

Abstract
A major metabolic aberration associated with cancer is a change in glucose metabolism. Isoform selection of the glycolytic enzyme pyruvate kinase has been implicated in the metabolic phenotype of cancer cells, and specific pyruvate kinase isoforms have been suggested to support divergent energetic and biosynthetic requirements of cells in tumors and normal tissues. PKM2 isoform expression has been closely linked to embryogenesis, tissue repair, and cancer. In contrast, forced expression of the PKM1 isoform has been associated with reduced tumor cell proliferation. Here, we discuss the role that PKM2 plays in cells and provide a historical perspective for how the study of PKM2 has contributed to understanding cancer metabolism. We also review recent studies that raise important questions with regard to the role of PKM2 in both normal and cancer cell metabolism.
Keywords: cancer metabolism, glycolysis, PKM2, pyruvate kinase
Subject Categories: Cancer, Metabolism
Glossary
- αKG
alpha‐ketoglutarate
- 3‐PG
3‐phosphoglycerate
- ACoA
acetyl coenzyme A
- ADP
adenosine diphosphate
- ATP
adenosine triphosphate
- F6P
fructose 6‐phosphate
- FBP
fructose 1,6‐bisphosphate
- FGFR1
fibroblast growth factor receptor type 1
- FH
fumarate hydratase
- G6P
glucose 6‐phosphate
- HCC
hepatocellular carcinoma
- HK
hexokinase
- IDH2
isocitrate dehydrogenase 2
- LDH
lactate dehydrogenase
- MLC2
myosin light chain 2
- PEP
phosphoenol pyruvate
- PFK
phosphofructokinase
- PGM
phosphoglycerate mutase
- PHGDH
phosphoglycerate dehydrogenase
- PKL
liver isoform of pyruvate kinase
- PKLR
liver and red blood cell pyruvate kinase
- PKM1
M1 isoform of pyruvate kinase
- PKM2
M2 isoform of pyruvate kinase
- PK
pyruvate kinase
- PKR
red blood cell isoform of pyruvate kinase
- PPP
pentose phosphate pathway
- SAICAR
succinylaminoimidazolecarboxamide ribose‐5′ phosphate
- SDH
succinate dehydrogenase
- TCA
tricarboxylic acid
Introduction
Cancer is a disease defined by uncontrolled cell proliferation. Many normal processes also involve rapid cell proliferation including embryonic development, immune responses to infection, injury repair, and cell turnover in tissues such as the intestine. While the latter are processes that are regulated by a complex genetic circuitry that often involves growth factor signaling, proliferation in the context of cancer is either independent of this circuitry or is reliant on the aberrant activation of the same pro‐growth signals that drive normal proliferation. Therefore, the challenge in treating cancer lies in being able to target abnormal proliferation while sparing normal proliferation, a feat that requires a comprehensive understanding of both the normal and malignant mechanisms that drive cell growth and proliferation.
The first time a cell‐intrinsic cause for cancer was proposed was in 1914, when in his essay, “Concerning the Origin of Malignant Tumors”, Theodor Boveri proposed a chromosomal cause for tumors 1. Approximately 10 years after the publication of Boveri's theory, Otto Warburg began to characterize the metabolism of tumors and made his seminal observations about cancer metabolism and the increased glucose fermentation exhibited by many tumors 2. Following some controversy about the mechanism responsible for the aberrant metabolism of tumors, however, the study of cancer metabolism faded to the background 3. In the meantime, in the 1970s and 1980s, various discoveries concerning the structure and function of DNA and the mechanism of transformation used by the Rous sarcoma virus culminated in the realization that mutations in cellular genes cause cancer 4. The genomic era of cancer research that followed has prompted a deeper understanding of how genetic mutations can activate the cellular signaling pathways that drive cancer growth and tumor progression. With this understanding comes an appreciation for the complexity of these processes and furthermore, the involvement of not just other cell types but also of other cell‐intrinsic factors. Evidence for the interconnectedness of oncogenic signaling and metabolic growth pathways has prompted a reemergence of cancer metabolism as a prominent area of cancer research.
On the origins of cancer metabolism: aerobic glycolysis
Otto Warburg, who, despite winning the Nobel Prize in 1931 for his work on “the respiratory enzyme” (cytochrome c oxidase) 2, is now best known for his experiments examining glucose fermentation and respiration in tumors. Warburg measured oxygen consumption (respiration) in tissue slices from rat liver, kidney, or transplanted seminal vesicle tumor and found very little difference in the respiration of tumor tissue slices compared to that of normal tissue slices. He then quantified lactate production as a measure of fermentation rate and found that under anaerobic conditions, all tissues exhibited increased production of lactate. In contrast, under aerobic conditions, the behavior of tumor tissue and normal tissue differed. While the tumor tissue continued to produce elevated levels of lactate, the normal tissue did not 5. This led to the definition of aerobic glycolysis, the fermentation of glucose even in the presence of abundant oxygen. In future work, Warburg went on to show that aerobic glycolysis is a common feature of tumors 2.
As the pathways involved in central carbon metabolism were elucidated over the subsequent decades, aerobic glycolysis in tumors was refined to involve the use of glycolytic pyruvate to generate lactate, rather than the incorporation of glycolytic pyruvate into the mitochondrial TCA cycle, a process that consumes oxygen and generates high amounts of ATP through oxidative phosphorylation. Because Warburg had also observed aerobic glycolysis in some normal proliferating tissues and noted that it was reversible in these normal tissues, he postulated that aerobic glycolysis was the consequence of irreversible damage to respiration in tumors 6. This hypothesis motivated Warburg and many of his contemporaries to search for the root cause of damaged respiration in tumors. Warburg's hypothesis, however, was not based on sound experimental evidence. Given that even Warburg himself had noted similar rates of respiration in tumors and in normal tissue, it is unlikely that tumors are caused by a general defect in respiration. In fact, Warburg's hypothesis was met with contention by some of his contemporaries, most famously Sidney Weinhouse, whose isotope tracing experiments in tumor cells provided conclusive evidence against the Warburg hypothesis 7.
In the decades that followed, the study of cancer as a genetic disease shifted to the forefront of cancer research while cancer metabolism was relegated to the background. Interestingly, in his 1976 review of the Warburg hypothesis, the main purpose of which was to underscore the evidence against it, Sidney Weinhouse made reference to some of his own research that suggested a link between the genetic and metabolic basis of cancer and that, over the course of another 50 years, would bring cancer metabolism back into mainstream cancer research 7. The review ended with a prescient observation that some liver tumors express the fetal rather than the adult isoforms of glycolytic enzymes (most notably pyruvate kinase) and that, “high aerobic glycolysis is not necessarily an intrinsic property of the cancer cell, but rather (…) a consequence of instability in the normally rigid mechanisms of gene regulation…”
PKM2 and cancer
Pyruvate kinase (PK) catalyzes the last and physiologically irreversible step in glycolysis, the conversion of phosphoenolpyruvate (PEP) to pyruvate through the transfer of a phosphate group to ADP 8 (Fig 1). In mammals, there are four PK isoforms encoded by two genes 9. The PKLR gene encodes PKL, expressed in the liver and some cells of the pancreas, intestine, and kidney, and PKR, expressed in erythrocytes. The PKM gene encodes PKM1 and PKM2 through alternative splicing of mutually exclusive exons that are identical in length but encode a 56‐amino acid region that differs at 22 residues 10 (Fig 2). Both PKM isoforms perform the same catalytic function. However, whereas PKM1 is a constitutively active tetrameric enzyme, the 22 amino acid differences in PKM2 create a fructose‐1,6‐bisphosphate (FBP) binding pocket that renders it dependent on the allosteric binding of FBP for formation of an active tetramer 11.
Figure 1. Overview of cancer metabolism.
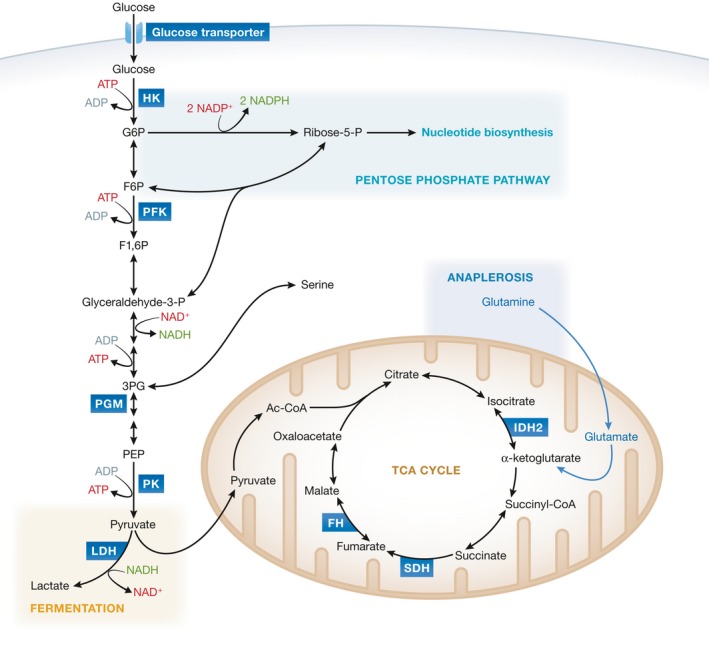
Schematic illustrating the relationship between some of the metabolic pathways altered in cancer cells. Metabolic enzymes implicated in cancer are depicted in blue boxes.
Figure 2. Pyruvate kinase: 2 genes, 4 isoforms.
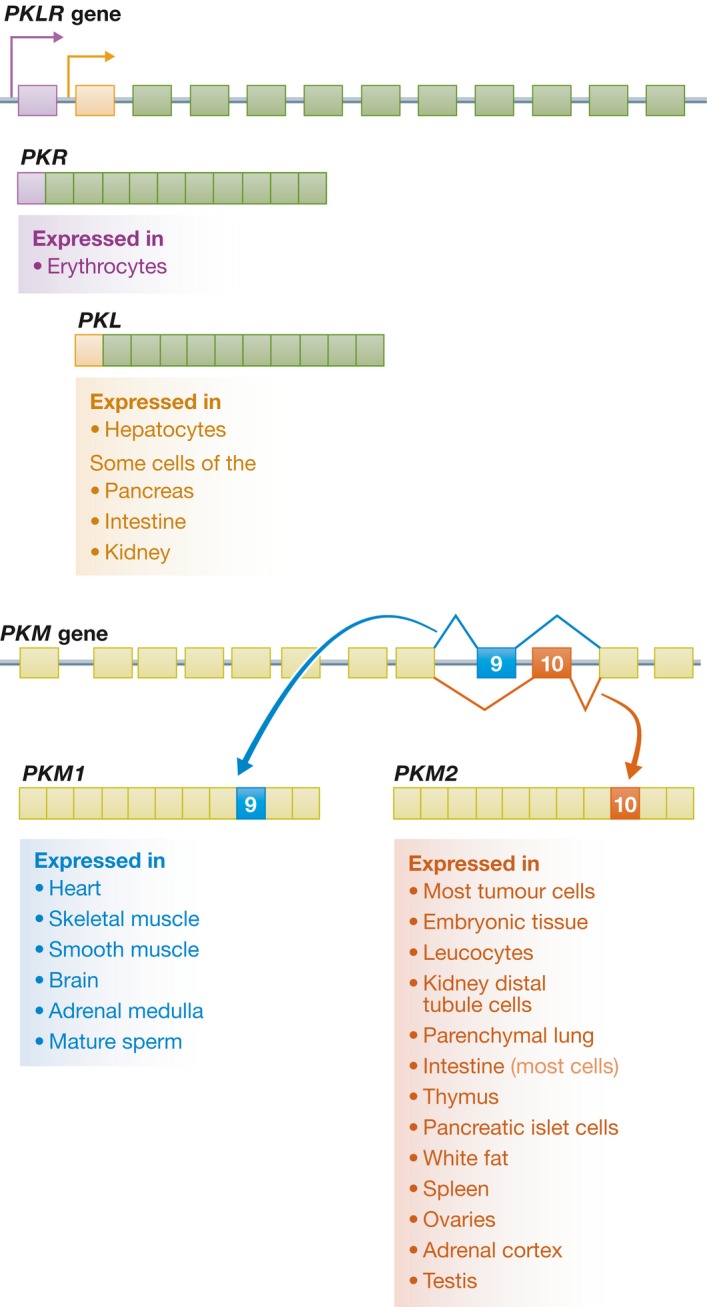
There are two pyruvate kinase genes, PKLR and PKM. PKLR encodes the PKL and PKR isoforms through tissue‐specific promoters (shown in purple for PKR and orange for PKL). PKM encodes the PKM1 and PKM2 isoforms through alternative splicing of the mutually exclusive exons 9 and 10 (shown in blue and red, respectively). Each of the pyruvate kinase isoforms shows a distinct pattern of tissue expression.
PKM2 is universally expressed during embryogenesis, regeneration, and cancer, which suggests that the ability to regulate pyruvate kinase enzymatic activity is important in actively proliferating cells 12, 13. However, early work on PKM2 as well as recent work using mouse models and analysis of RNASeq datasets shows that PKM2 is also expressed in some differentiated tissues and non‐proliferating cells 14, 15, 16, 17. PKM2 can also play an important role in maintaining the metabolic program of cancer cells. When engineered to express PKM1 in place of PKM2, cancer cells converted from aerobic glycolysis to mitochondrial respiration and were unable to form tumors after xenotransplantation 13. PKM2 enzymatic activity can also be inhibited through binding to tyrosine phosphorylated proteins, thereby linking decreased PKM2 activity and phospho‐tyrosine‐mediated growth signaling 18. While the studies of PKM2 that ensued have broadened our view of PKM2 biology and its function in maintaining tumor growth, they have also unveiled both misconceptions about PKM isoform expression patterns and controversy about putative non‐canonical roles for PKM2. Therefore, a historical perspective of PKM2 biology is informative to understand the role that this enzyme has played in the field of cancer metabolism (Fig 3).
Figure 3. A timeline of key events in the study of PKM2.
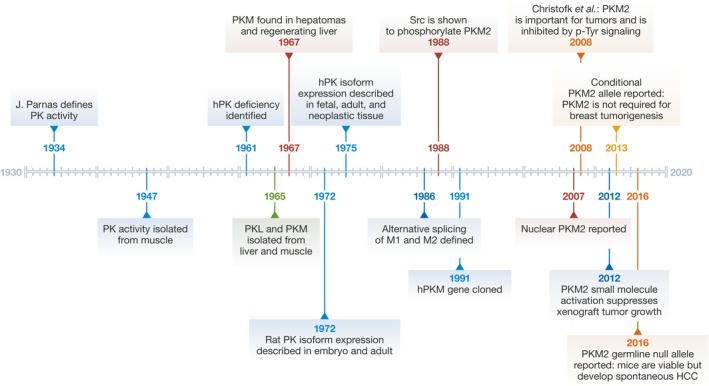
hPK, human pyruvate kinase.
Historical perspective
The glycolytic pathway was the first metabolic pathway to be elucidated, largely by sequencing known activities from cell lysates together into a series of reactions to provide a chemical route from glucose to lactate. Many of the principle discoveries that contributed to our modern view of glycolysis were made between 1927 and 1939 through the work of a number of scientists including Gustav Emden, who in 1933 provided the first cohesive outline of the pathway, in addition to Otto Meyerhof, Jacob Parnas, Dorothy Needham, Otto Warburg, Carl F. Cori, and Ulf von Euler 19. Emden's outline of the glycolytic pathway predicted much of what was discovered in the years following its publication and his sudden death that same year. One of the key advancements in understanding how the various reactions that had been described fit together was the recognition that ATP acts as an energy transfer system and one of the reactions that contributes to net ATP production in glycolysis is the reaction catalyzed by pyruvate kinase (PK), which was described for the first time by Jacob Parnas in 1934 20.
The PK enzyme was first isolated in 1947; however, it was not until the 1960s that different PK isoforms were identified 21. Although the PK isoform expressed in red blood cells (PKR) will not be discussed further in this review, it is notable that mutations in the PKR gene account for the most common cause of hereditary non‐spherocytic hemolytic anemia and it has been extensively studied in that context. PK deficiency in erythrocytes was identified in 1961 and the observation that this disease was mostly limited to erythrocytes contributed to the understanding that there were multiple PK genes 22. Aside from studies of PK deficiency in red blood cells, the majority of the initial work examining the properties of the PK enzymes was conducted in liver and muscle extracts. Initial studies in these tissues led to the identification by three separate groups of two PK isoforms, PKL in liver and PKM in muscle 23. Not long after, a PKM isoform corresponding to PKM2 was isolated from rat ascites hepatoma cells and regenerating liver 24. At the time, this isoform was equated with the muscle isoform and was referred to simply as PKM. A study the following year, however, first distinguished PKM1 and PKM2 through electrophoresis and reported their expression patterns in rat tissue as: PKM1 in brain, heart, and muscle; PKM2 in lung, spleen, testes, kidney, and a minor component in liver; and PKL in liver 25.
The last piece of the PK expression pattern puzzle was produced shortly thereafter in 1973 when two groups reported PK isoform patterns throughout development in rat liver, kidney, heart, and skeletal muscle 26. This was the first study to report expression of PKM2 in the embryo. Importantly, it was also the first study to report a shift in PK isoform expression from PKM2 in fetal heart and skeletal muscle to PKM1 in the same adult tissues. In liver and kidney, instead, the authors observed an increase in PKL accompanied by a decrease in PKM2 in the adult tissues. The transition from PKM1 in adult tissue to PKM2 in cancer was later reported (though somewhat overstated) in human tissues and tumors 27. Interestingly, the tissue types used in these studies consisted mostly of PKM1‐ or PKL‐expressing tissues. In fact, one might speculate that these studies unintentionally planted the seed that later led to confusion regarding whether or not PKM2 is normally expressed in adult tissues.
Progress in the study of PK continued with an initial focus on the differences in the enzymatic properties of the different PK isoforms. It was then that the allosteric binding of FBP to PKL and PKM2 was reported. Other allosteric effectors of each of these isoforms were also reported. The genetic study of PKM2 was made possible in 1981 when a mouse PKM2 gene was identified and localized to chromosome 9 28. Whether or not this same gene encoded both PKM1 and PKM2 was uncertain until 3 years later, when definitive evidence for this hypothesis was provided 29. In 1986, Noguchi et al 10 demonstrated that rat PKM1 and PKM2 were produced through alternative splicing of mutually exclusive exons, and soon after, the human gene was cloned and its alternative splicing to form PKM1 and PKM2 was reported 30.
Despite the widespread expression of PKM2 in several adult tissues, the majority of the subsequent studies on PKM2 were conducted in the context of tumors. In fact, studies of PKM2 expression in hepatomas, rhabdomyosarcomas, breast cancer, lung cancer, and Barrett's esophagus reported expression of PKM2 in these cancer types 27, 31, 32, 33. Importantly, for hepatomas and rhabdomyosarcomas, a switch from PKL or PKM1 in normal tissue, respectively, to PKM2 in the tumors was noted 34, 35. Thus, it was hypothesized that PKM2 must contribute to the process of malignant transformation in these tissues. Some researchers even speculated that the PKM2 expressed in tumors might have tumor‐specific properties that distinguished it from the PKM2 expressed in adult kidney and other non‐malignant tissues 33. This notion became more intriguing when reports that PKM2 could translocate to the nucleus in response to oncogenic signaling began to emerge 36, 37. The fact that PKM2 is expressed in tumors and in highly proliferating cells is striking; that PKM2 is also expressed in normal adult tissues, on the other hand, was of uncertain significance and subsequent work focused on roles PKM2 might play in cell proliferation.
PKM2 versus PKM1: Why less enzyme activity might be more permissive of proliferation
The expression patterns of PKM1 and PKM2 in normal tissue together with the switch in expression of PKL or PKM1 to PKM2 in liver tumors and rhabdomyosarcomas suggest that there are distinct functional requirements in different cell types for each of the four mammalian PK isoforms. Interestingly, the principal differences between PK isoforms can be traced to the way in which the enzymatic activity of each is regulated. Therefore, an understanding of the cellular signals that regulate one isoform but not the other is likely to provide insight into the contexts under which there might be a selective requirement for one isoform versus the other. Expression of PKL in hepatocytes provides a good example; PKL enzymatic activity is regulated by glucose, insulin, and hormonal signals, which altogether allow the liver to respond to a particular systemic energy state 38. PKL is also regulated transcriptionally by glucose and other nutrient signals 39.
With the notable exception of PKM1, which exists in a stable enzymatically active tetrameric state, all of the PK isoforms are allosterically regulated 40, 41, 42, 43, 44. Because PK enzymes are only active as tetramers, allosteric regulation is generally mediated through stabilization or destabilization of the intersubunit contacts that allow the enzyme to assemble into a homotetramer 42, 44, 45, 46. PKL, PKR, and PKM2 are activated by their substrate, PEP, and by the upstream glycolytic intermediate FBP. These PK isoforms are also inhibited by alanine and ATP 8 and both PKR and PKM2 are inhibited by the thyroid hormone T3 47. In addition, PKM2 has a number of unique allosteric effectors including, serine and SAICAR, an intermediate of de novo purine biosynthesis, which both activate PKM2 enzymatic activity 48, 49 (Fig 4). The only reported effector of PKM1 is phenylalanine, which reduces enzymatic activity through a mechanism that induces a conformational change in the tetramer and thereby reduces its affinity for PEP but otherwise maintains the tetramer formation. The same effect has also been observed for phenylalanine on PKM2 48, 50.
Figure 4. Overview of cellular signaling events that modulate PKM2 enzymatic activity.
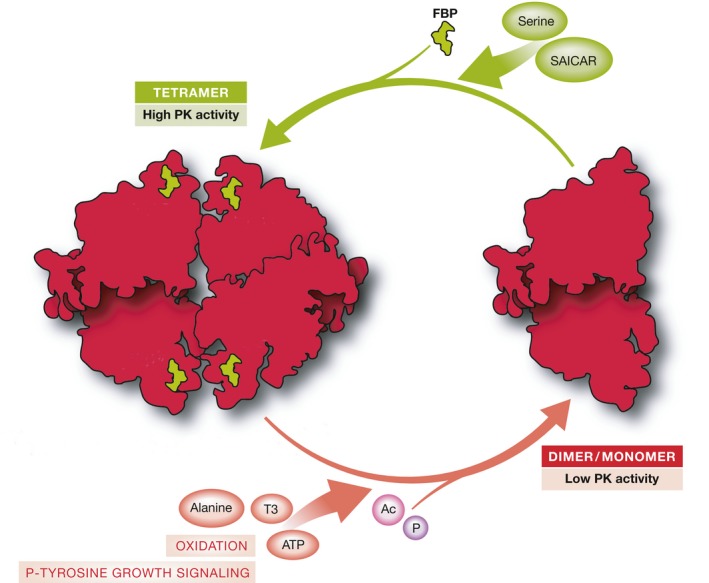
Schematic showing endogenous activators and inhibitors of PKM2 activity. PKM2 is only enzymatically active as a tetramer. Thus, allosteric regulation is achieved through stabilization or destabilization of the enzyme tetramer. PKM2 is activated by the upstream glycolytic intermediate FBP. It can also be activated by a number of unique allosteric effectors including serine and SAICAR. PKM2 activity can be inhibited by a number of endogenous inhibitors and cellular signaling events including alanine, ATP, and the thyroid hormone T3. In addition, PKM2 activity is inhibited by phospho‐tyrosine‐mediated release of FBP. Other post‐translational modifications that inhibit PKM2 activity include acetylation and oxidation.
In contrast to PKL, PKR, and PKM1, the enzymatic activity of PKM2 can be inhibited by a variety of mechanisms found in proliferating cells (Fig 4). PKM2 binds specifically to proteins that are targets of proliferation‐inducing tyrosine kinases and this binding induces the release of FBP and consequently a reduction in PKM2 enzymatic activity 18. PKM2 is also a direct substrate of these tyrosine kinases, and phosphorylation by FGFR1 on tyrosine residue 105 (Y105) can act to reduce activity of nearby PKM2 enzyme by this mechanism 51. PKM2 can also be phosphorylated by c‐Src and ERK1/2 52, 53. Acetylation of lysine 305 (K305) both reduces PKM2 activity and targets it for lysosomal‐dependent degradation 54. Finally, oxidation of cysteine residue 358 (C358) reduces PKM2 activity and renders it sensitive to the oxidative stress often associated with increased proliferation 55. Each of these mechanisms for inhibiting PKM2 activity has been shown to be important for proliferation in vitro and for the growth of xenograft tumors in mice, leading to the hypothesis that the ability to inhibit PK enzyme activity confers an advantage to proliferating cells 13, 51, 54, 55. The study of Pkm2 −/− mice supports this hypothesis and further argues that the ability to inhibit PKM2 activity is important for cells and tissues in vivo. PKM2‐expressing tissues isolated from Pkm2 −/− mice express PKM1 instead of PKM2 but have reduced levels of total PKM transcripts and display lower PK enzymatic activity than wild‐type (WT) tissues 14. This observation, in addition to the metabolic distress displayed by Pkm2 −/− mice with age, argues that changes in PK enzymatic activity levels can have profound consequences at the systemic level. Which specific regulatory events are most important in each context remains unknown.
The metabolic program of proliferating cells requires a rewiring of cellular metabolism toward increased biosynthesis and the selection for PKM2 in proliferating cells has been suggested to promote this metabolic program through reduced PK enzymatic activity. In particular, low PK enzymatic activity has been proposed to divert glycolytic intermediates toward biosynthetic pathways such as the pentose phosphate pathway (PPP) and serine biosynthesis 43, 56, 57. Indeed, small‐molecule activation of PKM2 in cells in culture leads to decreased levels of intracellular acetyl coenzyme A (ACoA), ribose phosphate, and serine; intermediates required for the production of palmitate, nucleotide biosynthesis, and proteins and lipids, respectively 46. A similar study using a different small molecular activator of PKM2 showed increased flux into the serine biosynthesis pathway without an increase in intracellular levels of serine 58. In agreement with both scenarios, decreased PKM2 activity has been shown to increase the flux of glucose to make serine 59. Altogether these data argue that modulating PKM2 enzymatic activity can impact the flux of glucose carbon into pathways downstream of pyruvate kinase itself. However, an exact mechanistic understanding of these observations is currently lacking and remains an important unanswered question for the field.
The link between PKM2 and the serine biosynthesis pathway is particularly interesting in that each has been shown independently to contribute to cancer cell growth 13, 60, 61, 62, 63, 64. As mentioned previously, serine can bind to and activate PKM2, which, based on the aforementioned experiments, could result in decreased serine biosynthesis. In turn, the increased serine biosynthesis elicited by decreased PKM2 activity could lead to a feedback loop where PKM2 is activated in response to increased serine concentrations. However, increased flux of glucose through the serine biosynthesis pathway does not necessarily imply increased dependency on serine production. In cell lines in which the serine biosynthesis pathway enzyme PHGDH is amplified, knockdown of PHGDH led to impaired proliferation that could not be rescued by serine supplementation 60. Although this observation is likely a consequence of the specific genetic context of the tested cancer cell lines, it highlights the potential and perhaps underappreciated importance of the intermediates generated by flux through a metabolic pathway as opposed to the end‐product metabolite of a pathway. Furthermore, the potential link between PKM2 enzymatic activity and serine biosynthesis underscores the interconnectedness of the metabolic network and how changes in a single node in a pathway can redistribute metabolic flux.
A comparison of the metabolic consequences of increased PKM2 enzymatic activity elicited by small molecular activators, to those elicited by forced expression of PKM1 points to the possibility that cells can adapt to the metabolic parameters imposed by chronic activation of PK enzymatic activity. The development of small‐molecule activators of PKM2 as potential therapeutic drugs for the treatment of cancer was prompted by the observation that forced expression of PKM1 in tumor cells leads to decreased tumor formation after xenotransplantation 13, 58. In fact, treatment of cancer cells with small‐molecule activators mimics the consequences of forced PKM1 expression with respect to xenotransplantation. Furthermore, decreased incorporation of glucose‐derived carbons into lipids has been observed in both settings, implying a decrease in some biosynthetic pathways. The acute PK enzymatic activation induced by small‐molecule activators, however, has been shown to differ from the chronic PK activation that results from forced expression of PKM1. Decreased levels of intracellular ACoA, ribose phosphate, and serine were observed in the context of small‐molecule PK activation but not in response to forced expression of PKM1 46. In fact, forced PKM1 expression may provoke compensatory metabolic adaptations that the transient activation of PKM2 through small‐molecule activators does not.
The notion that PKM2 expression is permissive to the metabolic requirements of proliferation is an enticing one that might explain the almost universal expression of PKM2 in rapidly proliferating cells. In addition, the fact that PKM2 enzymatic activity can be inhibited through endogenous mechanisms argues that the selection for PKM2 in proliferating cells confers metabolic flexibility by allowing PK enzymatic activity to be turned on or off in response to the environment. Expression of PKM1, on the other hand, likely renders cells less responsive to intercellular cues pertaining to energy state, nutrient availability, and growth. Given that the ability to respond to dynamic energy states is important for any cell type regardless of its proliferative state 65, it is likely that metabolic flexibility in PKM1 expressing cells is conferred at a different node in the metabolic network.
Non‐canonical functions of PKM2
In addition to its established role in metabolism, alternative non‐metabolic functions of PKM2 in tumor cells and other proliferating cells have been reported. PKM2 has been suggested to regulate gene expression by acting both as a protein transactivator along with Hif1α and β‐catenin 66, 67, 68, 69, and as a protein kinase 53, 70, 71. The studies of PKM2 acting as a protein kinase propose that, following translocation into the nucleus, PKM2 can use its metabolic substrate PEP as a phosphate donor for phosphorylation of a long list of target proteins that includes, Stat3 71, histone H3 69, myosin light chain 2 (MLC2) 72, Bub3 73, and ERK1/2 53. While each of these studies argued that the reported non‐canonical functions of PKM2 were important for tumor proliferation, recent evidence from mouse models in which genetic ablation of PKM2 did not abrogate tumor growth, argues that the requirement for PKM2 function, metabolic or not, might be context‐dependent 14, 74, 75. The phenotype of the PKM2 germline knockout mouse further calls into question the significance of non‐metabolic PKM2 functions for either normal or malignant cell proliferation 14. Furthermore, a recent study was unable to find evidence that PKM2 acts as a PEP‐dependent protein kinase and suggested that contaminating ATP‐dependent protein kinases and ADP might be responsible for the previously reported PEP‐dependent phosphorylation events attributed to PKM2 76.
Recent advances in PKM2 biology
Although there are clear lines of evidence to suggest that PKM2 expression is beneficial to the metabolic state of a proliferating cell, recent advances in the study of PKM2 have revealed a more complex relationship between PKM2 and proliferation and PKM2 and cancer than previously appreciated. The most surprising results have come from the use of a Pkm2 conditional allele (Pkm2 fl) that allows for Cre‐mediated deletion of the PKM2‐specific exon 10 74. Importantly, this Pkm2 fl allele does not abrogate expression of PKM1 and, as a result, deletion of PKM2 in this context allows for a switch in isoform expression as opposed to complete loss of the PKM protein.
The first use of this allele was reported in 2013 when the Pkm2 fl allele was crossed to a Cre‐driven autochthonous mouse model of breast cancer and it was found that deletion of PKM2 concomitant with tumor initiation accelerated tumor development. Interestingly, PKM2‐deleted breast tumors showed heterogeneous expression of PKM1; while PKM1 was expressed in non‐proliferating tumor cells, the proliferating tumor cells showed low or undetectable levels of PKM 74. While unexpected because they showed that PKM2 itself was not required for tumor growth in this model, these results are in line with the hypothesis that PKM1 expression is selected against in proliferating tumor cells. Similarly, when the Pkm2 fl allele was crossed to a leukemia model, PKM2 deletion concomitant with tumor initiation led to uniform expression of PKM1 across tumor cells and limited tumor progression 75. Further evidence to suggest that PKM1 is detrimental to proliferating cells is provided by the study of primary cells derived from Rosa26 CreER , Pkm2 fl/fl embryos. Acute deletion of PKM2 in primary cells resulted in forced expression of endogenous PKM1, which led to decreased de novo nucleotide biosynthesis and proliferative arrest 77. Notably, primary untransformed MEFs derived from embryos harboring a germline null allele of PKM2 (Pkm2 −) do not show any defects in proliferation (T.L. Dayton, unpublished observation), suggesting that the cellular response to acute as opposed to chronic absence of PKM2 is different.
This idea that acute loss of PKM2 differs from chronic absence of PKM2 might be one way to make sense of the recent observation that germline loss of PKM2 is compatible with normal embryonic development and adult organismic viability 14. Because there is a reduction in the levels of total PKM transcripts in Pkm2 −/− tissues that normally express PKM2, and because Pkm2 −/− tissues display lower PK enzymatic activity than wild‐type (WT) tissues, it is possible that the regulation of PK enzymatic activity that would normally be conferred through endogenous activators and inhibitors of the protein is, in the case of Pkm2 −/− tissues, conferred through changes in the levels of total Pkm expression.
Because PKM2 is not required for tumor formation in mouse models and is not required for embryonic development or viability of adult mice, the question arises, is there any requirement for PKM2 over PKM1? The analysis of Pkm2 −/− mice has started to answer this question but has also led to new questions. Unexpectedly, Pkm2 −/− mice develop late‐onset spontaneous hepatocellular carcinomas (HCC) accompanied by a progressive metabolic imbalance that becomes more pronounced with age and is associated with increased hepatocyte proliferation. Pkm2 −/− mice were also shown to be more sensitive to the effects of a high‐fat diet 14. Thus, while PKM2 is not required for development or overall viability, it does appear to play a key role in maintaining systemic metabolism and to the systemic response to metabolic stress associated with aging or a high‐fat diet.
It remains less clear how PKM2 expression might be contributing to other proliferative systems such as the immune system, where PKM2 is the primary isoform expressed, or adult stem cells, where in many cases, the exact PKM expression patterns have not been fully established. Because the progenitor and stem cell populations of the hematopoietic system are well defined, this proliferative system presents an interesting context for the study of PKM2. Conditional deletion of PKM2 in the hematopoietic system did not significantly affect hematopoiesis under homeostatic conditions, a finding supported by the lack of any overt hematopoietic deficiencies in Pkm2 −/− mice. Nonetheless, progenitor populations of Pkm2 −/− bone marrow displayed a competitive disadvantage following transplantation when compared to WT bone marrow 75, arguing that PKM2 might be required by hematopoietic progenitors under conditions of stress. Similarly, studies of PKM2 in different immune cell populations suggest that Pkm isoform selection plays a role in mediating the immune system response to triggers such as LPS and IL‐23 78, 79.
Summary
The study of PKM2 has contributed insight into the metabolic requirements of proliferating cells, and how changes in a single node can alter metabolic flux. These studies have also highlighted both the complexity of the metabolic network itself and the diverse mechanisms by which that network can be regulated or altered in different cellular contexts. The expression of PKM2 in cancer and other rapidly proliferating cells led to a focus on the role of PKM2 in mediating proliferative metabolism. Work showing that engineering cancer cells to express PKM1 in place of PKM2 impaired tumor formation further sparked interest in PKM2 as a potential therapeutic target for cancer treatment 13. However, subsequent work suggested this phenotype might be better explained by selection against PKM1, and the widespread expression of PKM2 in normal adult tissues implies PKM2 can play a role in cells other than supporting proliferation. Furthermore, the distinct cell‐type‐specific expression patterns of each the four PK isoforms, as well as their unique regulatory effectors, suggest non‐redundant functions of one isoform versus the other. The recent findings that PKM2 is dispensable for breast tumor formation and leukemogenesis in autochthonous mouse models together with the observation that germline loss of PKM2 is compatible with embryonic development and organismic viability underscore the potential for metabolic adaptability in response to a switch in PK isoform expression patterns 14, 74, 75. Nonetheless, two key observations: the limited progression of PKM2 null leukemias and spontaneous tumor formation in PKM2 null mice, suggest limitations to this metabolic adaptability and underscore how much we have yet to learn about PKM2 and the regulation of glucose metabolism. Importantly, these most recent findings paint a more complicated picture of the role played by PKM2 in proliferative and cancer metabolism than was suggested by earlier studies.
Conflict of interest
T.L.D. and T.J. have no conflicts to declare. M.G.V.H. is a consultant and scientific advisory board member for Agios Pharmaceuticals, a company that seeks to target pyruvate kinase for therapeutic benefit.
Sidebar A: In need of answers.
How does Pkm isoform selection (PKM2 versus PKM1) in normal adult tissue regulate systemic metabolism or the systemic response to metabolic stress?
How does decreased pyruvate kinase activity promote increased flux through pathways downstream of pyruvate kinase?
Will chronic long‐term treatment with PK activators mimic the consequences of germline loss of PKM2?
Is there a tumor or normal tissue context where PKM2 is required?
Is there a cellular sensor of PK enzymatic activity that allows for regulation at the levels of both Pkm gene expression and post‐translational modification?
What is the role of PKM1 in normal adult tissue?
EMBO Reports (2016) 17: 1721–1730
See the Glossary for abbreviations used in this article.
References
- 1. Kops G, Weaver B, Cleveland D (2005) On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer 5: 773–785 [DOI] [PubMed] [Google Scholar]
- 2. Koppenol WH, Bounds PL, Dang CV (2011) Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer 11: 325–337 [DOI] [PubMed] [Google Scholar]
- 3. Ferreira L, Hebrant A, Dumont JE (2012) Metabolic reprogramming of the tumor. Oncogene 31: 3999–4011 [DOI] [PubMed] [Google Scholar]
- 4. Ponder BA (2001) Cancer genetics. Nature 411: 336–341 [DOI] [PubMed] [Google Scholar]
- 5. Warburg O (1925) The metabolism of carcinoma cells. J Cancer Res 9: 148–163 [Google Scholar]
- 6. Warburg O (1956) On the origin of cancer cells. Science 123: 309–314 [DOI] [PubMed] [Google Scholar]
- 7. Weinhouse S (1976) The Warburg hypothesis fifty years later. Z Krebsforsch Klin Onkol 87: 115–126 [DOI] [PubMed] [Google Scholar]
- 8. Jurica M, Mesecar A, Heath PJ, Shi W, Nowak T, Stoddard BL (1998) Structure 2: 195–210 [DOI] [PubMed] [Google Scholar]
- 9. Takenaka M, Yamada K, Lu T, Kang R, Tanaka T, Noguchi T (1996) Alternative splicing of the pyruvate kinase M gene in a minigene system. Eur J Biochem 235: 366–371 [DOI] [PubMed] [Google Scholar]
- 10. Noguchi T, Inoue H, Tanaka T (1986) The M1‐ and M2‐type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J Biol Chem 261: 13807–13812 [PubMed] [Google Scholar]
- 11. Dombrauckas JD, Santarsiero BD, Mesecar AD (2005) Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry 44: 9417–9429 [DOI] [PubMed] [Google Scholar]
- 12. Garnett ME, Dyson RD, Dost FN (1974) Pyruvate kinase isozyme changes in parenchymal cells of regenerating rat liver. J Biol Chem 249: 5222–5226 [PubMed] [Google Scholar]
- 13. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC (2008) The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452: 230–233 [DOI] [PubMed] [Google Scholar]
- 14. Dayton TL, Gocheva V, Miller KM, Israelsen WJ, Bhutkar A, Clish CB, Davidson SM, Luengo A, Bronson RT, Jacks T et al (2016) Germline loss of PKM2 promotes metabolic distress and hepatocellular carcinoma. Genes Dev 30: 1020–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mazurek S (2010) Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol 43: 969–980 [DOI] [PubMed] [Google Scholar]
- 16. David CJ, Manley JL (2010) Alternative pre‐mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 24: 2343–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR (2010) The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci USA 107: 1894–1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC (2008) Pyruvate kinase M2 is a phosphotyrosine‐binding protein. Nature 452: 181–186 [DOI] [PubMed] [Google Scholar]
- 19. Cori CF (1983) Embden and the glycolytic pathway. Trends Biochem Sci 8: 257–259 [Google Scholar]
- 20. Cori CF (1974) Some highlights of the early period of bioenergetics. Mol Cell Biochem 5: 47–53 [DOI] [PubMed] [Google Scholar]
- 21. Barnett JA (2003) A history of research on yeasts 5: the fermentation pathway. Yeast 20: 509–543 [DOI] [PubMed] [Google Scholar]
- 22. Zanella A, Fermo E, Bianchi P, Valentini G (2005) Red cell pyruvate kinase deficiency: molecular and clinical aspects. Br J Haemotol 130: 11–25 [DOI] [PubMed] [Google Scholar]
- 23. Tanaka T, Harano Y, Morimura H, Mori R (1965) Evidence for the presence of two types of pyruvate kinase in rat liver. Biochem Biophys Res Commun 21: 55–60 [DOI] [PubMed] [Google Scholar]
- 24. Tanaka T, Harano Y, Sue F, Morimura H (1967) Crystallization, characterization and metabolic regulation of two types of pyruvate kinase isolated from rat tissues. J Biochem 62: 71–91 [DOI] [PubMed] [Google Scholar]
- 25. Susor WA, Rutter WJ (1968) Some distinctive properties of pyruvate kinase purified from rat liver. Biochem Biophys Res Commun 30: 14–20 [DOI] [PubMed] [Google Scholar]
- 26. Osterman J, Fritz PJ (1973) Pyruvate kinase isozymes: a comparative study in tissues of various mammalian species. Comp Biochem Physiol B 44: 1077–1085 [DOI] [PubMed] [Google Scholar]
- 27. Kamel R, Schwarzfischer F (1975) Pyruvate kinase isozyme patterns of human neoplastic, fetal and adult tissues. Humangenetik 28: 65–69 [DOI] [PubMed] [Google Scholar]
- 28. Peters J, Nash HR, Eicher EM, Bulfield G (1981) Polymorphism of kidney pyruvate kinase in the mouse is determined by a gene, Pk‐3, on chromosome 9. Biochem Genet 19: 757–769 [DOI] [PubMed] [Google Scholar]
- 29. Peters J, Andrews SJ (1984) The Pk‐3 gene determines both the heart, M1, and the kidney, M2, pyruvate kinase isozymes in the mouse; and a simple electrophoretic method for separating phosphoglucomutase‐3. Biochem Genet 22: 1047–1063 [DOI] [PubMed] [Google Scholar]
- 30. Takenaka M, Noguchi T, Sadahiro S, Hirai H, Yamada K, Matsuda T, Imai E, Tanaka T (1991) Isolation and characterization of the human pyruvate kinase M gene. Eur J Biochem 198: 101–106 [DOI] [PubMed] [Google Scholar]
- 31. Guguen‐Guillouzo C, Szajnert MF, Marie J, Delain D, Schapira F (1977) Differentiation in vivo and in vitro of pyruvate kinase isozymes in rat muscle. Biochimie 59: 65–71 [DOI] [PubMed] [Google Scholar]
- 32. Schneider J, Neu K, Velcovsky H‐G, Morr H, Eigenbrodt E (2003) Tumor M2‐pyruvate kinase in the follow‐up of inoperable lung cancer patients: a pilot study. Cancer Lett 193: 91–98 [DOI] [PubMed] [Google Scholar]
- 33. Ibsen KH, Orlando RA, Garratt KN, Hernandez AM, Giorlando S, Nungaray G (1982) Expression of multimolecular forms of pyruvate kinase in normal, benign, and malignant breast tissue. Cancer Research 42: 888–892 [PubMed] [Google Scholar]
- 34. Farina FA, Shatton JB, Morris HP, Weinhouse S (1974) Isozymes of pyruvate kinase in liver and hepatomas of the rat. Cancer Res 34: 1439–1446 [PubMed] [Google Scholar]
- 35. Farron F, Hsu HH, Knox WE (1972) Fetal‐type isoenzymes in hepatic and nonhepatic rat tumors. Cancer Res 32: 302–308 [PubMed] [Google Scholar]
- 36. Siwko S, Mochly‐Rosen D (2007) Use of a novel method to find substrates of protein kinase C delta identifies M2 pyruvate kinase. Int J Biochem Cell Biol 39: 978–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hoshino A, Hirst JA, Fujii H (2007) Regulation of cell proliferation by interleukin‐3‐induced nuclear translocation of pyruvate kinase. J Biol Chem 282: 17706–17711 [DOI] [PubMed] [Google Scholar]
- 38. Yamada K, Noguchi T (1999) Nutrient and hormonal regulation of pyruvate kinase gene expression. Biochem J 337: 1–11 [PMC free article] [PubMed] [Google Scholar]
- 39. Pilkis SJ, El‐Maghrabi MR (1988) Hormonal regulation of hepatic gluconeogenesis and glycolysis. Ann Rev Biochem 57: 755–783 [DOI] [PubMed] [Google Scholar]
- 40. Larsen TM, Laughlin LT, Holden HM, Rayment I, Reed GH (1994) Structure of rabbit muscle pyruvate kinase complexed with Mn2+, K+, and pyruvate. Biochemistry 33: 6301–6309 [DOI] [PubMed] [Google Scholar]
- 41. Ibsen KH (1977) Interrelationships and functions of the pyruvate kinase isozymes and their variant forms: a review. Cancer Res 37: 341–353 [PubMed] [Google Scholar]
- 42. Ikeda Y, Tanaka T, Noguchi T (1997) Conversion of non‐allosteric pyruvate kinase isozyme into an allosteric enzyme by a single amino acid substitution. J Biol Chem 272: 20495–20501 [DOI] [PubMed] [Google Scholar]
- 43. Israelsen WJ, Vander Heiden MG (2015) Pyruvate kinase: function, regulation and role in cancer. Semin Cell Dev Biol 43: 43–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ikeda Y, Noguchi T (1998) Allosteric regulation of pyruvate kinase M2 isozyme involves a cysteine residue in the intersubunit contact. J Biol Chem 273: 12227–12233 [DOI] [PubMed] [Google Scholar]
- 45. Yamada K, Noguchi T (1999) Regulation of pyruvate kinase M gene expression. Biochem Biophys Res Commun 256: 257–262 [DOI] [PubMed] [Google Scholar]
- 46. Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A et al (2012) Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol 8: 839–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ashizawa K, McPhie P, Lin KH, Cheng SY (1991) An in vitro novel mechanism of regulating the activity of pyruvate kinase M2 by thyroid hormone and fructose 1, 6‐bisphosphate. Biochemistry 30: 7105–7111 [DOI] [PubMed] [Google Scholar]
- 48. Morgan HP, O'Reilly FJ, Wear MA, O'Neill JR, Fothergill Gilmore LA, Hupp T, Walkinshaw MD (2013) M2 pyruvate kinase provides a mechanism for nutrient sensing and regulation of cell proliferation. Proc Natl Acad Sci USA 110: 5881–5886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Keller KE, Tan IS, Lee YS (2012) SAICAR stimulates pyruvate kinase isoform M2 and promotes cancer cell survival in glucose‐limited conditions. Science 338: 1069–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Williams R, Holyoak T, McDonald G, Gui C, Fenton A (2006) Differentiating a ligand's chemical requirements for allosteric interactions from those for protein binding. Phenylalanine inhibition of pyruvate kinase. Biochemistry 45: 5421–5429 [DOI] [PubMed] [Google Scholar]
- 51. Hitosugi T, Kang S, Vander Heiden MG, Chung T‐W, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ et al (2009) Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal 2: ra73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Presek P, Reinacher M, Eigenbrodt E (1988) Pyruvate kinase type M2 is phosphorylated at tyrosine residues in cells transformed by Rous sarcoma virus. FEBS Lett 242: 194–198 [DOI] [PubMed] [Google Scholar]
- 53. Keller KE, Doctor ZM, Dwyer ZW, Lee Y‐S (2014) SAICAR induces protein kinase activity of PKM2 that is necessary for sustained proliferative signaling of cancer cells. Mol Cell 53: 700–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y et al (2011) Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone‐mediated autophagy and promotes tumor growth. Mol Cell 42: 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang J‐K, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS et al (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334: 1278–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Eigenbrodt E, Reinacher M, Scheefers‐Borchel U, Scheefers H, Friis R (1992) Double role for pyruvate kinase type M2 in the expansion of phosphometabolite pools found in tumor cells. Crit Rev Oncog 3: 91–115 [PubMed] [Google Scholar]
- 57. Eigenbrodt E, Reinacher M, Scheefers‐Borchel U, Scheefers H, Friis R (1992) Double role for pyruvate kinase type M2 in the expansion of phosphometabolite pools found in tumor cells. Crit Rev Oncog 3: 91–115 [PubMed] [Google Scholar]
- 58. Kung C, Hixon J, Choe S, Marks K, Gross S, Murphy E, Delabarre B, Cianchetta G, Sethumadhavan S, Wang X et al (2012) Small molecule activation of PKM2 in cancer cells induces serine auxotrophy. Chem Biol 19: 1187–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chaneton B, Hillmann P, Zheng L, Martin AC, Maddocks OD, Chokkathukalam A, Coyle JE, Jankevics A, Holding FP, Vousden KH et al (2012) Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 491: 458–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK et al (2011) Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476: 346–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H et al (2011) Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet 43: 869–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kalhan SC, Hanson RW (2012) Resurgence of serine: an often neglected but indispensable amino Acid. J Biol Chem 287: 19786–19791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Maddocks ODK, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH (2013) Serine starvation induces stress and p53‐dependent metabolic remodelling in cancer cells. Nature 493: 542–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. DeNicola GM, Chen P‐H, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE et al (2015) NRF2 regulates serine biosynthesis in non‐small cell lung cancer. Nat Genet 47: 1475–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Olson KA, Schell JC, Rutter J (2016) Pyruvate and metabolic flexibility: illuminating a path toward selective cancer therapies. Trends Biochem Sci 41: 219–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Luo W, Hu H, Chang R, Zhong J, Knabel M, O'meally R, Cole RN, Pandey A, Semenza GL (2011) Pyruvate kinase M2 is a PHD3‐stimulated coactivator for hypoxia‐inducible factor 1. Cell 145: 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang H‐J, Hsieh Y‐J, Cheng W‐C, Lin C‐P, Lin Y‐S, Yang S‐F, Chen C‐C, Izumiya Y, Yu J‐S, Kung H‐J et al (2014) JMJD5 regulates PKM2 nuclear translocation and reprograms HIF‐1α‐mediated glucose metabolism. Proc Natl Acad Sci USA 111: 279–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K, Lu Z (2011) Nuclear PKM2 regulates β‐catenin transactivation upon EGFR activation. Nature 480: 118–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC, Lu Z (2012) ERK1/2‐dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol 14: 1295–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gao X, Wang H, Yang JJ, Liu X, Liu Z‐R (2012) Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell 45: 598–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gao X, Wang H, Yang JJ, Chen J, Jie J, Li L, Zhang Y, Liu Z‐R (2013) Reciprocal regulation of protein kinase and pyruvate kinase activities of pyruvate kinase M2 by growth signals. J Biol Chem 288: 15971–15979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jiang Y, Wang Y, Wang T, Hawke DH, Zheng Y, Li X, Zhou Q, Majumder S, Bi E, Liu DX et al (2014) PKM2 phosphorylates MLC2 and regulates cytokinesis of tumour cells. Nat Commun 5: 5566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jiang Y, Li X, Yang W, Hawke DH, Zheng Y, Xia Y, Aldape K, Wei C, Guo F, Chen Y et al (2014) PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Mol Cell 53: 75–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JW et al (2013) PKM2 isoform‐specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 155: 397–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang Y‐H, Israelsen WJ, Lee D, Yu VWC, Jeanson NT, Clish CB, Cantley LC, Vander Heiden MG, Scadden DT (2014) Cell‐state‐specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 158: 1309–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hosios AM, Fiske BP, Gui DY, Vander Heiden MG (2015) Lack of evidence for PKM2 protein kinase activity. Mol Cell 59: 850–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lunt SY, Muralidhar V, Hosios AM, Israelsen WJ, Gui DY, Newhouse L, Ogrodzinski M, Hecht V, Xu K, Acevedo PNM et al (2015) Pyruvate kinase isoform expression alters nucleotide synthesis to impact cell proliferation. Mol Cell 57: 95–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Palsson‐McDermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE, van den Bosch MWM, Quinn SR, Domingo‐Fernandez R, Johnston DGW et al (2015) Pyruvate kinase M2 regulates hif‐1alpha; activity and IL‐1beta; induction and is a critical determinant of the Warburg effect in LPS‐activated macrophages. Cell Metab 21: 65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lochmatter C, Fischer R, Charles PD, Yu Z, Powrie F, Kessler BM (2016) Integrative phosphoproteomics links IL‐23R signaling with metabolic adaptation in lymphocytes. Sci Rep 6: 24491 [DOI] [PMC free article] [PubMed] [Google Scholar]