

Abstract
PRLs (phosphatases of regenerating liver) are frequently overexpressed in human cancers and are prognostic markers of poor survival. Despite their potential as therapeutic targets, their mechanism of action is not understood in part due to their weak enzymatic activity. Previous studies revealed that PRLs interact with CNNM ion transporters and prevent CNNM4‐dependent Mg2+ transport, which is important for energy metabolism and tumor progression. Here, we report that PRL‐CNNM complex formation is regulated by the formation of phosphocysteine. We show that cysteine in the PRL catalytic site is endogenously phosphorylated as part of the catalytic cycle and that phosphocysteine levels change in response to Mg2+ levels. Phosphorylation blocks PRL binding to CNNM Mg2+ transporters, and mutations that block the PRL‐CNNM interaction prevent regulation of Mg2+ efflux in cultured cells. The crystal structure of the complex of PRL2 and the CBS‐pair domain of the Mg2+ transporter CNNM3 reveals the molecular basis for the interaction. The identification of phosphocysteine as a regulatory modification opens new perspectives for signaling by protein phosphatases.
Keywords: cysteine, magnesium, phosphatase, phosphorylation, X‐ray crystallography
Subject Categories: Membrane & Intracellular Transport, Signal Transduction, Structural Biology
Introduction
The phosphatases of regenerating liver (PRLs) are highly oncogenic proteins and strongly associated with the transition to metastatic disease 1, 2. The PRL family belongs to the PTP (protein tyrosine phosphatase) superfamily and is comprised of three closely related proteins: PRL1, PRL2, and PRL3 3. The family was first identified as immediate‐early genes up‐regulated in liver following partial resection and in mitogen‐stimulated cells 4. Several years later, PRL3 was identified as a potent oncogene 300‐fold overexpressed in metastatic liver lesions from colorectal cancer patients 2.
Clinically, PRL phosphatases are valuable prognostic markers of poor outcome and shortened patient survival (reviewed in 5, 6). Many animal studies have shown that PRLs are oncogenic, yet the mechanisms linking PRLs and cancer have remained unclear. PRLs have been suggested to contribute to cell proliferation and metabolic reprogramming via the PI‐3K/Akt/mTOR pathway; however, it has been difficult to reconcile these models with very poor catalytic activity of PRLs in vitro. The rate‐limiting step in catalysis is the hydrolysis of a phosphoenzyme intermediate. Like other PTPs, the PRL catalytic site contains a conserved cysteine residue that forms a phosphocysteine intermediate as part of the catalytic cycle 7, 8. In the case of PRLs, this intermediate has a long half‐life (> 1 h) and is rate‐limiting for dephosphorylation 9. As a result, PRLs are very poor enzymes, and no confirmed physiological substrates are known 3, 10.
Recently, PRLs were shown to interact physically with members of the CNNM (cyclin M/ancient conserved domain protein) family to regulate Mg2+ transport (Fig 1A) 11, 12. In mammalian cells, CNNM proteins mediate Mg2+ efflux 13. Four CNNM proteins in humans are known. They are highly conserved but have distinct functions and tissue distributions. CNNMs contain an N‐terminal integral membrane domain followed by a cystathionine‐β‐synthase (CBS) pair domain and a C‐terminal region of 250 residues or more of unknown structure 14, 15. Phylogenetic analysis shows co‐evolution of PRLs and CNNMs suggesting a functional relationship between the two protein families 12.
Figure 1. The PRL‐CNNM pathway.
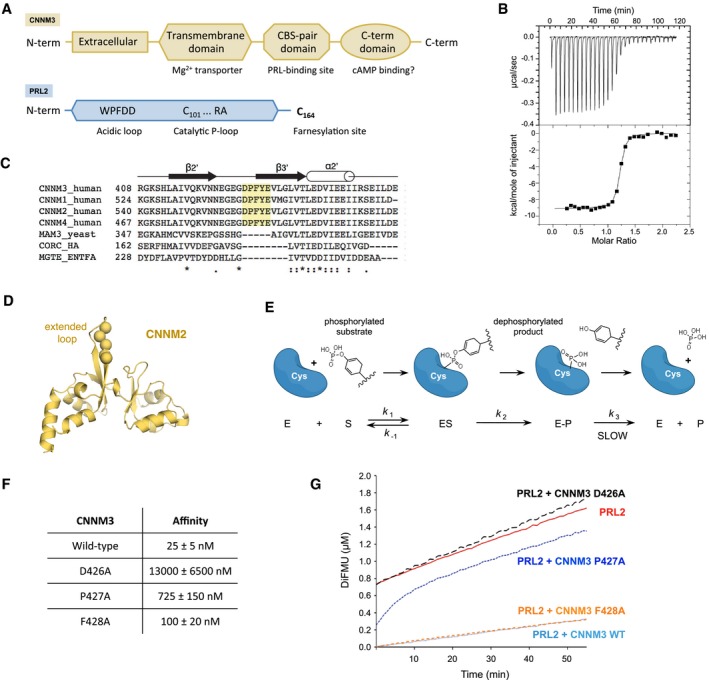
- Domain organization of CNNM magnesium transporters and PRL phosphatases.
- ITC experiment shows that the CBS‐pair domain of CNNM3 binds PRL2 with a nanomolar K d and one‐to‐one stoichiometry.
- Sequence alignment of the extended loop in the CBS‐pair domains of CNNM proteins. The loop is unique to the four human CNNM family members compared to related CBS‐pair domain proteins: MAM3, CorC efflux protein from Haemophilus influenzae, and the MgtE Mg2+ transporter from Enterococcus faecalis. Loop residues unique to CNNM proteins are highlighted in yellow.
- Structure of CBS‐pair domain of CNNM2 (PDB 4IY0) with the residues DPFYE indicated.
- Schematic of catalytic cycle of PRL phosphatases. Catalysis proceeds through a long‐lived phosphocysteine intermediate. Turnover of the intermediate is the rate‐limiting step.
- ITC data of CBS‐pair domain mutants binding to PRL2 identify aspartic acid 426 as essential for binding.
- Kinetics of hydrolysis of a synthetic substrate by PRL2 shows burst kinetics. Addition of the wild‐type CBS‐pair domain but not the D426A mutant inhibits phosphatase activity.
Mg2+ is the most abundant divalent cation in cells and required for many essential cellular functions. Mg2+ binds polyphosphate compounds such as ATP to proteins and is required for the activity of many enzymes associated with energy metabolism and nucleic acid synthesis and degradation 16. Cellular Mg2+ homeostasis is maintained through the action of a large number of different Mg2+ transporters present in different cellular membranes. Functional links to diseases have been established for TRPM6, claudin 16, and CNNM2, which cause hypomagnesemia when mutated, and for MagT1, SLC41A1, and CNNM4, which cause other conditions 16, 17. A link between elevated Mg2+ levels and cancer has long been suspected due to the importance of Mg2+ in cell proliferation, apoptotic signals, genetic stability, and metabolic reprogramming 18, 19. CNNM4 was shown to be a tumor suppressor in a mouse model of colon cancer 11, and the interaction between PRLs and CNNMs is required for PRL oncogenicity 12, 20.
Here, we report that phosphocysteine plays a role in Mg2+ homeostasis. We observe that the interaction between PRLs and CNNM is negatively regulated by phosphorylation of the PRL catalytic cysteine. We observe that endogenous PRL phosphatases are significantly phosphorylated in cultured cells and the phosphorylation levels decrease in response to Mg2+ deprivation. The crystal structure of a PRL phosphatase and fragment of a CNNM transporter reveals the molecular basis for the interaction and its regulation by cysteine phosphorylation. The study offers new insight into the function of PRLs and their long‐lived phosphocysteine intermediate.
Results
Interaction between PRL and CNNM proteins
We used isothermal titration calorimetry (ITC) to confirm the association between PRL and CNNM proteins. Previous immunoprecipitation and deletion analysis had shown that the interaction is mediated by the CBS‐pair domain of CNNM proteins 11, 12. We measured one‐to‐one stoichiometry and a K d of 25 nM between PRL2 and the CNNM3 CBS‐pair domain (Fig 1B).
We used analysis of sequence conservation within the CNNM proteins to identify potential residues involved in binding PRL2 21. The CBS‐pair domains contain an enlarged loop that has been proposed to bind PRL phosphatases 12. We mutated three residues in the loop, which are conserved across all CNNM family members (Fig 1C and D) 22. ITC experiments showed mutation of Asp426 in the loop decreased binding 500‐fold and confirmed the extended loop as the PRL‐binding site (Fig 1F). Mutation of the adjoining proline and phenylalanine residues had more moderate effects and decreased the affinity respectively 25‐fold and threefold. While this manuscript was in preparation, a more complete set of mutations in the same loop were shown to prevent PRL2 binding 20.
PRLs bind CNNMs as pseudosubstrates
We next tested the effect of the CBS‐pair domain on PRL catalytic activity. PTPs are characterized by the presence of a conserved active‐site motif HC(X)5R[S/T] with a key cysteine residue that acts as a nucleophile in catalysis (Fig 1A). This leads to formation of a phosphoenzyme intermediate, which is then resolved through the action of serine or threonine in the active‐site motif and an aspartic acid on the neighboring acidic loop. In PRLs, the serine/threonine residue is replaced by an alanine residue, which leads to unusual, burst kinetics due to the slow turnover of phosphocysteine intermediate 9 (Fig 1E).
We carried out enzymatic kinetic assays with PRL2 using the fluorescent substrate, DiFMUP. As previously reported for PRL3, PRL2 shows burst kinetics. The rapid turnover of one molecule of substrate is followed by a slow steady‐state rate that is determined by the hydrolysis of the phosphocysteine. In the absence of the CNNM3 CBS‐pair domain, PRL2 released 0.75 μM of product in the dead time of the experiment, followed by a linear increase of 1 μM per hour (Fig 1G).
In the presence of the CNNM3 CBS‐pair domain, the initial burst of activity was completely inhibited. The CNNM3 mutants showed inhibition that paralleled their binding affinities for PRL2. The D426A mutant, which is strongly impaired in its ability to bind PRL2, showed no inhibition. The F428A, which binds with near wild‐type affinity, strongly inhibited catalysis, and the P427A mutant, which has intermediate affinity, moderately inhibited PRL2 phosphatase activity (Fig 1G). These results are consistent with CNNM3 binding to PRL2 as a pseudosubstrate and hindering access of substrates to the active site. The decrease in the initial rate of catalysis reflects the amount of free PRL2.
The PRLs' phosphocysteine intermediates are unable to bind CNNMs
The possibility that the CBS‐pair domain bound as a substrate mimic suggested that the catalytic intermediate might inhibit the binding of CNNM proteins. To test this, we prepared phosphorylated PRL2 using a second synthetic substrate, 3‐O‐methylfluorescein phosphate (OMFP). Generation of the phosphocysteine intermediate can be conveniently monitored by SDS–PAGE using the Phos‐tag reagent that retards the migration of phosphorylated proteins 23. As shown in Fig 2A, addition of OMFP to PRL2 led to full phosphorylation, while PRL2 C101S, which lacks the catalytic cysteine, was not affected. Phosphocysteine is chemically unstable and PRL2 could be dephosphorylated by boiling in SDS loading buffer prior to electrophoresis or by incubation for several hours at room temperature (Fig EV1A). Phosphorylation of PRL2 by OMFP treatment was confirmed by mass spectrometry. The mass of recombinant PRL2 was increased by 80 amu following treatment with OMFP which allows us to conclude that only one phosphorylation site is present (Fig 2B). Addition of OMFP to the C101A mutant had no effect on the mass spectrum. Formation of the phosphocysteine intermediate could also be observed using 31P NMR spectroscopy (Fig 2C). The phosphocysteine chemical shift, 14.8 ppm, is similar to that reported for EIIMtl 24. A time series of NMR spectra showed that addition of OMFP to PRL2 led to the rapid formation of phosphocysteine, followed by its decay and generation of free phosphate (Fig EV1B).
Figure 2. Cysteine phosphorylation of PRLs prevents binding to CNNM proteins.
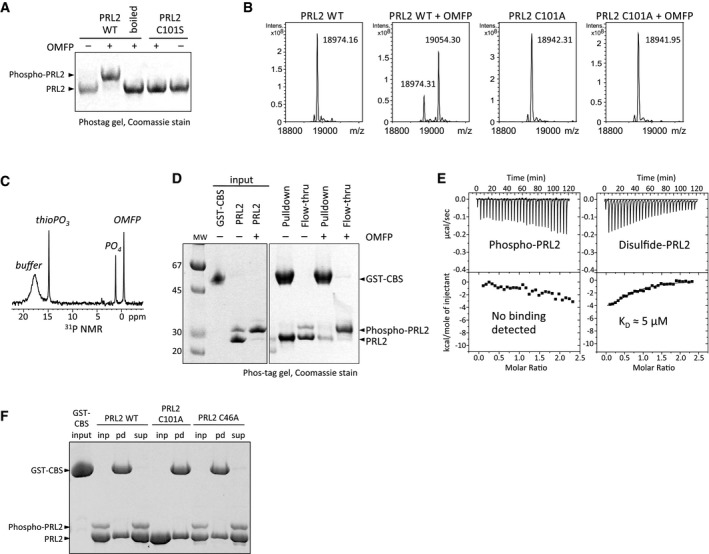
- Treatment of PRL2 with a synthetic substrate leads to complete phosphorylation of the catalytic residue, cysteine 101. Recombinant PRL2 was treated with 3‐O‐methylfluorescein phosphate (OMFP) and analyzed by SDS–PAGE with the Phos‐tag reagent. Phosphocysteine is hydrolyzed by boiling.
- Detection of phosphorylated PRL2 by electrospray mass spectrometry. OMFP treatment increased the mass of the intact protein by 79.99 amu but had no effect on the C101A mutant.
- 31P NMR spectrum of the thioPO3 signal from phosphocysteine in PRL2. The broad peak is TCEP in the buffer.
- Cysteine‐phosphorylated PRL2 does not bind to CNNM3. Recombinant PRL2 was phosphorylated by treatment with OMFP and subjected to pulldown by the CBS‐pair domain of CNNM3 as a GST fusion. Only unphosphorylated PRL2 was observed in the pulldown lanes, while phosphorylated PRL2 was in the flow‐through lanes.
- ITC measurement of the affinity of PRL2 for the CNNM3 GST‐CBS‐pair domain. Phosphorylation by OMFP completely blocked binding, while oxidation of PRL2 by diamide decreased the affinity 200‐fold.
- Pulldown assays with purified proteins show cysteine residues in the PRL2 active site are not required for binding the CNNM3 CBS‐pair domain. Input (inp), pulldown (pd), and supernatant (sup) fractions were analyzed on a Coomassie‐stained phos‐tag gel.
Figure EV1. PRL2 spontaneously dephosphorylates and regains its ability to bind the CNNM3 CBS‐pair domain.
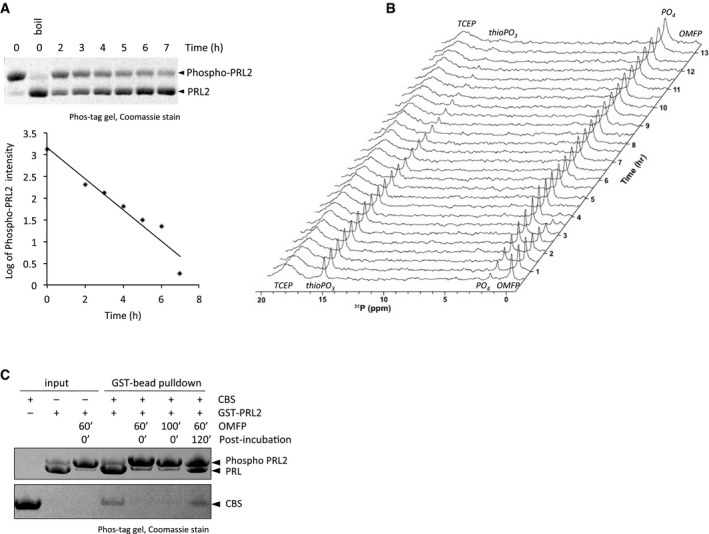
- Time course of spontaneous dephosphorylation of PRL2. PRL2 (C95A, C96A, C119A) was phosphorylated with OMFP (3‐O‐methylfluorescein phosphate) for 15 min and OMFP removed using a 10‐kDa centrifugal filter prior to incubation at room temperature. Samples were removed at times indicated and frozen in SDS loading buffer prior to analysis by SDS–PAGE with the Phos‐tag reagent (upper panel). Band intensities were quantified using ImageJ and the rate of decay of phospho‐PRL2 plotted on a logarithmic scale (lower panel). The initial rate of dephosphorylation fit with a half‐life of 2 h.
- Time course of phosphorus NMR spectra of PRL2 following addition of twofold excess of OMFP. The thioPO3 peak at 15 ppm arises from phosphocysteine on PRL2. Free phosphate (PO4) is generated from hydrolysis of phosphocysteine. Following depletion of OMFP, the thioPO3 peak decayed with a half‐life of approximately 3 h at 25°C.
- Phosphorylated GST‐PRL2 is unable to retain the CNNM3 CBS‐pair domain but regains binding activity after 2‐h incubation. GST‐PRL2 was untreated (−) or phosphorylated by OMFP (+) and bound to glutathione beads. CNNM3 CBS‐pair domain was added to the beads and the retained material analyzed by Phos‐tag SDS–PAGE.
We tested binding of phosphorylated PRL2 to the CNNM3 CBS‐pair domain using pulldown and ITC assays. In GST‐pulldown experiments, only the non‐phosphorylated form of PRL2 was retained by the GST‐CBS‐pair domain (Fig 2D and E). Conversely, phosphorylated PRL2 was only observed in the flow‐through fractions. Identical results were observed with the reverse pulldown experiments in which GST‐PRL2 was used to retain the CBS‐pair domain of CNNM3. Binding was again blocked by treatment of PRL2 with OMFP (Fig EV1C). Incubation following OMFP treatment led to partial dephosphorylation of PRL2 and a restoration of CNNM3 CBS‐pair binding. We also confirmed the lack of binding using ITC experiments. Phosphorylation of PRL2 by OMFP treatment prevented binding to the CNNM3 CBS‐pair (Fig 2E). To our knowledge, this is the first example in eukaryotes of regulation of a protein–protein interaction by phosphocysteine.
In addition to formation of phosphocysteine, the catalytic cysteine of PTP family phosphatases can be oxidized to a variety of forms 25. We treated PRL2 with diamide, a weak oxidizing agent, and performed ITC (Fig 2E). Diamide treatment significantly decreased the affinity of PRL2 for the CNNM3 CBS‐pair domain as previously observed in pulldown assays with intact CNNM proteins 11, 12. The catalytic cysteine per se was not required for binding. Mutation of the catalytic cysteine and the adjoining cysteine that can form a disulfide bond did not prevent PRL2 binding to the CNNM3 CBS‐pair domain (Fig 2F). Intriguingly, this differs from results obtained with the full‐length CNNM proteins where both cysteines were required in cultured cells 11, 12.
The PRL‐CNNM interaction regulates magnesium efflux in cells
We next tested whether the previously described CNNM mutations inhibit the interaction in cells. For functional assays, we chose to use PRL3 and CNNM4 because of the previously published work from our laboratory that showed PRL3 inhibition of CNNM4 Mg2+ efflux 11. CNNM3 is inactive in the cell Mg2+ efflux assay 26. The CBS domains of CNNM3 and CNNM4 are highly similar and are 100% identical in the regions that interact with PRLs (Fig 1C). We prepared the loop mutations in CNNM4 and expressed the proteins in COS7 cells along with Myc‐tagged PRL3 (Fig 3A). As observed with CNNM3 and PRL2, mutation of the aspartic acid residue (D485A) in the CBS‐pair domain blocked the co‐IP of CNNM4 and PRL3. Mutation of the other two residues in CBS loop (P486A and F487A) showed smaller effects on the co‐IP in agreement with the results observed with the purified CNNM3 and PRL2 proteins.
Figure 3. The PRL‐CNNM interaction inhibits magnesium efflux.
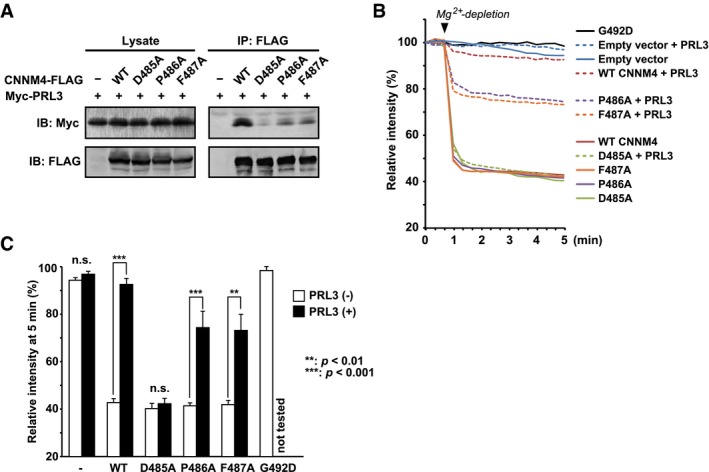
-
AMutations in the CBS‐pair loop of CNNM4 prevent binding to PRL3. Lysates of COS7 cells transfected with the indicated constructs were subjected to immunoprecipitation (IP) and immunoblotting (IB) with the indicated antibodies.
-
B, CWild‐type CNNM4‐dependent magnesium efflux is inhibited by co‐transfection with PRL3, but the efflux by the CNNM4 D485A mutant, which is unable to bind PRL3, is resistant to inhibition. HEK293 cells transfected with the indicated constructs were loaded with Magnesium Green and Mg2+ removed from the medium at the indicated time point (arrowhead). The means of relative fluorescence intensities of 10 cells are graphed as a function of time in (B) and the mean and s.e.m. at 5 min plotted in (C). P‐values were calculated using one‐way ANOVA followed by Bonferroni's multiple‐comparison test. n.s., not significant.
CNNM4 has previously been shown to mediate Mg2+ efflux and that this is inhibited by PRL3 11. We repeated this assay to assess the functional consequences of CBS‐pair mutations. Immunofluorescence microscopy was used to confirm the localization of the transfected proteins (Fig EV2). As previously observed, CNNM4 expression in HEK293 cells led to a large decrease in intracellular Mg2+ levels as measured by Magnesium Green fluorescence when Mg2+ was removed from the extracellular milieu (Fig 3B). The drop in intracellular Mg2+ levels was completely blocked when PRL3 was co‐expressed with CNNM4 (Fig 3C).
Figure EV2. Localization of CNNM4‐FLAG expressed in HEK293 cells.
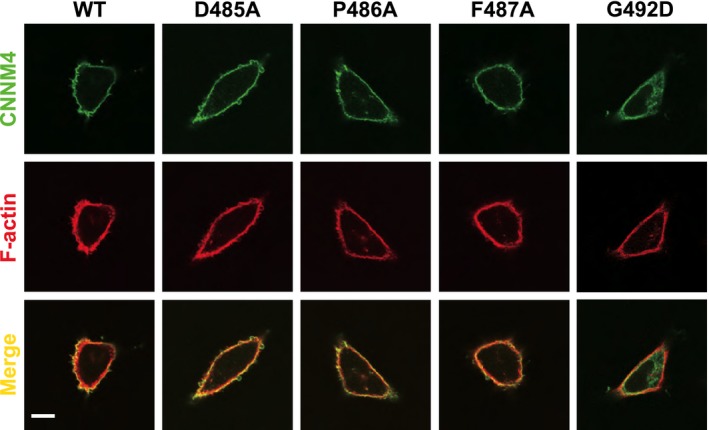
The wild‐type (WT), D485A, P496A, and F487A mutants localize at the plasma membrane, while G492D mainly localizes at the intracellular membraneous structures. Scale bar, 10 μm.
We tested the effect of the mutations in CNNM4 that blocked PRL3 binding. The mutations in the CBS loop, D485A, P486A, and F487A, had no effect on the efflux activity. All three mutant CNNM4 proteins were as active as the wild‐type. Mutation of Gly492 in CNNM4, which in the context of CNNM3 had been reported to affect PRL2 binding 12, led to its improper localization in the cell and a loss of activity in the magnesium efflux assay (Figs EV2 and 3C).
We next examined the effect of PRL3 co‐expression on the functional CNNM4 mutants (Fig 3C). The CNNM4 D485A mutant was completely resistant to inhibition by PRL3. CNNM4 P486A and F487A showed an intermediate level of inhibition in line with their intermediate affinities for binding PRL3 in the pulldown assay (Fig 3A). We conclude that the PRL‐binding site in the extended loop of CNNM Mg2+ transporters is essential for their regulation by PRL phosphatases.
Structural basis for the PRL‐CNNM interaction
To understand how PRL phosphorylation and oxidation regulate CNNM binding, we determined the structure of a complex of PRL2 and the CBS‐pair domain of CNNM3 (Fig 4; Table EV1). In the structure, the CBS‐pair domain is present as a dimer in the head‐to‐head arrangement that is typical for other CBS‐pair domains. The CBS‐pair domain is composed of two tandem modules, CBS1 (residues 324–378) and CBS2 (residues 390–446), related by an approximate twofold symmetry (Fig 4A). As previously observed for CNNM2 and CNNM4 21, 27, the CNNM3 CBS‐pair domain contains a long extended β‐stranded loop that extends away from the dimerization interface. Each of the extended loops binds one PRL2 molecule.
Figure 4. Crystal structures of PRL2 in complex with the CNNM3 CBS‐pair domain.
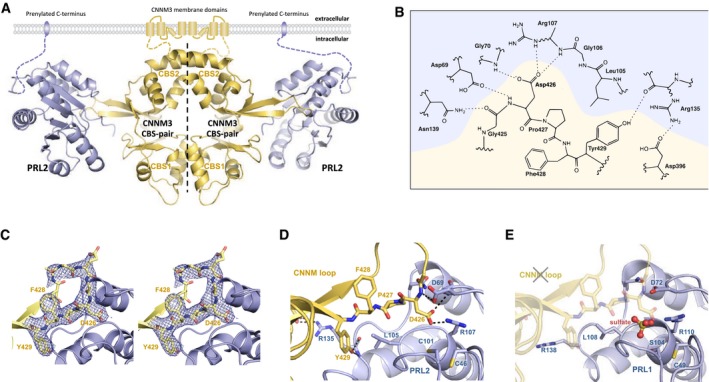
- The CBS‐pair domain (yellow) forms a dimer and binds two molecules of PRL2 via an extended loop. The positions of the PRL2 C‐terminal CAAX prenylation motif 42 and CNNM3 N‐terminus suggest that the membrane would sit above the complex.
- Contacts between the CNNM3 CBS2 loop and the PRL2 active site.
- Stereoview of the 2FO‐FC omit map of the PRL2 binding loop of CNNM3 contoured at 1 σ.
- Detail of complex with reduced PRL2, showing how CNNM3 Asp426 inserts into the pocket adjacent to the catalytic cysteine Cys101.
- Model of phosphocysteine preventing CNNM binding. Crystal structure of PRL1 with a bound sulfate shows that phosphocysteine would prevent Asp426 binding. Residues are numbered according to the PRL1 structure (PDB 1XM2).
The structure of PRL2 is typical of dual‐specificity phosphatases and comprised of a central β‐sheet flanked by α‐helices (Fig 4A). PRL phosphatase structures have previously been reported for PRL1 28, 29 and PRL3 9, 30. The catalytic site of PRL2 is a shallow pocket formed by the two highly conserved, catalytic motifs: W65PFDD (the acidic loop) and C101…RA (the P‐loop). Superimposition of PRL2 with the crystal structure of PRL1 (PDB code: 1XM2) gave a root‐mean‐square deviation (RMSD) of 0.98 Å, suggesting that there were no major conformational changes associated with CNNM binding.
PRL1 has been observed to form trimers both in protein crystals and weakly in solution 28, 29. It is thought that the membrane association through the PRL C‐terminal prenylation favors trimerization in cells. While we have not observed any evidence of PRL2 or PRL3 trimers, formation of the PRL‐CNNM complex is compatible with PRL trimers if small adjustments are allowed in the orientation of the PRL‐binding extended loop (Movie EV1).
Complex formation is mediated by the CNNM loop (residues 418–430) which contacts the PRL2 catalytic site (Fig 4B–D). In agreement with the mutagenesis studies, Asp426 of CNNM3 plays a key role in binding PRL2. The aspartic acid side chain sits in the pocket formed by the phosphatase P‐loop and WPFDD motif and likely mimics the negatively charged phosphate of a bound substrate. In addition to the many polar contacts, Pro427 and Tyr429 provide a hydrophobic surface that contacts Leu105 of PRL2. The relatively small surface area between PRL2 and CNNM3 highlights the importance of the many electrostatic interactions. The structure is similar to the model of the complex based on interaction surfaces identified by mutagenesis 20.
The structure of PRL1 with a sulfate ion mimicking phosphocysteine explains why phosphorylation blocks CNNM binding (Fig 4E). The phosphate prevents CNNM binding through the steric and electrostatic repulsion of Asp426. We obtained the same result when we used the coordinates of the phosphocysteine in PTB1B 31 to model the structure of phosphorylated PRL.
PRL phosphatases are endogenously cysteine‐phosphorylated in response to Mg2+ levels
We next asked whether PRL proteins are cysteine‐phosphorylated in vivo. Analysis of cell lysates of HEK293 cells expressing FLAG‐PRL proteins showed two bands for each PRL on Phos‐tag gels but only a single band using standard SDS–PAGE (Fig 5A). Treatment of the lysates with OMFP led to an increase in intensity of the upper band, which we attribute to the phosphorylated form of the protein. As observed with purified PRL2, boiling of the lysates prior to electrophoresis hydrolyzed phosphocysteine and led to complete loss of the upper band. We also confirmed that the catalytic cysteine was required for formation of the phosphorylated form of the protein. Mutation of the catalytic cysteine (C104) but not the adjacent cysteine (C49) in FLAG‐PRL1 prevented formation of the phosphocysteine intermediate (Fig 5B).
Figure 5. PRLs in cells are cysteine‐phosphorylated in response to Mg2+ .
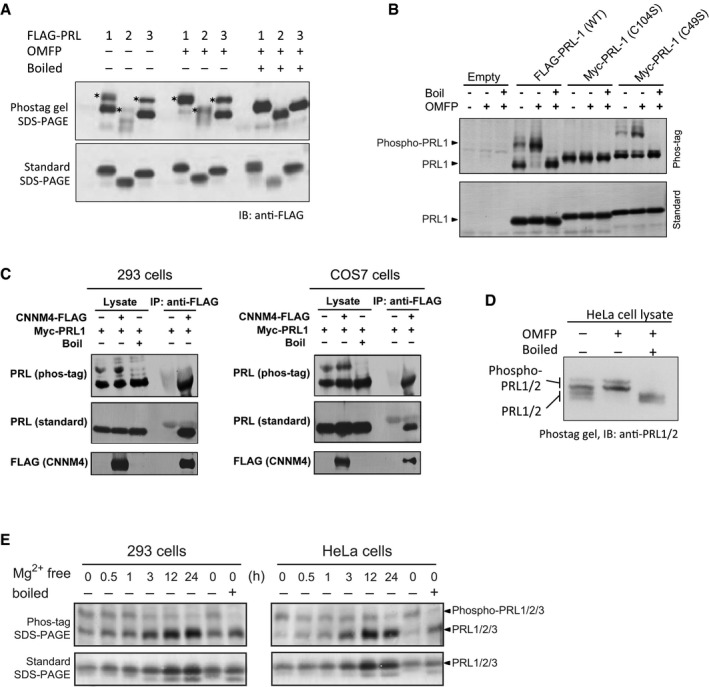
- PRLs expressed in HEK293 cells are intrinsically phosphorylated on cysteine. Phos‐tag analysis of cell lysates shows that close to 50% of transiently expressed FLAG‐PRL1, FLAG‐PRL2, and FLAG‐PRL3 are phosphorylated. The phosphorylated PRL bands are marked by an asterisk.
- Immunoblots of PRL1 expressed in HEK293 cells show that phosphorylation requires the catalytic cysteine (C104) but not the adjacent cysteine (C49) in the active site.
- Co‐immunoprecipitation of Myc‐PRL by FLAG‐CNNMs shows that only the unphosphorylated forms of PRL1 bind CNNM4.
- Endogenous PRLs in HeLa cells are cysteine‐phosphorylated.
- Levels of unphosphorylated PRL increase in cultured cells in response to removal of magnesium from the cultured medium.
Source data are available online for this figure.
A co‐immunoprecipitation experiment confirmed that phosphorylation of PRL1 blocked binding to CNNM4 in both 293 and COS7 cells (Fig 5C). CNNM4‐FLAG immunoprecipitates were analyzed by Phos‐tag and standard SDS–PAGE and probed to detect Myc‐PRL1. Both phosphorylated and unphosphorylated PRL1 were observed in the lysate, but only the unphosphorylated form co‐immunoprecipitated with CNNM4.
We then examined the phosphorylation status of endogenous PRL proteins in HeLa cells using a PRL antibody that recognizes PRL1 and PRL2. Western blotting of a Phos‐tag gel revealed four bands in the cell lysate that could be reduced to the two upper bands by treatment with OMFP (Fig 5D). Boiling prior to electrophoresis eliminated the upper bands and increased the staining of the two lower bands. Based on this, we conclude that PRL1 and PRL2 are significantly and endogenously cysteine‐phosphorylated in HeLa cells.
Finally, we asked whether the level of phosphorylation of PRL proteins varied in response to environmental conditions. We analyzed cell lysates of HEK293 and HeLa cells following a switch to a growth medium without Mg2+ (Fig 5E). Before Mg2+ depletion (at time 0), a significant amount of endogenous PRL was phosphorylated as detected on Phos‐tag gels using an antibody that recognizes all three PRL isoforms. Based on the immunostaining, most of the PRL proteins in HeLa cells and roughly half in HEK293 cells were phosphorylated. When subjected to Mg2+ depletion for 24 h, there was a very clear decrease in the ratio of phosphorylated to unphosphorylated PRLs in both cell lines (compare the 0‐ and 24‐h time points in the right most lanes). We also observed a large increase in the total level of PRL proteins as also evident in the SDS–PAGE gels without the Phos‐tag reagent. These results support a model in which unphosphorylated PRLs bind CNNM transporters to maintain intracellular Mg2+ levels.
Discussion
Cysteine phosphorylation is a rare protein modification only known to occur in a few contexts. It was first observed in bacteria as part of the phosphorylation cascade for bacterial sugar uptake 32. More recently, cysteine phosphorylation was observed in the regulation of bacterial transcriptional signaling 33. In eukaryotes, cysteine phosphorylation has long been known to occur as an enzymatic intermediate for protein phosphatases but not in a regulatory role 34. The chemical instability of phosphocysteine makes it difficult to detect. The sulfur–phosphorus bond is sensitive to low pH and to heating, so typical conditions for the preparation of samples for LC–MS and SDS–PAGE lead to its degradation. The techniques developed here, particularly the use of Phos‐tag gels, are likely to lead to the observation of phosphocysteine in new contexts. Specifically, it is possible that other PTPs such as DUSP19 that have the same alanine substitution in the catalytic site may be regulated by formation of a long‐lived phosphocysteine intermediate.
Phosphorylation of PRLs is not the direct result of kinase activity. PRLs are autophosphorylated in a reaction that depends on the availability of substrate and the phosphatase redox state. Phosphatidylinositol phosphates and a number of proteins have been proposed as PRL substrates, but as yet there is no clear consensus 3. We observed that recombinant PRL2 purified from E. coli is partially phosphorylated in the absence of added substrate (Fig 2D and F, PRL2 input lanes). The phosphorylation of PRL2 in E. coli rules out the requirement of a eukaryote‐specific substrate for its phosphorylation. The dephosphorylation rate appears to be an intrinsic property of the phosphatases. The rate measured in vitro with PRL2 is similar to the rate observed in cells, although the cell‐based experiment did not distinguish between new protein synthesis, ongoing phosphorylation, and dephosphorylation.
PRL phosphorylation is likely controlled in part by oxidation of catalytic site. Like protein tyrosine phosphatases, PRLs are prone to oxidation to a variety of catalytically inactive forms 9, 25. In vitro, PRLs can only be phosphorylated when fully reduced. Some of the forms such as the catalytic cysteine disulfide are reversible, while others are not. Widely considered a mechanism for regulating phosphatase activity, oxidation of the PRL active site also leads to a decrease in CBS‐pair domain binding. We observed a 200‐fold decrease in affinity following diamide treatment in agreement with pulldown experiments with H2O2 11, 12. The loss of affinity is likely due to a conformational change since, in our crystal structure, the catalytic cysteine does not directly contact the CBS‐pair domain.
Magnesium transport
The importance of PRL‐CNNM binding in cellular Mg2+ homeostasis has been shown by two groups, but the direction of Mg2+ transport and mechanism differ. Hardy and colleagues observed that shRNA knockdown of PRL2 in HEK293 cells decreased Mg2+ influx and concluded that PRL2 activates CNNM activity to increase cellular Mg2+ 12. In contrast, Funato and colleagues observed Mg2+ efflux and PRL3 inhibition of CNNM4 activity 11. The groups nonetheless concurred in the conclusion that a functional interplay between PRL and CNNM regulates Mg2+ levels.
Our studies support the model of inhibition of CNNM activity by PRL binding. Mutations in CNNM4 that specifically block PRL binding are active in Mg2+ efflux but prevent the inhibition by PRL3. The G433D CNNM3 mutation previously reported to block the interaction 12 likely unfolds the CBS‐pair domain as the glycine residue is buried in the structure. Attempts to express the recombinant CBS‐pair domain with the G433D mutation were unsuccessful (not shown), and the equivalent mutation in CNNM4, G492D, was inactive for Mg2+ efflux, likely due to its mislocalization (Fig EV2).
We observed a decrease in PRL phosphorylation in response to depletion of Mg2+ in the cell culture medium, which further supports a model in which PRLs inhibit CNNM function (Fig 5E). We additionally observed that Mg2+ depletion led to a large increase in total PRL protein as previously reported 12. Thus, while there are differences in the nature of the regulation, stimulatory or inhibitory, there is clear consensus that the PRL‐CNNM interaction plays a role in cellular Mg2+ homeostasis.
Role of PRLs in cancer
The identification of CNNM proteins as tumor suppressors and their inhibition by PRL proteins provides an explanation for the strong association of PRLs with metastatic disease 25. Through direct binding, PRLs inhibit CNNM‐dependent Mg2+ efflux to raise intracellular Mg2+ and promote proliferation. As shown here, PRLs can act as pseudophosphatases specifically for CNNM proteins. The aspartic acid of CNNM proteins binds to the active site and mimics the phosphate group of a substrate. The binding of pseudophosphatases is typically regulated by the phosphorylation of the binding partner 35, 36, but in the case of PRLs, the binding is regulated by phosphorylation and oxidation of the active site. Unlike other pseudophosphatases, PRLs retain their catalytic activity and, in fact, their catalytic activity regulates CNNM binding.
Guo et al 37 first showed the importance of the phosphatase activity in tumor promotion based on mutagenesis studies. They observed that an inactive PRL3 mutant in which the catalytic cysteine was replaced with serine failed to promote tumor formation. This same mutation in PRL2 prevents CNNM3 binding 12, and the mutation in PRL3 prevents CNNM4 binding 11. Surprisingly, pulldown assays with purified PRL2 and the isolated CBS‐pair domain from CNNM3 show that the catalytic cysteine is not essential for CNNM binding (Fig 2F). The difference between our in vitro assays and results in cultured cells is indicative of additional complexity in the PRL‐CNNM interaction. Of interest, cysteine 49 in PRL3, which can form a disulfide bond with the catalytic cysteine, is not required for catalytic activity but is required for PRL3 oncogenicity 11. The biological consequences of disulfide bond formation, cysteine phosphorylation, and PRL catalytic activity remain key questions for future investigation.
Materials and Methods
Protein expression and mutagenesis
Plasmids for bacterial expression of human PRL2 with a His‐tag and the human CNNM3 CBS‐pair domain as a GST fusion protein were described previously 12. Proteins were expressed in Escherichia coli BL21 and purified by affinity and size‐exclusion chromatography. The final buffer was 20 mM HEPES and 100 mM NaCl, pH 7. For crystallization, the GST tag was cleaved from the CNNM3 CBS‐pair domain using PreScission protease (GE Healthcare). Plasmids for mammalian cell expression of murine PRL1 and PRL3 and human PRL2 (amino acid sequence is identical to murine PRL2) and CNNM4 were described previously 11. Amino acid‐substituted mutants of CNNM and PRL proteins were generated using the QuikChange Site‐Directed Mutagenesis Kit (Agilent). To avoid spurious disulfide bond formation, a mutant of PRL2 (C95A, C96A, C119A, ΔCCVQ) was prepared with Cys95, Cys96 and Cys119 mutated to alanine and four C‐terminal amino acids deleted.
Isothermal titration calorimetry measurements
Experiments were carried out on a MicroCal VP‐ITC titration calorimeter (GE Healthcare) at 295 K in 20 mM HEPES and 100 mM NaCl, pH 7.0. Injections of 160 μM PRL2 were done into a stirred reaction cell filled with 16 μM of wild‐type or mutant CNNM3 CBS‐pair domain as a GST fusion protein. The calorimetric data were processed using the software package ORIGIN (version 7.0). Oxidized PRL2 was obtained using 5 mM diamide. Phosphorylated PRL2 was obtained by incubation with 3‐O‐methylfluorescein phosphate (OMFP) in the presence of 5 mM tris(2‐carboxyethyl)phosphine (TCEP).
Phosphatase assays
Assays were carried out using 6,8‐difluoro‐4‐methylumbelliferyl phosphate (DiFMUP) as a fluorescent substrate (Life Technologies). Experiments were done at 22°C in 20 mM HEPES and 100 mM NaCl, pH 7, with the addition of 10 mM DTT as a reducing agent. The concentration of DiFMUP was 100 μM, and the concentrations of the PRL enzymes and the CNNM3 CBS‐pair domain (WT and mutants) were 3 and 6 μM, respectively. Hydrolysis was initiated by the addition of substrate to the reaction mixture, and measurements were collected using a SpectraMax M5e (Molecular Devices) plate reader. The delay before the first measurement was approximately 3 min.
Mass spectrometry
150 μg of PRL2 WT or PRL2 C101A (both without the CCVQ prenylation motif) was untreated or treated with 2 nmol of OMFP for 45 min at 30°C. The protein samples were desalted using Amicon Ultra 0.5‐ml centrifugal filters with a 10‐kDa molecular weight cutoff. Then, the samples were dissolved in 50% acetonitrile, 0.1% formic acid for direct injection into ESI–MS using the Bruker amaZon speed ETD. Deconvolution of resulting mass spectra was performed using Compass software (Bruker).
NMR spectroscopy
1.6 mM of PRL2 triple mutant (C95A, C96A, C119A, ΔCCVQ) was mixed with 1.5 equivalents of OMFP in 20 mM HEPES, 100 mM NaCl, and 5 mM TCEP, pH 7.0. After the addition of 10% D2O, 30 spectra were recorded over 15 h at 25°C on a Bruker NMR spectrometer with a frequency of 243 MHz for 31P. Inverse‐gated 1H composite pulse decoupling was used with a 65° excitation pulse and 1.6‐s relaxation delay. 31P chemical shifts were internally referenced to the HDO signal.
Phos‐tag SDS–PAGE
Phos‐tag reagent was purchased from Wako Chemicals, Japan. For Phos‐tag gels, 20–40 μM Phos‐tag and 40–80 μM manganese chloride were added to the separating gel before polymerization. We used a Tris–tricine SDS–PAGE system with 17.5% acrylamide final concentration. For Western blots, gel acrylamide concentration was 12.5–15%, and the gel was soaked in transfer buffer with 1 mM EDTA before transfer to remove the Mn2+ ions.
In vitro pulldown assays
Recombinant GST‐CBS‐pair was bound to glutathione‐Sepharose beads and the beads washed twice with phosphate‐buffered saline. A threefold molar excess of OMFP‐treated or untreated PRL2 was added to the resin‐immobilized GST‐CBS‐pair domain. After incubation, a portion of the ensuing supernatant was reserved for analysis. The beads were washed in 20 mM HEPES, 100 mM NaCl, 0.02% IGEPAL, and 0.04% Triton X‐100, pH 7.0, and the bound proteins were analyzed by Phos‐tag or normal SDS–PAGE.
Cell culture, transfection, and immunofluorescence analysis
HeLa, COS7, and HEK293 cells were cultured in DMEM supplemented with 10% FBS and antibiotics. Expression plasmids were transfected in each cell with Lipofectamine 2000 (Invitrogen). Studies of CNNM4 localization were performed as previously described 13 using a rabbit anti‐FLAG antibody (Sigma) diluted in blocking buffer for CNNM4‐FLAG visualization and rhodamine‐phalloidin (Invitrogen) for F‐actin visualization. Cells were imaged with a FLUOVIEW FV1000 confocal scanning laser microscope (Olympus).
Mg2+ imaging analyses with Magnesium Green
Mg2+ imaging analyses with Magnesium Green were performed as described previously 13, with slight modifications. The cells were incubated with Mg2+ loading buffer (78.1 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 40 mM MgCl2, 5.5 mM glucose, and 5.5 mM HEPES–KOH, pH 7.4), including 2 μM Magnesium Green‐AM (Invitrogen), and were viewed using a microscope (IX81; Olympus) equipped with an ORCA‐Flash 4.0 CMOS camera (Hamamatsu) and a USH‐1030L mercury lamp (Olympus). Fluorescence was measured (excitation at 470–490 nm and emission at 505–545 nm) using MetaMorph software (Molecular Devices). Then, the buffer was changed to buffer without Mg2+ (MgCl2 was replaced with 60 mM NaCl). The data are presented as line plots (mean of 10 cells). After imaging analyses, cells were fixed with PBS containing 3.7% formaldehyde and subjected to immunofluorescence microscopy to confirm protein expression.
Crystallization and structure determination
Crystals of the PRL2/CBS‐pair complex were obtained with 2.5 mg/ml of PRL2 with an equimolar amount of CNNM3 CBS‐pair domain and 5 mM TCEP using 0.8 M succinic acid pH 8.0 supplemented with 30% w/v glucose. Diffraction data were collected at a wavelength of 0.63 Å on beamline A1 at the Cornell High‐Energy Synchrotron Source (CHESS). Data processing and scaling were performed with HKL‐2000 38.
Initial phases were acquired using molecular replacement with the CBS‐pair domain structure of the magnesium and cobalt efflux protein from Bordetella pertussis (PDB code 3JTF) and the structure of human protein tyrosine phosphatase PRL1 (PDB code 1RXD) as search models in Phaser 39. The model was extended manually using Coot 40 and improved by several cycles of refinement using Refmac 41 and translation–libration–screw (TLS) refinement. Structure figures were produced using PyMOL (http://www.pymol.org). The atomic coordinates and structure factors have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
Immunoprecipitation and immunoblotting
We obtained mouse and rabbit anti‐FLAG antibodies from Sigma, mouse and rabbit anti‐Myc antibodies from Santa Cruz, and mouse anti‐PRL2 monoclonal antibody from Millipore. Anti‐PRL polyclonal antibody that recognizes all three PRL isoforms equally was generated in the previous study 11. Transfected COS7 or HEK293 cells were washed with PBS and harvested with lysis buffer (20 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.5% Triton X‐100, 2 mM EDTA, and 1 mM phenylmethylsulfonyl fluoride). The lysates were centrifuged, and the supernatants were incubated with agarose bead‐conjugated anti‐FLAG antibody (Wako). The immunoprecipitated proteins were separated by SDS–PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane. After blocking, membranes were incubated with primary antibodies and then with alkaline phosphatase‐conjugated anti‐rabbit IgG (Promega) for chromogenic detection with 5‐bromo‐4‐chloro‐3‐indolyl phosphate/nitro blue tetrazolium.
For immunoprecipitation and detection of endogenous PRL2, HeLa cells were lysed in 50 mM HEPES, 150 mM NaCl, and 1% Triton X‐100, pH 7.5 with protease inhibitors (Roche). After centrifugation, supernatants were incubated with anti‐PRL2 antibody (Millipore), and this mixture was then incubated with Protein A Dynabeads (ThermoFisher). The complex was finally eluted from the beads using strong SDS loading dye (70 mM Tris–HCl, 10% SDS, 7.5% glycerol, 10% β‐mercaptoethanol, and 0.02% bromophenol blue, pH 6.8) without boiling. The eluted proteins were separated by Phos‐tag SDS–PAGE and transferred to a PVDF membrane. The membrane was blocked and first incubated with mouse anti‐PRL2 (Millipore), then with HRP‐conjugated anti‐mouse secondary antibody (Jackson ImmunoResearch 115‐035‐071 1:5,000). Chemiluminescent detection on film was done using the Amersham ECL Prime detection kit (GE Healthcare).
Accession numbers
The PDB code of PRL2‐CNNM3 CBS‐pair complex is 5K22.
Author contributions
IG, YF, HW, and GK performed experiments and data analysis. IG, MY, HM, and KG interpreted the data and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Movie EV1
Review Process File
Source Data for Figure 5
Acknowledgements
We thank Michel Tremblay for the PRL2 and CNNM3 plasmids. This work was supported by the Japan Society for the Promotion of Science (KAKENHI) grant 26460364 to Y. Funato and 26291042, 26111007, and 16K14723 to H. Miki, and the Canadian Institutes of Health Research grant MOP‐14219 to K. Gehring.
EMBO Reports (2016) 17: 1890–1900
Contributor Information
Hiroaki Miki, Email: hmiki@biken.osaka-u.ac.jp.
Kalle Gehring, Email: kalle.gehring@mcgill.ca.
References
- 1. Tonks NK (2013) Protein tyrosine phosphatases–from housekeeping enzymes to master regulators of signal transduction. FEBS J 280: 346–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Saha S, Bardelli A, Buckhaults P, Velculescu VE, Rago C, St Croix B, Romans KE, Choti MA, Lengauer C, Kinzler KW et al (2001) A phosphatase associated with metastasis of colorectal cancer. Science 294: 1343–1346 [DOI] [PubMed] [Google Scholar]
- 3. Rios P, Li X, Kohn M (2013) Molecular mechanisms of the PRL phosphatases. FEBS J 280: 505–524 [DOI] [PubMed] [Google Scholar]
- 4. Diamond RH, Cressman DE, Laz TM, Abrams CS, Taub R (1994) PRL‐1, a unique nuclear protein tyrosine phosphatase, affects cell growth. Mol Cell Biol 14: 3752–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bessette DC, Qiu D, Pallen CJ (2008) PRL PTPs: mediators and markers of cancer progression. Cancer Metastasis Rev 27: 231–252 [DOI] [PubMed] [Google Scholar]
- 6. Kobayashi M, Chen S, Gao R, Bai Y, Zhang ZY, Liu Y (2014) Phosphatase of regenerating liver in hematopoietic stem cells and hematological malignancies. Cell Cycle (Georgetown, Tex.) 13: 2827–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Denu JM, Dixon JE (1995) A catalytic mechanism for the dual‐specific phosphatases. Proc Natl Acad Sci USA 92: 5910–5914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang ZY, VanEtten RL (1991) Pre‐steady‐state and steady‐state kinetic analysis of the low molecular weight phosphotyrosyl protein phosphatase from bovine heart. J Biol Chem 266: 1516–1525 [PubMed] [Google Scholar]
- 9. Kozlov G, Cheng J, Ziomek E, Banville D, Gehring K, Ekiel I (2004) Structural insights into molecular function of the metastasis‐associated phosphatase PRL‐3. J Biol Chem 279: 11882–11889 [DOI] [PubMed] [Google Scholar]
- 10. McParland V, Varsano G, Li X, Thornton J, Baby J, Aravind A, Meyer C, Pavic K, Rios P, Kohn M (2011) The metastasis‐promoting phosphatase PRL‐3 shows activity toward phosphoinositides. Biochemistry 50: 7579–7590 [DOI] [PubMed] [Google Scholar]
- 11. Funato Y, Yamazaki D, Mizukami S, Du L, Kikuchi K, Miki H (2014) Membrane protein CNNM4‐dependent Mg2+ efflux suppresses tumor progression. J Clin Invest 124: 5398–5410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hardy S, Uetani N, Wong N, Kostantin E, Labbe DP, Begin LR, Mes‐Masson A, Miranda‐Saavedra D, Tremblay ML (2015) The protein tyrosine phosphatase PRL‐2 interacts with the magnesium transporter CNNM3 to promote oncogenesis. Oncogene 34: 986–995 [DOI] [PubMed] [Google Scholar]
- 13. Yamazaki D, Funato Y, Miura J, Sato S, Toyosawa S, Furutani K, Kurachi Y, Omori Y, Furukawa T, Tsuda T et al (2013) Basolateral Mg2+ extrusion via CNNM4 mediates transcellular Mg2+ transport across epithelia: a mouse model. PLoS Genet 9: e1003983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang CY, Shi JD, Yang P, Kumar PG, Li QZ, Run QG, Su YC, Scott HS, Kao KJ, She JX (2003) Molecular cloning and characterization of a novel gene family of four ancient conserved domain proteins (ACDP). Gene 306: 37–44 [DOI] [PubMed] [Google Scholar]
- 15. de Baaij JH, Stuiver M, Meij IC, Lainez S, Kopplin K, Venselaar H, Muller D, Bindels RJ, Hoenderop JG (2012) Membrane topology and intracellular processing of cyclin M2 (CNNM2). J Biol Chem 287: 13644–13655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Baaij JH, Hoenderop JG, Bindels RJ (2015) Magnesium in man: implications for health and disease. Physiol Rev 95: 1–46 [DOI] [PubMed] [Google Scholar]
- 17. Stuiver M, Lainez S, Will C, Terryn S, Gunzel D, Debaix H, Sommer K, Kopplin K, Thumfart J, Kampik NB et al (2011) CNNM2, encoding a basolateral protein required for renal Mg2+ handling, is mutated in dominant hypomagnesemia. Am J Hum Genet 88: 333–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rubin H (1975) Central role for magnesium in coordinate control of metabolism and growth in animal cells. Proc Natl Acad Sci USA 72: 3551–3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wolf FI, Trapani V (2012) Magnesium and its transporters in cancer: a novel paradigm in tumour development. Clin Sci (London, England: 1979) 123: 417–427 [DOI] [PubMed] [Google Scholar]
- 20. Kostantin E, Hardy S, Valinsky WC, Kompatscher A, de Baaij JH, Zolotarov Y, Landry M, Uetani N, Martinez‐Cruz LA, Hoenderop JG et al (2016) Inhibition of the PRL‐2/CNNM3 Protein Complex Formation Decreases Breast Cancer Proliferation and Tumor Growth. J Biol Chem 291: 10716–10725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Corral‐Rodriguez MA, Stuiver M, Abascal‐Palacios G, Diercks T, Oyenarte I, Ereno‐Orbea J, de Opakua AI, Blanco FJ, Encinar JA, Spiwok V et al (2014) Nucleotide binding triggers a conformational change of the CBS module of the magnesium transporter CNNM2 from a twisted towards a flat structure. Biochem J 464: 23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gulerez I (2015) Structural Characterization of the Complex between the Oncogenic Phosphatase PRL2 and the CBS pair domain of the Magnesium Transporter CNNM3. MSc Dissertation. McGill University, Montreal, Canada: [Google Scholar]
- 23. Kinoshita E, Kinoshita‐Kikuta E, Takiyama K, Koike T (2006) Phosphate‐binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics 5: 749–757 [DOI] [PubMed] [Google Scholar]
- 24. Pas HH, Meyer GH, Kruizinga WH, Tamminga KS, van Weeghel RP, Robillard GT (1991) 31phospho‐NMR demonstration of phosphocysteine as a catalytic intermediate on the Escherichia coli phosphotransferase system EIIMtl. J Biol Chem 266: 6690–6692 [PubMed] [Google Scholar]
- 25. Funato Y, Miki H (2014) Reversible oxidation of PRL family protein‐tyrosine phosphatases. Methods 65: 184–189 [DOI] [PubMed] [Google Scholar]
- 26. Hirata Y, Funato Y, Takano Y, Miki H (2014) Mg2+‐dependent interactions of ATP with the cystathionine‐beta‐synthase (CBS) domains of a magnesium transporter. J Biol Chem 289: 14731–14739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ereno‐Orbea J, Oyenarte I, Martinez‐Cruz LA (2013) CBS domains: Ligand binding sites and conformational variability. Arch Biochem Biophys 540: 70–81 [DOI] [PubMed] [Google Scholar]
- 28. Sun JP, Wang WQ, Yang H, Liu S, Liang F, Fedorov AA, Almo SC, Zhang ZY (2005) Structure and biochemical properties of PRL‐1, a phosphatase implicated in cell growth, differentiation, and tumor invasion. Biochemistry 44: 12009–12021 [DOI] [PubMed] [Google Scholar]
- 29. Jeong DG, Kim SJ, Kim JH, Son JH, Park MR, Lim SM, Yoon TS, Ryu SE (2005) Trimeric structure of PRL‐1 phosphatase reveals an active enzyme conformation and regulation mechanisms. J Mol Biol 345: 401–413 [DOI] [PubMed] [Google Scholar]
- 30. Kim KA, Song JS, Jee J, Sheen MR, Lee C, Lee TG, Ro S, Cho JM, Lee W, Yamazaki T et al (2004) Structure of human PRL‐3, the phosphatase associated with cancer metastasis. FEBS Lett 565: 181–187 [DOI] [PubMed] [Google Scholar]
- 31. Pannifer AD, Flint AJ, Tonks NK, Barford D (1998) Visualization of the cysteinyl‐phosphate intermediate of a protein‐tyrosine phosphatase by x‐ray crystallography. J Biol Chem 273: 10454–10462 [DOI] [PubMed] [Google Scholar]
- 32. Pas HH, Robillard GT (1988) S‐phosphocysteine and phosphohistidine are intermediates in the phosphoenolpyruvate‐dependent mannitol transport catalyzed by Escherichia coli EIIMtl. Biochemistry 27: 5835–5839 [DOI] [PubMed] [Google Scholar]
- 33. Sun F, Ding Y, Ji Q, Liang Z, Deng X, Wong CC, Yi C, Zhang L, Xie S, Alvarez S et al (2012) Protein cysteine phosphorylation of SarA/MgrA family transcriptional regulators mediates bacterial virulence and antibiotic resistance. Proc Natl Acad Sci USA 109: 15461–15466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guan KL, Dixon JE (1991) Evidence for protein‐tyrosine‐phosphatase catalysis proceeding via a cysteine‐phosphate intermediate. J Biol Chem 266: 17026–17030 [PubMed] [Google Scholar]
- 35. Kharitidi D, Manteghi S, Pause A (2014) Pseudophosphatases: methods of analysis and physiological functions. Methods 65: 207–218 [DOI] [PubMed] [Google Scholar]
- 36. Reiterer V, Eyers PA, Farhan H (2014) Day of the dead: pseudokinases and pseudophosphatases in physiology and disease. Trends Cell Biol 24: 489–505 [DOI] [PubMed] [Google Scholar]
- 37. Guo K, Li J, Tang JP, Koh V, Gan BQ, Zeng Q (2004) Catalytic domain of PRL‐3 plays an essential role in tumor metastasis: formation of PRL‐3 tumors inside the blood vessels. Cancer Biol Ther 3: 945–951 [DOI] [PubMed] [Google Scholar]
- 38. Otwinowski Z, Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- 39. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- 41. Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ (1999) Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr D Biol Crystallogr 55: 247–255 [DOI] [PubMed] [Google Scholar]
- 42. Cates CA, Michael RL, Stayrook KR, Harvey KA, Burke YD, Randall SK, Crowell PL, Crowell DN (1996) Prenylation of oncogenic human PTP(CAAX) protein tyrosine phosphatases. Cancer Lett 110: 49–55 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Movie EV1
Review Process File
Source Data for Figure 5