

Abstract
The centrosome is crucial for neuronal migration and polarisation, processes that are disrupted in a number of neurodevelopmental disorders including schizophrenia. Mutation of DISC1, associated with increased risk of schizophrenia and psychiatric illness, has been shown to affect the centrosome, but the mechanisms involved have not been elucidated. In this issue of EMBO Reports, Fukuda and colleagues demonstrate that a DISC1‐interacting protein, CAMDI, suppresses the activity of the histone deacetylase HDAC6, thereby promoting centrosome stability and consequently neuronal migration 1. Loss of CAMDI leads to cortical migration defects and behavioural phenotypes that model autism spectrum disorders and which can be rescued by inhibition of HDAC6. The study provides novel mechanistic insight into centrosome regulation in neurodevelopment.
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Molecular Biology of Disease; Neuroscience
The centrosome is the main microtubule organising centre in the cell and hence important for cytoskeleton function. Studies of centrosomal protein mutations have revealed roles of the centrosome in correct neuronal migration and cell polarisation, key steps in the patterning of the developing nervous system. Noteworthy, mutations in proteins located at the centrosome have been identified in patients with psychiatric disorders.
The disrupted‐in‐schizophrenia 1 (DISC1) gene has been identified as a strong risk factor for schizophrenia and psychiatric disorders. DISC1 is localised throughout the cell, including at the centrosome, primary cilia, microtubules, mitochondria and nucleus. Previous studies have demonstrated that DISC1 is part of the microtubule‐associated dynein motor complex, important for cargo transport along microtubules in the cell. DISC1 also maintains this complex at the centrosome 2.
DISC1 interacts with a number of proteins at the centrosome (Fig 1A), including the other components of the microtubule‐associated dynein motor complex Ndel1, LIS1 and BBS4. DISC1 dysfunction has also been shown to disrupt cortical patterning, with centrosome impairment appearing to play a role 2, 3. Interestingly, a previous study identified coiled‐coil protein associated with myosin II and DISC1 (CAMDI) as a DISC1 interactor that depends on DISC1 binding for its translocation to the centrosome 4. In vivo studies demonstrated that knockdown of CAMDI led to defects in neuronal migration and centrosome mispositioning in cortical development, but the underlying mechanism was unclear.
Figure 1. CAMDI represses HDAC6 activity to promote centrosome stability and neuronal migration.
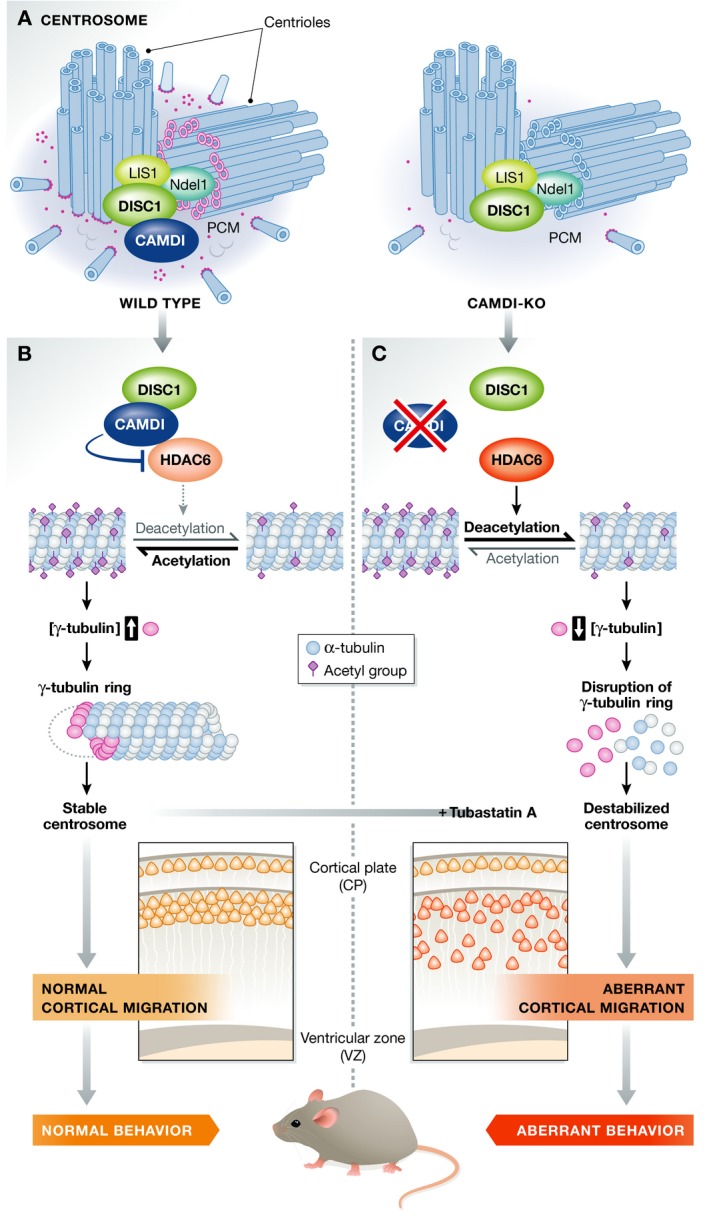
(A) Left: In wild‐type mice, the DISC1‐interacting protein CAMDI is located at the centrosome, along with DISC1 and other DISC1 interactors key for microtubule transport. Right: The absence of CAMDI possibly reduces γ‐tubulin ring formation. (B) When CAMDI is present, DISC1 binds to CAMDI, recruiting it to the centrosome. CAMDI in turn binds to HDAC6 and inhibits its deacetylase activity. Consequently, acetylated α‐tubulin is increased, which allows γ‐tubulin to accumulate in the centrosome and form γ‐tubulin rings. The centrosome is stabilised. This results in normal migration of cortical neurons and normal mouse behaviour. (C) In CAMDI KO mice, loss of CAMDI protein results in HDAC6 overactivation and deacetylation of α‐tubulin. This blocks the formation of γ‐tubulin rings and destabilises the centrosome. Cortical migration is delayed and mouse behaviour impaired. Addition of Tubastatin A can reverse the effects of CAMDI KO on cortical migration.
Loss of CAMDI leads to […] phenotypes that model autism spectrum disorders and which can be rescued by inhibition of HDAC6
In this issue of EMBO Reports, Fukuda et al 1 characterise the role of CAMDI in centrosome function and neuronal migration, and link the function of CAMDI to psychiatric behaviours. They identify histone deacetylase 6 (HDAC6) as a novel interactor of CAMDI. HDAC6 is a cytoplasmically localised deacetylase, responsible for removal of acetyl groups from non‐histone proteins such as α‐tubulin, Hsp90 and cortactin. Fukuda et al demonstrate that CAMDI inhibits the deacetylase activity of HDAC6 at the centrosome, hence increasing acetylation of α‐tubulin. This effect appears to be specific to HDAC6‐mediated deacetylation of α‐tubulin, as acetylation of Hsp90, another HDAC6 substrate, was not affected. Acetylation of α‐tubulin allows accumulation of γ‐tubulin at the centrosome, so that when HDAC6 is overactivated, the increase in α‐tubulin deacetylation inhibits formation of the γ‐tubulin ring at the centrosome. Indeed, in CAMDI knockout (KO) mice, reduced γ‐tubulin is observed in centrosomal fractions. The authors demonstrate that cortical migration is delayed in CAMDI KO mice and after RNAi knockdown of CAMDI 4. Moreover, CAMDI KO results in abnormal cortico‐striatal projections of callosal neurons, a key circuit for the regulation of motivated behaviours.
Interestingly, HDAC6‐mediated deacetylation of α‐tubulin and cortactin was recently reported to play a role in ciliary disassembly 5. DISC1 and other centrosomal proteins are also located at the base of primary cilia, and these collective findings suggest similar mechanisms of acetylation are required for both processes. It remains to be seen, however, if CAMDI also localises to primary cilia.
Characterisation of the behaviour phenotype of CAMDI KO mice revealed hyperactivity, increased anxiety, increased behavioural despair, increased repetitive behaviours and reduced social interaction, suggesting a strong neuropsychiatric behavioural profile in CAMDI KO mice, with many features common to models of autism spectrum disorders. To examine whether the cortical migration and behavioural defects in CAMDI KO mice are due HDAC6 overactivation, Fukuda et al tested whether the HDAC6 inhibitor Tubastatin A could rescue these defects. Tubastatin A is a novel HDAC6‐specific inhibitor that has recently been demonstrated to reverse deacetylation of α‐tubulin in a model of stroke, with neuroprotective effects 6. Prenatal administration of Tubastatin A rescued the cortical migration defects and partially rescued the behavioural phenotypes of CAMDI KO mice. Hyperactivity and increased repetitive behaviours could be reversed in juvenile and adult CAMDI KO mice, but the reduction in social interaction could not be rescued with in utero Tubastatin A.
This mechanism of CAMDI regulation of centrosomal integrity and its effect on neuronal migration and behaviour, together with previous studies on DISC1's involvement in centrosome regulation, suggest a new mechanism by which DISC1 regulates centrosomal function. By binding to CAMDI, DISC1 could be involved in suppressing HDAC6 activity, resulting in increased acetylation of α‐tubulin and stabilisation of the centrosome leading to correct cortical development (Fig 1B). In the absence of CAMDI, the CAMDI‐HDAC6 interaction is lost, and HDAC6 is overactivated, leading to increased α‐tubulin deacetylation, disrupting centrosome stability and consequently neuronal migration (Fig 1C).
Such a mechanism could well explain previous reports of misorientated neurons upon DISC1 knockdown 2. Indeed, another study has shown that in migrating interneurons depleted of DISC1, acetylated tubulin is reduced in the leading process of the neuron 7. It will be of interest to investigate whether reduced acetylation of α‐tubulin is also seen in the centrosome of neurons in DISC1 knockdown or knockout models, and whether Tubastatin A treatment can also reverse the corticogenesis defects in DISC1 mutation models. This study on the effects of Tubastatin A in the CAMDI KO suggests a promising route for investigating therapeutic alleviation of symptoms related to hyperactivity and repetitive behaviours that are linked to centrosome dysregulation.
See also: T Fukuda et al (December 2016)
References
- 1. Fukuda T, Nagashima S, Abe T et al (2016) EMBO Rep 17: 1785–1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kamiya A, Kubo K, Tomoda T et al (2005) Nat Cell Biol 7: 1167–1178 [DOI] [PubMed] [Google Scholar]
- 3. Shimizu S, Matsuzaki S, Hattori T et al (2008) Biochem Biophys Res Commun 377: 1051–1056 [DOI] [PubMed] [Google Scholar]
- 4. Fukuda T, Sugita S, Inatome R et al (2010) J Biol Chem 285: 40554–40561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ran J, Yang Y, Li D et al (2015) Sci Rep 5: 12917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang Z, Leng Y, Wang J et al (2016) Sci Rep 6: 19626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Steinecke A, Gampe C, Nitzsche F et al (2014) Front Cell Neurosci 8: 190 [DOI] [PMC free article] [PubMed] [Google Scholar]