

Abstract
The DISC1‐interacting protein CAMDI has been suggested to promote radial migration through centrosome regulation. However, its physiological relevance is unclear. Here, we report the generation and characterization of CAMDI‐deficient mice. CAMDI‐deficient mice exhibit delayed radial migration with aberrant neural circuit formation and psychiatric behaviors including hyperactivity, repetitive behavior, and social abnormality typically observed in autism spectrum disorder patients. Analyses of direct targets of CAMDI identify HDAC6 whose α‐tubulin deacetylase activity is inhibited by CAMDI at the centrosome. CAMDI deficiency increases HDAC6 activity, leading to unstable centrosomes with reduced γ‐tubulin and acetylated α‐tubulin levels. Most importantly, psychiatric behaviors as well as delayed migration are significantly rescued by treatment with Tubastatin A, a specific inhibitor of HDAC6. Our findings indicate that HDAC6 hyperactivation by CAMDI deletion causes psychiatric behaviors, at least in part, through delayed radial migration due to impaired centrosomes.
Keywords: CAMDI, HDAC6, psychiatric behaviors, radial migration
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Molecular Biology of Disease; Neuroscience
Introduction
Neuronal migration is a critical phase in nervous system development, and the regulation of centrosome stability underlies the directed migration of cortical neurons 1, 2. The identification of genes involved in human neuronal migration disorders has advanced our understanding of the regulatory mechanisms underlying neuronal migration 3, 4. The classical example is disrupted‐in‐schizophrenia 1 (DISC1), which is located at the breakpoint of a balanced translocation identified in a large Scottish family with psychiatric disorders 5. DISC1 has been proposed to at least in part control centrosome function, notably by interacting with NUDEL, LIS1, and dynein 6, 7, 8. Indeed, DISC1 dysfunction disrupts cortical architecture at least in part through centrosome impairment 9, 10, 11. Thus, the collapse of centrosome regulation is closely involved in the pathogenesis of psychiatric disorders.
We previously identified CAMDI (coiled‐coil protein associated with myosin II and DISC1) as a DISC1‐interacting protein 12. The second coiled‐coil domain of CAMDI interacts with the second coiled‐coil domain of DISC1. In utero electroporation assay revealed that CAMDI is required for radial migration via centrosome regulation during brain development 12. The CAMDI gene is located at 2q31.2, where genetic linkage regions of autism spectrum disorder (ASD) are mapped 13, 14, 15, 16, suggesting CAMDI dysfunction is closely associated with psychiatric disorders. Accordingly, in order to understand the physiological relevance of CAMDI, we analyzed mutant mice lacking the CAMDI gene.
In the present study, we showed that CAMDI regulates neuronal migration through the modulation of HDAC6 and that HDAC6 inhibitor rescues delayed neuronal migration and psychiatric behaviors in CAMDI‐deficient mice. Our findings may provide new insights into the pathogenesis of psychiatric disorders and raise the possibility of a new approach using HDAC6 inhibitor to treat psychiatric disorders associated with centrosome dysfunction.
Results
Delayed cortical migration in CAMDI‐KO mice
We generated mutant mice lacking the CAMDI gene (Fig EV1A–C). Homozygous CAMDI‐knockout (CAMDI‐KO) mice were born at the expected Mendelian frequency and were viable and fertile; they had normal body weight at birth and during the juvenile stage (Fig EV1D). Adult CAMDI‐KO mice had a normal appearance and exhibited no obvious changes in overall brain weight or morphology (data not shown). Thus, CAMDI is not required for fundamental brain development or survival.
Figure EV1. Generation of CAMDI‐KO mice.
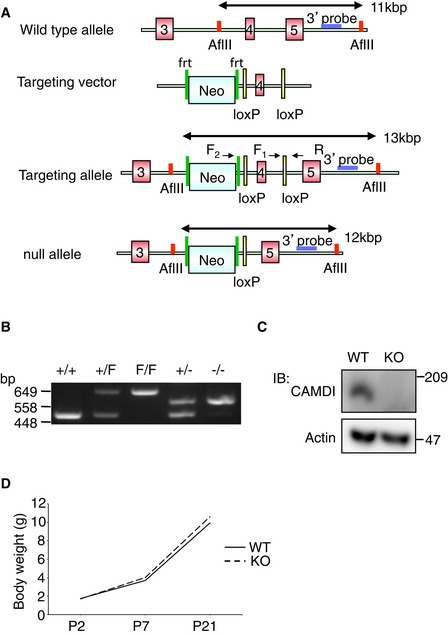
- Targeting strategy for CAMDI‐KO mice. The 3′ probes are used for Southern blot analysis. F1, F2, and R mark the primers used for genomic PCR.
- Genotypes of CAMDI mutant mice were determined by PCR on tail DNAs.
- Western blot analysis of E16 mouse brain lysate probed with anti‐CAMDI‐specific antibody to demonstrate an absence of CAMDI protein by CAMDI KO.
- Body weight analysis revealed normal body weight at birth and during juvenile age. n = 8 mice for each genotype.
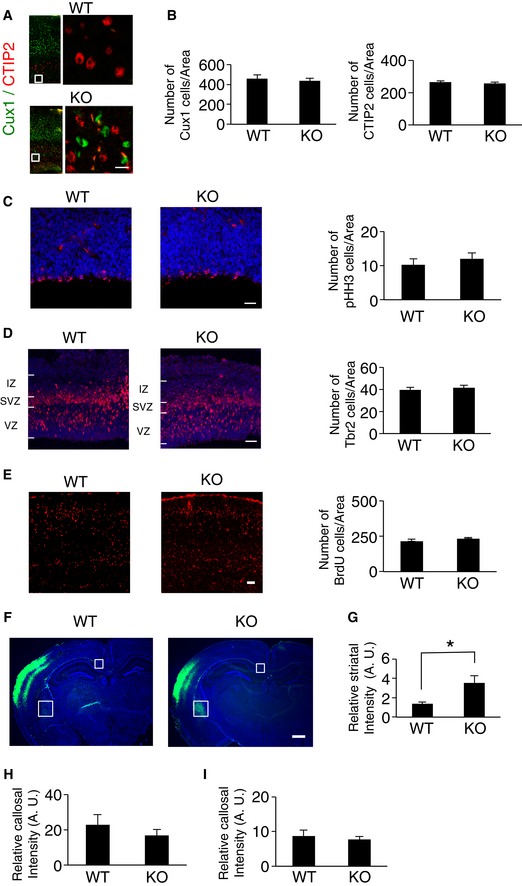
To examine the role of CAMDI in cortical migration in vivo, we compared the expression pattern of Cux1, a molecular marker of the superficial layers of the somatosensory cortex (layers II–IV) between wild‐type (WT) and CAMDI‐KO mice at postnatal day (P) 2. Comparative analysis of the distribution pattern of Cux1‐positive cells in the cerebral cortex revealed that some migrating neurons were significantly delayed in CAMDI‐KO mice compared to WT mice (Fig 1A and B). To corroborate this, histological analyses of radial migration in CAMDI‐KO mice were performed by in utero electroporation assay. In WT mice, almost all EGFP+ cells electroporated at embryonic day (E) 14.5 migrated to layers II/III of the cerebral cortex at P21, when cortical migration is essentially complete. In contrast, several EGFP+ cells in CAMDI‐KO mice remained in the lower cortical layers (Fig 1C and D). Mislocalization of neurons to the lower cortical layers in CAMDI‐KO mice at P21 was further confirmed by other markers such as Cux1 and CTIP2 (Fig EV2A). These results corroborate our previous observation that CAMDI is required for cortical migration during neuronal development. The inhibitory effect of CAMDI KO on cortical migration appears to be milder than shCAMDI‐mediated knockdown by in utero electroporation 12; this finding suggests the existence of a compensatory pathway for CAMDI deficiency during brain development in CAMDI‐KO mice. The BrdU incorporation assay for labeling newborn neurons suggested that the overall proliferation rate did not change due to CAMDI KO (Fig EV2E). Consistently, the total numbers of Cux1‐, CTIP2‐, pHH3‐, and TBR2‐positive neurons did not change due to CAMDI KO (Fig EV2B–D). Thus, we conclude that delayed migration by CAMDI KO is not due to alterations in cell proliferation and cell fate determination.
Figure 1. Abnormal neuronal migration in CAMDI‐KO mice.
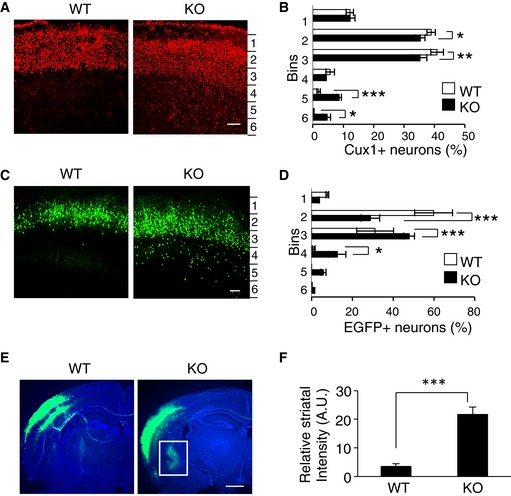
- Abnormal distribution of Cux1‐positive neurons in CAMDI‐KO mice. Expression of Cux1 in the somatosensory cortex was compared between P2 wild‐type (WT) and CAMDI‐KO (KO) mice. Scale bar, 100 μm.
- Quantification of the number of Cux1‐positive neurons. Note the abnormal distribution of neurons in deep cortical layers of CAMDI‐KO mice. n = 3 mice/genotype (WT = 788 cells, KO = 2,139 cells). *P < 0.05, **P < 0.01, ***P < 0.001; two‐way ANOVA followed by Scheffe's post hoc test. Data are presented as mean ± SEM.
- Delay in neuronal migration by CAMDI deletion. Coronal sections through the somatosensory cortex of P21 WT and CAMDI‐KO mice were analyzed following in utero electroporation of EGFP plasmid at E14.5. Scale bar, 100 μm.
- Quantification of the number of EGFP‐positive neurons. Note the abnormal distribution of neurons in lower cortical layers of CAMDI‐KO mice. n = 3 for WT mice (546 cells), n = 4 for KO mice (680 cells). *P < 0.05, ***P < 0.001. Two‐way ANOVA followed by Scheffe's post hoc test. Data are presented as mean ± SEM.
- Aberrant axonal projection and terminal arbors by CAMDI deletion. Coronal sections through the somatosensory cortex of P21 WT and CAMDI‐KO mice were analyzed following in utero electroporation of EGFP plasmid at E14.5. Representative pictures show an aberrant axonal projection of corticostriatal neurons in CAMDI‐KO mice. Sections were stained with Hoechst 33258 and anti‐EGFP antibody. Scale bar, 1 mm.
- Quantification of the striatal intensity. n = 3 mice/genotype. ***P < 0.001; one‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM.
Figure EV2. No significant difference in progenitor cell proliferation, cell fate, and callosal projection by CAMDI deletion.
-
AAbnormal distribution of Cux1‐positive neurons in CAMDI‐KO mice at P21. Expressions of Cux1 and CTIP2 in the somatosensory cortex were compared between P21 WT and CAMDI‐KO mice. Scale bar, 20 μm.
-
BQuantification of the number of Cux1‐ and CTIP2‐positive neurons of P21. n = 3 for WT mice (Cux1: 2,309 cells, CTIP2: 4,421 cells) and n = 4 for KO mice (Cux1: 2,324 cells, CTIP2: 4,393 cells). One‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM.
-
CQuantification of the number of phospho‐histone H3‐positive cells at E16. n = 3 mice/genotype (WT = 96 cells, KO = 150 cells). One‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM. Scale bar, 50 μm.
-
DQuantification of the number of TBR2‐positive cells at E16. n = 3 mice/genotype (WT = 316 cells, KO = 327 cells). One‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM. Scale bar, 100 μm.
-
EQuantification of the number of BrdU‐positive cells at E18. BrdU injection was performed at E14. n = 3 mice/genotype (WT = 1,953 cells, KO = 2,103 cells). One‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM. Scale bar, 100 μm.
-
FCoronal sections through the somatosensory cortex of P56 WT and CAMDI‐KO mice were analyzed following in utero electroporation of EGFP plasmid at E14.5. Scale bar, 1 mm.
-
GQuantification of the striatal intensity at P56. n = 3 mice/genotype. *P < 0.05; one‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM.
-
H, IQuantification of the callosal intensity at P56 (H) and P21 (I). n = 3 mice/genotype. One‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM.
Callosal projection neurons connect to the contralateral hemisphere in order to develop proper neural circuits. Axons of upper layer II/III cells project into the contralateral side via the corpus callosum, while axons of lower layer V/VI cells project into the striatum of the ipsilateral side 17, 18. The results show that CAMDI knockout causes abnormal corticostriatal projection of callosal neurons. When EGFP expression plasmid was electroporated into the cerebral ventricle of E14.5 mouse embryos to label layer II/III neurons, CAMDI‐KO mice exhibited not only contralateral projection, but also several axons projecting into the ipsilateral striatum both at P21 and P56 (Figs 1E and EV2F). Quantitative analysis indicated a significant increase in the striatal projection of CAMDI‐KO mice (Figs 1F and EV2G–I). This indicates that the layer‐specific innervation patterns of callosal axons are disrupted in CAMDI‐KO mice. As delayed neurons in layer V of CAMDI‐KO mice had the properties of layer II/III neurons, these axons are considered to aberrantly project into the striatum. This finding also corroborates the finding that CAMDI knockout delays neuronal migration.
Psychiatric behaviors in CAMDI‐KO mice
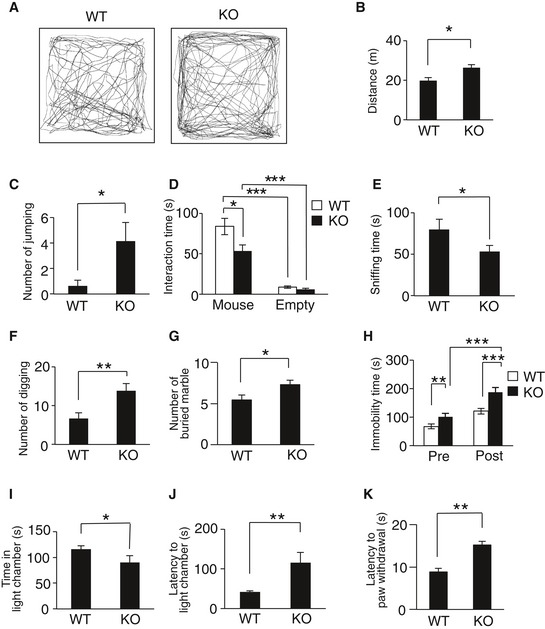
Delayed cortical migration and aberrant axonal projection detected in CAMDI‐KO brains are associated with the pathogenesis of psychiatric disorders. Therefore, adult CAMDI‐KO mice were submitted to behavioral tests. The open‐field test, a widely used measure of exploratory behavior and general locomotive activity in mice, revealed CAMDI‐KO mice were hyperactive as measured by the total distance traveled (Fig 2A and B). CAMDI‐KO mice showed an increase in travel time without a velocity change (Fig EV3A and B) and spent less time in the center of the field than WT mice (Fig EV3C), suggesting that CAMDI‐KO mice are more hyperactive and anxious than WT mice. Interestingly, the behavioral differences between WT and CAMDI‐KO mice become reduced in the adult stage. Similarly, the grooming time was comparable between WT and adult CAMDI‐KO mice (Fig EV3D), suggesting progressive and age‐dependent behavioral alterations. Meanwhile, there were no significant differences between the WT and CAMDI‐KO mice in the olfaction or cage‐top hang test (Fig EV3E and F), suggesting the observed hyperlocomotion is not due to disturbances of olfactory function or motor units strength. Consistently, the number of repetitive jumps in an open field was significantly greater in CAMDI‐KO mice than the WT mice (Fig 2C). We next determined whether CAMDI‐KO mice exhibit disordered social behavior. In the three‐chamber social interaction test, CAMDI‐KO mice spent significantly less time interacting with the strange mouse than WT mice (Figs 2D and EV3G), suggesting reduced social interaction behavior. Notably, in the resident–intruder test, CAMDI‐KO mice exhibited significantly less sniffing time during interactions with intruder mice (Fig 2E) but no obvious change in aggressive behavior (data not shown). In addition, the frequencies of times digging and marble burying were higher in CAMDI‐KO mice than the WT mice (Fig 2F and G). CAMDI‐KO mice exhibited repetitive behavior in the marble‐burying test, which is considered to reflect repetitive and perseverative behavior 19. We subsequently submitted the mice to the forced swimming test, which evaluates behavioral response to inescapable stress. As mice exhibit a response to escape from stressful situations, we measured immobility time, which was interpreted as “behavioral depression”. Compared to the WT mice, CAMDI‐KO mice exhibited greater immobility in the forced swimming test, indicating depression‐like behavior (Fig 2H). Consistent with the result from the open‐field test, the light–dark test revealed an anxiety behavior in CAMDI‐KO mice (Fig 2I and J). The hot plate test revealed sensory hyposensitivity in CAMDI‐KO mice (Fig 2K). Taken together, CAMDI‐deficient mice exhibited psychiatric behaviors characteristic of ASD, including hyperactivity, repetitive behavior, anxiety, and social abnormality.
Figure 2. Psychiatric behaviors in CAMDI‐KO mice.
-
A, BRepresentative traces from adult control (WT) and CAMDI KO (KO) in open‐field test. Locomotor activity (A) and distance traveled (B) for 15 min. n = 8 for WT mice, n = 7 for KO mice. *P < 0.05; one‐way ANOVA with Bonferroni's post hoc test (F(1, 13) = 5.53). Data are presented as mean ± SEM.
-
CRepetitive jumping behavior in open‐field test. n = 8 for WT mice, n = 7 for KO mice. *P < 0.05; one‐way ANOVA with Bonferroni's post hoc test (F(1, 13) = 5.09). Data are presented as mean ± SEM.
-
DThree‐chamber social interaction test. The time spent sniffing with stranger mouse. n = 19 for WT mice, n = 15 for KO mice. *P < 0.05, ***P < 0.001; two‐way ANOVA followed by Scheffe's post hoc test (main effect of genotype F(1, 63) = 5.79, main effect of chamber F(1, 63) = 90.47, interaction F(1, 63) = 5.29). Data are presented as mean ± SEM.
-
EResident–intruder test (social sniffing). n = 7 for WT mice, n = 8 for KO mice. *P < 0.05; one‐way ANOVA with Bonferroni's post hoc test (F(1, 13) = 5.15). Data are presented as mean ± SEM.
-
FResident–intruder test (digging). n = 7 for WT mice, n = 8 for KO mice. **P < 0.01; one‐way ANOVA with Bonferroni's post hoc test (F(1, 13) = 10.97). Data are presented as mean ± SEM.
-
GMarble‐burying test. n = 9 for WT mice, n = 25 for KO mice. *P < 0.05; one‐way ANOVA with Bonferroni's post hoc test (F(1, 32) = 5.39). Data are presented as mean ± SEM.
-
HForced swimming test. n = 10 for WT mice, n = 13 for KO mice. **P < 0.01, ***P < 0.001; two‐way ANOVA followed by Scheffe's post hoc test (main effect of genotype F(1, 42) = 84.30, main effect of session F(1, 42) = 33.88, interaction F(1, 42) = 4.92). Data are presented as mean ± SEM.
-
I, JLight–dark test. Time in light chamber (I) and latency to light chamber (J). n = 8 for WT mice, n = 7 for KO mice. *P < 0.05, **P < 0.01; one‐way ANOVA with Bonferroni's post hoc test, [(I) F(1, 13) = 8.75, (J) F(1, 13) = 9.73]. Data are presented as mean ± SEM.
-
KHot plate test. n = 8 for WT mice, n = 12 for KO mice. **P < 0.01; one‐way ANOVA with Bonferroni's post hoc test (F(1, 18) = 17.51). Data are presented as mean ± SEM.
Figure EV3. The behavioral differences between WT and CAMDI‐KO mice at juvenile and adult stages.
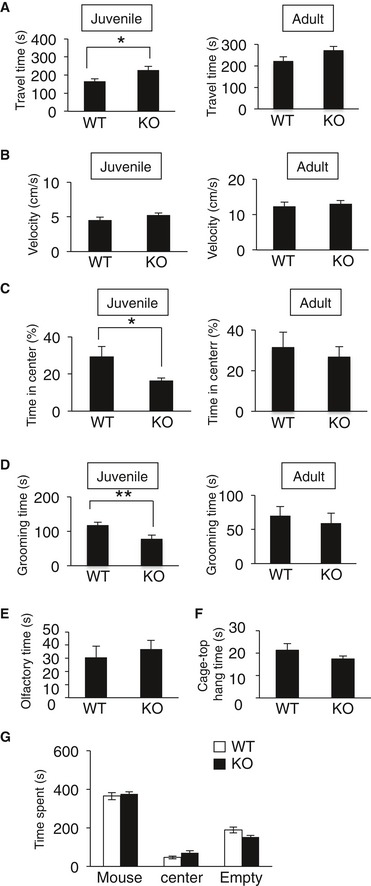
- Travel time on open‐field test at juvenile and adult stages. For juvenile, n = 13 for WT mice and n = 13 for KO mice. For adult, n = 8 for WT mice and n = 7 for KO mice. *P < 0.05; one‐way ANOVA with Bonferroni's post hoc test (juvenile: F(1, 24) = 5.87, adult: F(1, 13) = 2.55). Data are presented as mean ± SEM.
- The velocity in the open‐field test at juvenile and adult stages. For juvenile, n = 13 for WT mice and n = 13 for KO mice. For adult, n = 8 for WT mice and n = 7 for KO mice. One‐way ANOVA with Bonferroni's post hoc test (juvenile: F(1, 24) = 1.74, adult: F(1, 13) = 0.17). Data are presented as mean ± SEM.
- Time in center (%) on open‐field test at juvenile and adult stages. For juvenile, n = 13 for WT mice and n = 13 for KO mice. For adult, n = 8 for WT mice and n = 7 for KO mice. *P < 0.05; one‐way ANOVA with Bonferroni's post hoc test (juvenile: F(1, 24) = 5.75, adult: F(1, 13) = 0.63). Data are presented as mean ± SEM.
- Grooming time in open‐field test at juvenile and adult stages. For juvenile, n = 13 for WT mice and n = 13 for KO mice. For adult, n = 8 for WT mice and n = 7 for KO mice. **P < 0.01; one‐way ANOVA with Bonferroni's post hoc test (juvenile: F(1, 24) = 7.84, adult: F(1, 13) = 0.31). Data are presented as mean ± SEM.
- Olfactory function test at adult. n = 13 for WT mice and n = 21 for KO mice. One‐way ANOVA with Bonferroni's post hoc test (F(1, 32) = 0.21). Data are presented as mean ± SEM.
- Cage‐top hang test at adult. n = 10 for WT mice and n = 16 for KO mice. One‐way ANOVA with Bonferroni's post hoc test (F(1, 24) = 0.32). Data are presented as mean ± SEM.
- Three‐chamber social interaction test at adult. Total time spent in each chamber. n = 19 for WT mice and n = 20 for KO mice. Two‐way ANOVA followed by Scheffe's post hoc test (main effect of genotype F(1, 96) = 0.E+0, main effect of chamber F(2, 96) = 282, interaction F(2, 96) = 3.26). Data are presented as mean ± SEM.
CAMDI interacts with and inhibits HDAC6 α‐tubulin deacetylase
To elucidate how CAMDI deficiency leads to delayed cortical migration and psychiatric behaviors, we performed a yeast two‐hybrid screen of a mouse brain cDNA library using the N‐terminal spectrin domain of CAMDI as a bait. The results identified a fragment containing amino acids 372–1,149 of HDAC6 α‐tubulin deacetylase. Co‐immunoprecipitation assay demonstrated a specific interaction of CAMDI with HDAC6 both in vitro and in vivo (Fig 3A and B). To determine the region of CAMDI that interacts with HDAC6, various FLAG‐tagged CAMDI fragments were generated. Co‐immunoprecipitation assay revealed that the N‐terminal region (amino acids 1–250) of CAMDI specifically recognizes HDAC6 (Fig 3C). On the other hand, CAMDI interacted with the HDAC6 fragment containing the second HDAC domain (Fig 3D). Thus, the N‐terminus of CAMDI directly interacts with the second HDAC domain of HDAC6 (Fig 3E).
Figure 3. CAMDI interacts with HDAC6 α‐tubulin deacetylase.
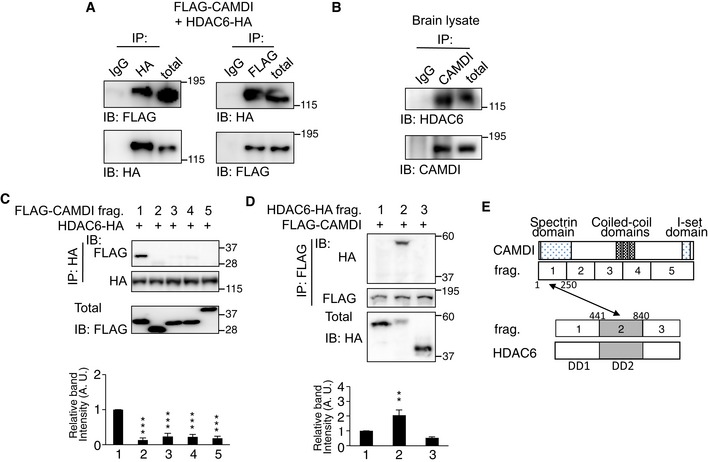
-
ACo‐expressed FLAG‐CAMDI and HDAC6‐HA interacted with each other in SH‐SY5Y cells. n = 3 independent experiments.
-
BEndogenous interaction of CAMDI with HDAC6 in the mouse brain. Co‐IP assay was performed with the indicated antibodies using E16 mouse brain. n = 3 independent experiments.
-
C, DN‐terminal region of CAMDI recognizes the second HDAC domain of HDAC6. SH‐SY5Y cells were co‐transfected with HDAC6‐HA and each FLAG‐CAMDI fragment (C) (fragment 1, 1–250; 2, 251–500; 3, 501–750; 4, 751–1,000; 5, 1,001–1,451 amino acids) or co‐transfected with FLAG‐CAMDI and each HDAC6‐HA fragment (D) (fragment 1 (DD1), 1–440; 2 (DD2), 441–840; 3, 841–1,215 amino acids). Co‐IP assay was performed with the indicated antibodies. n = 3 independent experiments. **P < 0.01, ***P < 0.001; one‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM.
-
ESchematic illustration of interaction between CAMDI and HDAC6.
Immunofluorescence and line‐scan analyses revealed CAMDI overexpression enhanced α‐tubulin acetylation in HeLa cells, which are generally used for the analyses of the cell cycle and centrosome (Fig 4A), suggesting CAMDI inhibits HDAC6 activity. Accordingly, to examine the effect of CAMDI on HDAC6 enzymatic activity, the centrosomal fraction was isolated from cell lysates expressing CAMDI and/or HDAC6, and the Ac‐tubulin level was assessed by immunoblot analysis. CAMDI overexpression alone increased the Ac‐tubulin level, and co‐expression of CAMDI blocked the HDAC6‐induced deacetylation of α‐tubulin (Fig 4B). Expression of the CAMDI N‐terminal domain alone substantially increased the Ac‐tubulin level by inhibiting HDAC6 activity (Fig 4C). Consistently, deletion of the CAMDI N‐terminal domain resulted in the loss of HDAC6 inhibitory activity (Fig 4D). Thus, the results indicate CAMDI inhibits HDAC6 via its N‐terminal domain. To further verify the specificity, we examined the effect of CAMDI on tubulin acetylation in the condition of HDAC6 knockdown. As expected, CAMDI had no effect on tubulin acetylation in the condition of HDAC6 knockdown (Fig EV4A and B). We next examined whether CAMDI was a general inhibitor of HDAC6 or specific to Ac‐tubulin. We assessed the level of acetylated HSP90, which is known to be a substrate for HDAC6 20. The immunoblot analysis demonstrated that the Ac‐HSP90 level was not changed by CAMDI KO (Fig EV4C), suggesting that CAMDI specifically inhibits HDAC6‐mediated catalysis of Ac‐tubulin.
Figure 4. HDAC6 inhibition by CAMDI regulates Ac‐tubulin level at the basal bodies.
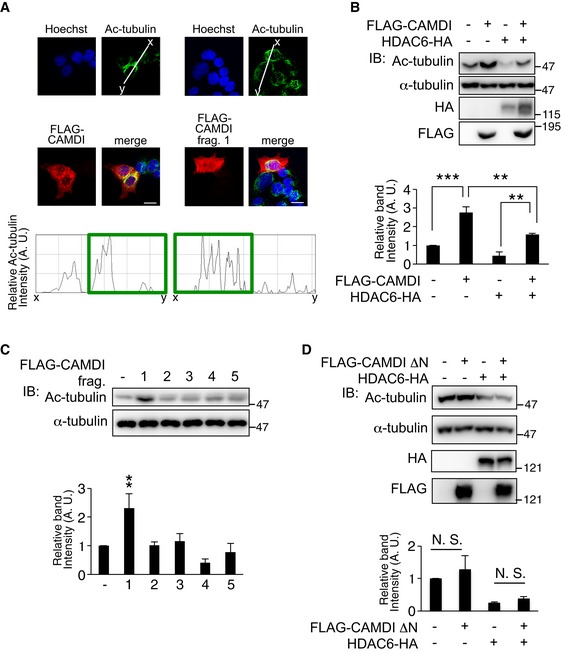
-
ACAMDI and CAMDI fragment 1 overexpression enhanced α‐tubulin acetylation. HeLa cells overexpressing FLAG‐CAMDI and FLAG‐CAMDI fragment 1 were immunostained with anti‐FLAG or anti‐Ac‐tubulin antibodies. Ratio line scans along the line (from x‐ to y‐axis) from images. Scale bar, 10 μm.
-
B, CCAMDI blocked HDAC6‐mediated α‐tubulin deacetylation. (B) Centrosomal fraction was isolated from cell lysates of SH‐SY5Y cells expressing each construct, and Ac‐tubulin level was assessed by immunoblot analysis. n = 3 independent experiments. **P < 0.01, ***P < 0.001; two‐way ANOVA followed by Scheffe's post hoc test. Data are presented as mean ± SEM. (C) CAMDI N‐terminal domain alone increased Ac‐tubulin level. n = 3 independent experiments. **P < 0.01; one‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM.
-
DDeletion of CAMDI N‐terminal domain (1–250 amino acids) failed to inhibit HDAC6. n = 3 independent experiments. N.S., not significant. Two‐way ANOVA followed by Scheffe's post hoc test. Data are presented as mean ± SEM.
Figure EV4. Effects of HDAC6 overexpression on the γ‐tubulin at centrosome.
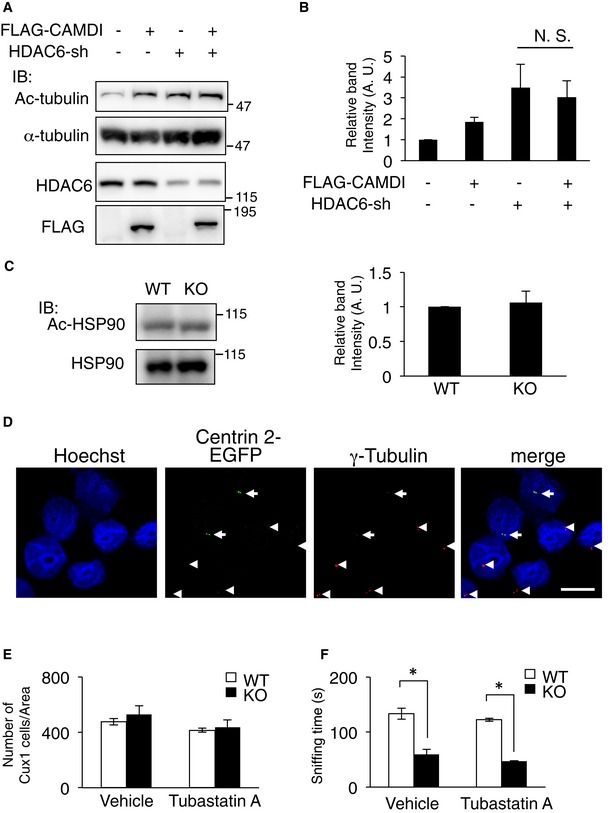
-
A, BCAMDI overexpression has no effect on tubulin acetylation in the condition of HDAC6 knockdown by shRNA. SH‐SY5Y cells were transfected with indicated vectors, and IP and IB assays were performed using the indicated antibodies. n = 3 independent experiments. N. S., not significant. Two‐way ANOVA followed by Scheffe's post hoc test. Data are presented as mean ± SEM.
-
CNo significant change in Ac‐HSP90 level in CAMDI‐KO cerebral cortex. Ac‐HSP90 level was assessed by immunoblot analysis. n = 3 independent experiments. One‐way ANOVA followed with Bonferroni's post hoc test. Data are presented as mean ± SEM.
-
DHeLa cells were transfected with centrin 2‐EGFP and HDAC6‐HA and immunostained with anti‐EGFP (green) and anti‐γ‐tubulin antibodies (red). DNA was stained with Hoechst 33258. Arrow: HDAC6‐HA and centrin 2‐EGFP transfected cells. Arrowhead: non‐transfected cells. Scale bar, 10 μm.
-
EQuantification of the number of Cux1‐positive neurons at P2. n = 3 mice/genotype (WT (vehicle) = 1,426 cells, KO (vehicle) = 2,116 cells, WT (Tubastatin A) = 1,245 cells, KO (Tubastatin A) = 1,311 cells). Two‐way ANOVA followed by Scheffe's post hoc test. Data are presented as mean ± SEM.
-
FThree‐chamber social interaction test. The time spent sniffing with stranger mouse. n = 8 for WT (vehicle) mice, n = 6 for KO (vehicle) mice, n = 5 for WT (Tubastatin A) mice, and n = 3 for KO (Tubastatin A) mice. *P < 0.05; two‐way ANOVA followed by Scheffe's post hoc test (main effect of genotype F(1, 18) = 11.11, main effect of drug F(1, 18) = 0.24, interaction F(1, 18) = 0.064). Data are presented as mean ± SEM.
HDAC6 inhibition by CAMDI regulates γ‐tubulin localization to the centrosome
We subsequently examined whether CAMDI inhibits the HDAC6‐mediated deacetylation of α‐tubulin at the centrosome. Immunofluorescence analysis revealed that the intensity of acetylated α‐tubulin at centrin 2‐positive foci was significantly reduced by HDAC6 overexpression; this effect was abolished by CAMDI co‐expression (Fig 5A and B). Similarly, the intensity of γ‐tubulin at centrin 2‐positive foci was also significantly reduced by HDAC6 overexpression; this effect was also abolished by CAMDI co‐expression (Figs 5C and D, and EV4D). Immunoblot analysis of the centrosomal fraction also indicated HDAC6 overexpression drastically decreased γ‐tubulin and Ac‐tubulin; CAMDI co‐expression canceled this effect (Fig 5E). In addition, in utero electroporation assay revealed that CAMDI knockdown reduced γ‐tubulin intensity in vivo (Fig 5F). Furthermore, immunoblot analysis showed CAMDI‐KO mouse brains had reduced levels of γ‐tubulin and acetylated α‐tubulin in the centrosomal fraction (Fig 5G). These results suggest that the centrosomal accumulation of γ‐tubulin is suppressed by α‐tubulin deacetylation by HDAC6 and that CAMDI regulates γ‐tubulin stability through HDAC6 inhibition. Therefore, HDAC6 overactivation by CAMDI deletion inhibits the formation of γ‐tubulin ring complexes at the centrosome in CAMDI‐KO mice.
Figure 5. HDAC6 inhibition by CAMDI regulates γ‐tubulin localization to the centrosome.
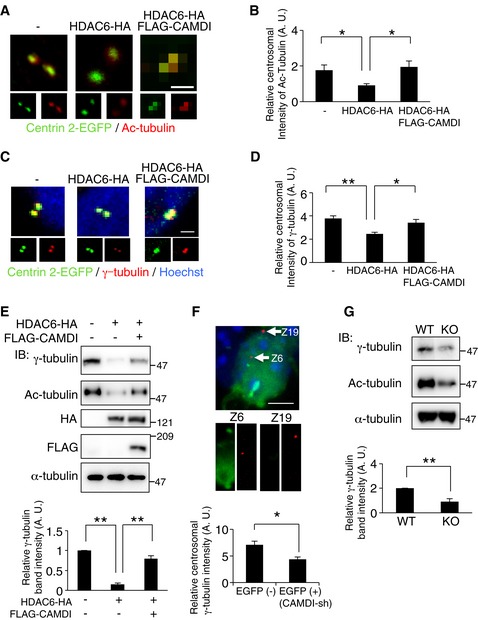
- Effect of HDAC6 overexpression or CAMDI co‐expression on the level of acetylated α‐tubulin in the centrosome. HeLa cells co‐transfected with centrin 2‐EGFP with/without HDAC6‐HA and FLAG‐CAMDI were immunostained with anti‐EGFP (green) and anti‐acetylated α‐tubulin antibodies (red). Scale bar, 1 μm.
- Quantification of intensity of acetylated α‐tubulin at centrosome. n = 25, 30, 30 cells. *P < 0.05; one‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM.
- Effects of HDAC6 overexpression or CAMDI co‐expression on the centrosomal localization of γ‐tubulin. HeLa cells co‐transfected with centrin 2‐EGFP with/without HDAC6‐HA and FLAG‐CAMDI were immunostained with anti‐EGFP (green) and anti‐γ‐tubulin antibodies (red). DNA was stained with Hoechst 33258. Scale bar, 1 μm.
- Quantification of intensity of γ‐tubulin at centrosome. n = 39, 16, 17 cells. *P < 0.05, **P < 0.01; one‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM.
- CAMDI stabilizes centrosomal γ‐tubulin through HDAC6 inhibition. Centrosomal fraction was isolated from cell lysates of SH‐SY5Y cells expressing each construct, and γ‐tubulin level was assessed by immunoblot analysis. n = 3 independent experiments. **P < 0.01; one‐way ANOVA followed with Bonferroni's post hoc test. Data are presented as mean ± SEM.
- Effects of CAMDI knockdown on the centrosomal localization of γ‐tubulin in vivo. Coronal sections through the somatosensory cortex of E17.5 were analyzed following in utero electroporation of EGFP and CAMDI‐sh plasmid at E14.5. The sections were immunostained with anti‐EGFP (green) and anti‐γ‐tubulin antibodies (red). Representative images of EGFP at cortical plate neuron expressing CAMDI‐sh stained for endogenous γ‐tubulin and with Hoechst (blue). Z‐stack number 6 indicates centrosome of EGFP‐positive CAMDI‐sh‐expressing neuron. Z‐stack number 19 indicates centrosome of a nearby non‐electroporated neuron. Quantification of γ‐tubulin intensity. EGFP (−) = 12 cells, EGFP (+) (CAMDI‐sh) = 12 cells. *P < 0.05; one‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM. Scale bar, 5 μm.
- Reduced γ‐tubulin in the centrosome fraction of CAMDI‐KO brain. The centrosomal fraction was isolated from brain lysates of WT and CAMDI‐KO mice and γ‐tubulin level was assessed by immunoblot analysis. n = 3 independent experiments. **P < 0.01; one‐way ANOVA followed with Bonferroni's post hoc test. Data are presented as mean ± SEM.
HDAC6 inhibitor rescues delayed cortical migration and psychiatric behaviors
We hypothesized that HDAC6 overactivation results in the abnormal cortical migration and psychiatric behaviors of CAMDI‐KO mice, raising the possibility that the CAMDI‐KO phenotype can be rescued by treatment with HDAC6 inhibitors. Accordingly, we used Tubastatin A, a specific HDAC6 inhibitor 21. Immunohistochemical analysis revealed that delayed cortical migration in the CAMDI‐KO somatosensory cortex was rescued by a prenatal injection of Tubastatin A (Fig 6A and B). As expected, immunoblot analysis revealed enhanced α‐tubulin acetylation by Tubastatin A (Fig 6C and D). Although HDAC6 has been shown to be involved in cell proliferation 22, there was no significant difference in the number of Cux1‐positive neurons after treatment with or without Tubastatin A (Fig EV4E), suggesting that the rescue of delayed migration by Tubastatin A was not due to the change in cell proliferation. Juvenile CAMDI‐KO mice at P21 also exhibited psychiatric behaviors including hyperactivity, repetitive jumping, and decreased grooming time. These psychiatric behaviors were significantly rescued by Tubastatin A injection into pregnant CAMDI‐KO mice from E12.5–17.5 (Fig 7A–D). Similarly, abnormal behaviors of adult CAMDI‐KO mice were also rescued by prenatal Tubastatin A treatment (Fig 7E and F). However, the time spent sniffing was not altered by Tubastatin A treatment (Fig EV4F). These results suggest that Tubastatin A can improve hyperactivity, impulsive behavior, and diminished environmental adaptation, but not social behavior. The results collectively indicate that HDAC6 overactivation by CAMDI deletion causes psychiatric behaviors at least in part through delayed radial migration due to centrosome impairment.
Figure 6. Rescue of abnormal migration by HDAC6 inhibition.
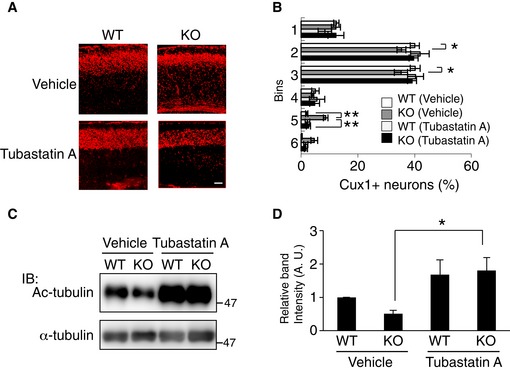
- Rescue of a delayed cortical migration in CAMDI‐KO mice by Tubastatin A. Immunohistochemical analysis of Cux1 expression in the somatosensory cortex at P2 after intraperitoneal injections of Tubastatin A into pregnant CAMDI‐KO mice from E12.5–17.5. Scale bar, 100 μm.
- Quantification of the number of Cux1‐positive neurons. Note the abnormal distribution of neurons in deep cortical layers of CAMDI‐KO mice. n = 3 mice/genotype (WT (vehicle) = 3,628 cells, KO (vehicle) = 2,816 cells, WT (Tubastatin A) = 1,847 cells, KO (Tubastatin A) = 2,364 cells). *P < 0.05, **P < 0.01; two‐way ANOVA followed by Tukey HSD test. Data are presented as mean ± SEM.
- Enhanced α‐tubulin acetylation at P0 by Tubastatin A. Ac‐tubulin level was assessed by immunoblot analysis.
- Quantification of the α‐tubulin acetylation. n = 3 independent experiments. *P < 0.05; two‐way ANOVA followed by Scheffe's post hoc test. Data are presented as mean ± SEM.
Figure 7. Rescue of psychiatric behaviors by HDAC6 inhibition.
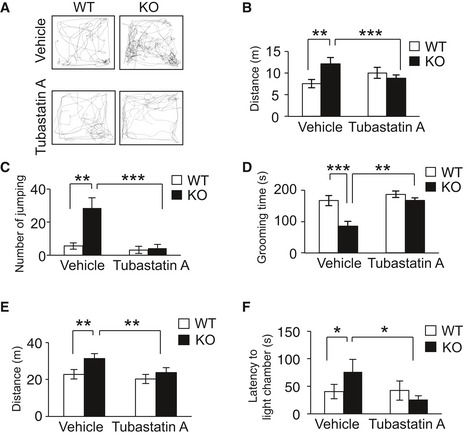
- Representative traces from control (WT) and CAMDI‐KO (KO) mice in the open‐field test at P21. Sample traces of locomotor activity were shown.
- Distance traveled for 10 min. n = 13 for WT (vehicle) mice, n = 13 for KO (vehicle) mice, n = 5 for WT (Tubastatin A) mice, and n = 16 for KO (Tubastatin A) mice. **P < 0.01, ***P < 0.001. Two‐way ANOVA followed by Scheffe's post hoc test (main effect of genotype F(1, 43) = 6.25, main effect of drug F(1, 43) = 2.14, interaction F(1, 43) = 12.68). Data are presented as mean ± SEM.
- Repetitive jumping behavior. n = 13 for WT (vehicle) mice, n = 27 for KO (vehicle) mice, n = 5 for WT (Tubastatin A) mice, and n = 18 for KO (Tubastatin A) mice. **P < 0.01, ***P < 0.001. Two‐way ANOVA followed by Scheffe's post hoc test (main effect of genotype F(1, 59) = 5.04, main effect of drug F(1, 59) = 10.13, interaction F(1, 59) = 4.98). Data are presented as mean ± SEM.
- Grooming time in small open‐field test. n = 13 for WT (vehicle) mice, n = 13 for KO (vehicle) mice, n = 5 for WT (Tubastatin A) mice, and n = 16 for KO (Tubastatin A) mice. **P < 0.01, ***P < 0.001. Two‐way ANOVA followed by Scheffe's post hoc test (main effect of genotype F(1, 43) = 11.18, main effect of drug F(1, 43) = 7.23, interaction F(1, 43) = 9.34). Data are presented as mean ± SEM.
- Distance traveled for 15 min. n = 10 for adult WT (vehicle) mice, n = 5 for KO (vehicle) mice, n = 6 for WT (Tubastatin A) mice, and n = 11 for KO (Tubastatin A) mice. **P < 0.01; two‐way ANOVA followed by Scheffe's post hoc test (main effect of genotype F(1, 28) = 1.73, main effect of drug F(1, 28) = 3.96, interaction F(1, 28) = 11.80). Data are presented as mean ± SEM.
- Light–dark test. Time in latency to the light chamber (latency). n = 10 for adult WT (vehicle) mice, n = 5 for KO (vehicle) mice, n = 6 for WT (Tubastatin A) mice, and n = 11 for KO (Tubastatin A) mice. *P < 0.05; two‐way ANOVA followed by Scheffe's post hoc test (main effect of genotype F(1, 28) = 2.34, main effect of drug F(1, 28) = 3.82, interaction F(1, 28) = 10.64). Data are presented as mean ± SEM.
Discussion
We previously reported that the rapid downregulation of CAMDI by in utero electroporation impairs radial migration with disorientated centrosome. However, it remained unknown whether CAMDI is essential for neuronal migration in vivo, because losses of target genes can often be compensated by other genes in knockout mice. For instance, DISC1‐KO mice exhibit no significant delay in radial migration, suggesting a compensatory mechanism in these mice 23.
In the present study, radial migration was delayed in CAMDI‐KO mice, although the extent of the delay of neuronal migration by CAMDI knockout was much milder than that induced by CAMDI knockdown, suggesting insufficient compensation in CAMDI‐KO mice (Fig 1A–D). As a result of delayed migration, an aberrant neural circuit formation was observed in CAMDI‐KO cerebrum (Fig 1E). Indeed, corpus callosum thinning was observed in patients with ASD 24, 25, suggesting that long‐distance disconnection is a pathophysiological state in ASD 26, 27. Therefore, the results indicate CAMDI plays a critical role in neuronal migration in vivo.
DISC1 has been shown to be involved in neuronal migration via centrosome regulation 9, 28, 29, 30. Indeed, cortical architecture is disrupted in in vivo analyses of DISC1 mutant mice and when DISC1 or DISC1‐associated proteins are downregulated by shRNA 31, 32, 33, 34. On the other hand, postmortem studies on the brains of patients with schizophrenia or autism as well as in mice with the responsible genes knocked out demonstrate abnormal neuronal migration 35, 36, 37, 38, 39, 40, indicating a causal relationship between psychiatric disorders and aberrant neuronal migration. In the present study, CAMDI‐KO mice exhibited psychiatric behaviors including hyperactivity, repetitive behavior, anxiety, and social abnormality. The psychiatric behaviors in CAMDI‐KO mice are considered to be at least in part due to delayed radial migration. To examine whether CAMDI‐KO mice show few other brain phenotypes relevant to psychiatric disorders, we performed qRT–PCR to address the expression levels of TNF‐α, IL‐1β, IL‐6 (inflammation related molecules), and myelin basic protein (oligodendrocyte marker), and assessed the number of parvalbumin‐positive cells. The TNF‐α expression was increased in CAMDI‐KO mice, but other parameters were comparable to those of WT mice. (Fig EV5). Hence, we conclude CAMDI‐KO mice are suitable model for studying the molecular mechanisms underlying psychiatric disorders induced by aberrant neuronal migration.
Figure EV5. Analysis of biological alterations in CAMDI‐KO brain relevant to psychiatric disorders.
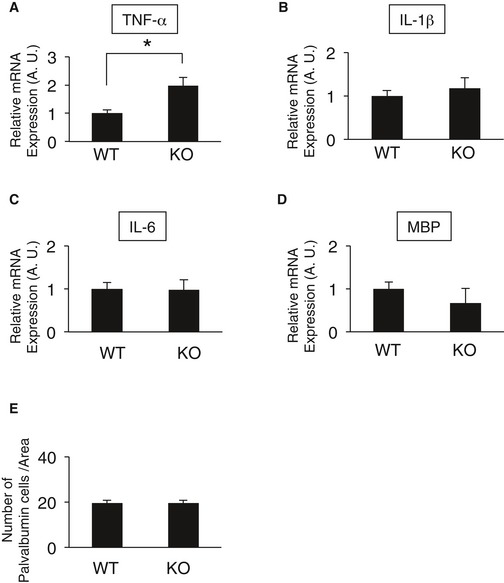
- Real‐time PCR quantification of the transcript for TNF‐α. n = 3 mice/genotype. *P < 0.05; one‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM.
- Real‐time PCR quantification of the transcript for IL‐1β. n = 3 mice/genotype. One‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM.
- Real‐time PCR quantification of the transcript for IL‐6. n = 3 mice/genotype. One‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM.
- Real‐time PCR quantification of the transcript for MBP (myelin basic protein). n = 3 mice/genotype. One‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM.
- Quantification of the number of Parvalbumin‐positive neurons. Note the abnormal distribution of neurons in deep cortical layers of CAMDI‐KO mice. n = 3 mice/genotype (WT = 816 cells, KO = 438 cells). One‐way ANOVA with Bonferroni's post hoc test. Data are presented as mean ± SEM.
During radial migration, the centrosome rapidly moves toward the cortical plate and away from the nucleus in the leading process; it is subsequently immobilized by the microtubules and actomyosin cytoskeleton, which can support subsequent nuclear movement 41, 42, 43. Therefore, centrosome stability may be regulated in conjunction with neuronal migration, possibly by a similar mechanism responsible for centrosome regulation in proliferating cells. Centrosomes comprise two orthogonally arranged centrioles surrounded by the pericentriolar material, which contains proteins responsible for microtubule nucleation and anchoring, including γ‐tubulin and pericentrin. Interestingly, acetylated α‐tubulin is localized at the centrosome in mammalian cells 44, 45, 46. The expression of K40A α‐tubulin mutant, a dominant‐negative α‐tubulin form that cannot be acetylated, disrupts redial migration during development 47, suggesting that disrupted migration is partially due to centrosome impairment by this mutant. Therefore, acetylated α‐tubulin is likely required not only for microtubule stabilization, but also for centrosome function. Furthermore, a microtubule‐associated α‐tubulin deacetylase, HDAC6, localizes at the centrosome 48, 49, 50, 51, suggesting that HDAC6 is a key enzyme that regulates centrosome function via the deacetylation of acetylated α‐tubulin and that HDAC6 inhibition at the centrosome is a pivotal step for stabilizing the centrosome. However, the regulatory mechanism underlying HDAC6 activity in the centrosome is unclear. The present study demonstrates for the first time that CAMDI inhibits HDAC6 in the centrosome and that HDAC6 inhibitor restores migration delayed by CAMDI deletion. Hence, CAMDI regulates radial migration via centrosome stability by HDAC6 inhibition. DISC1 has also been shown to accumulate in the centrosome and regulate centrosomal function as well as neuronal migration. Since we previously suggested that CAMDI translocates to the centrosome in a DISC1‐dependent manner, it is possible that CAMDI and DISC1 coordinately regulate the centrosome stability during cortical migration.
As no fundamental therapies are available, psychiatric disorders are mainly treated by symptom‐targeting therapy using drugs to correct imbalances in neurotransmitter levels (e.g., dopamine and serotonin). In the present study, we identified HDAC6 as a target molecule of CAMDI and found that microtubule stability is increased by HDAC6 suppression in vivo. Furthermore, treatment of CAMDI‐KO fetuses with the HDAC6‐specific inhibitor Tubastatin A, which is reported to prevent neuronal cell death 21, rescued abnormal psychiatric behaviors such as hyperactivity and impaired adjustment to a new environment. Tubastatin A could rescue some behaviors, but not all behavioral defects such as social behaviors in CAMDI‐KO mice. In this study, Tubastatin A was administered to pregnant mice only during E12.5–E17.5. Since social behaviors are generally acquired through various experiences after birth, they may not be rescued by recovered migration during the prenatal period. If Tubastatin A was administered continuously after birth, all behaviors may have been rescued. Alternatively, besides HDAC6 inhibition, CAMDI may have other function(s) such as dendrite formation and spine maturation in the adult stage, because CAMDI interacts with myosin regulatory light chain 2a. If so, not all behaviors might be rescued even if Tubastatin A was administered continuously after birth. Taken together, HDAC6 inhibitors may be effective for some psychiatric diseases caused by centrosome dysfunction. Moreover, the results suggest HDAC6 dysregulation causes abnormal neuronal development and psychiatric diseases. Therefore, we propose normalization of centrosome impairment by HDAC6 inhibition as a therapeutic strategy for psychiatric disorders on the basis of a new perspective of their pathogeneses.
Materials and Methods
Mice
CAMDI flox/flox mice (Accession No. CDB0658K: http://www.cdb.riken.jp/arg/mutant%20mice%20list.html) were crossed with CAG‐Cre mice 52. The recombination occurs in all over the body. The CAMDI‐knockout mice are viable and fertile. The CAMDI‐KO mice were backcrossed for at least 10 generations onto C57B/6N mice (Japan SLC). The day of vaginal plug detection was designated as E0.5 and the day of birth as P0. Genotype was confirmed by tail tipping mice at around 28 days. Mice were genotyped for knockout mice using PCR primers F1, 5′‐GTG ATT CTC AGA GAG GCA GCG CTT GGA G‐3′; F2, 5′‐GCC CCA GAT TCT TGT GGC GAA GAC TGA C‐3′; and R, 5′‐GGC CAG CTC ATT CCT CCC ACT CAT GAT C‐3′. All animals were maintained under the university guidelines for the care and use of animals. The experiments were performed after securing Tokyo University of Pharmacy and Life Sciences Animal Use Committee Protocol approval. In pharmacological experiments, mice received intraperitoneal injections with either cone oil or the Tubastatin A (25mg/kg body weight, Active Biochem) daily from E12.5 through E17.5 53, 54.
Southern blotting
Genomic DNA was digested with AflII and hybridized with a 762‐bp‐long probe by PCR using the primer, forward, 5′‐GCACCAATCCCTTGGTCACAGG and reverse, 5′‐GGCAGGAACCAGCAGGAGATGC.
Antibodies
Anti‐CAMDI antibody was described previously 12. Anti‐FLAG M2 monoclonal, anti‐acetyl‐tubulin, anti‐α‐tubulin, anti‐actin, and anti‐γ‐tubulin monoclonal antibodies were obtained from Sigma‐Aldrich. Anti‐HA high affinity antibody was obtained from Roche Applied Science. Anti‐GFP rabbit polyclonal antibody and secondary antibodies conjugated with Alexa Fluor 350, 488, and 594 were obtained from Invitrogen. Anti‐GFP mouse monoclonal and polyclonal antibodies were purchased from Clontech. Anti‐Cux1 (CDP) rabbit polyclonal antibody was obtained from Santa Cruz Biotechnology. Anti‐HDAC6 rabbit monoclonal antibody, anti‐TBR2 rabbit polyclonal antibody, and anti‐CTIP2 rat polyclonal antibody were obtained from Abcam. Anti‐pHH3 rabbit polyclonal antibody was obtained from Merck Millipore. Anti‐BrdU mouse monoclonal antibody was obtained from DAKO. Anti‐HSP90 and anti‐Ac‐HSP90 rabbit polyclonal antibodies were obtained from Rockland. Hoechst 33258 was obtained from Nacalai Tesque.
Plasmids
The rat full‐length CAMDI was previously described 12. The mouse HDAC6 was derived from mouse brain total RNA by RT–PCR and cloned to pCDNA3.1 plasmid. CAMDI‐sh plasmid was established previously 12. HDAC6 shRNA target sequences are shown below (with an order of sense, loop (underlined), antisense): 5′‐gccaaggatataccatcaatttcaagagaattgatggtatatccttggc‐3′.
Yeast two‐hybrid screening
The Matchmaker Two‐Hybrid System kit (Clontech) was used for detecting specific proteins interacting with CAMDI, according to the information supplied by the manufacturer.
Cell culture and transfections
SH‐SY5Y and HeLa cells were obtained from the American Type Culture Collection. The cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum at 37°C, in 5% CO2, in a humidified chamber. Transfection was carried out using Lipofectamine 2000 (Invitrogen).
Centrosome isolation
Centrosomes were isolated from cells as previously described 55. We obtained a single cell suspension from papain‐treated P0 brain. This preparation included treatment of cells with microtubule‐ and actin filament‐disrupting reagents (10 μg/ml nocodazole, 5 μg/ml cytochalasin D for 60 min) prior to lysis at low ionic strength in the presence of a neutral detergent. Centrosomes are harvested by centrifugation through a discontinuous (70, 50 and 40%) sucrose gradient.
Immunoprecipitation and western blotting
Culture cells were lysed in Nonidet P‐40 lysis buffer (20 mM Tris–HCl, pH 7.2, 2 mM EDTA, 0.5% Nonidet P‐40, 8% sucrose, 80 mM dithiothreitol). The lysate was clarified by centrifugation at 15,000 g for 10 min and immunoprecipitated with the appropriate antibody. Immunoprecipitates were washed three times with lysis buffer. After boiling for 3 min, equal protein amounts of the lysates were subjected to SDS–PAGE and transferred to polyvinylidene difluoride membranes (Immobilon P; Millipore). Membranes were blocked for 1 h at room temperature in 5% skim milk in PBST with gentle shaking and incubated with primary antibodies overnight at 4°C. After washing the membranes three times with PBST, they were incubated with secondary antibody conjugated to horseradish peroxidase for 1 h at room temperature. The blotted membranes were developed using the Immobilon Western Chemiluminescent HRP Substrate (Millipore) according to the manufacturer's instructions.
Immunofluorescence
Cells were fixed for 20 min in PBS containing 4% paraformaldehyde or cold methanol and permeabilized with 0.2% Triton X‐100. After incubation in PBS containing 1% bovine serum albumin for 30 min, the cells were reacted with first antibody for overnight at 4°C, followed by incubation with secondary antibody. The staining was analyzed by confocal microscope (Olympus FV1000‐D). The images were analyzed using ImageJ (National Institutes of Health) software. The intensity of acetylated α‐tubulin and γ‐tubulin in the centrosome was analyzed by measuring the intensity of a circle around the centrosome with a fixed area of 32 pixels. The centrosomal region was determined by the pericentrin co‐staining. Background value is measured from same‐sized circle as a circle including the centrosome in an adjacent region.
Immunohistochemistry
For immunohistochemical analysis, brains were fixed by immersion in 4% paraformaldehyde in 0.1 M PBST (phosphate‐buffered saline containing 0.1% Tween‐20), cryoprotected in 20% sucrose, frozen in OCT compound, and stored at −80°C until sectioning. Coronal sections (20 μm) were cut with a cryostat and stored at −20°C prior to use. Tissue sections were blocked for 1 h at room temperature in PBS containing 5% horse serum and then incubated overnight at 4°C with primary antibody. For analysis of neuronal morphology and dendrite spine number in vivo, three‐dimensional reconstructions of each EGFP‐positive neuron were produced by Z‐series stacks of confocal images. The projection images were automatically traced with ImageJ software (National Institutes of Health).
In utero electroporation
In utero electroporation was previously described 12. Briefly, in utero electroporation to dorsal neocortex was performed by injecting the DNA plasmid solution (5 mg/ml) plus 1% Fast Green using a glass capillary into the E14.5 mouse ventricle. DNA mixture was 2–3‐fold higher than that of the EGFP plasmid, which was the electroporation marker. Electroporation was performed using a CUY‐21 electroporator (NEPA GENE) and the following parameters: four 50‐ms‐long pulse separated by 950‐ms‐long intervals at 33 V.
Behavioral studies
Male mice between the ages of 8–10 weeks and P21 (juvenile test) were used for behavior studies. Mice were tested in the wire hang, food hidden, open field, novel cage, marble burying, hot plate, three‐chamber social interaction, light–dark test, resident–intruder test, and forced swim test. The experimenters were blind of the genotype of the tested animals for data collection and analyses. The number of mice in each behavioral test was different because different groups of mice were used for the behavioral tests.
Cage‐top hang test
The strength of the motor units was examined in a basic cage‐top hang test. Mice at 8 weeks of age were placed on a wire cage lid, which was then inverted that the mouse gripped the wire. Mice are subjected to a 60 s.
Olfactory test
Mice were chow food deprived for 24 h. A piece of biscuit was placed in a randomly chosen area on the cage floor with clean bedding. The experimenter manually scored the latency to find the biscuit was recorded during 15 min.
Open‐field test
Locomotor activity was measured using an open‐field test (40 × 40 × 25 cm). Mice were placed in the center of apparatus, and mouse movements were recorded with a video camera for 15 min: total distance traveled, vertical activity, time spent in the center (20 × 20 cm), and the stereotyped jumping behaviors. The arena was cleaned with 70% ethanol solution between the subjects.
Marble burying
Empty home cages were filled with 5 cm of bedding and 9 black glass marbles were evenly positioned in the cage. Mice were placed in the cage freely for 15 min, and the number of marbles buried was counted. A marble was defined as “buried” by < 25% of the marble visible.
Hot plate sensitivity
The hot plate sensitivity was conducted as described previously 56. Mice were placed on a black, anodized, constant temperature plate of 55°C (HP‐4530, As One). Latency to lick any paw was measured. The cutoff time was set at 30 s.
Three‐chamber social interaction test
A subject mouse was placed into the middle chamber of the divided (45 × 45 × 30 cm) apparatus. An empty, inverted wire cup was placed in each of the two outer compartments. After habituation, a mouse was placed in the central chamber of the apparatus and was given the choice to interact with either an empty wire cup (located in one side chamber) or a similar wire cup with an unfamiliar C57BL/6N mouse inside (located in the opposite chamber) for 10 min. The time spent in each compartment was collected in video file and sniffing time was measured.
Light–dark test
Mice were placed into the dark side of a two‐chambered apparatus allowed to move freely explore for 5 min between the darkened and lighted chambers. The score behaviors comprised the latency to enter the lighted chamber, time spent in the light chamber, and number of crossings between lighted and darkened chambers.
Resident–intruder test
Social response to unfamiliar mouse (intruder) was assessed by the resident–intruder test. Wild‐type and CAMDI‐KO mice were housed individually for 2 weeks before the test to establish dominance in their home cage. C57BL/6N mice were served as intruder. In the test, an intruder mouse was introduced into the home cage of a test mouse (resident). For 10 min, behaviors of the resident mouse were video‐recorded and examined in terms of the time spent in investigation of digging.
Forced swim test
Mice were gently placed in 2‐l glass beakers filled with tap water (25°C) to a depth at which the tail could not contact the bottom of the beaker, and they remained in the beakers for 5 min (pretest). After 24 h, test was performed (posttest). The duration of immobility, which was defined as floating in an upright position without additional activity other than that necessary for the animal to keep its head above water, was recorded. Mice were videotaped and scored manually by an observer blind to genotype. The total time spent immobile was measured by the observer.
Juvenile test
Mice at age P21 were placed in a cage on self‐directed behaviors, After 10‐min habituation, its behavior was video‐recorded for 10 min. We recorded the movement distance, repetitive jumping, and the time spent self‐grooming.
Quantitative reverse transcriptase–PCR
Total RNA was extracted from adult brain by using RNeasy kit (Qiagen). For RT–PCR, total RNA was reverse‐transcribed to cDNA by using ReverTra Ace qPCR RT Kit (TOYOBO) following the manufacturer's protocol. PCR was performed using a THUNDERBIRD SYBR qPCR Mix (TOYOBO). The following program was used for RT–PCR: 95°C for 1 min followed by 40 cycles at 95°C for 15 s, 58°C for 30 s, 72°C for 40 s. The following primers were used: TNF‐α, sense, 5′‐GACCCTCACACTCAGATCTTCT‐3′, antisense, 5′‐CCACTTGGTGGTTTGCTAGCA‐3′; IL‐1β, sense, 5′‐GGACCCATATGAGCTGAAAGC‐3′, antisense, 5′‐TCGTGGCTTGGTTCTCCTTGT‐3′; IL‐6, sense, 5′‐TGGAGTCACaGAAGGAGTGGCTAAG‐3′, antisense, 5′‐TCTGACCACAGTGAGGAATGTCAA‐3′; myelin binding protein (MBP), sense, 5′‐CCTGCCCCAGAAGTCGC‐3′, antisense, 5′‐CTTGGGATGGAGGTGGTGTTC‐3′; and GAPDH, sense, 5′‐AACTTTGGCATTGTGGAAGG‐3′, antisense, 5′‐GGATGCAGGGATGATGTTCT‐3′.
Statistical analyses
All results are expressed as mean ± SEM. For cell quantifications and behavioral tests, either one‐way ANOVA with Bonferroni's post hoc test or two‐way ANOVA with repeated measures followed by Scheffe's post hoc test or Tukey HSD test was used.
Author contributions
TF and SY designed and carried out the experiments. TF analyzed the data. TA and HK generated CAMDI flox/flox mice. SN and RI supported to establish CAMDI‐KO mice. TF and SY wrote the manuscript with contribution from RI.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Kana Yanagisawa, Hiroko Yamazaki, Katsuya Saito, and Tomohiko Takahashi for technical assistance. This study was supported in part by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology and the Japan Society for the Promotion of Science (to T.F, S.N, R.I, and S.Y) and MEXT‐Supported Program for the Strategic Research Foundation at Private Universities (to S.N and S.Y).
EMBO Reports (2016) 17: 1785–1798
See also: SC Borrie & C Bagni (December 2016)
Contributor Information
Toshifumi Fukuda, Email: tfukuda@toyaku.ac.jp.
Shigeru Yanagi, Email: syanagi@toyaku.ac.jp.
References
- 1. Higginbotham HR, Gleeson JG (2007) The centrosome in neuronal development. Trends Neurosci 30: 276–283 [DOI] [PubMed] [Google Scholar]
- 2. Kuijpers M, Hoogenraad CC (2011) Centrosomes, microtubules and neuronal development. Mol Cell Neurosci 48: 349–358 [DOI] [PubMed] [Google Scholar]
- 3. Manzini MC, Walsh CA (2011) What disorders of cortical development tell us about the cortex: one plus one does not always make two. Curr Opin Genet Dev 21: 333–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reiner O, Sapir T (2009) Polarity regulation in migrating neurons in the cortex. Mol Neurobiol 40: 1–14 [DOI] [PubMed] [Google Scholar]
- 5. Millar JK, Wilson‐Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, Devon RS, Clair DM, Muir WJ, Blackwood DH et al (2000) Disruption of two novel genes by a translocation co‐segregating with schizophrenia. Hum Mol Genet 9: 1415–1423 [DOI] [PubMed] [Google Scholar]
- 6. McKenney RJ, Vershinin M, Kunwar A, Vallee RB, Gross SP (2010) LIS1 and NudE induce a persistent dynein force‐producing state. Cell 141: 304–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hirotsune S (2008) [Molecular mechanism of lissencephaly–how LIS1 and NDEL1 regulate cytoplasmic dynein?]. Brain Nerve 60: 375–381 [PubMed] [Google Scholar]
- 8. Wynshaw‐Boris A, Gambello MJ (2001) LIS1 and dynein motor function in neuronal migration and development. Genes Dev 15: 639–651 [DOI] [PubMed] [Google Scholar]
- 9. Kamiya A, Kubo K, Tomoda T, Takaki M, Youn R, Ozeki Y, Sawamura N, Park U, Kudo C, Okawa M et al (2005) A schizophrenia‐associated mutation of DISC1 perturbs cerebral cortex development. Nat Cell Biol 7: 1167–1178 [DOI] [PubMed] [Google Scholar]
- 10. Bradshaw NJ, Ogawa F, Antolin‐Fontes B, Chubb JE, Carlyle BC, Christie S, Claessens A, Porteous DJ, Millar JK (2008) DISC1, PDE4B, and NDE1 at the centrosome and synapse. Biochem Biophys Res Commun 377: 1091–1096 [DOI] [PubMed] [Google Scholar]
- 11. Shimizu S, Matsuzaki S, Hattori T, Kumamoto N, Miyoshi K, Katayama T, Tohyama M (2008) DISC1‐kendrin interaction is involved in centrosomal microtubule network formation. Biochem Biophys Res Commun 377: 1051–1056 [DOI] [PubMed] [Google Scholar]
- 12. Fukuda T, Sugita S, Inatome R, Yanagi S (2010) CAMDI, a novel disrupted in Schizophrenia 1 (DISC1)‐binding protein, is required for radial migration. J Biol Chem 285: 40554–40561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shao Y, Raiford KL, Wolpert CM, Cope HA, Ravan SA, Ashley‐Koch AA, Abramson RK, Wright HH, DeLong RG, Gilbert JR et al (2002) Phenotypic homogeneity provides increased support for linkage on chromosome 2 in autistic disorder. Am J Hum Genet 70: 1058–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buxbaum JD, Silverman JM, Smith CJ, Kilifarski M, Reichert J, Hollander E, Lawlor BA, Fitzgerald M, Greenberg DA, Davis KL (2001) Evidence for a susceptibility gene for autism on chromosome 2 and for genetic heterogeneity. Am J Hum Genet 68: 1514–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Manolakos E, Vetro A, Kefalas K, Thomaidis L, Aperis G, Sotiriou S, Kitsos G, Merkas M, Sifakis S, Papoulidis I et al (2011) Deletion 2q31.2‐q31.3 in a 4‐year‐old girl with microcephaly and severe mental retardation. Am J Med Genet A 155A: 1476–1482 [DOI] [PubMed] [Google Scholar]
- 16. Philippe A, Martinez M, Guilloud‐Bataille M, Gillberg C, Rastam M, Sponheim E, Coleman M, Zappella M, Aschauer H, Van Maldergem L et al (1999) Genome‐wide scan for autism susceptibility genes. Paris Autism Research International Sibpair Study. Hum Mol Genet 8: 805–812 [DOI] [PubMed] [Google Scholar]
- 17. Berlucchi G (2012) Frontal callosal disconnection syndromes. Cortex 48: 36–45 [DOI] [PubMed] [Google Scholar]
- 18. Fame RM, MacDonald JL, Macklis JD (2011) Development, specification, and diversity of callosal projection neurons. Trends Neurosci 34: 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thomas A, Burant A, Bui N, Graham D, Yuva‐Paylor LA, Paylor R (2009) Marble burying reflects a repetitive and perseverative behavior more than novelty‐induced anxiety. Psychopharmacology 204: 361–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP (2005) HDAC6 regulates Hsp90 acetylation and chaperone‐dependent activation of glucocorticoid receptor. Mol Cell 18: 601–607 [DOI] [PubMed] [Google Scholar]
- 21. Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP (2010) Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, Tubastatin A. J Am Chem Soc 132: 10842–10846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li Y, Zhang X, Polakiewicz RD, Yao TP, Comb MJ (2008) HDAC6 is required for epidermal growth factor‐induced beta‐catenin nuclear localization. J Biol Chem 283: 12686–12690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuroda K, Yamada S, Tanaka M, Iizuka M, Yano H, Mori D, Tsuboi D, Nishioka T, Namba T, Iizuka Y et al (2011) Behavioral alterations associated with targeted disruption of exons 2 and 3 of the Disc1 gene in the mouse. Hum Mol Genet 20: 4666–4683 [DOI] [PubMed] [Google Scholar]
- 24. Just MA, Cherkassky VL, Keller TA, Kana RK, Minshew NJ (2007) Functional and anatomical cortical underconnectivity in autism: evidence from an FMRI study of an executive function task and corpus callosum morphometry. Cereb Cortex 17: 951–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Frazier TW, Keshavan MS, Minshew NJ, Hardan AY (2012) A two‐year longitudinal MRI study of the corpus callosum in autism. J Autism Dev Disord 42: 2312–2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Courchesne E, Pierce K (2005) Why the frontal cortex in autism might be talking only to itself: local over‐connectivity but long‐distance disconnection. Curr Opin Neurobiol 15: 225–230 [DOI] [PubMed] [Google Scholar]
- 27. Geschwind DH, Levitt P (2007) Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol 17: 103–111 [DOI] [PubMed] [Google Scholar]
- 28. Steinecke A, Gampe C, Valkova C, Kaether C, Bolz J (2012) Disrupted‐in‐Schizophrenia 1 (DISC1) is necessary for the correct migration of cortical interneurons. J Neurosci 32: 738–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tomita K, Kubo K, Ishii K, Nakajima K (2011) Disrupted‐in‐Schizophrenia‐1 (Disc1) is necessary for migration of the pyramidal neurons during mouse hippocampal development. Hum Mol Genet 20: 2834–2845 [DOI] [PubMed] [Google Scholar]
- 30. Meyer KD, Morris JA (2009) Disc1 regulates granule cell migration in the developing hippocampus. Hum Mol Genet 18: 3286–3297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Okamoto M, Iguchi T, Hattori T, Matsuzaki S, Koyama Y, Taniguchi M, Komada M, Xie MJ, Yagi H, Shimizu S et al (2015) DBZ regulates cortical cell positioning and neurite development by sustaining the anterograde transport of Lis1 and DISC1 through control of Ndel1 dual‐phosphorylation. J Neurosci 35: 2942–2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Singh KK, De Rienzo G, Drane L, Mao Y, Flood Z, Madison J, Ferreira M, Bergen S, King C, Sklar P et al (2011) Common DISC1 polymorphisms disrupt Wnt/GSK3beta signaling and brain development. Neuron 72: 545–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Singh KK, Ge X, Mao Y, Drane L, Meletis K, Samuels BA, Tsai LH (2010) Dixdc1 is a critical regulator of DISC1 and embryonic cortical development. Neuron 67: 33–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kamiya A, Tomoda T, Chang J, Takaki M, Zhan C, Morita M, Cascio MB, Elashvili S, Koizumi H, Takanezawa Y et al (2006) DISC1‐NDEL1/NUDEL protein interaction, an essential component for neurite outgrowth, is modulated by genetic variations of DISC1. Hum Mol Genet 15: 3313–3323 [DOI] [PubMed] [Google Scholar]
- 35. Wegiel J, Kuchna I, Nowicki K, Imaki H, Marchi E, Ma SY, Chauhan A, Chauhan V, Bobrowicz TW, de Leon M et al (2010) The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol 119: 755–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hori K, Nagai T, Shan W, Sakamoto A, Taya S, Hashimoto R, Hayashi T, Abe M, Yamazaki M, Nakao K et al (2014) Cytoskeletal regulation by AUTS2 in neuronal migration and neuritogenesis. Cell Rep 9: 2166–2179 [DOI] [PubMed] [Google Scholar]
- 37. Insolera R, Shao W, Airik R, Hildebrandt F, Shi SH (2014) SDCCAG8 regulates pericentriolar material recruitment and neuronal migration in the developing cortex. Neuron 83: 805–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hutsler JJ, Love T, Zhang H (2007) Histological and magnetic resonance imaging assessment of cortical layering and thickness in autism spectrum disorders. Biol Psychiatry 61: 449–457 [DOI] [PubMed] [Google Scholar]
- 39. Beckmann H (1999) Developmental malformations in cerebral structures of schizophrenic patients. Eur Arch Psychiatry Clin Neurosci 249(Suppl 4): 44–47 [DOI] [PubMed] [Google Scholar]
- 40. Akbarian S, Kim JJ, Potkin SG, Hetrick WP, Bunney WE Jr, Jones EG (1996) Maldistribution of interstitial neurons in prefrontal white matter of the brains of schizophrenic patients. Arch Gen Psychiatry 53: 425–436 [DOI] [PubMed] [Google Scholar]
- 41. Kawauchi T (2015) Cellullar insights into cerebral cortical development: focusing on the locomotion mode of neuronal migration. Front Cell Neurosci 9: 394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cooper JA (2013) Cell biology in neuroscience: mechanisms of cell migration in the nervous system. J Cell Biol 202: 725–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Govek EE, Hatten ME, Van Aelst L (2011) The role of Rho GTPase proteins in CNS neuronal migration. Dev Neurobiol 71: 528–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Piperno G, LeDizet M, Chang XJ (1987) Microtubules containing acetylated alpha‐tubulin in mammalian cells in culture. J Cell Biol 104: 289–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Farina F, Gaillard J, Guerin C, Coute Y, Sillibourne J, Blanchoin L, Thery M (2016) The centrosome is an actin‐organizing centre. Nat Cell Biol 18: 65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ling H, Peng L, Seto E, Fukasawa K (2012) Suppression of centrosome duplication and amplification by deacetylases. Cell Cycle 11: 3779–3791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Creppe C, Malinouskaya L, Volvert ML, Gillard M, Close P, Malaise O, Laguesse S, Cornez I, Rahmouni S, Ormenese S et al (2009) Elongator controls the migration and differentiation of cortical neurons through acetylation of alpha‐tubulin. Cell 136: 551–564 [DOI] [PubMed] [Google Scholar]
- 48. Kim AH, Puram SV, Bilimoria PM, Ikeuchi Y, Keough S, Wong M, Rowitch D, Bonni A (2009) A centrosomal Cdc20‐APC pathway controls dendrite morphogenesis in postmitotic neurons. Cell 136: 322–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA (2007) HEF1‐dependent Aurora A activation induces disassembly of the primary cilium. Cell 129: 1351–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ran J, Yang Y, Li D, Liu M, Zhou J (2015) Deacetylation of alpha‐tubulin and cortactin is required for HDAC6 to trigger ciliary disassembly. Sci Rep 5: 12917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sanchez de Diego A, Alonso Guerrero A, Martinez AC, van Wely KH (2014) Dido3‐dependent HDAC6 targeting controls cilium size. Nat Commun 5: 3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Matsumura H, Hasuwa H, Inoue N, Ikawa M, Okabe M (2004) Lineage‐specific cell disruption in living mice by Cre‐mediated expression of diphtheria toxin A chain. Biochem Biophys Res Commun 321: 275–279 [DOI] [PubMed] [Google Scholar]
- 53. Selenica ML, Benner L, Housley SB, Manchec B, Lee DC, Nash KR, Kalin J, Bergman JA, Kozikowski A, Gordon MN et al (2014) Histone deacetylase 6 inhibition improves memory and reduces total tau levels in a mouse model of tau deposition. Alzheimers Res Ther 6: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. d'Ydewalle C, Krishnan J, Chiheb DM, Van Damme P, Irobi J, Kozikowski AP, Vanden Berghe P, Timmerman V, Robberecht W, Van Den Bosch L (2011) HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1‐induced Charcot‐Marie‐Tooth disease. Nat Med 17: 968–974 [DOI] [PubMed] [Google Scholar]
- 55. Mitchison TJ, Kirschner MW (1986) Isolation of mammalian centrosomes. Methods Enzymol 134: 261–268 [DOI] [PubMed] [Google Scholar]
- 56. Blundell J, Tabuchi K, Bolliger MF, Blaiss CA, Brose N, Liu X, Sudhof TC, Powell CM (2009) Increased anxiety‐like behavior in mice lacking the inhibitory synapse cell adhesion molecule neuroligin 2. Genes Brain Behav 8: 114–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File