

Abstract
Multi‐disciplinary research into the causes of autism reveals how a complex interplay of genes and diverse environmental factors can affect neuronal development during early childhood.

Subject Categories: Molecular Biology of Disease, Neuroscience, S&S: Health & Disease
“A developmental disability that hinders the normal functioning of the brain, affecting, in varying degrees, communication skills and social interaction. Repetitive behaviours, and different ways of learning, paying attention, or reacting to things are often distinctive signs”. This standard definition of autism fails to describe the complexity of a condition that ranges in its manifestations from severe intellectual impairment to superior cognitive skills, like in the Asperger syndrome. To comprise such diversity, autism disorders are now covered under the umbrella term “autism spectrum disorder” (ASD). In most cases, ASD manifests during the first 5 years of life, with boys significantly more likely to be diagnosed than girls. ASD usually goes together with several other problems that frequently include anxiety, sleep disorders, or epilepsy. No cure exists; treatment, such as speech therapy, just attempts to alleviate specific deficits of autistic patients.
Nothing is simple in autism. Even the real number of people affected is uncertain.
Nothing is simple in autism. Even the real number of people affected is uncertain. The US CDC estimates that about 1 in 68 (or 1.5%) of children in the USA are living with ASD (http://www.cdc.gov/ncbddd/autism/data.html). The WHO has a more conservative estimate, last revised in January this year, of 1 in 160 children, based on a larger set of epidemiological surveys (http://www.who.int/mediacentre/factsheets/autism-spectrum-disorders/en/). Needless to say, most studies were conducted in developed countries, and the prevalence of ASD in many low‐ and middle‐income countries remains largely unknown.
Along the years, many potential causes have been indicated, including genetic and environmental factors, exposure to toxins during pregnancy, wide gaps between parent ages, and so on
Although the general consensus is that prevalence rates are increasing globally, this point is debated too. Some analyses indicate that a large percentage of the increase in ASD owes to improved awareness and changes in diagnostic criteria (http://www.forbes.com/sites/tarahaelle/2015/01/05/majority-of-autism-increase-due-to-diagnostic-changes-finds-new-study/#518cbb50737c). Others maintain that these factors cannot fully account for the rise and some other causes must be at work (https://www.autismspeaks.org/science/science-news/shift-diagnosis-only-partly-explains-rise-autism-prevalence).
In search of causal factors
The real problem, though, is that no one understands the origins of autism. Along the years, many potential causes have been indicated, including genetic and environmental factors, exposure to toxins during pregnancy, wide gaps between parent ages, and so on. However, just a small percentage of ASD in children might be causally linked to any of these factors, while the etiology of the vast majority of cases remains obscure. The picture that is emerging from a new wave of studies is that, just as there is no one type of autism, there is probably no single cause either. A combination of genetic, neurological, and metabolic elements influence early brain development in children with ASD, affecting communication between neurons and rewiring the brain in a substantially different way.
Initial efforts to find the cause of autism have been mainly directed at identifying mutations. However, it early appeared that, although autism is recognized as one of the most heritable neurodevelopmental disorders, its genetic landscape is exceedingly heterogeneous. Single mutations—often de novo events that occur early during development or in a germ cell—and copy number variations can cause ASD symptoms, but this happens only in a minority of affected individuals, while in most cases a diverse array of variants with low‐ to medium‐risk effects is common. “After comparison of the currently available data for genetic association with ASD, the data fit a model in which the largest component of genetic risk derives from common genetic variants of an additive effect with a smaller, although clearly important, contribution from de novo and rare inherited variation”, wrote Daniel Geschwind and colleagues on our current understanding of autism genetics 1.
there is a strong interplay between common and rare variants […], the combination of which determines the risk whether a child develops the disorder and the severity of its manifestations
In other terms, there is a strong interplay between common and rare variants in ASD, the combination of which determines the risk whether a child develops the disorder and the severity of its manifestations 2. A strong genetic background, with a low burden of inherited common risk alleles, will buffer the effect of even rare, new disruptive mutations making ASD less probable; at the other extreme, genetic buffering will be poor in children with a high burden of common risk variants and ASD could develop more easily as a consequence of rare deleterious mutations (Fig 1).
Figure 1. The interplay between rare mutations and genetic background.
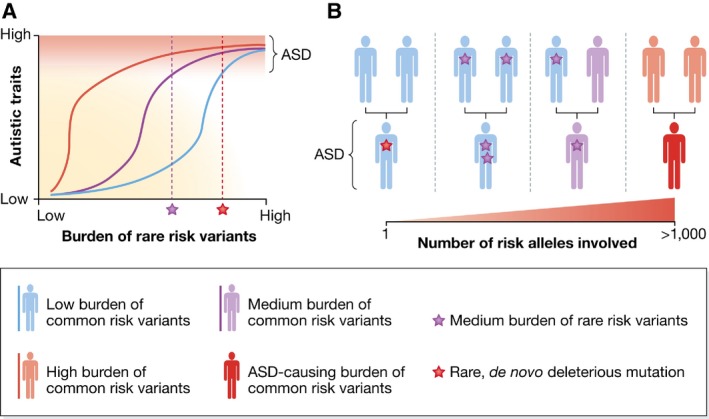
(A) For an individual with a high load of common genetic autism spectrum disorder (ASD) risk variants, a small burden of these rare risk variants can already cause ASD. In contrast, in an individual with a low burden of common genetic risk variants, only a high burden of deleterious mutations will cause ASD. (B) The transmission of ASD may occur through one of various paths. A de novo highly penetrant mutation can cause ASD even in individuals with a high genetic buffer for ASD (far left). ASD may also arise in children if both parents have a medium burden of rare variants (left‐of‐middle) or if one parent has a medium load of common risk variants for ASD and one has medium burden of rare risk variants (right‐of‐middle). Finally, children might develop ASD if both parents have a high load of common risk variants (far right). Reproduced from 2, with permission.
Synaptic pruning gone wrong?
A number of key studies have converged on singling out synaptic plasticity and connectivity as central for the development of ASD and as a possible target for new treatments. “Many mutations associated with ASD are predicted to influence the structure and the turnover of synapses at different levels because they encode proteins involved in chromatin remodelling and transcription, protein synthesis and degradation, actin cytoskeleton dynamics or synaptic transmission”, wrote Thomas Bourgeron reviewing the topic 2. Based on extensive work on animal models, specific biological pathways are now being identified at the crossroads between the causes and the mechanism of ASD (Fig 2).
Figure 2. Convergent neurobiological mechanisms in ASD .
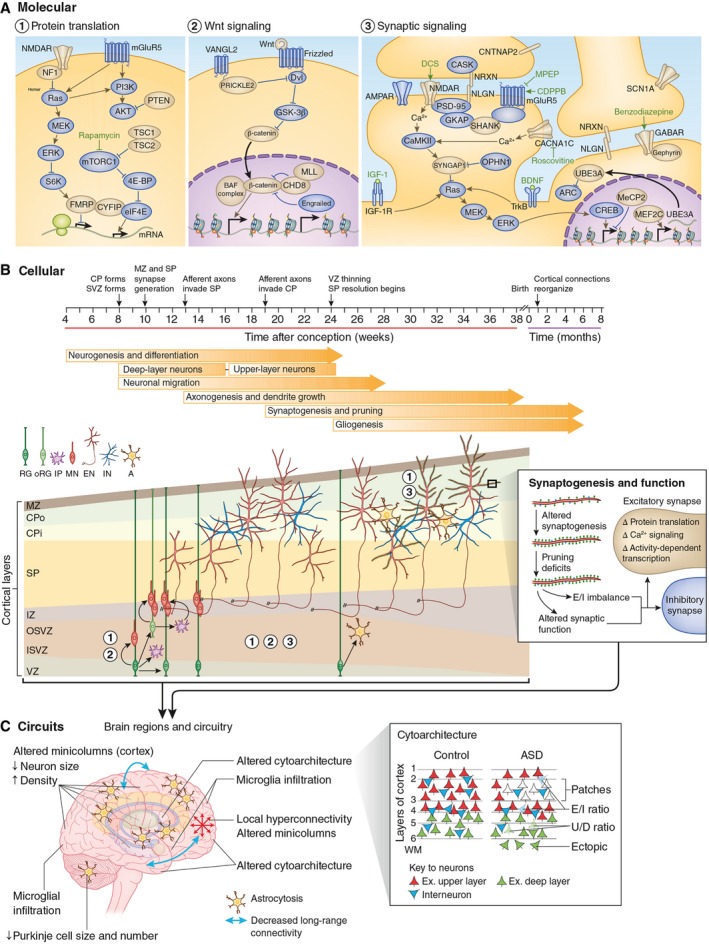
Normal brain development requires the generation and positioning of the correct number and type of cells, and the formation of the precise number and type of synapses. (A) These events are regulated by molecular pathways in development. Genes within these pathways for which there is genetic evidence for a link to ASD are colored in gold. Chemical compounds that reverse behavioral or cellular ASD phenotypes in model systems are indicated in green font near their predicted site of action. (B) The cellular events leading to changes in the higher‐order organization of the brain, including disruption of fetal cortical development and synaptic function. The cortical laminae are depicted from early fetal to neonatal stages (not to scale). The numbers indicate the molecular pathways important at each stage of development. (C) The widespread pathology and functional phenotypes observed in ASD, including altered brain growth trajectories, altered cortical cytoarchitecture (red triangles indicate excitatory upper‐layer neurons; green triangles are excitatory deep‐layer neurons; blue triangles are interneurons; numbers indicate cortical layers; WM, white matter) and connectivity, may arise from combined deficits in neurogenesis, cell fate, neuronal migration, and morphogenesis during fetal development and dysregulated synaptic function, possibly in combination with reactive microglia infiltration and astrocytosis. RG, radial glia; oRG, outer radial glia; IP, intermediate progenitor; MN, migrating neuron; EN, excitatory neuron; IN, interneuron; A, astrocyte; E/I, excitatory or inhibitory neuron; U/D, upper‐layer or deep‐layer neuron; MPEP, 2‐methyl‐6‐(phenylethynyl)‐pyridine; CDPPB, 3‐cyano‐N‐(1,3‐diphenyl‐1H‐pyrazol‐5‐yl)benzamide; DCS, D‐cycloserine; IGF1, insulin‐like growth factor 1; VZ, ventricular zone; ISVZ, inner subventricular zone; OSVZ, outer subventricular zone; IZ, intermediate zone; SP, subplate; CPi, inner cortical plate; CPo, outer cortical plate; MZ, marginal zone. Reproduced from 1, with permission.
Synaptic pruning, in particular, is receiving considerable attention. Rapid learning during birth to early childhood is reflected by a dramatic increase of brain connections. After puberty, the number of synapses is strongly reduced—both in humans and rodents—a process which is considered crucial for establishing proper coordination between neuronal circuits and correct integration of sensory information. This essential maturation step goes awry in ASD, because of an altered mTOR pathway and the inhibition of autophagy required for normal synaptic pruning during development 3.
Of late, a team led by Sheryl Smith at SUNY Downstate Medical Center in New York, USA, investigated the role of α4βδ, a GABAA receptor expressed in the brain, which has been implicated in ASD 4. “We showed in a mouse model that synaptic pruning during adolescence is triggered by this receptor. In its absence, synaptic pruning is prevented. In addition, mice which do not express this receptor are able to learn spatial locations, but are not able to re‐learn additional locations, suggesting that cognitive flexibility is impaired”, Smith explained. Since a lack of synaptic pruning and impaired cognitive flexibility are hallmarks of autism, and reduced expression of the α4βδ GABAA receptor is associated with both features, enhancing the function of this receptor with selective drugs may enhance the pruning of unnecessary synapses and thereby improve cognition in autistic patients. “Although autism emerges in early childhood, the adolescent period is associated with loss of cognitive improvements achieved before puberty”, Smith said. “This suggests that adolescence may be a time when drug therapies targeting this receptor can lead to improved cognition in autism”.
Further research reveals that autism may not be solely a disease of the brain. Working with mice expressing ASD‐related mutations only in peripheral sensory neurons, a team from the Harvard Medical School in Boston, USA, recently found that these animals display deficits in tactile perception together with heightened anxiety and impaired ability to interact socially, all features that are typical of ASD 5. Tampering with touch‐processing ASD‐associated mutations alters the way how the environment is perceived and how this sensory information is communicated to the brain, which could eventually lead to anxiety and poor social interactions; yet, the neurobiological mechanisms behind these causal connections are so far unknown.
Gut microbiota
Other work on the causes of autism expands radially in many other directions, often seemingly being influenced by the “scientific craze” of the moment. Whether all these avenues will advance our understanding of ASD is uncertain, but this is the reality of autism research: many hypotheses and few certainties. Lately, the idea that perturbations of the microbiota–gut–brain axis might be an important factor in the generation of autistic behaviors is gaining momentum. “My theory rests on the fact that gut microbes are involved in early brain development”, wrote microbiologist Martin Blaser, Director of the Human Microbiome Program at New York University, in his recent book, Missing Microbes 6. Blaser recalls the intense cross talk between microbes in the gut, the enteric nervous system—whose neuroendocrine cells produce some 80% of the serotonin of the entire body—and the brain. The gut microbiome is also deeply involved in the synthesis of gangliosides, key components of neuronal cell membranes.
… this is the reality of autism research: many hypotheses and few certainties
Blaser mainly puts the blame on the overuse of antibiotics. “If the composition of the microbes that lead to the production of gangliosides and serotonin is perturbed, the brain will be perturbed”, he wrote. “There might still be a conversation among the microbes, gut wall, and brain, but it might be in the wrong language. In an adult, this might not make a big difference, but in a newborn baby or young child whose brain is developing rapidly?” Evidence that ASD‐related neurodevelopmental deficits co‐occur with gastrointestinal symptoms in a significant number of patients seems to support Blaser's hypothesis 7. In addition to antibiotics, other factors, including infection, diet, stress or disease, that influence the composition and activity of the gut microbiota, may also contribute. Mice infected with the parasite Trichuris muris, which causes colonic inflammation, develop anxiety‐like behaviors, with altered central nervous system biochemistry 8.
Another possible mechanism through which the gut microbiome can modulate cerebral function is via the production of short‐chain fatty acids (SCFA). These substances, present in diet and produced by gut bacteria following fermentation of dietary carbohydrates, can affect host immune and inflammatory responses. They are absorbed and transported by the bloodstream and eventually cross the blood–brain barrier to directly influence brain activity 7. Alterations in the gut microbiome and in its production of SCFA might therefore contribute to ASD in predisposed individuals (Fig 3).
Figure 3. The microbiota–gut–brain axis and its potential role in ASD .
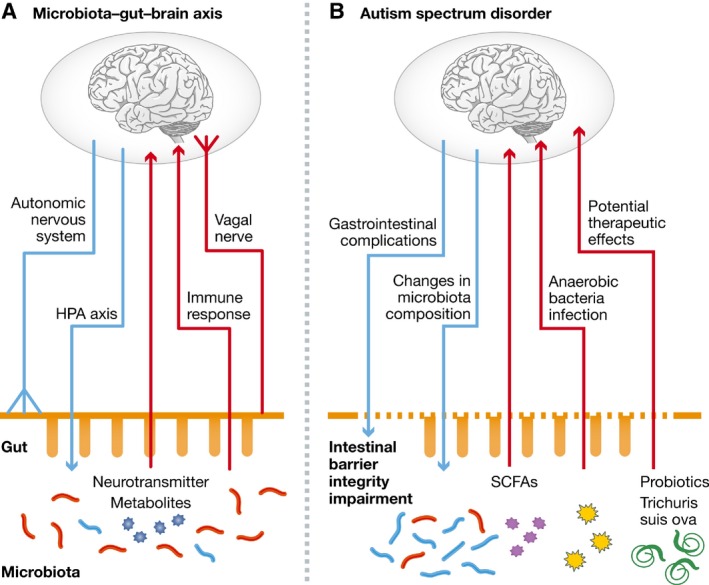
(A) The bidirectional communication within the microbiota–gut–brain axis occurs mainly through the autonomic nervous system (ANS), HPA axis, neuroendocrine, and neuroimmune pathways. (B) The manifestations of disturbances within the microbiota–gut–brain axis in the pathology of ASD. Autistic deficits are associated with gastrointestinal symptoms and compositional changes in gut microbiota. Conversely, noxious metabolites derived from gut microbiota and intestinal anaerobic bacteria infection may participate in the development of ASD. Modulators of microbiota–gut–brain axis, such as the probiotics and Trichuris suis ova (TSO), have shown potential therapeutic effects in ASD. HPA, hypothalamus–pituitary–adrenal; SCFAs, short‐chain fatty acids. Reproduced from 7, with permission.
Gene–environment interactions
Beyond the gut microbiota, antibiotics, and diet, other environmental factors might also influence the risk of ASD, which makes the search for its causes roots even more formidable. Gene–environment interactions in human diseases are per se nothing new, but establishing such connections for ASD's genetic architecture proved challenging. Identified and supposed environmental determinants range from exposure to air pollutants and endocrine‐disrupting chemicals to fertility treatments, to smoking habits and alcohol consumption. Kimberly Keil and Pamela Lein from the University of California at Davis, USA, have highlighted the role played by the epigenome, and in particular DNA methylation, in mediating the effects of environmental risk factors on the developing brain 9. In fact, epidemiological and experimental evidence has shown that various environmental chemicals, including polychlorinated biphenyls (PCBs), lead, and bisphenol A, affect DNA methylation and could thereby increase the risk of developing ASD. However, much of the relevant work has been performed on animal models, and clinical research showing a cause and effect relationship on neuronal development is still lacking in most cases.
Nonetheless, the concept that chemicals interfere with brain development by modifying the epigenome is appealing, because it suggests a mechanism by which early exposure can influence multiple genes that control behaviors that do not manifest until later in life, according to Lein. “Understanding the cause–effect relationship between chemical effects on the epigenome and increased risk for neurodevelopmental disorders will require both mechanistic studies, to delineate how environmental chemicals modify the brain epigenome at the molecular level, and preclinical and epidemiological studies that assess the effects of environmental chemicals not on global changes in the epigenome, but also on the epigenetic landscape of specific genes that regulate brain development”, she said. This will be a big challenge for epidemiological studies, Lein explained, given the current state of uncertainty as to whether epigenetic changes in peripheral tissues “mirror” or reflect epigenetic changes in the brain. “Answers to these questions may provide insight as to therapeutic approaches for altering the epigenome in a gene‐specific manner to alleviate the conditions of children with ASD”, Lein commented.
A new, more detailed picture of autism has begun to emerge from the mist thanks to better understanding of the genetic, epigenetic, metabolic and environmental factors involved
To reconcile this variety of hypotheses, putative causes, and known risk factors, Sarah Crawford from the Southern Connecticut State University in New Haven, USA, has sketched out a unifying theory of autism's origins. Her Quantitative Threshold Exposure (QTE) hypothesis attempts to explain the “cumulative effects of risk factor exposure in both the causation of ASD and its dramatic increase over the past 30 years” 10. ASD would emerge from pre‐ and/or post‐natal exposure to an array of endogenous and environmental factors that may act synergistically as antigens during critical developmental windows of both the immune and central nervous system. When the combined level of exposure reaches a given threshold, it affects brain maturation in genetically predisposed children. The model serves as a quantitative framework to evaluate risk factors with respect to their combined relative impact on ASD, but it does not assess the importance of any specific risk factor in the causation of ASD.
A new, more detailed picture of autism has begun to emerge from the mist thanks to better understanding of the genetic, epigenetic, metabolic, and environmental factors involved. Broader and more comprehensive epidemiological surveys, a sort of “Large Autism Womb‐to‐Adulthood Project”, could probably help to solve the riddle of what causes ASD (Box 1). However, the image is a fractured one as so many different causes combine to determine risk, and it could be wise to delve into the underlying biology of the distinct autism subtypes. Divide et impera, the ancient Roman strategy based on breaking down enemy forces to defeat isolated units more easily, might prove to be very modern.
Box 1: Standing on the shoulders of big numbers.
Pinning down the cause(s) of ASD has revealed extremely difficult. Given the multifactorial nature of the condition, a large‐scale population screening might help linking ASD to specific combinations of genetic and environmental settings. This is the rationale behind so called “autism biobanks”, a goldmine for epidemiological research.
One such initiative is being fostered by the Kaiser Permanente Division of Research in Oakland, CA, with the goal to gather information from 5,000 affected children and their biological parents (http://autismfamilybiobank.kaiser.org/). Collection of blood and saliva samples will permit statistically significant research on a number of aspects, for example, the search for new and better markers for ASD for early diagnosis. In addition, detailed surveys on family history and social environment are conducted to put the genetic information in a broader context.
A similar project has been launched last year in Australia, involving some 1,200 families where a family member has autism (http://www.autismcrc.com.au/creation-first-australian-autism-biobank). “It isn't just about collecting the biological data, it is also about making sense of this information and there are literally millions upon millions of different biological pathways that may be related to autism”, commented Andrew Whitehouse, the project's leading scientist from the Telethon Kids Institute, University of Western Australia, in a press release (http://www.autismcrc.com.au/news/autism-crc-biobank-launched).
Although such databases could add significantly to the toolbox of autism research, these efforts could still fall short of expectations because of a critical size defect. “Lessons from other common neuropsychiatric disorders suggest that large cohorts (> 50,000 subjects) are needed to identify predicted common variants”, recently warned Daniel Geschwind and colleagues about the need for more extensive efforts in ASD genetic discovery 1.
References
- 1. de la Torre‐Ubieta L, Won H, Stein JL, Geschwind DH (2016) Advancing the understanding of autism disease mechanisms through genetics. Nat Med 22: 345–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bourgeron T (2015) From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci 16: 551–563 [DOI] [PubMed] [Google Scholar]
- 3. Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A et al (2014) Loss of mTOR‐dependent macroautophagy causes autistic‐like synaptic pruning deficits. Neuron 83: 1131–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Afroz S, Parato J, Shen H, Smith SS (2016) Synaptic pruning in the female hippocampus is triggered at puberty by extrasynaptic GABAA receptors on dendritic spines. eLife 5: e15106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Orefice LL, Zimmerman AL, Chirila AM, Sleboda SJ, Head JP, Ginty DD (2016) Peripheral mechanosensory neuron dysfunction underlies tactile and behavioral deficits in mouse models of ASDs. Cell 166: 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blaser MJ (2014) Missing microbes: how the overuse of antibiotics is fueling our modern plagues. New York, NY: Henry Holt and Company; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li Q, Zhou JM (2016) The microbiota‐gut‐brain axis and its potential therapeutic role in autism spectrum disorder. Neuroscience 324: 131–139 [DOI] [PubMed] [Google Scholar]
- 8. Bercik P, Verdu EF, Foster JA, Macri J, Potter M, Huang X, Malinowski P, Jackson W, Blennerhassett P, Neufeld KA et al (2010) Chronic gastrointestinal inflammation induces anxiety‐like behavior and alters central nervous system biochemistry in mice. Gastroenterology 139: 2102–2112 [DOI] [PubMed] [Google Scholar]
- 9. Keil KP, Lein PJ (2016) DNA methylation: a mechanism linking environmental chemical exposures to risk of autism spectrum disorders? Environ Epigenet 2: dvv012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Crawford S (2015) On the origins of autism: the quantitative threshold exposure hypothesis. Med Hypotheses 85: 798–806 [DOI] [PubMed] [Google Scholar]