autophosphorylation (Fig 6J). Consistently, flag‐S109D increases TBK1 phosphorylation more efficient than wild‐type RKIP, providing further evidences that S109 phosphorylation of RKIP by TBK1 is prerequisite for RKIP to associate with TBK1 and enhance TBK1 activation. Taken together, these data demonstrate that RKIP and TBK1 form a positive feedback loop, which plays an important role in type I interferon production in innate immunity.
Discussion
TBK1 plays a central role in type I interferon production in innate immunity. Albeit been intensively studied, how TBK1 is activated and regulated remains unclear. In the current study, we observed that RKIP deficiency inhibits, and RKIP overexpression enhances virus‐induced TBK1 activation. Consistently, RKIP deficiency significantly inhibits viruses, including VSV, SeV, and HSV, and also bacteria LM‐induced type I interferon production. RKIP directly binds to TBK1 and promotes TBK1 activation upon virus stimulation. These results provide convincing evidence that RKIP is an important positive regulator of TBK1 activation and type I interferon production in innate immunity. Recently, GSK3β was reported to positively regulate viral infection‐induced TBK1 activation (Lei et al, 2010). We found that GSK3β positively regulates TBK1 activation in an RKIP‐dependent manner and RKIP directly potentiates TBK1 activation in a cell‐free system, suggesting that RKIP is the pivotal molecule responsible for TBK1 activation. However, how RKIP promotes TBK1 activation remains unclear. RKIP is not an enzyme and does not promote TBK1 dimerization. It might interact with TBK1 and induce conformational change to regulate TBK1 activation.
Since the discovery as a phosphatidylethanolamine‐binding protein, RKIP has emerged as a critical molecule that modulates and controls crucial intracellular signaling networks, including the signaling cascades of Raf/MEK/ERK, NF‐kB, GSK3β, and GPCR (Yeung et al, 2001; Thornton et al, 2008; Al‐Mulla et al, 2011), thus regulates a variety of physiological processes such as differentiation, cell cycle, apoptosis, and contractile activity of cardiomyocytes (Al‐Mulla et al, 2013). Our data demonstrate that RKIP functions as anti‐viral molecular by directly promoting TBK1 activation. These data not only extend the physiological functions of RKIP, but also add a new dimension to the pleiotropic actions of RKIP, as RKIP mediates kinase inhibition through disrupting its interactions with substrates, instead of modulation of the catalytic activities (Lorenz et al, 2003).
How such a small protein of 21 kDa can coordinate its diverse signaling functions is still a matter of discussion. Thus far, protein kinase C controls alternate binding of RKIP to its targets through phosphorylating RKIP at Ser153, is the only well‐characterized regulatory mechanism. This phosphorylation switches RKIP from Raf1 to G‐protein‐coupled receptor kinase‐2, a negative regulator of GPCRs (Lorenz et al, 2003). Our data provide strong evidence showing that RKIP controls a different kinase via post‐translational modification: Phosphorylation at S109 of RKIP elevates the binding affinity between RKIP and TBK1 and enhances the autophosphorylation of TBK1, which is critical for RKIP to promote TBK1 activation and anti‐viral response. Based on the crystal structure of RKIP, we hypothesize that the phosphorylation of S109 induces a conformational switch in RKIP. It is worth to further explore how this single phosphorylation can cause such a dramatic change of binding specificity in such a small protein.
Meanwhile, our findings identify a novel mechanism of TBK1 autoregulation under virus‐infected condition. Upon viral infection, TBK1 mediates RKIP S109 phosphorylation and the phosphorylated RKIP in turn interacts with TBK1 and promotes TBK1 activation, the fully activated TBK1 then phosphorylates IRF3 and ultimately leads to the production of type I interferon to clear the invading virus and bacterium (Fig 7). Thus, the phosphorylation at S109 of RKIP by TBK1 triggers a positive feedback on TBK1 activity, which is essential for type I interferon production in anti‐viral innate immunity. Except for its critical role in innate immunity, TBK1 is an important node protein for multiple other signaling pathways including autophagy, cell proliferation, and growth and insulin signaling (Helgason et al, 2013). RKIP has been reported to regulate these physiological processes (Al‐Mulla et al, 2013). Whether the feedback of TBK1 and RKIP is involved in other physiological or pathological processes is worth to be investigated.
Figure 7. A positive feedback loop between RKIP and TBK1 promotes type I interferon production in innate immunity.
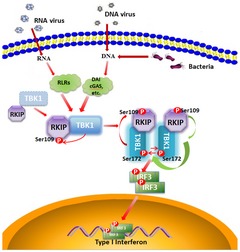
Upon viral infection, TBK1 phosphorylates RKIP at S109. The phosphorylation of RKIP enhances its interaction with TBK1 and in turn promotes TBK1 autophosphorylation, thus triggering a positive feedback control of TBK1 activation, which is essential for type I interferon production in innate immunity.
In summary, we have shown that RKIP is specifically phosphorylated at S109 by TBK1 upon viral infection and RKIP regulates TBK1 activation through a positive feedback mechanism. These findings not only provide insight into the positive regulation of TBK1‐mediated anti‐viral innate immune responses, but also provide a novel post‐translational modification mechanism that allows RKIP to act as an anti‐virus agent.
Materials and Methods
Cell culture and reagents
HEK293T, RAW264.7, peritoneal macrophage, and bone marrow‐derived macrophage cells were maintained in DMEM (Gibco) or RPMI‐1640 medium (Invitrogen) containing 10% heat‐inactivated FBS (Biology Industries). TBK1 −/− MEFs were gift from Dr. Genhong Cheng (University of California, Los Angeles). Poly(I:C) was obtained from Sigma‐Aldrich. cGAMP was from InvivoGen. Recombination GST‐TBK1 (P01) was from Abnova. Anti‐flag (M2) was from Sigma‐Aldrich. The polyclonal antibody against mouse phospho‐RKIP‐Ser109 was generated by immunizing rabbit (Abmart) with peptides: C‐SDYVG(pS)GPPSG. Antibodies are listed in Appendix Table S1.
Bone marrow transplantation
Bone marrow cells (1 × 107) from RKIP +/+ or RKIP −/− mice were transplanted into lethally irradiated wild‐type C57BL/6J mice (cumulative dose, 7.8 Gy) by injection into the tail vein. After 8 weeks, the expression of RKIP on hematopoietic cells was analyzed by PCR.
Virus infection
Virus was generated as described previously (Gu et al, 2014). Cells were infected with VSV (MOI 1), SEV (MOI 10), and HSV (MOI 5) for the indicated times. Cytokine production was analyzed at the indicated time. For in vivo studies, age‐ and sex‐matched groups of littermate mice were infected with VSV (1 × 107 pfu/g, i.p., 12 h) for the determination of IFN‐β production in serum or organs and the determination of viral titer in organs. For in vivo survival studies, age‐ and sex‐matched groups of littermate mice were infected intravenously with VSV (1 × 108 pfu/g, i.v.). For HSV infection in vivo, HSV (1 × 106 pfu/mouse, i.v.) was injected intravenously into age‐ and sex‐matched mice for the determination of IFN‐β production in serum (6 h) or peritoneal macrophages (24 h) and viral load in brain (3 days).
Histological analysis
Livers or lungs from control or virus‐infected mice were dissected, fixed in 4% paraformaldehyde, embedded into paraffin, cut into sections, stained with hematoxylin and eosin solution, and examined by light microscopy for histology changes. Immunohistochemical staining was performed using standard procedures.
GST pull‐down assay
The fusion proteins of GST‐GFP, GST‐his‐RKIP, and GST‐his‐S109D were expressed in E. coli and purified according to standard protocols with pre‐equilibrated glutathione–Sepharose beads (GE Healthcare). Flag‐TBK1 was transiently overexpressed in 293T cells and purified using anti‐flag (M2) agarose beads according to the manufacturer's protocol. For GST pull‐down assay, approximately 1 μg GST‐fusion proteins mix with 20 μl pre‐cleared agarose beads in 800 μl reaction medium, followed by adding 1 μg flag‐TBK1 and incubating at 4°C for 3 h with gentle rotation. Precipitates were extensively washed and then eluted with 2× loading buffer, boil and then run SDS–PAGE gel.
Immunoblot analysis and immunoprecipitation
Total cells were washed two times with ice‐cold PBS and lysed with cell lysis buffer (Cell Signaling Technology). Protein concentration was determined by the BCA protein assay kit (Thermo). Cell lysates (20–50 μg) were separated by SDS–PAGE, transferred onto PVDF membranes (Millipore), and probed with a primary antibody against target protein. The results were analyzed by Adobe Photoshop software and normalized to actin. For complex coimmunoprecipitation, cell extracts were prepared by using lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, and 0.5% (vol/vol) Nonidet P‐40, 1 mM EDTA) supplemented with a protease inhibitor cocktail (Roche). Lysates were incubated with the anti‐flag (M2)‐agarose or antibody‐coupled beads for 3 h at 4°C.The immunoprecipitates were washed three times with the same buffer and subjected to immunoblot analysis.
In vitro kinase assay
In vitro kinase assay was performed as described previously (Cheung et al, 2003; Ouyang et al, 2014). Briefly, flag‐TBK1 was transiently overexpressed in 293T cells and purified using anti‐flag (M2)‐agarose beads according to the manufacturer's protocol. GST‐his‐RKIP, GST‐his‐S109D, GST, or GST‐GFP was expressed in E. coli BL21 and purified using glutathione beads (Thermore) according to the manufacturers' protocols. The in vitro kinase assay was carried out in 10 μl of reaction mixture containing 50 mM Tris–HCl, pH 7.5, 2 mM ATP, 5 mM MgCl2, 100 μM GST‐RKIP/GST‐S109D or 100 μM GST‐GFP, and 5 μM flag‐TBK1 or 50 μM GST‐TBK1. The mixture was incubated at 30°C for 30 min and analyzed by immunoblotting with the indicated antibodies.
Statistical analysis
Statistical analysis was performed with Prism v5.0 (Graphpad Software). Data are expressed as mean ± SEM by a Student's t‐test. For in vivo experiments, values are expressed as the mean ± SEM of n animals. Mouse survival curves and statistics were analyzed with the Mantel–Cox test. The level of statistically significant difference was defined as P < 0.05.
Structural modeling
The crystal structure of RKIP in complex with a phosphotyrosine was downloaded from Protein Data Bank (PDB ID: 2QYQ), analyzed, and modeled using Coot (Emsley & Cowtan, 2004). Based on this structure, S109 was changed to phosphoserine and relocated to the nearby phosphate binding site. Other residues were also manually modeled correspondingly. The geometries of the modeled structure were refined using REFMAC (Collaborative Computational Project Number 4, 1994). All structural illustrations were prepared with PyMOL (http://www.pymol.org).
Author contributions
XW designed research; MG, ZL, SL, RL, WL, CO, and SY performed research; HH contributed the reagents; XW, MG, and SY analyzed data and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank Dr. Aiping Wu for providing evolutionary characteristic of RKIP and Dr. Huzhang An and Feng Ma for critical reading of the manuscript. This work was supported by grants from the National Basic Research Program of China (973) (2014CB542101), the National Natural Science Foundation of China (31570864, 81230014), and the Natural Science Foundation of Zhejiang Province (LR13C080001).
The EMBO Journal (2016) 35: 2553–2565
References
- Al‐Mulla F, Bitar MS, Al‐Maghrebi M, Behbehani AI, Al‐Ali W, Rath O, Doyle B, Tan KY, Pitt A, Kolch W (2011) Raf kinase inhibitor protein RKIP enhances signaling by glycogen synthase kinase‐3beta. Cancer Res 71: 1334–1343 [DOI] [PubMed] [Google Scholar]
- Al‐Mulla F, Bitar MS, Taqi Z, Yeung K (2013) RKIP: much more than Raf kinase inhibitory protein. J Cell Physiol 228: 1688–1702 [DOI] [PubMed] [Google Scholar]
- Ausubel FM (2005) Are innate immune signaling pathways in plants and animals conserved? Nat Immunol 6: 973–979 [DOI] [PubMed] [Google Scholar]
- Barbalat R, Ewald SE, Mouchess ML, Barton GM (2011) Nucleic acid recognition by the innate immune system. Annu Rev Immunol 29: 185–214 [DOI] [PubMed] [Google Scholar]
- Bernier I, Jolles P (1984) Purification and characterization of a basic 23 kDa cytosolic protein from bovine brain. Biochim Biophys Acta 790: 174–181 [DOI] [PubMed] [Google Scholar]
- Chen G, Shaw MH, Kim YG, Nunez G (2009) NOD‐like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol 4: 365–398 [DOI] [PubMed] [Google Scholar]
- Cheung PC, Campbell DG, Nebreda AR, Cohen P (2003) Feedback control of the protein kinase TAK1 by SAPK2a/p38alpha. EMBO J 22: 5793–5805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Corbit KC, Trakul N, Eves EM, Diaz B, Marshall M, Rosner MR (2003) Activation of Raf‐1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J Biol Chem 278: 13061–13068 [DOI] [PubMed] [Google Scholar]
- Dangi‐Garimella S, Yun J, Eves EM, Newman M, Erkeland SJ, Hammond SM, Minn AJ, Rosner MR (2009) Raf kinase inhibitory protein suppresses a metastasis signalling cascade involving LIN28 and let‐7. EMBO J 28: 347–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Gack MU (2014) Mechanisms of RIG‐I‐like receptor activation and manipulation by viral pathogens. J Virol 88: 5213–5216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geijtenbeek TB, Gringhuis SI (2009) Signalling through C‐type lectin receptors: shaping immune responses. Nat Rev Immunol 9: 465–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M, Zhang T, Lin W, Liu Z, Lai R, Xia D, Huang H, Wang X (2014) Protein phosphatase PP1 negatively regulates the Toll‐like receptor‐ and RIG‐I‐like receptor‐triggered production of type I interferon by inhibiting IRF3 phosphorylation at serines 396 and 385 in macrophage. Cell Signal 26: 2930–2939 [DOI] [PubMed] [Google Scholar]
- Hacker H, Karin M (2006) Regulation and function of IKK and IKK‐related kinases. Sci STKE 2006: re13 [DOI] [PubMed] [Google Scholar]
- Helgason E, Phung QT, Dueber EC (2013) Recent insights into the complexity of Tank‐binding kinase 1 signaling networks: the emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett 587: 1230–1237 [DOI] [PubMed] [Google Scholar]
- Honda K, Takaoka A, Taniguchi T (2006) Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 25: 349–360 [DOI] [PubMed] [Google Scholar]
- Ikeda F, Hecker CM, Rozenknop A, Nordmeier RD, Rogov V, Hofmann K, Akira S, Dotsch V, Dikic I (2007) Involvement of the ubiquitin‐like domain of TBK1/IKK‐i kinases in regulation of IFN‐inducible genes. EMBO J 26: 3451–3462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen S, Thomsen AR (2012) Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol 86: 2900–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Kim YJ (2007) Signaling pathways downstream of pattern‐recognition receptors and their cross talk. Annu Rev Biochem 76: 447–480 [DOI] [PubMed] [Google Scholar]
- Lei CQ, Zhong B, Zhang Y, Zhang J, Wang S, Shu HB (2010) Glycogen synthase kinase 3beta regulates IRF3 transcription factor‐mediated antiviral response via activation of the kinase TBK1. Immunity 33: 878–889 [DOI] [PubMed] [Google Scholar]
- Lin R, Heylbroeck C, Pitha PM, Hiscott J (1998) Virus‐dependent phosphorylation of the IRF‐3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome‐mediated degradation. Mol Cell Biol 18: 2986–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Ma C, Su F, Jiang Y, Lai R, Zhang T, Sun K, Fan L, Cai Z, Li Z, Huang H, Li J, Wang X (2016) Raf kinase inhibitor protein mediates intestinal epithelial cell apoptosis and promotes IBDs in humans and mice. Gut doi:10.1136/gutjnl‐2015‐310096 [DOI] [PubMed] [Google Scholar]
- Loo YM, Gale M Jr (2011) Immune signaling by RIG‐I‐like receptors. Immunity 34: 680–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz K, Lohse MJ, Quitterer U (2003) Protein kinase C switches the Raf kinase inhibitor from Raf‐1 to GRK‐2. Nature 426: 574–579 [DOI] [PubMed] [Google Scholar]
- Morgan C, LaCourse EJ, Rushbrook BJ, Greetham D, Hamilton JV, Barrett J, Bailey K, Brophy PM (2006) Plasticity demonstrated in the proteome of a parasitic nematode within the intestine of different host strains. Proteomics 6: 4633–4645 [DOI] [PubMed] [Google Scholar]
- Ouyang C, Nie L, Gu M, Wu A, Han X, Wang X, Shao J, Xia Z (2014) Transforming growth factor (TGF)‐beta‐activated kinase 1 (TAK1) activation requires phosphorylation of serine 412 by protein kinase A catalytic subunit alpha (PKACalpha) and X‐linked protein kinase (PRKX). J Biol Chem 289: 24226–24237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paludan SR, Bowie AG (2013) Immune sensing of DNA. Immunity 38: 870–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VA, Fitzgerald KA (2011) Innate immune sensing of DNA viruses. Virology 411: 153–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reumer A, Bogaerts A, Van Loy T, Husson SJ, Temmerman L, Choi C, Clynen E, Hassan B, Schoofs L (2009) Unraveling the protective effect of a Drosophila phosphatidylethanolamine‐binding protein upon bacterial infection by means of proteomics. Dev Comp Immunol 33: 1186–1195 [DOI] [PubMed] [Google Scholar]
- Serre L, Pereira de Jesus K, Zelwer C, Bureaud N, Schoentgen F, Benedetti H (2001) Crystal structures of YBHB and YBCL from Escherichia coli, two bacterial homologues to a Raf kinase inhibitor protein. J Mol Biol 310: 617–634 [DOI] [PubMed] [Google Scholar]
- Shu C, Sankaran B, Chaton CT, Herr AB, Mishra A, Peng J, Li P (2013) Structural insights into the functions of TBK1 in innate antimicrobial immunity. Structure 21: 1137–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ (2013) Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140: 805–820 [DOI] [PubMed] [Google Scholar]
- Thornton TM, Pedraza‐Alva G, Deng B, Wood CD, Aronshtam A, Clements JL, Sabio G, Davis RJ, Matthews DE, Doble B, Rincon M (2008) Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation. Science 320: 667–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakul N, Menard RE, Schade GR, Qian Z, Rosner MR (2005) Raf kinase inhibitory protein regulates Raf‐1 but not B‐Raf kinase activation. J Biol Chem 280: 24931–24940 [DOI] [PubMed] [Google Scholar]
- Vance RE, Isberg RR, Portnoy DA (2009) Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe 6: 10–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welchman RL, Gordon C, Mayer RJ (2005) Ubiquitin and ubiquitin‐like proteins as multifunctional signals. Nat Rev Mol Cell Biol 6: 599–609 [DOI] [PubMed] [Google Scholar]
- Werling D, Jann OC, Offord V, Glass EJ, Coffey TJ (2009) Variation matters: TLR structure and species‐specific pathogen recognition. Trends Immunol 30: 124–130 [DOI] [PubMed] [Google Scholar]
- Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, Sedivy JM, Kolch W (1999) Suppression of Raf‐1 kinase activity and MAP kinase signalling by RKIP. Nature 401: 173–177 [DOI] [PubMed] [Google Scholar]
- Yeung KC, Rose DW, Dhillon AS, Yaros D, Gustafsson M, Chatterjee D, McFerran B, Wyche J, Kolch W, Sedivy JM (2001) Raf kinase inhibitor protein interacts with NF‐kappaB‐inducing kinase and TAK1 and inhibits NF‐kappaB activation. Mol Cell Biol 21: 7207–7217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Imamoto A, Rosner MR (2008) Raf kinase inhibitory protein (RKIP): a physiological regulator and future therapeutic target. Expert Opin Ther Targets 12: 1275–1287 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6