

Abstract
Homologous recombination (HR) is a key pathway that repairs DNA double‐strand breaks (DSBs) and helps to restart stalled or collapsed replication forks. How HR supports replication upon genotoxic stress is not understood. Using in vivo and in vitro approaches, we show that the MMS22L–TONSL heterodimer localizes to replication forks under unperturbed conditions and its recruitment is increased during replication stress in human cells. MMS22L–TONSL associates with replication protein A (RPA)‐coated ssDNA, and the MMS22L subunit directly interacts with the strand exchange protein RAD51. MMS22L is required for proper RAD51 assembly at DNA damage sites in vivo, and HR‐mediated repair of stalled forks is abrogated in cells expressing a MMS22L mutant deficient in RAD51 interaction. Similar to the recombination mediator BRCA2, recombinant MMS22L–TONSL limits the assembly of RAD51 on dsDNA, which stimulates RAD51‐ssDNA nucleoprotein filament formation and RAD51‐dependent strand exchange activity in vitro. Thus, by specifically regulating RAD51 activity at uncoupled replication forks, MMS22L–TONSL stabilizes perturbed replication forks by promoting replication fork reversal and stimulating their HR‐mediated restart in vivo.
Keywords: DNA repair, DNA replication, DNA replication stress, homologous recombination
Subject Categories: DNA Replication, Repair & Recombination
Introduction
All cells must replicate their DNA before each round of cell division. In humans, genome duplication is critically dependent on homologous recombination (HR), which protects stalled replication forks or restores integrity of collapsed forks (San Filippo et al, 2008). Recombination mechanisms are largely studied in the context of DSB repair, yet endogenous two‐ended DSBs are relatively rare and usually repaired by a non‐homologous end‐joining pathway in mammals. In contrast, HR is required for the completion of replication even in the absence of exogenous DNA damage, as recombination‐deficient human cells undergo only a very limited number of DNA replication rounds. However, how recombination supports replication remains mostly undefined (Petermann & Helleday, 2010; Neelsen & Lopes, 2015). Furthermore, mutations in several regulatory HR genes including breast cancer susceptibility protein 2 (BRCA2) or RAD51C predispose affected individuals to cancer, demonstrating that aberrant HR results in genome instability that drives tumorigenesis (Wooster et al, 1995; Meindl et al, 2010).
A key player in the HR‐mediated processes is the RAD51 recombinase, which assembles on ssDNA upon nuclease‐mediated resection of the broken DNA ends. RAD51 nucleoprotein filament then invades homologous DNA, which serves as the repair template. The assembly of RAD51 filaments on ssDNA is regulated at multiple levels. First, resected ssDNA is rapidly coated and protected by RPA, which coordinates DNA damage signaling and repair. However, as RPA also competes with RAD51 for binding to ssDNA, proteins termed recombination mediators are required to load RAD51 on the RPA‐coated ssDNA. This overcomes the inhibitory effect of RPA on RAD51‐mediated DNA strand exchange and represents an important regulatory step that allows recombination on appropriate DNA substrates. These mediators including BRCA2 and the five RAD51 paralogues RAD51B/C/D and XRCC2/3 (San Filippo et al, 2008) help to form the RAD51‐ssDNA filament in the presence of RPA, and particularly BRCA2 also prevents assembly of RAD51 on dsDNA (Sigurdsson et al, 2001b; Jensen et al, 2010; Liu et al, 2010; Thorslund et al, 2010; Taylor et al, 2015). However, although the recombination mediators appear to be key to understand how HR is fine‐tuned, their exact roles and interplay at DNA damage sites are still poorly understood.
The human MMS22L–TONSL complex and its budding yeast counterpart Mms22 play essential roles during DNA replication stress but their precise function remains elusive (Luke et al, 2006; Duro et al, 2008, 2010; O'Connell et al, 2010; O'Donnell et al, 2010; Piwko et al, 2010). Yeast Mms22 was proposed to be a putative substrate adaptor of the Rtt101 cullin E3 ubiquitin ligase functioning downstream of the histone H3‐K56 acetylation pathway, which marks newly replicated DNA and is required for genome stability maintenance (Collins et al, 2007; Zaidi et al, 2008). Human cells expressing low levels of MMS22L or TONSL as well as yeast mms22Δ mutants are hypersensitive to drugs such as camptothecin (CPT) inducing replication fork reversal or DSBs at replication forks, but not to ionizing radiation, suggesting specific defects in dealing with replication fork stalling or collapse (Luke et al, 2006; Duro et al, 2008, 2010; O'Connell et al, 2010; O'Donnell et al, 2010; Piwko et al, 2010; Ray Chaudhuri et al, 2012; Buser et al, 2016). Cells lacking MMS22L were unable to restart replication forks stalled upon treatment with CPT and were defective in RAD51 foci formation at collapsed replication forks and DSBs, and thus, MMS22L was suggested to act as a recombination mediator (Duro et al, 2010; O'Donnell et al, 2010). The TONSL subunit is a reader of unmethylated histone H4 tails that mark newly replicated DNA, which was proposed to facilitate recombination when newly replicated DNA is available as a template for HR‐based repair (Saredi et al, 2016). However, evidence for a direct involvement of MMS22L–TONSL in recombination has been missing. To this point, Arabidopsis thaliana TONSL homologue TONSOKU/BRU1 was implicated in reestablishing the chromatin state after DNA replication and TONSOKU mutants are characterized by loss of a transcriptional silencing phenotype closely resembling the phenotype exhibited with defects in chromatin assembly factor 1 (CAF‐1) (Takeda et al, 2004). Moreover, both yeast Mms22 and human MMS22L–TONSL were found in complexes with histones and chromatin remodeling factors, raising the possibility that they may act indirectly by regulating the chromatin milieu or the mobility of broken DNA (Duro et al, 2010; O'Connell et al, 2010; O'Donnell et al, 2010; Piwko et al, 2010; Buser et al, 2016).
Here, we report that MMS22L–TONSL is present at replication forks under unperturbed conditions and its presence is enhanced at collapsed replication forks that had undergone extensive resection. MMS22L–TONSL recruitment to replication fork stall sites is facilitated via a direct interaction with RPA‐coated ssDNA. Its activity at stalled replication forks leads to replication fork protection by RAD51‐dependent fork reversal. We demonstrate that purified MMS22L–TONSL directly binds the RAD51 recombinase and favors the interaction of RAD51 with ssDNA, which promotes the formation of RAD51 presynaptic filaments and invasion of homologous DNA. These data demonstrate a direct involvement of MMS22L–TONSL in RAD51‐dependent recombination and establish the heterodimer as a key factor that functions as a recombination regulator at stalled replication forks.
Results
MMS22L is recruited to stalled and collapsed replication forks
MMS22L–TONSL accumulates as nuclear foci upon induction of DSBs in human cells (Duro et al, 2010; O'Donnell et al, 2010). To determine whether MMS22L recruitment to DSBs is cell cycle‐regulated, we treated synchronized HeLa cells with the Topo II inhibitor etoposide. As expected, etoposide induced DSBs in all cell cycle phases, demonstrated by γH2AX foci accumulation (Nitiss, 2009), but only S‐phase cells showed MMS22L foci (Figs 1A and B, and EV1A–C). When replication was inhibited by the polymerase‐alpha inhibitor aphidicolin, the number of etoposide‐induced MMS22L foci in S‐phase cells was strongly reduced (Fig EV1D and E), suggesting that MMS22L is not generally recruited to DNA breaks but rather acts preferentially in the context of replication forks. Since Saccharomyces cerevisiae mms22Δ cells are severely impaired in HR specifically induced by drugs that inhibit DNA replication (Duro et al, 2008), the function of MMS22L in promoting replication‐linked recombination appears conserved in evolution.
Figure 1. MMS22L foci form during S phase of the cell cycle and colocalize with HR proteins.
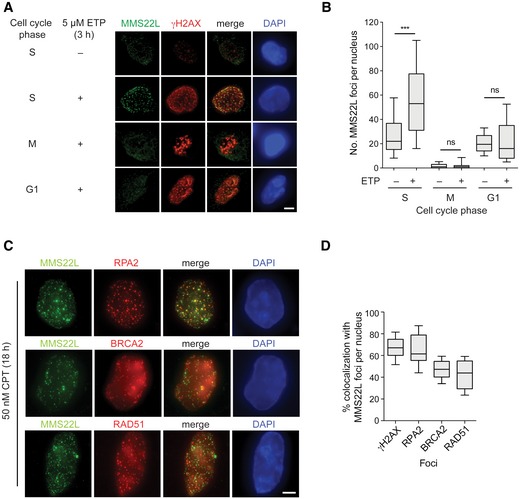
- Immunofluorescence images of MMS22L foci formation at etoposide (ETP)‐induced DSBs during S, M, and G1 phases of the cell cycle in HeLa cells. Scale bar: 5 μm.
- Quantification of a representative experiment from (A) shows number of MMS22L foci per nucleus during indicated cell cycle phases (n = 2; n nuclei ≥ 60).
- Immunofluorescence images of CPT‐induced MMS22L, RPA2, BRCA2, and RAD51 foci in U2OS cells. Scale bar: 5 μm.
- Quantification of a representative experiment (n = 3) from (C) shows percentage of γH2AX, RPA2, BRCA2, and RAD51 foci colocalizing with MMS22L foci (n nuclei = 30; nuclei with ≥ 25 MMS22L foci were analyzed).
Figure EV1. MMS22L foci formation during replication stress depends on RPA recruitment to resected ssDNA.
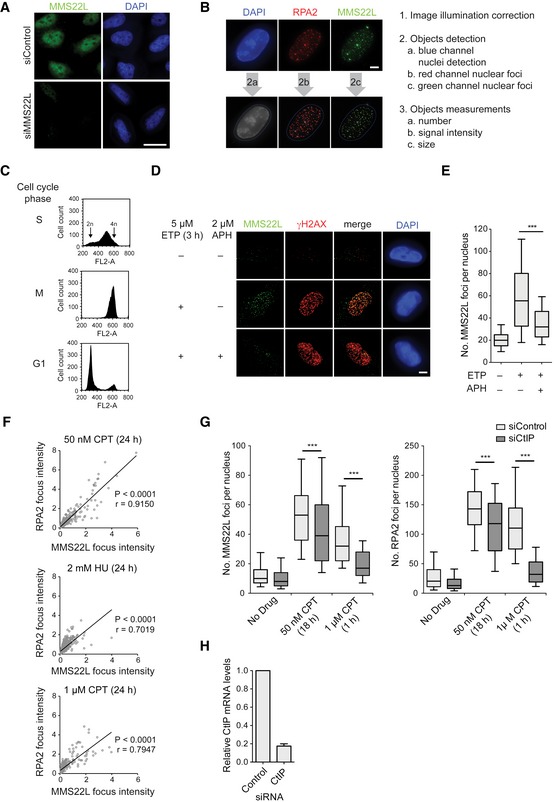
- Immunofluorescence images of control and siMMS22L‐treated HeLa cells demonstrating specificity of anti‐MMS22L antibody. Scale bar: 20 μm.
- Example images (left) and workflow (right) of the CellProfiler‐based automated image processing pipeline to quantify nuclear speckles from immunofluorescence microscopy images used in this study. Scale bar: 5 μm.
- Related to the experiment in Fig 1A and B; DNA content analysis of synchronized HeLa cells using propidium iodide staining and flow cytometry.
- Immunofluorescence images of MMS22L foci formation in S phase‐synchronized HeLa cells treated with or without ETP and APH. APH, polymerase‐alpha inhibitor aphidicolin. Scale bar: 5 μm.
- Quantification of a representative experiment from (D); n = 2. Graph represents distribution of foci numbers per nuclei, n nuclei ≥ 177.
- Graphs show the signal intensities of colocalizing MMS22L and RPA2 foci from U2OS cells treated with the indicated replication stress agents. Foci intensities were quantified using CellProfiler and their correlation assessed by the Pearson method. r, Pearson correlation coefficient; n = 3.
- Graphs show dependence of MMS22L foci formation on CtIP‐mediated DNA end resection in U2OS cells treated with indicated replication stress agents. Immunofluorescence images were analyzed using CellProfiler, and cells were in silico enriched in S phase based on increased nuclear RPA2 signal intensity. Boxplots show quantifications of MMS22L and RPA2 foci from a representative experiment, n = 3.
- Related to experiment in (G). qRT–PCR analysis demonstrating CtIP depletion efficiency; error bars, SEM. n = 3.
Key HR players such as RPA, recombination mediator BRCA2, and the strand exchange protein RAD51 accumulate as nuclear foci at DSBs (Raderschall et al, 1999; Tarsounas et al, 2004). To better understand the nature of replication stress‐induced MMS22L foci, we investigated their colocalization and dependence on RPA, BRCA2, and RAD51 in human osteosarcoma U2OS cells. We observed that 47% BRCA2 and 43% RAD51 foci colocalized with MMS22L at stalled replication fork sites upon prolonged treatment with the Topo I inhibitor camptothecin (CPT) (Fig 1C and D), indicating that MMS22L cooperates or functions alongside BRCA2 in the assembly of RAD51 presynaptic filaments at these sites. Colocalization of MMS22L foci was greatest with RPA2 (~63%) and the RPA2 and MMS22L foci signal intensity correlated with each other upon various replication stress conditions (Fig EV1F). Moreover, disrupting RPA accumulation upon replication stress by the depletion of the DNA end resection factor CtIP strongly reduced the number of MMS22L foci (Sartori et al, 2007; O'Donnell et al, 2010) (Fig EV1G and H), suggesting that RPA may recruit MMS22L–TONSL to stalled replication forks.
MMS22L directly binds RPA‐coated ssDNA
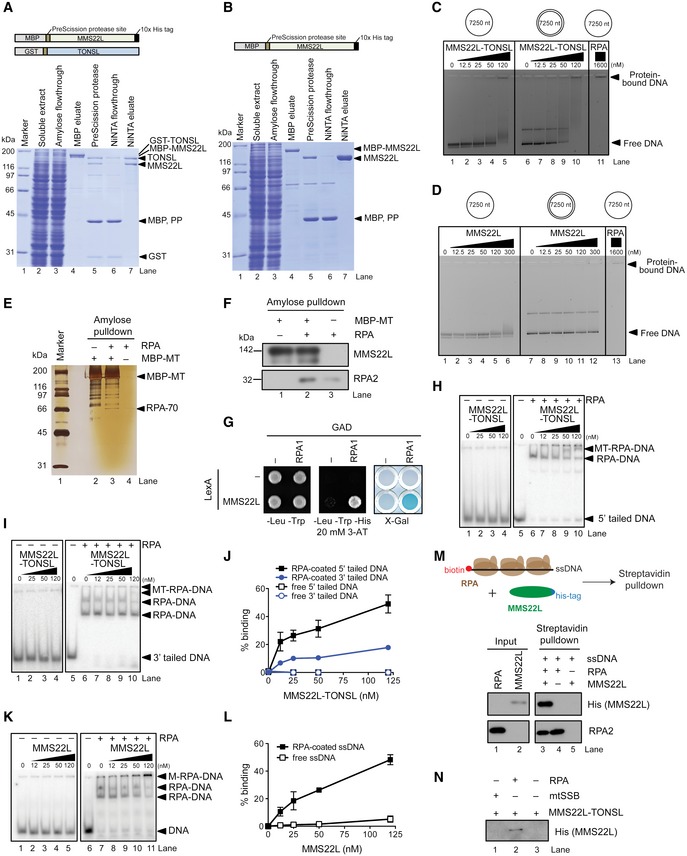
To understand the recruitment of the complex to replication stress sites at the molecular level, we assessed whether MMS22L–TONSL directly interacts with ssDNA and RPA. We purified the full‐length recombinant heterodimer and the MMS22L subunit from Sf9 cells (Fig EV2A and B). TONSL expressed without MMS22L was rapidly degraded and could not be obtained. On its own, recombinant MMS22L–TONSL was incapable to stably bind oligonucleotide‐based DNA (data not shown, see also below), and we could only observe binding to plasmid‐length DNA, which likely reflects its low DNA binding affinity (Fig EV2C). This limited DNA binding largely depends on the TONSL subunit, as the DNA‐binding capacity of MMS22L was even lower than that of the heterodimer (Fig EV2D). Additionally, no difference between MMS22L–TONSL binding to ssDNA and dsDNA was observed (Fig EV2C). In solution, MMS22L–TONSL bound recombinant RPA directly but with a weak affinity (Fig EV2E and F), which could also be recapitulated in a yeast two‐hybrid assay (Fig EV2G; note that the MMS22L subunit interacts directly with RPA). However, when we pre‐bound RPA to ssDNA and introduced MMS22L–TONSL (Fig 2A and B, lanes 6–10, and Fig EV2H–L), we observed a novel band likely corresponding to the ternary MMS22L–TONSL‐RPA‐ssDNA complex, since MMS22L–TONSL does not bind oligonucleotide‐based ssDNA in the absence of RPA (Figs 2A and EV2H, I and K, lanes 1–5). To verify these findings, we performed streptavidin pulldowns of biotinylated ssDNA incubated with RPA and His‐tagged MMS22L–TONSL. We detected MMS22L–TONSL only when RPA was present in the reaction (Fig 2C, compare lanes 3 and 5). Likewise, in NiNTA pulldowns, more RPA was obtained in reactions containing ssDNA (Fig 2C, compare lanes 6 and 7). Thus, MMS22L–TONSL forms a stable complex with RPA bound to ssDNA, which is dependent on the MMS22L subunit (Fig EV2K–M). MMS22L–TONSL failed to form a complex with ssDNA coated with human mitochondrial ssDNA‐binding protein (mtSSB), indicating that the binding to ssDNA‐RPA is specific (Fig EV2N). Pulldowns with MMS22L fragments mapped the RPA interaction site to the N‐terminal part of MMS22L (Fig EV3A), which could be further narrowed by yeast two‐hybrid assay to a MMS22L region between residues 384 and 522 (Fig 2D). This site corresponds to the MMS22L sequence that we previously identified as the region with strongest homology to yeast Mms22 protein (Piwko et al, 2010), implying that the RPA‐binding domain is evolutionarily conserved. While the MMS22L Δ384–522 mutant protein was unable to bind RPA, it still interacted with TONSL (Fig EV3B). Importantly, the MMS22L Δ384–522 mutant deficient in the interaction with RPA was incapable to localize to CPT‐induced γH2AX foci in U2OS cells (Fig 2E), consistent with results using transiently overexpressed truncated MMS22L variants lacking this region (Fig EV3C). We conclude that the RPA interaction facilitates MMS22L–TONSL recruitment to ssDNA present at or behind stalled and collapsed replication forks to promote their HR‐mediated restart.
Figure EV2. Recombinant MMS22L–TONSL interacts with RPA‐coated ssDNA.
-
A, BSchematic representation of the recombinant constructs (top) and polyacrylamide gels (bottom) showing samples from MMS22L–TONSL (A) and MMS22L (B) purifications. MBP, maltose‐binding protein; GST, glutathione S‐transferase; PP, PreScission protease.
-
C, DIndicated concentrations of the MMS22L–TONSL heterodimer (C) or MMS22L (D) were incubated with bacteriophage M13‐based DNA and the formed complexes were separated by agarose gel electrophoresis. The gels were stained with GelRed.
-
E, FAmylose pulldown of MBP‐tagged MMS22L–TONSL (MBP‐MT) and RPA. Proteins in the eluates were analyzed by silver staining (E) or Western blotting (F).
-
GYeast two‐hybrid analysis of the interaction between MMS22L fused to LexA DNA‐binding domain and RPA1 fused to GAL4 activating domain (GAD) assessed by survival on media containing 3‐aminotriazole (3‐AT) in the absence of histidine and β‐galactosidase activity (X‐Gal) assays.
-
H–JElectrophoretic mobility shift assays with 5′‐tailed (H), 3′‐tailed DNA (I) and MMS22L–TONSL and/or RPA. The assays were quantified and shown as average (J), n = 2; error bars, SEM.
-
K, LElectrophoretic mobility shift assay with MMS22L and ssDNA, with or without RPA. Panel (K) shows a representative experiment. The assays were quantified and shown as average (L), n = 2; error bars, SEM.
-
MSchematic (top) and Western blot (bottom) of streptavidin pulldown of biotinylated ssDNA incubated with MMS22L and/or RPA.
-
NStreptavidin pulldown of biotinylated ssDNA pre‐incubated with either human mitochondrial SSB (mtSSB, 100 nM) or human RPA (100 nM) and subsequently incubated with MMS22L–TONSL.
Figure 2. MMS22L–TONSL directly interacts with RPA‐coated ssDNA.
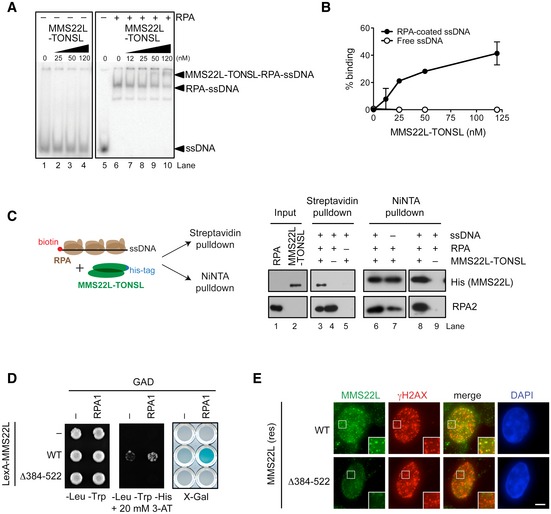
- Electrophoretic mobility shift assay monitoring MMS22L–TONSL binding to ssDNA without RPA and with RPA‐coated ssDNA.
- Quantification of data as in (A) shows percent MMS22L–TONSL binding to ssDNA with or without RPA (n = 2; error bars, SEM).
- Schematic (left) and Western blots (right) of streptavidin and NiNTA pulldowns of His‐tagged MMS22L–TONSL, RPA, and biotinylated ssDNA.
- Yeast two‐hybrid analysis shows the effect of deletion of the amino acids 384–522 of MMS22L on the interaction with RPA. The interaction between indicated MMS22L variants fused to LexA DNA‐binding domain and RPA1 fused to GAL4‐activating domain (GAD) is assessed by survival on media containing 3‐aminotriazole (3‐AT) in the absence of histidine and by β‐galactosidase activity (X‐Gal) assays.
- Images of MMS22L foci formed in U2OS cells stably expressing wild‐type (WT) or Δ384–522 variants of siRNA‐resistant HSS‐MMS22L, depleted of endogenous MMS22L by RNAi and treated with 50 nM CPT for 18 h. Scale bar: 5 μm.
Figure EV3. Mapping the RPA‐binding sites on MMS22L.
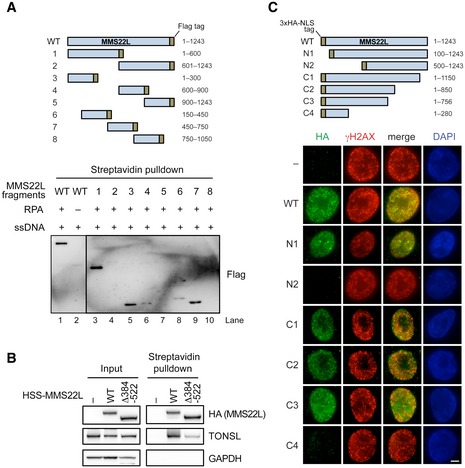
- Schematic of recombinant truncated MMS22L constructs (top) used in streptavidin pulldowns (bottom) of RPA‐coated ssDNA. Various fragments of MMS22L were expressed as MBP and FLAG fusions. The MBP tag was cleaved, and the FLAG‐tagged constructs (50 nM) were incubated with biotinylated RPA‐coated ssDNA in a buffer containing 50 mM Tris–HCl (pH 7.5), 0.1% Triton X‐100, 0.1 mg/ml bovine serum albumin, 120 mM NaCl. The MMS22L fragments in the streptavidin pulldowns were detected by Western blotting using an anti‐FLAG antibody.
- Streptavidin pulldown of HSS‐tagged MMS22L variants stably integrated and overexpressed in U2OS cells. Input and coprecipitated proteins were detected by immunoblotting.
- Schematic (top) and representative immunofluorescence images (bottom) of MMS22L truncation constructs used to investigate recruitment to CPT‐induced DNA damage sites. HeLa cells were transiently transfected with the indicated constructs fused to the 3× HA tag and the SV40 NLS sequence (to assure nuclear localization). One day after DNA transfection, cells were treated with 50 nM CPT for 18 h, then subjected to pre‐extraction to analyze chromatin‐bound fraction, and processed for immunofluorescence staining using indicated antibodies. Fragments N2 and C4, which lack putative RPA‐binding region, are not recruited to CPT‐induced DNA damage sites (γH2AX foci). Scale bar: 5 μm
MMS22L–TONSL promotes RAD51 loading to stalled replication forks
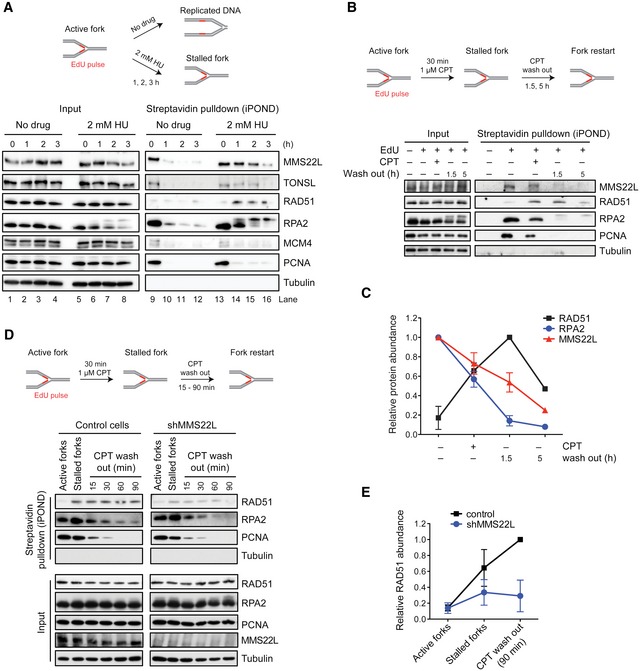
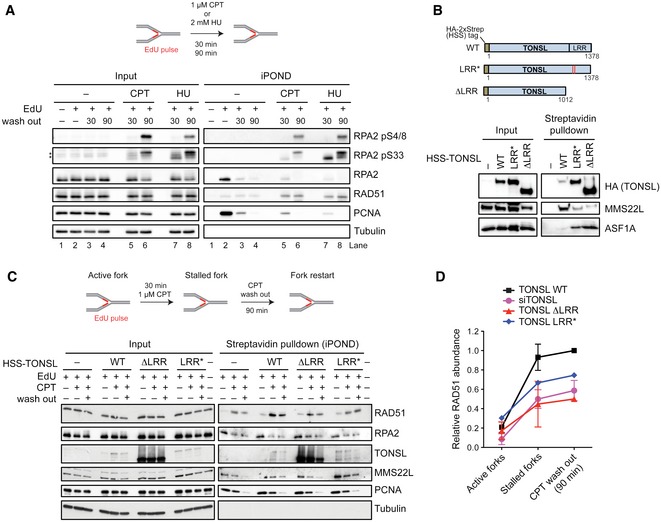
We next wanted to understand whether MMS22L–TONSL acts directly at DNA replication forks. To assess MMS22L–TONSL dynamics at replication forks, we employed the isolation of proteins on nascent DNA (iPOND) assay (Sirbu et al, 2011) based on affinity purification of DNA pulse‐labeled during replication with the thymidine analog ethynyl‐deoxyuridine (EdU). We found both MMS22L and TONSL proteins at active forks with unperturbed conditions, and binding was strongly reduced to fully replicated chromatin (Fig 3A; compare lane 9 with lanes 10–12). Hydroxyurea (HU) addition to stalled forks led to a prolonged MMS22L–TONSL retention on the nascent DNA (Fig 3A; lanes 13–16), suggesting that the complex functions directly at stalled replication forks. These results are in agreement with a recent study using an iPOND technology coupled to mass spectrometry analysis, which also detected MMS22L and TONSL at replication forks (Dungrawala et al, 2015). We next stalled forks with a 30‐min CPT treatment, followed by drug washout to monitor the recovery process over time. We observed gradual RPA2 reduction along with loading of the RAD51 recombinase directly to forks (Figs 3B and C, and EV4A), and the MMS22L dynamics followed exchange of RPA for RAD51. As the MMS22L–TONSL complex is required for RAD51 foci formation upon DNA damage (Duro et al, 2010; O'Donnell et al, 2010), we next tested whether MMS22L–TONSL is required to recruit RAD51 to stalled forks. Using the CPT‐recovery assay with cells depleted for MMS22L or TONSL, we observed a strong RAD51 reduction at CPT‐stalled forks in the absence of either MMS22L (Fig 3D and E) or TONSL (Fig EV4C and D). Moreover, ectopic expression of TONSL mutants defective in MMS22L interaction reduced RAD51 recruitment to stalled forks (Fig EV4B–D), indicating that an association of both components of the complex is required to promote RAD51 loading. These data strongly suggest that MMS22L–TONSL is involved in efficient recruitment and/or maintenance of the RAD51 recombinase directly on DNA replication forks immediately upon stalling.
Figure 3. MMS22L–TONSL localizes to active and stalled replication forks.
-
AAbundance of MMS22L–TONSL at active and HU‐stalled replication forks, and fully replicated DNA.
-
B, CMMS22L dynamics at stalled replication forks. Quantification (C) shows average relative abundance of indicated proteins detected in immunoblots (n = 3 except at time point 5 h, n = 1; error bars, SEM).
-
D, EEffect of MMS22L depletion on RAD51 levels at stalled replication forks. Quantification (E) shows average relative RAD51 abundance (n = 2; error bars, SEM).
Figure EV4. MMS22L–TONSL regulates RAD51 recruitment at stalled replication forks.
-
AiPOND assay schematic (top) and immunoblots (bottom) show effects of CPT and HU treatments on the RPA2 phosphorylation status in HeLa cells. Note that 30‐min treatment with 1 μM CPT stalls forks without inducing fork breakage, as the phosphorylation status of RPA2 using specific antibodies indicates the presence of pS33 associated with ssDNA rather than pS4/8 associated with DSBs (induced at later time point; 90 min). Asterisks indicate cross‐reactive bands.
-
BSchematic representation (top) of TONSL, TONSL LRR*: E1089K and D1104N mutations within the LRR domain identified in ovarian cancer patient (COSMIC sample OCC06PT (Forbes et al, 2015)) and TONSL ΔLRR: C‐terminal TONSL truncation deleting the LRR domain; LRR, leucine‐rich repeats. Streptavidin pulldown (bottom) of indicated stably integrated HSS‐tagged‐TONSL variants overexpressed in HeLa cells. Input and coprecipitated proteins were detected by immunoblotting. ASF1A, known to interact with the N‐terminal region of TONSL (Duro et al, 2010; O'Donnell et al, 2010), was included as a control.
-
C, DThe presence of the indicated proteins at replication forks was analyzed by the iPOND‐CPT‐release assay in cells expressing either HSS‐tagged wild‐type TONSL, or TONSL LRR* or TONSL ΔLRR mutants and depleted for endogenous TONSL by siRNA targeting 3′UTR. The experiments were quantified and shown as average; n = 2 except for LRR* n = 1; error bars, SEM. TONSL and its interaction with MMS22L are required for normal levels of RAD51 at stalled replication forks.
MMS22L promotes replication fork reversal
Stalled replication forks either may collapse resulting in single‐ended DSBs or may be transiently stabilized through fork reversal (Neelsen & Lopes, 2015). Micromolar CPT concentrations that induce replication‐associated DSBs resulted in early MMS22L foci (Fig EV5A–C). In contrast, nanomolar CPT or HU that stall replication forks but do not lead to early DSBs (Petermann et al, 2010; Ray Chaudhuri et al, 2012) did not result in detectable MMS22L foci at similar time points (Fig EV5A–C). MMS22L foci number and intensity were strongly increased in HU‐treated cells upon ATR kinase inhibition, which leaves stalled forks unprotected leading to fork collapse (Cimprich & Cortez, 2008) (Fig EV5D and E). These observations suggest that MMS22L foci detectable by conventional microscopy mainly form upon DNA breakage, and thus also implicate MMS22L–TONSL in the recombination‐mediated repair of collapsed replication forks (Fig 1). However, although MMS22L–TONSL is also recruited to stalled forks early to support replication fork restart (Fig 3A), this process does not create visible MMS22L foci (Fig EV5A–C).
Figure EV5. MMS22L forms foci upon replication fork collapse.

-
A–CRepresentative immunofluorescence images (A) and boxplots of endogenous MMS22L (B) and RPA2 (C) foci distribution in U2OS cells treated with the indicated replication stress‐inducing agents. Cells in silico enriched in S phase based on increased nuclear RPA2 signal were analyzed. Quantification of a representative experiment is shown; n = 3. Scale bar: 5 μm.
-
D, ERepresentative immunofluorescence images of MMS22L and RPA foci in U2OS cells upon collapse of HU‐stalled replication forks by treatment with the ATR inhibitor (ATRi). Cells in silico enriched in S phase based on increased nuclear RPA2 signal were analyzed. Scale bar: 5 μm. Statistical analysis: Mann–Whitney U‐test; ***P ≤ 0.0001. n = 2.
-
FWestern blot analysis of cell extracts from cells used in the experiments described in Fig 4A and B. Tubulin serves as a loading control.
-
GWestern blot analysis of cell extracts from cells used in a representative EM experiment described in Fig 4C–F. GAPDH serves as a loading control.
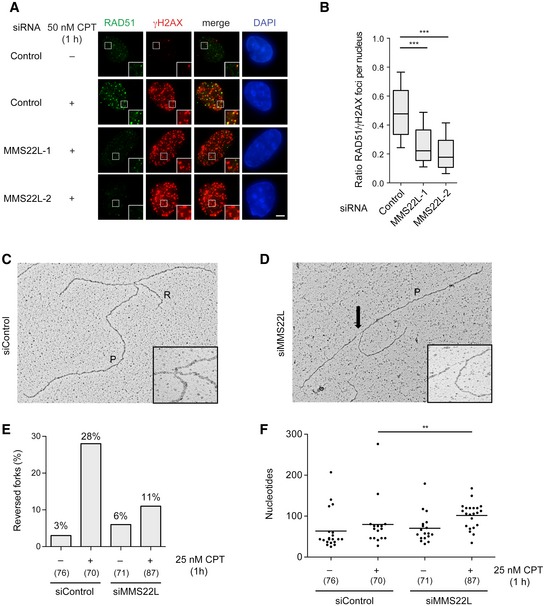
Recently, RAD51 activity at CPT‐stalled forks has been linked to fork reversal in human cells (Zellweger et al, 2015). This first response to fork stalling allows transient fork pausing and was proposed as a mechanism to avoid collision with lesions or gaps on template DNA, thus preventing fork collapse (Neelsen & Lopes, 2015). Importantly, lack of MMS22L prevents RAD51 foci formation as early as 1 h after nanomolar CPT treatment (Figs 4A and B, and EV5F). Electron microscopy analysis of in vivo psoralen‐cross‐linked replication intermediates from MMS22L‐depleted U2OS cells revealed a marked reduction (~60%) in the frequency of CPT‐induced reversed forks (Figs 4C–E and EV5G), similar to defects recently reported upon depletion of RAD51 (Zellweger et al, 2015). Moreover, forks in CPT‐treated MMS22L‐depleted cells displayed significantly longer ssDNA regions (Fig 4D and F), strongly suggesting that MMS22L–TONSL is recruited to extended ssDNA gaps on template DNA, thereby assisting fork reversal by loading RAD51 at these sites.
Figure 4. MMS22L is required to promote reversal of uncoupled replication forks.
-
ARepresentative immunofluorescence images of foci induced by short CPT treatment (50 nM, 1 h) in U2OS cells treated with indicated siRNAs showing the effect of MMS22L depletion on the RAD51 foci. Scale bar: 5 μm.
-
BBoxplot quantification of representative experiment in (A) (n = 3; n nuclei ≥ 78). Boxes indicate the 25–75 percentile and whiskers the 10–90 percentile; horizontal lines mark the medians. Nuclei with ≥ 25 γH2AX foci (marker of replication stress sites) were analyzed. Statistical analysis: Mann–Whitney U‐test; ***P ≤ 0.0001.
-
C, DElectron micrographs of a representative reversed fork (C) and a fork with extended ssDNA region at the junction (D) (arrow). P, parental duplex; R, regressed arm.
-
EFrequency of reversed replication forks detected by EM. Similar results were obtained in one independent experiment (n = 2). In brackets, the total number of analyzed molecules is given.
-
FDistribution of ssDNA length at replication forks isolated from U2OS cells transfected with the indicated siRNAs and treated with 25 nM CPT for 1 h. In brackets, the total number of analyzed molecules is given. Statistical analysis according to Mann–Whitney U‐test; **P ≤ 0.01, n = 2.
MMS22L directly interacts with RAD51
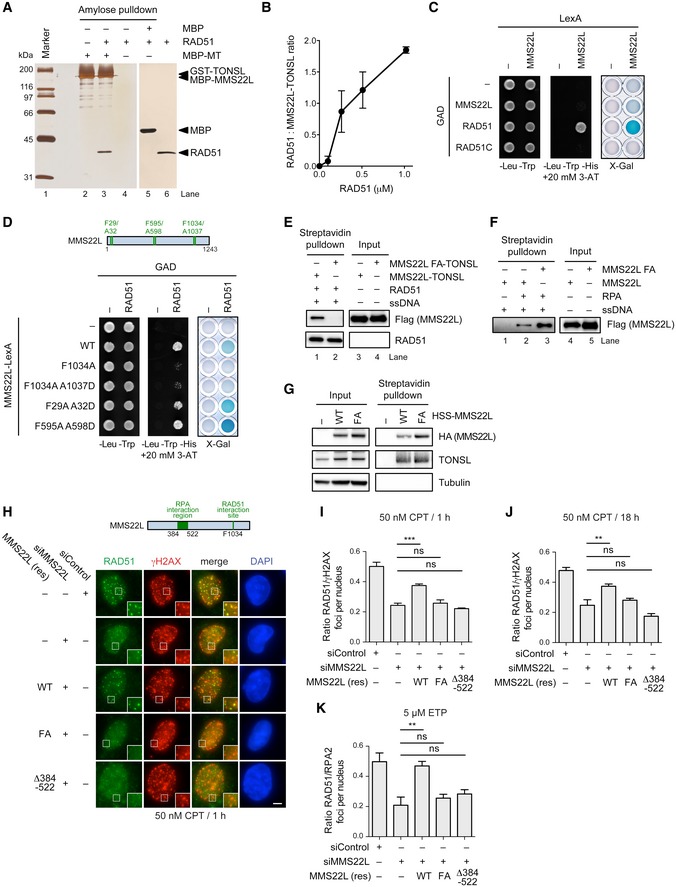
The functional association of MMS22L–TONSL with RAD51 prompted us to test for a direct physical interaction between RAD51 and the heterodimer. We immobilized MBP‐tagged MMS22L–TONSL or MMS22L on amylose resin and performed pulldowns with recombinant RAD51. We found that MMS22L–TONSL directly interacts with RAD51 (Fig 5A), with up to two RAD51 molecules tightly bound by the heterodimer (we estimate the affinity in the nanomolar range, Figs 5B and EV6A). MMS22L–TONSL binds RAD51 with similar affinities as the MMS22L subunit (Fig EV6B–D), suggesting that the interaction is mediated by MMS22L. This direct physical interaction between MMS22L and RAD51 was confirmed by yeast two‐hybrid assays (Fig 5C). To define the RAD51 interaction region in MMS22L, we searched for the previously described FXXA motif present in the BRC repeats of BRCA2, RECQ5L, and other RAD51‐interacting proteins (Islam et al, 2012) and found three of these motifs in MMS22L (Fig 5D). Whereas mutating either of the first two motifs had no effect on the interaction with RAD51, F1034A A1037D mutation dramatically reduced the capacity of MMS22L to interact with RAD51 in yeast two‐hybrid assays, and an identical result was observed upon mutation of a single phenylalanine residue at position F1034 (Fig 5D). Consistent with these results, purified recombinant MMS22L F1034A (FA)‐TONSL heterodimer failed to interact with RAD51 in vitro (Fig 5E), while the MMS22L FA mutation did not affect the interaction with RPA (Fig 5F) or TONSL in vivo or in vitro (Figs 5G and EV6E). To determine the effects of MMS22L mutations on the response to replication fork stalling by CPT or DSBs induced by ETP in vivo, we created stable U2OS cell lines expressing siRNA‐resistant HSS (HA‐2x Strep)‐tagged RAD51 interaction‐deficient MMS22L FA or RPA interaction‐deficient MMS22L Δ384–522 variants under the control of doxycycline‐inducible promoter. Upon downregulation of endogenous MMS22L by RNAi, the mutant proteins were expressed as the sole variant of MMS22L in these cells. We noted that the CPT‐induced foci of MMS22L FA mutant are weaker than the ones formed by the wild‐type protein (Fig EV6F), suggesting that binding of MMS22L–TONSL to RAD51 (presumably nucleofilaments) stabilizes the heterodimer at DNA damage sites. Strikingly, whereas the wild‐type MMS22L was capable to restore DNA damage‐dependent RAD51 foci upon all tested conditions, the two MMS22L variants failed to do so (Figs 5H–K and EV6G–J). Together, these results demonstrate a direct physical and functional interaction between the C‐terminal region of the MMS22L and RAD51.
Figure 5. MMS22L interacts physically and functionally with RAD51.
-
AAmylose pulldown of MBP (control) or MBP‐tagged MMS22L–TONSL and RAD51. Eluate proteins were detected by silver staining. MT: MBP‐MMS22L‐GST‐TONSL.
-
BQuantitative analysis of RAD51 binding to MMS22L–TONSL such as shown in Fig EV6A. Averages shown, n = 2; error bars, SEM.
-
C, DYeast two‐hybrid analysis of interactions between LexA DNA‐binding domain‐tagged MMS22L and indicated proteins fused to GAL4‐activating domain (GAD) assessed by survival on media containing 3‐AT in the absence of histidine and by X‐Gal assays.
-
E, FStreptavidin pulldowns of indicated recombinant proteins and biotinylated ssDNA. The proteins in the input and eluates were detected by Western blotting.
-
GStreptavidin pulldown of HSS‐tagged MMS22L variants stably integrated and overexpressed in U2OS cells. Input and coprecipitated proteins were detected by immunoblotting.
-
HRepresentative immunofluorescence images of CPT‐induced foci in stable U2OS cell lines depleted of endogenous MMS22L and overexpressing indicated siRNA‐resistant variants of HSS‐MMS22L treated with 50 nM CPT for 1 h. Scale bar: 5 μm.
-
I–KQuantification of the effect of indicated MMS22L mutations on RAD51 foci in U2OS cells treated with 50 nM CPT for 1 h (I; n = 3), 50 nM CPT for 18 h (J; n = 3) or 5 μM ETP for 1 h followed by 3‐h incubation without the drug (K; n = 3). Graphs represent averaged median values; error bars indicate SEM. Statistical analysis according to one‐way ANOVA with Bonferroni post‐test; ***P ≤ 0.001; **P ≤ 0.01; ns, not significant. Representative immunofluorescence images are shown in (H), Fig EV6G and H, respectively.
Figure EV6. MMS22L interacts physically and functionally with RAD51.

-
AQuantitative analysis of RAD51 binding to MMS22L–TONSL. Proteins were mixed in the indicated ratio, the MMS22L–TONSL complex was retained on amylose beads, and proteins in the eluate were analyzed by silver staining. Lane 6, the same amount of RAD51 was used as in lane 5, but no MMS22L–TONSL (MT) was added to control for non‐specific binding of RAD51 to the resin.
-
BNiNTA pulldown of his‐tagged MMS22L and RAD51. Proteins in the eluate were detected by silver staining.
-
C, DQuantitative analysis of RAD51 binding to MMS22L. Proteins were mixed with the indicated ratio, MMS22L immobilized on NiNTA beads and the eluate analyzed by silver staining. Lane 6: the same amount of RAD51 was used as in lane 5, but no MMS22L (No M) was added to control for non‐specific binding of RAD51 to the resin. Proteins in the eluates were quantified by comparing with known amounts of MMS22L or RAD51, respectively. Averages shown, n = 2; error bars, SEM.
-
EPurification of the MMS22L FA‐TONSL complex. The MMS22L FA mutant was co‐expressed with TONSL and the complex purified from Sf9 cells using the same protocol as for wild‐type MMS22L–TONSL. Although wild‐type and mutant heterodimers purified with approximately 1:1 stoichiometry, the yield of the mutant heterodimer was dramatically lower.
-
F–HRepresentative microscopy images of DNA damage‐induced foci in stable U2OS cell lines depleted of endogenous MMS22L and overexpressing indicated siRNA‐resistant variants of HSS‐MMS22L treated with 50 nM CPT for 18 h (F, G) or 5 μM ETP for 1 h followed by 3‐h incubation without the drug (H). Proteins are stained using indicated antibodies. Scale bars: 5 μm.
- I, J
MMS22L–TONSL facilitates RAD51‐dependent DNA strand exchange at high RAD51 concentrations
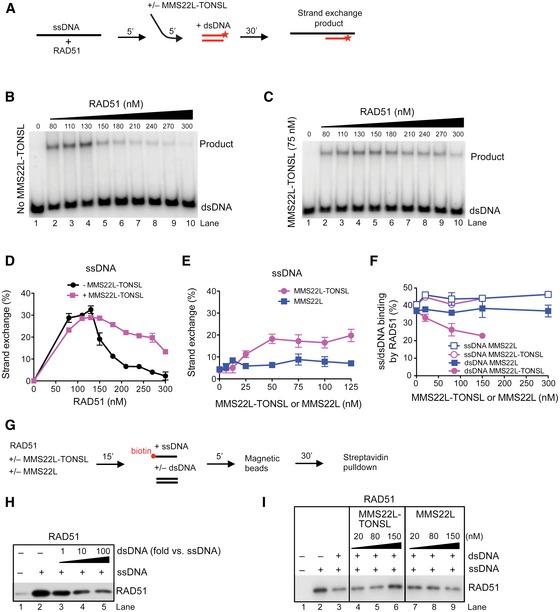
To determine whether MMS22L–TONSL directly promotes RAD51‐dependent recombination, we employed in vitro DNA strand exchange assays (Fig 6A). While ssDNA‐bound RAD51 filaments are the active species capable to search and invade homologous DNA, RAD51 bound to dsDNA is inhibitory for recombination (Benson et al, 1994; Sigurdsson et al, 2001a). Reconstituted strand exchange reactions thus show a very narrow RAD51 concentration optimum. After RAD51 fully saturates ssDNA (~130 nM, Fig 6B), further increase in RAD51 concentration results in its binding to dsDNA, which inhibits DNA strand exchange (Benson et al, 1994). When supplemented with MMS22L–TONSL (75 nM), the reaction optimum was reached at a similar RAD51 concentration (~130 nM, Fig 6C), but strikingly the inhibition at higher RAD51 concentrations was partially alleviated (Fig 6C and D). A comparable behavior was observed in strand exchange reactions with both 3′‐ and 5′‐tailed as well as gapped DNA (Fig EV7A–C), showing that the stimulatory effect of MMS22L–TONSL is not limited to a specific DNA structure and does not require a terminus, in keeping with the fork reversal model described above. All reactions were ATP dependent and required RAD51 (Fig EV7D and E), implying that MMS22L–TONSL is not capable of DNA strand exchange alone, but functions via promoting RAD51‐dependent reactions (Sung, 1994). We then used a high RAD51 concentration (270 nM) and titrated MMS22L–TONSL or MMS22L into the reactions. The heterodimer promoted strand exchange in a concentration‐dependent manner showing saturation at ~50 nM (Fig 6E), while the MMS22L subunit alone was largely inactive (Figs 6E and EV7F and G). The fact that both subunits of the MMS22L–TONSL complex are required for the stimulatory effect at high RAD51 concentrations (Fig 6E), while MMS22L is sufficient for the interaction with RAD51 (Figs 5D and EV6C), implies that the stimulatory effect of MMS22L–TONSL is not a simple result of chelating and reducing the free RAD51 pool. Together, these results demonstrate that MMS22L–TONSL promotes DNA strand exchange at high RAD51 concentrations.
Figure 6. MMS22L–TONSL promotes DNA strand exchange by reducing RAD51 binding to dsDNA.
-
ASchematic representation of the DNA strand exchange assay with ssDNA.
-
B, CStrand exchange assay with ssDNA and RAD51, and with (C) or without (B) the addition of MMS22L–TONSL (75 nM).
-
DQuantification of (B, C) shows averages, n = 2; error bars, SEM.
-
EAverages of quantified strand exchange assays with ssDNA, RAD51 (270 nM), and varying concentrations of MMS22L–TONSL or MMS22L. n = 2; error bars, SEM.
-
FQuantification of electrophoretic mobility shift assays with dsDNA or ssDNA, RAD51, and MMS22L–TONSL or MMS22L. Averages shown, n = 2; error bars, SEM.
-
GA scheme of streptavidin pulldown assay performed in (H, I).
-
HA representative immunoblot showing the effect of increasing amounts of dsDNA on RAD51 binding to ssDNA (1 nM).
-
IA representative immunoblot showing effects of MMS22L–TONSL and MMS22L titration into reactions containing RAD51, ssDNA (1 nM), and dsDNA (10 nM), as indicated.
Figure EV7. MMS22L–TONSL promotes DNA strand exchange by RAD51.

-
A–CQuantification of DNA strand exchange with RAD51, with or without MMS22L–TONSL (75 nM) using a 3′‐tailed (A), 5′‐tailed (B), or gapped (C) DNA substrate. Averages are shown, n = 2; error bars, SEM.
-
DA representative DNA strand exchange experiment with ssDNA and RAD51, as indicated with or without ATP. See scheme in Fig 6A.
-
EA representative DNA strand exchange experiment with ssDNA, RAD51, MMS22L–TONSL (MT), as indicated with or without ATP. See scheme in Fig 6A.
-
F, GRepresentative DNA strand exchange experiments with a fixed concentration of RAD51 (270 nM) and varying concentrations of MMS22L–TONSL (F) or MMS22L (G). See scheme in Fig 6A.
-
H, IRepresentative electrophoretic mobility shift assays with MMS22L–TONSL, MMS22L, RAD51, and either dsDNA (H) or ssDNA (I). Note that MMS22L–TONSL reduces RAD51 binding to dsDNA, but does not affect binding of RAD51 to ssDNA.
-
JA representative DNA strand exchange experiment with RAD51 (120 nM), ssDNA, RPA (30 nM), and MMS22L–TONSL. Note that MMS22L–TONSL is not capable to alleviate the inhibitory effect of RPA on DNA strand exchange.
-
KMMS22L (50 nM), MMS22L–TONSL (50 nM), RAD51 (500 nM), and RPA (100 nM) were incubated with biotinylated ssDNA in a buffer containing 20 mM Tris–HCl (pH 7.5), 0.1% Triton X‐100, 0.1 mg/ml bovine serum albumin, 120 mM NaCl, 2 mM CaCl2, 10 mM magnesium acetate, 1 mM dithiothreitol with or without ATP (2 mM). MMS22L–TONSL, but not MMS22L, forms a ternary complex with ssDNA‐bound RAD51 in the presence of ATP.
-
LSimilar assay as in (K), but in a buffer lacking calcium, magnesium, and ATP. Under conditions that do not favor RAD51 filament formation, MMS22L–TONSL preferentially binds RPA‐coated ssDNA.
MMS22L–TONSL reduces affinity of RAD51 to dsDNA
Our results presented in Fig 6A–E infer that the heterodimer might affect RAD51‐binding capacity to single‐ or double‐stranded DNA. Specifically, MMS22L–TONSL might function similar to the BRCA2 tumor suppressor by reducing RAD51 affinity for dsDNA (Jensen et al, 2010; Liu et al, 2010; Thorslund et al, 2010). To directly test whether MMS22L–TONSL affects DNA‐binding capacity of RAD51, we performed electrophoretic mobility shift assays with RAD51 and the MMS22L–TONSL heterodimer. Indeed, MMS22L–TONSL reduced RAD51 binding to dsDNA in a concentration‐dependent manner, but had no effect on RAD51 binding to ssDNA (Figs 6F and EV7H and I), which we also confirmed by DNA footprinting assays with DNaseI (data not shown). This activity was specific to the heterodimer, as the MMS22L subunit did not alter binding to either ssDNA or dsDNA (Fig EV7H and I). In contrast, MMS22L–TONSL was incapable to promote DNA strand exchange in the presence of RPA (Fig EV7J), which is another key capacity of BRCA2 and the RAD51 paralogues (Sigurdsson et al, 2001b; Jensen et al, 2010; Liu et al, 2010; Thorslund et al, 2010; Taylor et al, 2015). This shows that MMS22L–TONSL is incapable to relieve inhibition of DNA strand exchange imposed by RPA, and infers that it may function with additional partners. Finally, we used competition pulldown assays to test whether the reduction in RAD51‐binding capacity to dsDNA might promote its affinity for ssDNA (Fig 6G–I). As expected (Jensen et al, 2010), increasing the amount of dsDNA resulted in correspondingly less RAD51 bound to ssDNA (Fig 6H). Conversely, titration of MMS22L–TONSL into reactions containing both ssDNA and dsDNA resulted in more RAD51 bound to ssDNA, an effect not observed with the MMS22L subunit (Fig 6I). Finally, we observed that MMS22L–TONSL but not MMS22L was capable to bind an active RAD51 nucleoprotein filament in the presence of ATP (Fig EV7K). Under reaction conditions that are optimal for RAD51 filament formation, the binding of MMS22L–TONSL to ssDNA was stronger than binding to RPA‐ssDNA (Fig EV7K and L). These results suggest that the capacity of MMS22L to bind RAD51 filaments might facilitate the retention of the heterodimer at DNA damage sites, consistent with the reduced DNA damage‐dependent MMS22L foci in cells expressing the MMS22L FA variant incapable of interacting with RAD51 (Fig EV6F). Together, these results demonstrate that the MMS22L–TONSL heterodimer reduces the RAD51 capacity to bind dsDNA, which likely facilitates RAD51 assembly on stretches of ssDNA at perturbed replication forks, and thereby promotes HR‐mediated events.
Discussion
The integrity and repair of stalled or collapsed replication forks is assured by homologous recombination through largely undefined mechanisms. Previous reports indicated that the human MMS22L–TONSL heterodimer plays a crucial role in this process. Here, we demonstrate that MMS22L–TONSL directly promotes DNA recombination upon replication stress. We show that MMS22L–TONSL is present at nascent DNA under unperturbed conditions, and MMS22L specifically interacts with RPA‐coated ssDNA and the strand exchange protein RAD51. These interactions are required for the accumulation of MMS22L–TONSL at DNA damage sites during replication stress, as well as for efficient RAD51 foci formation upon DNA damage. Mechanistically, MMS22L–TONSL promotes replication fork reversal upon stalling at DNA damage sites, which helps to prevent replication fork collapse. Our data suggest that MMS22L–TONSL directly stimulates invasion of homologous DNA by limiting the capacity of RAD51 to interact with dsDNA. This activity promotes recombination leading to replication fork reversal, which permits fork restoration or repair (Fig 7).
Figure 7. MMS22L–TONSL functions during replication stress.
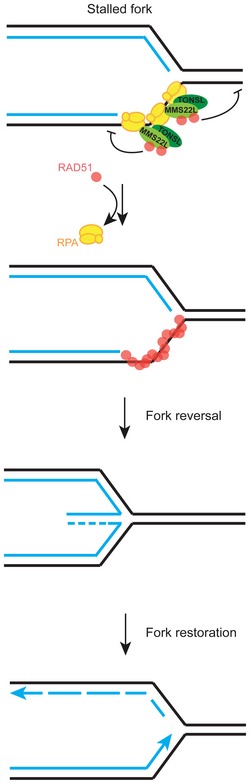
Model of MMS22L–TONSL function at stalled replication forks. MMS22L–TONSL is present near active replication forks and promotes fork reversal upon stalling. Our results suggest that MMS22L–TONSL heterodimers control fork metabolism by reducing the capacity of RAD51 to bind dsDNA, which facilitates the formation of RAD51 filaments on stretches of ssDNA and initiates fork reversal. This prevents fork collapse and allows subsequent replication fork restoration.
Why is MMS22L–TONSL particularly important for the repair of replication forks? A unique feature of MMS22L is its ability to preferentially bind RPA‐coated ssDNA. This interaction may allow MMS22L–TONSL to dynamically associate with ssDNA produced by DNA unwinding during DNA replication or with extended ssDNA tracts that accumulate upon replication fork stalling. Indeed, in our iPOND experiments MMS22L–TONSL and RPA2 abundances correlate at replication forks (Fig 3). The interaction with RPA‐coated ssDNA also enables the enrichment of MMS22L at collapsed replication forks that had undergone extensive resection. In addition, TONSL has been shown to directly bind histones (Duro et al, 2010) and the specific interaction with histone H4 unmethylated at K20 contributes to the recruitment of the MMS22L–TONSL complex to newly replicated DNA, when a homologous template is available for repair (Saredi et al, 2016). This interaction could also facilitate the access of MMS22L–TONSL to post‐replicative ssDNA gaps. Thus, the MMS22L–TONSL complex seems ideally equipped to participate in the repair of stalled and collapsed replication forks that accumulate stretches of ssDNA. Since the basal fork localization of the complex precedes the activation of the S‐phase checkpoint, MMS22L–TONSL may be among the first factors helping to nucleate RAD51 filaments not only upon replication stress but also upon endogenous fork pausing conditions. In support of this, depletion of human MMS22L or TONSL, as well as the deletion of yeast MMS22 gene, leads to accumulation of spontaneous DNA breaks during unperturbed S phase (Luke et al, 2006; Duro et al, 2010; O'Connell et al, 2010; O'Donnell et al, 2010; Piwko et al, 2010). This is in agreement with our observations that MMS22L–TONSL promotes fork reversal (Fig 4), which emerged as an important global response to replication stress and can occur in the absence of DNA breaks (Ray Chaudhuri et al, 2012; Neelsen & Lopes, 2015; Zellweger et al, 2015). Given that MMS22L‐ or TONSL‐depleted cells are hypersensitive to low nanomolar CPT treatments that induce robust fork reversal but not to ionizing radiation causing DSBs (Duro et al, 2010), it is tempting to speculate that promotion of RAD51‐dependent fork reversal represents the essential activity of the MMS22L–TONSL complex. Interestingly, a recent study found that BRCA2‐deficient cells still load RAD51 on nascent DNA on stressed replication forks, which is sufficient to promote sister chromatid exchange (Chaudhuri et al, 2016). This infers BRCA2‐independent mechanisms to load RAD51 on DNA and promote recombination upon replication stress. It will be of particular interest to investigate the role of MMS22L–TONSL heterodimer under these conditions. Our data suggest that MMS22L–TONSL‐facilitated nucleation of RAD51 on extended RPA‐coated ssDNA prevents DSB formation by assisting fork reversal, but we cannot exclude that it may also function at a subset of forks that unavoidably collapsed, as MMS22L–TONSL‐depleted cells are also impaired in homologous recombination reporter assays initiated by a dsDNA break (Duro et al, 2010; O'Connell et al, 2010; O'Donnell et al, 2010).
How does MMS22L–TONSL promote recombination? We demonstrate that recombinant MMS22L directly binds the RAD51 recombinase, and provide evidence for a direct involvement of MMS22L–TONSL in promoting DNA strand exchange by RAD51 (Fig 6). The activity of the complex favors interaction of RAD51 with ssDNA over dsDNA, therefore promoting the formation of RAD51‐ssDNA presynaptic nucleoprotein filaments at high RAD51 concentrations. This process is a prerequisite for the invasion of homologous DNA and further steps in the HR pathway. MMS22L–TONSL thus acts as a regulator by a mechanism similar to human BRCA2 (Jensen et al, 2010). However, unlike BRCA2, MMS22L–TONSL cannot catalyze the assembly of RAD51 filaments on ssDNA coated with RPA, even though it is directly recruited to such substrates in vitro and in vivo. This suggests that MMS22L–TONSL does not regulate RAD51 on its own, but the process likely requires the interplay with additional factors. We hypothesize that the MMS22L–TONSL heterodimer might functionally interact with other recombination mediators such as the RAD51 paralogues in the efficient assembly of RAD51 filaments upon replication stress in vivo. Recently, it has been demonstrated that the yeast RAD51 paralogues Rad55‐Rad57 protect Rad51‐ssDNA filaments from disruption by the antirecombinase Srs2 (Liu et al, 2011), while the RAD51 paralogues RFS‐1/RIP‐1 from Cenorabditis elegans are able to directly remodel and stabilize open conformation of ssDNA‐RAD‐51 filaments (Taylor et al, 2015). Human RAD51 paralogues have functions that partially overlap with those of BRCA2 (Sigurdsson et al, 2001b). Our data establish that MMS22L–TONSL belongs to the growing group of factors that regulate recombination, but in contrast to the other mediators, its function seems highly specific to stalled or collapsed replication forks. Elucidating the precise relationship between the various recombination mediators and the MMS22L–TONSL complex at stalled replication forks represents an exciting future challenge.
The strong connections between defects in HR and tumorigenesis suggest that mutations in the MMS22L and TONSL genes might be associated with cancer development. It has been demonstrated that defects in recombination mediators including BRCA2, RAD51B, and RAD51C frequently associate with breast and ovarian cancers (Wooster et al, 1995; Meindl et al, 2010; Golmard et al, 2013). Indeed, cancer genome sequencing projects identified several somatic mutations in the MMS22L and TONSL genes (Forbes et al, 2015), but at present none of these alterations has been confirmed as tumorigenic. Here, we demonstrate that the two point mutations in TONSL (E1089K D1104N) identified in a genome of an ovarian cancer patient could potentially be tumorigenic (Forbes et al, 2015), as they abolish the interaction with MMS22L and negatively influence the recruitment of RAD51 to distressed replication forks (Fig EV4). Moreover, we identified several additional cancer patient‐derived point mutations in both MMS22L and TONSL that affect protein stability, suggesting that misregulation of the MMS22L–TONSL‐dependent processes might be involved in promoting carcinogenesis. We therefore speculate that genome instability in some types of ovarian and possibly breast cancers might be promoted by MMS22L or TONSL mutations.
Materials and Methods
Cell culture conditions and reagents
U2OS (R. Aebersold, ETH Zurich) and HeLa (S. Taylor, University of Manchester) cell lines were grown in a humidified incubator at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM, Gibco, Invitrogen) supplemented with 10% fetal bovine serum (FBS, PAA), 0.2 mM L‐glutamine, and standard antibiotics. The FRT‐TetR‐HeLa cell line (kindly provided by S. Taylor, University of Manchester) for creating stable cell lines using the Flp‐In system (Invitrogen) was maintained as described previously (Tighe et al, 2008). For transient overexpression and stable cell line generation, DNA plasmid transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The stable HeLa cell line for doxycycline‐inducible downregulation of MMS22L was obtained by transfection of FRT‐TetR‐HeLa cells with the shMMS22L‐expressing vector based on pSUPERIOR.puro (OligoEngine) and maintained in the presence of 0.5 μg/ml puromycin (Gibco). The stable cell lines for doxycycline‐inducible expression of HSS‐tagged MMS22L and TONSL constructs were obtained by transfecting FRT‐TetR U2OS or HeLa cell lines with the pcDNA5 (Invitrogen)‐based constructs of the respective MMS22L/TONSL variants and maintained in the presence of 100 μg/ml hygromycin B (Invitrogen). Stable cell lines for MMS22L expression were treated with 2.5 mM sodium butyrate (Sigma) for 48 h to revert silencing of the CMV promoter and subsequently maintained in the medium containing 10% tetracycline‐free FBS (Bioconcept) to prevent resilencing of the integrated constructs. To induce expression in the FRT‐TetR system, 1 μg/ml doxycycline (Sigma‐Aldrich) was used. Thymidine block and release experiments were performed as described previously (Piwko et al, 2010). In brief, cells were synchronized at the G1/S transition by 16‐h incubation in medium containing 2 mM thymidine (Sigma‐Aldrich), washed twice with pre‐warmed DMEM, followed by incubation for 9 h. Subsequently, thymidine was added and cells were incubated for another 16 h. Next, cells were washed twice with DMEM and incubated for 2 h to obtain S‐phase cells and 11 h to obtain M (including 5 h of treatment with 0.23 μM nocodazole) and G1‐synchronized cells. Subsequently, cells were treated with 5 μM etoposide for 3 h prior to fixation.
Cells were treated with the following drugs diluted in growth medium: Topo II inhibitor etoposide (5 μM; Sigma‐Aldrich), Topo I inhibitor camptothecin (Sigma‐Aldrich), hydroxyurea (Sigma‐Aldrich), polymerase‐alpha inhibitor aphidicolin (2 μM; Sigma‐Aldrich), and ATR inhibitor VE‐821 (3 μM; Selleck Chemicals).
Cloning and DNA manipulations
The pCDNA5‐HSS‐MMS22L and pCDNA5‐HSS‐TONSL plasmids for stable cell line generation by the Flip‐In system (Invitrogen) were generated previously (Piwko et al, 2010). TONSL ΔLRR truncation was generated by PCR using primers: 5′‐GACTGGATCCAATGAGCCTGGAGCGCGAGCTTC‐3′ and 5′‐AGTCCTCGAGTCACAGGTCCCACGAAGTCACC‐3′ and sub‐cloned into pcDNA5‐HSS vector. MMS22L mutants were obtained by “round‐the‐horn” PCR‐based mutagenesis (OpenWetWare) using following oligo pairs:
| MMS22L mutant | Oligo pair |
|---|---|
| si‐resistant | 5′‐GGCATCTCATGTTTTAGACCTCCTGAATTTCC‐3′ |
| 5′‐ACGTCCTCTACCTCTGCAACAGCTGCTAAC‐3′ | |
| Δ384–522 | 5′‐AACAACCTAACTGAAGTTGGTCTACAGAACTT‐3′ |
| 5′‐TACAAAGTTCCAATTTGATTCCACTTTTC‐3′ | |
| F1034A | 5′‐CCAGCTTCACCATATGTTTCAGATC‐3′ |
| 5′‐AAGGGCTCGCCCAATATACTGCTCAATAAC‐3′ | |
| F1034A A1037D | 5′‐CCAGACTCACCATATGTTTCAGATCTTGGAC‐3′ |
| 5′‐AAGGGCTCGCCCAATATACTGCTCAATAAC‐3′ | |
| F29A A32D | 5′‐TGTGACGTTGACAACAGAGGAGGAGGAAAAC‐3′ |
| 5′‐AGAGGCGTAAGGAGGTTTGCACCATTCCG‐3′ | |
| F595A A598D | 5′‐TGTGACTTCCGGGAGAAAGCAAAGGAATTC‐3′ |
| 5′‐TGAGGCTTTCTCAGCCAAAACACCAATGTCC‐3′ |
TONSL LRR* mutation (double point mutation E1089K D1104N in the LRR domain) was obtained by QuikChange site‐directed mutagenesis method (Stratagene) by two PCRs using oligonucleotide pairs: 5′‐GACAAGTGTGTGGCTAAGCTGGTGGCTGCC‐3′ and 5′‐GGCAGCCACCAGCTTAGCCACACACTTGTC‐3′, and 5′‐GCCTGGCCCTCCTTAACCTCTCCTCCAATC‐3′ and 5′‐GATTGGAGGAGAGGTTAAGGAGGGCCAGGC‐3′. The plasmid for expressing shMMS22L was obtained from synthesized (Microsynth, Switzerland) annealed DNA fragments: 5′‐GATCCCGGTTAGGTCTTCTGAATCATTCAAGAGATGATTCAGAAGACCTAACCTTTTTA‐3′ and 5′‐AGCTTAAAAAGGTTAGGTCTTCTGAATCATCTCTTGAATGATTCAGAAGACCTAACCGG‐3′ cloned into pSUPERIOR.puro linearized with BglII and HindIII. The oligonucleotides corresponding to the sh‐target sequence of MMS22L are 5′‐GGTTAGGTCTTCTGAATCA‐3′. All obtained constructs were verified by DNA sequencing (Microsynth, Switzerland).
siRNA transfections
For RNAi experiments, HeLa or U2OS cells were transfected with 30 nM siRNAs using Lipofectamine RNAiMAX (Invitrogen) and incubated for 3 days prior analysis. Expression of si‐resistant HSS‐MMS22L variants in siMMS22L complementation experiments in Fig 5 was induced with 1 μg/ml doxycycline added to medium at the beginning of siRNA transfection. All siRNA duplexes were purchased from Microsynth (Switzerland) with the exception of AllStars Negative Control siRNA, which was obtained from Qiagen. The target sequences of siRNA oligonucleotides used in this study were as follows: MMS22L (5′‐GGUAGAAGAUGUUGCAAGU‐3′) (Piwko et al, 2010), MMS22L‐2 (5′‐CCCUUAAUGAUACGACGAA‐3′) (Piwko et al, 2010), CtIP (5′‐GCUAAAACAGGAACGAAUC‐3′) (Sartori et al, 2007), TONSL‐3′UTR (pooled oligonucleotides 5′‐CGACAGACCGAGACUCCGU‐3′ and 5′‐CCUAAUAAAUGAAGCUGCU‐3′). Downregulation of the targets was confirmed by immunoblotting and immunofluorescence using specific antibodies or quantitative RT–PCR.
Immunofluorescence microscopy
Cultured cells grown on coverslips were pre‐extracted with ice‐cold 0.2% Triton X‐100 in phosphate‐buffered saline (PBS) for 45 s at room temperature before fixation with 3% formaldehyde in PBS for 12 min at room temperature, except for cells in Fig EV1A, which were directly fixed with formaldehyde and subsequently permeabilized by incubation with 0.2% Triton X‐100 for 5 min. Permeabilized cells were then incubated for 1 h at room temperature with blocking buffer (1.5% bovine serum albumin, 0.01% Triton X‐100 in PBS), followed by additional 1‐h incubation with antibodies diluted in blocking buffer. Secondary antibodies were labeled with Alexa Fluor 488 or 568 (Invitrogen). 0.2 μg/ml DAPI was added to visualize DNA. Samples were mounted using Immu‐Mount (Thermo), and images were acquired on fully automated inverted epifluorescence microscopes (Ti‐Eclipse, Nikon) with 60× or 100× oil objectives. A motorized XY‐stage and piezo drive was used to acquire z‐stacks (26 steps at 0.235 μm) and multiple fields of view per sample. Images in Figs 1A and EV1D were deconvolved using the Huygens deconvolution software (SVI). Endogenous and ectopically expressed MMS22L was stained using previously described antibody (Piwko et al, 2010). The following commercial antibodies were used for immunofluorescence: anti‐γH2AX (05–636, Millipore, 1:750), anti‐γH2AX (NB100–384, Novus Biologicals, 1:1,500), anti‐RPA2 (9H8, Imgenex, 1:500), anti‐BRCA2 (Clone 2B, Millipore, 1:300), anti‐RAD51 (H‐92, Santa Cruz Biotech, 1:400), and anti‐RAD51 (14B4, Abcam, 1:500).
Image analysis
Non‐deconvolved z‐stack images were subjected to a maximum intensity projection using ImageJ software. CellProfiler (Carpenter et al, 2006) was used to analyze microscopy images (see Fig EV1B). After image illumination correction, global image thresholding using Otsu's method was applied to the DAPI channel to segment nuclei. Subsequently, nuclear foci in red and green channels were detected using Otsu's method and their signal intensities were quantified from illumination corrected images.
EM analysis
The procedure was essentially performed as previously described (Zellweger et al, 2015). Asynchronous subconfluent cultures of U2OS cells were treated with 25 nM CPT for 1 h. 4,5′,8‐trimethylpsoralen (10 μg/ml final concentration) followed by irradiation pulses with UV 365‐nm monochromatic light (UV Stratalinker 1800; Agilent Technologies) was used for in vivo psoralen cross‐linking of DNA. For DNA extraction, the cells were first lysed with cell lysis buffer (buffer C1: 1.28 M sucrose, 40 mM Tris–HCl [pH 7.5], 20 mM MgCl2, and 4% Triton X‐100; Qiagen) and then digested in digestion buffer (Qiagen buffer G2: 800 mM guanidine–HCl, 30 mM Tris–HCl [pH 8.0], 30 mM EDTA [pH 8.0], 5% Tween‐20, and 0.5% Triton X‐100) and 1 mg/ml proteinase K at 50°C for 2 h. Chloroform/isoamylalcohol (24:1) was used to collect DNA via phase separation through centrifugation at 11,300 g for 20 min, followed by DNA precipitation by adding 0.7× volume of isopropanol. About 70% EtOH was used to wash DNA. After air‐drying, the DNA was resuspended in 200 μl TE (Tris–EDTA) buffer. 100 U restriction enzyme (PvuII high fidelity, New England Biolabs) was used to digest 12 μg mammalian genomic DNA for 4–5 h. Replication intermediates enrichment was performed by Poly‐Prep chromatography columns. Benzoylated naphthoylated DEAE–cellulose granules were resuspended in 10 mM Tris–HCl (pH 8.0) and 300 mM NaCl to a final concentration of 0.1 g/ml. The columns were washed and equilibrated with 10 mM Tris–HCl (pH 8.0), followed by 1 M NaCl and 10 mM Tris–HCl (pH 8.0), and 300 mM NaCl, respectively. DNA was then loaded and incubated for 30 min. The columns were then washed with high NaCl solution (10 mM Tris–HCl [pH 8.0] and 1 M NaCl) and eluted in caffeine solution (10 mM Tris–HCl [pH 8.0], 1 M NaCl, and 1.8% [wt/vol] caffeine). To purify and concentrate the DNA, Amicon size‐exclusion column was used. DNA was then resuspended in TE buffer. DNA was then spread by the “BAC method” and loaded on carbon‐coated 400‐mesh copper grids. We used a High Vacuum Evaporator MED 020 (BalTec) to coat DNA with platinum. Microscopy was performed with a transmission electron microscope (Tecnai G2 Spirit; FEI; LaB6 filament; high tension ≤ 120 kV) and acquired with a side mount charge‐coupled device camera (2,600 × 4,000 pixels; Orius 1000; Gatan, Inc.). To perform statistics, at least 70 replication fork molecules were analyzed for each experimental condition. DigitalMicrograph version 1.83.842 (Gatan, Inc.) and ImageJ (National Institutes of Health) were used to process and analyze the images.
Immunoblotting, pulldowns, and antibodies
For immunoblotting, proteins were resolved by SDS–PAGE and transferred to PVDF immobilon‐P membrane (Millipore). Blocking and antibody incubations were carried out in 5% low‐fat milk (Migros, Switzerland) in PBS‐T (PBS, 0.1% Tween‐20), and washings in PBS‐T. Blots were developed using Immun‐Star™ HRP (Bio‐Rad) and quantified using ImageJ software.
For HSS (HA‐2 × Strep)‐tagged‐TONSL pulldown experiments from HeLa cell extracts, expression of indicated genes was induced by addition of doxycycline to medium for 24 h. For HSS‐MMS22L pulldowns from stable U2OS cell lines, expression was induced by addition of doxycycline and 2.5 mM sodium butyrate to medium for 24 h. Cell extracts were prepared as described previously (Piwko et al, 2010) except that lysis buffer was supplemented with 20 μg/ml avidin (IBA). Streptavidin MyOne T1 magnetic beads (Life Technologies) were used to capture Strep‐tagged TONSL. The beads were washed four times with lysis buffer, bound proteins eluted by incubating with 2.5 mM D‐biotin (Sigma‐Aldrich), boiled at 95°C, and analyzed by immunoblotting.
Rabbit polyclonal antibodies against the 6× His‐tagged amino‐terminal fragment (1–325) of MMS22L were described previously (Piwko et al, 2010). Additional rabbit polyclonal antibodies against the carboxy‐terminal peptide (GQMGQDEMQRLENDNT) of MMS22L (used in Figs EV5F and EV6I and J) were raised (Eurogentec, Belgium). Rabbit polyclonal antibodies against TONSL were raised (Animal Facility, University of Zurich, Switzerland) and affinity‐purified against a 6× His‐tagged fusion protein encoding amino acids 934–1,012 of TONSL. The following commercial antibodies were used: anti‐RAD51 (H‐92, Santa Cruz Biotech, 1:2,000), anti‐PCNA (PC10, Santa Cruz Biotech, 1:5,000), anti‐RPA2 (9H8, Imgenex, 1:2,000), anti‐RPA2 pS33 (A300–246A, Bethyl Laboratories, 1:1,000), anti‐RPA2 pS4/8 (A300–245A, Bethyl Laboratories, 1:1,000), anti‐MCM4 (clone B01P, Abnova, 1:1,000), anti‐α‐tubulin (DM1A, Sigma‐Aldrich, 1:10,000), anti‐HA.11 (16B12, Covance, 1:2,000), anti‐ASF1A (C6E10, Cell Signaling, 1:5,000), anti‐GAPDH (MAB374, Millipore, 1:5,000), and peroxidase‐conjugated anti‐mouse or anti‐rabbit antibodies (Pierce, 1:5,000).
Quantitative real‐time PCR
Total RNA was isolated from cells using the RNeasy Mini Kit (Qiagen). Reverse transcription was carried out using random primers and SuperScript II Reverse Transcriptase (Invitrogen) according to the manufacturer's protocol. qRT–PCR was carried out using the LightCycler 480 (Roche Life Science) with the LightCycler 480 SYBR Green I Master (Roche) hot start reaction mix. Experiments were carried out in technical triplicates and normalized to GAPDH. The following primers were used: CtIP forward 5′‐GTGAGGAAGACGTTATTCCAG‐3′ and reverse 5′‐TTTGCACACACAGAGTGCTC‐3′; GAPDH forward 5′‐GAAGGTGAAGGTCGGAGTC‐3′ and reverse 5′‐GAAGATGGTGATGGGATTTC‐3′.
Flow cytometry cell cycle analysis
Synchronized HeLa cells were collected by trypsinization and fixed in 70% ethanol overnight at −20°C. Cells were washed in PBS and incubated with 50 mg/ml propidium iodide and 20 mg/ml ribonuclease A for 30 min at room temperature. Flow cytometry was performed with a FACScalibur flow cytometer (BD Biosciences) using CellQuest software. Data analysis was performed using FlowJo software.
Preparation of recombinant proteins
The genes coding for MMS22L or TONSL were amplified from cDNA (Piwko et al, 2010) by PCR using primers hMMS22L‐F (5′‐GGCTAGCTGGGCCCGCTAGCGGATCCATGGAGAACTGTTCTGCT‐3′) and hMMS22L‐R (5′‐CGCAAATCCTCGAGCCCGGGAGTATTATCATTTTCCAGTCTCT‐3′) for MMS22L and hTONSL2‐F (5′‐GGCTAGCTGGGCCCGCTAGCGGATCCATGAGCCTGGAGCGCGAGCTTC‐3′) and hTONSL‐R (5′‐CGCAAATCCTCGAGCCCGGGTCAGAGGCGCCGAAAGAAGAGC‐3′) for TONSL. The PCR products of MMS22L were digested with ApaI and XhoI restriction endonucleases and cloned into ApaI and XhoI sites of pFB‐MBP‐Mlh3‐his vector (Ranjha et al, 2014), generating pFB‐MBP‐MMS22L‐his. The PCR products of TONSL were digested with NheI and XhoI sites into pFB‐GST‐Top3 vector (Cejka & Kowalczykowski, 2010), generating pFB‐GST‐TONSL. The MMS22L–TONSL complex was expressed in Sf9 insect cells upon co‐infection with MMS22L and TONSL baculoviruses according to the manufacturer's recommendation (Invitrogen). The proteins were extracted from Sf9 cell pellets with 325 mM NaCl as described previously for Sgs1 (Cejka & Kowalczykowski, 2010). The protein was then bound to amylose resin (New England Biolabs) via the MBP tag on the MMS22L subunit. Both MBP and GST tags were then cleaved off by PreScission protease. The GST‐tag was not used in our final purification protocol due to low binding efficiency of the recombinant construct to glutathione sepharose resin. The final protein complex was obtained by affinity purification on NiNTA agarose (Qiagen) via a His‐tag on the C‐terminus of MMS22L. The NaCl concentration was kept at 200 mM during the washing steps on both amylose and NiNTA resins to avoid disassembly of the complex. The MMS22L protein was prepared similarly, except 1 M NaCl was used during the washing steps. The final sample was dialyzed against dialysis buffer containing 50 mM Tris–HCl (pH 7.5), 5 mM β‐mercaptoethanol, 100 mM NaCl, 10% glycerol, and 0.5 mM phenylmethanesulfonyl fluoride. The MMS22L F1034A‐TONSL heterodimer was expressed and purified as the wild‐type heterodimer, but the yield of the variant was much lower.
RAD51 was expressed from a pTXB3 vector (New England Biolabs) where it was fused to an intein tag at its C‐terminus. Human RAD51 ORF was amplified by PCR from the plasmid pFB530 (Benson et al, 1994) using the following primers: 5′‐GGGAATTCCATGGCAATGCAGATGCAGCTT‐3′ and 5′‐AAGGCCGCTCTTCCGCAGTCTTTGGCATCTCCCACTCC‐3′. The resulting PCR product was digested with NcoI and SapI and inserted in pTXB3 (New England Biolabs), creating a fusion of RAD51 with a self‐cleavable affinity tag containing a chitin‐binding domain. The fusion construct was expressed in E. coli BL21 cells and purified on chitin‐binding resin (New England Biolabs) according to the manufacturer's recommendation. The cleavage of the intein tag was carried out in a cleavage buffer containing 50 mM Tris–HCl (pH 8.0), 500 mM NaCl, 1 mM EDTA, 10% glycerol, and 100 mM dithiothreitol for 32 h at 4°C. The cleaved RAD51 was eluted with the same buffer without dithiothreitol, diluted with 50 mM Tris–HCl (pH 8.0) to a final concentration of 200 mM NaCl, and loaded onto pre‐equilibrated HiTrap Q HP column (GE Healthcare). The column was washed with R buffer containing 20 mM Tris–HCl (pH 8.0), 1 mM EDTA, 0.5 mM dithiothreitol, 10% glycerol supplemented with 150 mM NaCl. RAD51 was eluted with 10 ml gradient of 150–700 mM NaCl in R buffer supplemented with 1 M NaCl. The final sample was dialyzed against dialysis buffer containing 20 mM Tris–HCl (pH 8.0), 20% glycerol, 100 mM NaCl, 1 mM dithiothreitol. Human RPA was purified as described previously (Thangavel et al, 2015).
Electrophoretic mobility shift assays
The binding reactions with RPA and MMS22L–TONSL (or MMS22L) were carried out in a 15‐μl volume in 25 mM Tris acetate (pH 7.5), 2 mM magnesium acetate, 1 mM dithiothreitol, 0.1 mg/ml bovine serum albumin (New England Biolabs), 32P‐labeled DNA substrate (1 nM, ssDNA, 5′‐tailed DNA, or 3′‐tailed DNA), and recombinant proteins as indicated for 30 min at 37°C. Where indicated, RPA (8 nM for ssDNA, 64 nM for 3′‐ and 5′‐tailed DNA) was pre‐bound to the DNA on ice for 5 min; subsequently, MMS22L–TONSL or MMS22L was added and the reaction was further incubated for 30 min at 37°C. 5 μl of loading dye containing 50% glycerol and 0.01% bromophenol blue was added and the reactions were separated at 4°C on 6% polyacrylamide gels in TBE buffer (89.1 mM Tris–base, 88.6 mM boric acid, 2 mM EDTA) or on 4% polyacrylamide gels in TBE buffer for reaction containing RPA at 4°C. The gels were dried, exposed to a Phosphorimager screen (GE Healthcare), and analyzed by Typhoon FLA 9500 (GE Healthcare). The ssDNA was oligonucleotide X12‐3, the 3′‐tailed DNA was prepared by annealing oligonucleotides X12‐3 SC and X12‐4 NC, and the 5′‐tailed DNA was prepared by annealing oligonucleotides X12‐3 and X12‐4 SC. The sequences of oligonucleotides were listed previously (Cejka & Kowalczykowski, 2010).
The binding assays (15 μl volume) with RAD51 and MMS22L–TONSL or MMS22L were performed similarly as described previously (Carreira et al, 2009). The binding buffer contained 20 mM Tris–HCl (pH 7.5), 10 mM magnesium acetate, 1 mM dithiothreitol, 0.1 mg/ml bovine serum albumin, 2 mM ATP, 2 mM calcium chloride, DNA substrate, and recombinant proteins. For ssDNA reactions, we used unlabeled 3 nM poly dA40 oligonucleotide and 1 nM 32P‐radiolabeled poly dA40. For dsDNA binding, the dA40 oligonucleotide was annealed with a complementary dT40. We used 29 nM unlabeled poly dA40:dT40 and 1 nM 32P‐radiolabeled poly dA40:dT40 dsDNA. The concentration of RAD51 was 150 nM in reactions with dsDNA and 640 nM in reactions with ssDNA. The reaction products were analyzed as described above.
Protein–protein pulldowns
Purified MMS22L–TONSL (with a MBP tag on the MMS22L subunit, 5.65 μg in a 150‐μl reaction volume) was mixed with a 5× excess of recombinant RAD51 or RPA in a binding buffer containing 50 mM Tris–HCl (pH 7.5), 2 mM beta‐mercaptoethanol, 50 mM NaCl, 10% glycerol, and 0.1% NP‐40. The reaction was mixed with 30 μl pre‐equilibrated amylose resin (New England Biolabs) and incubated at room temperature for 30 min with occasional stirring. The resin was then washed four times with 300 μl binding buffer. Bound proteins were eluted with 60 μl protein loading buffer (50 mM Tris [pH 6.8], 10% glycerol, 1.6% sodium dodecyl sulfate, 0.1 M dithiothreitol, 0.01% bromophenol blue) by boiling at 95°C for 3 min. The samples were analyzed on 10% SDS–PAGE and visualized by silver staining. Pulldowns with MMS22L (3.27 μg in a 210‐μl reaction volume) were carried out as above, except for the binding buffer also contained 10 mM imidazole and NiNTA agarose (Qiagen) was used instead of amylose resin, exploiting the C‐terminal His‐tag on MMS22L. The resin was then washed four times with 450 μl binding buffer. Quantitative analysis of bound RAD51 was performed by a comparison with known amounts of the recombinant RAD51 or MMS22L–TONSL/MMS22L that were loaded on the same gel and used as protein standards, as described previously (Jensen et al, 2010).
Streptavidin or NiNTA pulldowns with biotinylated DNA, RPA, and MMS22L–TONSL/MMS22L
In Figs 2C, EV2M and EV3A 5′ biotinylated oligonucleotide (B‐90‐mer; 5′‐CGGGTGTCGGGGCTGGCTTAACTATGCGGCATCAGAGCAGATTGTACTGAGAGTGCACCATATGCGGTGTGAAATACCGCACAGATGCGT‐3′; 20 nM) was premixed with RPA (100 nM) in a 50‐μl volume in a reaction buffer containing 50 mM Tris–HCl (pH 7.5), 120 mM NaCl, 0.1% (v/v) Triton X‐100, 0.1 mg/ml bovine serum albumin and incubated for 15 min at 37°C. Then, MMS22L–TONSL and MMS22L (both 50 nM) were added and incubated for further 15 min at 37°C. In reactions lacking RPA or MMS22L–TONSL/MMS22L, the reaction was mock‐incubated under the same experimental conditions. Reaction mixture was subsequently mixed with 15 μl pre‐equilibrated Streptavidin Dynabeads (Invitrogen, M‐280) or NiNTA agarose (Qiagen) and incubated at 4°C for 30 min. The mixture was stirred occasionally. The beads were washed four times with 150 μl wash buffer containing 50 mM Tris–HCl (pH 7.5), 120 mM NaCl, 0.1% (v/v) Triton X‐100 and eluted with 60 μl protein loading buffer (50 mM Tris [pH 6.8], 10% glycerol, 1.6% sodium dodecyl sulfate, 0.1 M dithiothreitol, 0.01% bromophenol blue) by boiling for 3 min at 95°C. Samples were resolved on 10% SDS–polyacrylamide gel. The proteins in the pulldowns were detected by Western blotting using anti‐RPA antibody (NA19L; Calbiochem; 1:500) or anti‐His‐tag antibody (A00186‐100; GenScript; 1:2,500) against His‐tagged MMS22L. Pulldown with mitochondrial SSB was carried out in the same way as with RPA.
Streptavidin pulldowns with biotinylated ssDNA and recombinant proteins
The assays (Fig 6G–I) were performed essentially as described previously (Jensen et al, 2010) in a 20‐μl reaction volume. RAD51 (200 nM) alone or with MMS22L–TONSL or MMS22L (20, 80, and 150 nM) was pre‐incubated at 37°C for 15 min in a binding buffer containing 20 mM Tris–HCl (pH 7.5), 120 mM NaCl, 0.1% (v/v) Triton X‐100, 2 mM calcium chloride, 10 mM magnesium acetate, 2 mM ATP, 1 mM dithiothreitol, and 0.1 mg/ml bovine serum albumin. Then, biotinylated B‐90‐mer (for sequence, see above; 1 nM) was added with or without cold dsDNA (X12‐3 annealed with X12‐4 C (Cejka & Kowalczykowski, 2010); 10 nM) and incubated for 5 min at 37°C. Reactions were further incubated upon adding pre‐equilibrated Streptavidin Dynabeads (15 μl, Invitrogen, M‐280) at 4°C for 30 min while stirring occasionally. The beads were washed four times with 150 μl wash buffer containing 20 mM Tris–HCl (pH 7.5), 120 mM NaCl, 0.1% (v/v) Triton X‐100, 2 mM calcium chloride, 10 mM magnesium acetate, 2 mM ATP, and 1 mM dithiothreitol. The protein was eluted by boiling with protein loading buffer (50 mM Tris [pH 6.8], 10% glycerol, 1.6% sodium dodecyl sulfate, 0.1 M dithiothreitol, 0.01% bromophenol blue) and 1 μl bovine serum albumin (10 mg/ml) at 95°C for 3 min and loaded onto 10% SDS–polyacrylamide gel. The RAD51 protein bound to ssDNA was detected by Western blotting using a polyclonal anti‐RAD51 antibody (Simandlova et al, 2013). The assays in Figs 5E and F, and EV7K and L were performed as above with the following modifications. Biotinylated oligonucleotide B‐90 (20 nM) was premixed with RPA (100 nM) in a 50‐μl volume in a reaction buffer as above and incubated for 10 min at 37°C. Then, MMS22L–TONSL, MMS22L FA‐TONSL, MMS22L, or MMS22L FA (all proteins 50 nM) was added either without or with RAD51 (500 nM) and further incubated for 10 min at 37°C. Next, pre‐equilibrated Streptavidin Dynabeads (5 μl) were added to the reaction and incubated at 4°C for another 30 min while stirring occasionally, washed, and eluted as above. Eluted proteins were detected by Western blotting with antibodies as indicated above or anti‐FLAG (Sigma, F3165, 1:2,000).
Strand exchange assays
The reactions (15 μl) were performed essentially as described previously (Jensen et al, 2010). First, non‐labeled DNA substrate (2.5 nM, either ssDNA, 5′‐tailed, 3′‐tailed, or gapped DNA) was pre‐incubated with the indicated amounts of RAD51 in a binding buffer containing 25 mM Tris acetate (pH 7.5), 3 mM magnesium acetate, 2 mM calcium chloride, 0.1 mg/ml bovine serum albumin, 1 mM ATP, and 1 mM DTT for 5 min at 37°C. Subsequently, MMS22L–TONSL or MMS22L was added to the reaction mixture and incubated for 5 min at 37°C. Next, 32P‐radiolabeled dsDNA (2.5 nM) was added and the reaction was further incubated for 30 min at 37°C. The reaction was terminated by the addition of 5 μl STOP buffer (150 mM EDTA, 2% sodium dodecyl sulfate, 30% glycerol, 0.1% bromophenol blue) and 1 μl proteinase K (14–24 mg/ml, Roche) for 10 min at 37°C. The reaction products were separated on 8% polyacrylamide gels in TBE buffer (89.1 mM Tris–base, 88.6 mM boric acid, 2 mM EDTA). Where indicated, RPA was pre‐bound to the DNA for 5 min at 37°C in the binding buffer containing 25 mM Tris acetate (pH 7.5), 0.66 mM magnesium acetate, 2 mM calcium chloride, 0.1 mg/ml bovine serum albumin, 1 mM ATP, and 1 mM DTT; subsequently MMS22L–TONSL or MMS22L was added and incubated for 5 min at 37°C. Upon addition of 32P‐radiolabeled dsDNA (2.5 nM), the reaction was further incubated for 30 min at 37°C. The gel was dried and exposed to a Phosphorimager screen (GE Healthcare) and analyzed by Typhoon FLA 9500 (GE Healthcare). The ssDNA was oligonucleotide RJ‐167‐mer, the 3′‐tailed DNA was prepared by annealing oligonucleotides RJ‐167‐mer and RJ‐PHIX‐42‐1, the 5′‐tailed DNA was prepared by annealing oligonucleotide RJ‐5′TAIL‐167‐mer and RJ‐PHIX‐42‐1, the gapped DNA was prepared by annealing oligonucleotide RJ‐167‐mer, RJ‐PHIX‐42‐1, and RJ‐END, and the dsDNA was prepared by annealing oligonucleotide RJ‐Oligo1 and RJ‐Oligo2. The sequences of oligonucleotides were described previously (Jensen et al, 2010), except RJ‐END (5′‐TAAATAAGATAAGGA‐3′).
iPOND assay
For iPOND (Sirbu et al, 2011), HeLa cells were incubated in DMEM containing 10 μM EdU (Invitrogen) for 10 min. For the CPT‐recovery assay, EdU‐labeled cells were first treated with 1 μM CPT for 30 min, then washed three times with 10 mM thymidine‐containing medium, and incubated for indicated times. For the experiment in Fig 3D and E, stably transfected HeLa cells with doxycycline‐inducible shMMS22L were treated with doxycycline for 48 h before performing CPT‐recovery iPOND. For the experiment in Fig EV4B and C, endogenous TONSL was depleted by siRNA targeting 3′UTR for 24 h and TONSL variants were expressed from a doxycycline‐inducible promoter for additional 24 h before the CPT‐recovery assay. To cross‐link proteins to DNA, cells were incubated in PBS containing 1% formaldehyde for 20 min at room temperature. To quench excess of cross‐linker, glycine was added to a final concentration of 125 mM for 10 min. Cells were washed three times with PBS and collected by centrifugation. For cell permeabilization, pellets were resuspended in PBS containing 0.25% Triton X‐100 and complete protease inhibitor cocktail (Roche) and incubated at 4°C for 20 min. Permeabilized cells were washed once with PBS containing 0.5% bovine serum albumin (Roche). Cells were incubated in Click reaction buffer (PBS containing 10 mM sodium ascorbate (Sigma‐Aldrich), 10 μM Azide‐PEG11‐Biotin Conjugate (Tocris Bioscience), and 2 μM CuSO4) for 90 min at room temperature. Cells were washed twice with PBS containing 0.5% bovine serum albumin and resuspended in lysis buffer (25 mM Tris–HCl [pH 7.0], 150 mM NaCl, 0.2% NP‐40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, complete protease inhibitor cocktail, Roche). Suspension was sonicated on ice using Branson 250 sonifier and then centrifuged for 30 min at 20,800 g. Supernatants were collected, and Streptavidin MyOne T1 magnetic beads (Life Technologies) was added and incubated for 1 h at 4°C. Beads were washed twice with lysis buffer, once with 1 M NaCl, and finally once with lysis buffer. Captured proteins were eluted by boiling for 30 min at 95°C and detected by Western blotting.
Yeast two‐hybrid assays
DNA sequence encoding MMS22L was fused to the LexA DNA‐binding domain by cloning into pLexA‐N/C plasmids (Dualsystems, Switzerland); human MMS22L, RAD51, RAD51C, and RPA1 gene sequences were fused to the GAL4 activation domain in plasmid pGAD‐HA (Dualsystems, Switzerland). Yeast strain L40 was transformed with each plasmid and plated onto SD–Leu–Trp medium and incubated at 30°C. For RPA1‐binding assays, myc‐RPA2 and flag‐RPA3 were cloned into YIpDCE1 plasmid (Stearman et al, 1998), subsequently integrated in L40 to allow co‐expression of all RPA subunits. For survival assays, equal numbers of cells were spotted on SD–Leu–Trp control plates and SD–Leu–Trp–His plates containing 20 mM 3‐aminotriazole (3‐AT; Sigma) and incubated at 30°C for 3 days. For monitoring β‐galactosidase expression, pellets of yeast cells grown in SD–Leu–Trp medium corresponding to OD600 = 1.5 was washed with 1× PBS, resuspended in 100 μl X‐Gal reagent (0.1% (w/v) X‐Gal (Sigma‐Aldrich), 0.05% (v/v) β‐mercaptoethanol, 0.1% (w/v) SDS in 1× PBS) and transferred to 96‐well plate. Suspensions were incubated at room temperature until the reaction color developed and scanned.
Statistical analysis
Statistical analyses were performed. Standard error of the mean values was calculated using Prism software (GraphPad). Mann–Whitney U‐test, Pearson's correlation test, and one‐way ANOVA with Bonferroni post‐test methods were employed to verify statistical significance.
Author contributions
WP, DS, and MMLB performed cell biology experiments, and WP and MP analyzed the data; AS prepared expression constructs, LJM and LR performed biochemistry experiments, and LJM, LR, and PC analyzed the data. KM and RZ performed EM analysis of reversed forks; AS and PJ provided the polyclonal anti‐RAD51 antibody and the modified pTXB3 expression vector; WP, LJM, KM, ML, MP, PJ, and PC participated in the experimental design; WP, LJM, MP, and PC wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank S. Taylor for the FRT‐TetR‐HeLa cell line; A. van Drogen and R. Aebersold for FRT‐TetR‐U2OS cell line; J. Tilma, O. Oros, S. S. Lee, F. van Drogen, and the ETH ScopeM microscopy facility for technical support; A. Smith for critical reading of the manuscript and members of the Peter and Cejka laboratories for helpful discussions and comments on the manuscript. This work was supported by Oncosuisse, ERC, the Swiss National Science Foundation (SNF), and the ETH Zurich.
The EMBO Journal (2016) 35: 2584–2601
Contributor Information
Wojciech Piwko, Email: wojciech.piwko@bc.biol.ethz.ch.
Matthias Peter, Email: matthias.peter@bc.biol.ethz.ch.
Petr Cejka, Email: petr.cejka@irb.usi.ch.
References
- Benson FE, Stasiak A, West SC (1994) Purification and characterization of the human Rad51 protein, an analogue of E. coli RecA. EMBO J 13: 5764–5771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buser R, Kellner V, Melnik A, Wilson‐Zbinden C, Schellhaas R, Kastner L, Piwko W, Dees M, Picotti P, Maric M, Labib K, Luke B, Peter M (2016) The replisome‐coupled E3 ubiquitin ligase Rtt101‐Mms22 counteracts Mrc1 function to tolerate genotoxic stress. PLoS Genet 12: e1005843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM (2006) Cell Profiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7: R100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira A, Hilario J, Amitani I, Baskin RJ, Shivji MK, Venkitaraman AR, Kowalczykowski SC (2009) The BRC repeats of BRCA2 modulate the DNA‐binding selectivity of RAD51. Cell 136: 1032–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P, Kowalczykowski SC (2010) The full‐length Saccharomyces cerevisiae Sgs1 protein is a vigorous DNA helicase that preferentially unwinds holliday junctions. J Biol Chem 285: 8290–8301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri AR, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, John S, Day A, Crespo AV, Shen B, Starnes LM, de Ruiter JR, Daniel JA, Konstantinopoulos PA, Cortez D, Cantor SB et al (2016) Replication fork stability confers chemoresistance in BRCA‐deficient cells. Nature 535: 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D (2008) ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 9: 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SR, Miller KM, Maas NL, Roguev A, Fillingham J, Chu CS, Schuldiner M, Gebbia M, Recht J, Shales M, Ding H, Xu H, Han J, Ingvarsdottir K, Cheng B, Andrews B, Boone C, Berger SL, Hieter P, Zhang Z et al (2007) Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 446: 806–810 [DOI] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, Cortez D (2015) The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol Cell 59: 998–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duro E, Vaisica JA, Brown GW, Rouse J (2008) Budding yeast Mms22 and Mms1 regulate homologous recombination induced by replisome blockage. DNA Repair (Amst) 7: 811–818 [DOI] [PubMed] [Google Scholar]
- Duro E, Lundin C, Ask K, Sanchez‐Pulido L, MacArtney TJ, Toth R, Ponting CP, Groth A, Helleday T, Rouse J (2010) Identification of the MMS22L‐TONSL complex that promotes homologous recombination. Mol Cell 40: 632–644 [DOI] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, Teague JW, Stratton MR, McDermott U, Campbell PJ (2015) COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 43: D805–D811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golmard L, Caux‐Moncoutier V, Davy G, Al Ageeli E, Poirot B, Tirapo C, Michaux D, Barbaroux C, d'Enghien CD, Nicolas A, Castera L, Sastre‐Garau X, Stern MH, Houdayer C, Stoppa‐Lyonnet D (2013) Germline mutation in the RAD51B gene confers predisposition to breast cancer. BMC Cancer 13: 484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MN, Paquet N, Fox D 3rd, Dray E, Zheng XF, Klein H, Sung P, Wang W (2012) A variant of the breast cancer type 2 susceptibility protein (BRC) repeat is essential for the RECQL5 helicase to interact with RAD51 recombinase for genome stabilization. J Biol Chem 287: 23808–23818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RB, Carreira A, Kowalczykowski SC (2010) Purified human BRCA2 stimulates RAD51‐mediated recombination. Nature 467: 678–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Doty T, Gibson B, Heyer WD (2010) Human BRCA2 protein promotes RAD51 filament formation on RPA‐covered single‐stranded DNA. Nat Struct Mol Biol 17: 1260–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Renault L, Veaute X, Fabre F, Stahlberg H, Heyer WD (2011) Rad51 paralogues Rad55‐Rad57 balance the antirecombinase Srs2 in Rad51 filament formation. Nature 479: 245–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke B, Versini G, Jaquenoud M, Zaidi IW, Kurz T, Pintard L, Pasero P, Peter M (2006) The cullin Rtt101p promotes replication fork progression through damaged DNA and natural pause sites. Curr Biol 16: 786–792 [DOI] [PubMed] [Google Scholar]
- Meindl A, Hellebrand H, Wiek C, Erven V, Wappenschmidt B, Niederacher D, Freund M, Lichtner P, Hartmann L, Schaal H, Ramser J, Honisch E, Kubisch C, Wichmann HE, Kast K, Deissler H, Engel C, Muller‐Myhsok B, Neveling K, Kiechle M et al (2010) Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet 42: 410–414 [DOI] [PubMed] [Google Scholar]
- Neelsen KJ, Lopes M (2015) Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol 16: 207–220 [DOI] [PubMed] [Google Scholar]
- Nitiss JL (2009) Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 9: 338–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell BC, Adamson B, Lydeard JR, Sowa ME, Ciccia A, Bredemeyer AL, Schlabach M, Gygi SP, Elledge SJ, Harper JW (2010) A genome‐wide camptothecin sensitivity screen identifies a mammalian MMS22L‐NFKBIL2 complex required for genomic stability. Mol Cell 40: 645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell L, Panier S, Wildenhain J, Tkach JM, Al‐Hakim A, Landry MC, Escribano‐Diaz C, Szilard RK, Young JT, Munro M, Canny MD, Kolas NK, Zhang W, Harding SM, Ylanko J, Mendez M, Mullin M, Sun T, Habermann B, Datti A et al (2010) The MMS22L‐TONSL complex mediates recovery from replication stress and homologous recombination. Mol Cell 40: 619–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Helleday T (2010) Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol 11: 683–687 [DOI] [PubMed] [Google Scholar]
- Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T (2010) Hydroxyurea‐stalled replication forks become progressively inactivated and require two different RAD51‐mediated pathways for restart and repair. Mol Cell 37: 492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piwko W, Olma MH, Held M, Bianco JN, Pedrioli PG, Hofmann K, Pasero P, Gerlich DW, Peter M (2010) RNAi‐based screening identifies the Mms22L‐Nfkbil2 complex as a novel regulator of DNA replication in human cells. EMBO J 29: 4210–4222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raderschall E, Golub EI, Haaf T (1999) Nuclear foci of mammalian recombination proteins are located at single‐stranded DNA regions formed after DNA damage. Proc Natl Acad Sci USA 96: 1921–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjha L, Anand R, Cejka P (2014) The Saccharomyces cerevisiae Mlh1‐Mlh3 heterodimer is an endonuclease that preferentially binds to Holliday junctions. J Biol Chem 289: 5674–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M (2012) Topoisomerase I poisoning results in PARP‐mediated replication fork reversal. Nat Struct Mol Biol 19: 417–423 [DOI] [PubMed] [Google Scholar]
- San Filippo J, Sung P, Klein H (2008) Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 77: 229–257 [DOI] [PubMed] [Google Scholar]
- Saredi G, Huang H, Hammond CM, Alabert C, Bekker‐Jensen S, Forne I, Reveron‐Gomez N, Foster BM, Mlejnkova L, Bartke T, Cejka P, Mailand N, Imhof A, Patel DJ, Groth A (2016) H4K20me0 marks post‐replicative chromatin and recruits the TONSL‐MMS22L DNA repair complex. Nature 534: 714–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP (2007) Human CtIP promotes DNA end resection. Nature 450: 509–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson S, Trujillo K, Song B, Stratton S, Sung P (2001a) Basis for avid homologous DNA strand exchange by human Rad51 and RPA. J Biol Chem 276: 8798–8806 [DOI] [PubMed] [Google Scholar]
- Sigurdsson S, Van Komen S, Bussen W, Schild D, Albala JS, Sung P (2001b) Mediator function of the human Rad51B‐Rad51C complex in Rad51/RPA‐catalyzed DNA strand exchange. Genes Dev 15: 3308–3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simandlova J, Zagelbaum J, Payne MJ, Chu WK, Shevelev I, Hanada K, Chatterjee S, Reid DA, Liu Y, Janscak P, Rothenberg E, Hickson ID (2013) FBH1 helicase disrupts RAD51 filaments in vitro and modulates homologous recombination in mammalian cells. J Biol Chem 288: 34168–34180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D (2011) Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25: 1320–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearman R, Dancis A, Klausner RD (1998) YIpDCE1 ‐ an integrating plasmid for dual constitutive expression in yeast. Gene 212: 197–202 [DOI] [PubMed] [Google Scholar]
- Sung P (1994) Catalysis of ATP‐dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science 265: 1241–1243 [DOI] [PubMed] [Google Scholar]
- Takeda S, Tadele Z, Hofmann I, Probst AV, Angelis KJ, Kaya H, Araki T, Mengiste T, Mittelsten Scheid O, Shibahara K, Scheel D, Paszkowski J (2004) BRU1, a novel link between responses to DNA damage and epigenetic gene silencing in Arabidopsis . Genes Dev 18: 782–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarsounas M, Davies AA, West SC (2004) RAD51 localization and activation following DNA damage. Philos Trans R Soc Lond B Biol Sci 359: 87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MR, Spirek M, Chaurasiya KR, Ward JD, Carzaniga R, Yu X, Egelman EH, Collinson LM, Rueda D, Krejci L, Boulton SJ (2015) Rad51 paralogs remodel pre‐synaptic Rad51 filaments to stimulate homologous recombination. Cell 162: 271–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA, Cejka P, Stewart S, Lopes M, Vindigni A (2015) DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 208: 545–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorslund T, McIlwraith MJ, Compton SA, Lekomtsev S, Petronczki M, Griffith JD, West SC (2010) The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single‐stranded DNA. Nat Struct Mol Biol 17: 1263–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tighe A, Staples O, Taylor S (2008) Mps1 kinase activity restrains anaphase during an unperturbed mitosis and targets Mad2 to kinetochores. J Cell Biol 181: 893–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G (1995) Identification of the breast cancer susceptibility gene BRCA2. Nature 378: 789–792 [DOI] [PubMed] [Google Scholar]
- Zaidi IW, Rabut G, Poveda A, Scheel H, Malmstrom J, Ulrich H, Hofmann K, Pasero P, Peter M, Luke B (2008) Rtt101 and Mms1 in budding yeast form a CUL4(DDB1)‐like ubiquitin ligase that promotes replication through damaged DNA. EMBO Rep 9: 1034–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M (2015) Rad51‐mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208: 563–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File