

Abstract
Despite being one of the most studied proteases in bacteria, very little is known about the role of ClpXP in mitochondria. We now present evidence that mammalian CLPP has an essential role in determining the rate of mitochondrial protein synthesis by regulating the level of mitoribosome assembly. Through a proteomic approach and the use of a catalytically inactive CLPP, we produced the first comprehensive list of possible mammalian ClpXP substrates involved in the regulation of mitochondrial translation, oxidative phosphorylation, and a number of metabolic pathways. We further show that the defect in mitoribosomal assembly is a consequence of the accumulation of ERAL1, a putative 12S rRNA chaperone, and novel ClpXP substrate. The presented data suggest that the timely removal of ERAL1 from the small ribosomal subunit is essential for the efficient maturation of the mitoribosome and a normal rate of mitochondrial translation.
Keywords: CLPP, ERAL1, mitochondrial ribosome assembly, OXPHOS deficiency
Subject Categories: Metabolism; Post-translational Modifications, Proteolysis & Proteomics; Protein Biosynthesis & Quality Control
Introduction
Proteases have an essential role in safeguarding cell survival by maintaining protein homeostasis, which is threatened by different internal and external insults. They also perform another, equally important role in cellular physiology, namely regulated proteolysis that ensures constant turnover of house‐keeping proteins as well as the elimination of specific proteins when they are no longer needed. The irreversible nature of this process demands rigorous selectivity in the choice of substrates and the degradation timing. In prokaryotes, regulated protein degradation is governed by energy‐dependent proteases, of which ClpXP is probably the best studied as it contributes to a large fraction of protein turnover in bacteria (Sauer & Baker, 2011). ClpXP is a highly conserved proteasome‐like machinery present in all bacterial species and is also found in the mitochondria and chloroplasts of eukaryotic cells (Sauer & Baker, 2011). In mammals, ClpXP protein substrates are recognized and unfolded by the hexameric AAA+ protein CLPX that delivers them to the CLPP protease for degradation (Sauer & Baker, 2011). The CLPP protease is formed by one or two heptameric rings and on its own can degrade small peptides but has no significant activity against proteins (Sauer & Baker, 2011). CLPX appears to be the only unfoldase present in mammalian mitochondria that interacts with the CLPP protease (Kang et al, 2002). Some eukaryotes, like Saccharomyces cerevisiae, lack CLPP but still possess the CLPX unfoldase that has specific roles, independent of the protease activity, like a recently shown function in the regulation of heme synthesis (Kardon et al, 2015).
ClpXP is important for the viability and growth, cell cycle progression, recovery from environmental insults, and sporulation in many bacterial species (Damerau & St John, 1993; Jenal & Fuchs, 1998). Much less is known about the role of mitochondrial ClpXP, and we just begin to understand the different aspects of its impact on mitochondrial physiology. In humans, CLPP deficiency causes Perrault syndrome characterized by sensorineural hearing loss and a premature ovarian failure (Jenkinson et al, 2013). Recently, it was shown that CLPP‐deficient mice faithfully replicate major physiological aspects of the human disease (Gispert et al, 2013). Remarkably, deletion of mitochondrial Clpp leads to improved health and increased life span in the filamentous fungus, Podospora anserina (Fischer et al, 2013).
The present study aimed to analyze the role of CLPP in mammalian mitochondria by creating Clpp knockout mice. Depletion of CLPP leads to the development of moderate respiratory deficiency caused by ineffective mitochondrial protein synthesis, due to decreased amounts of fully assembled mitochondrial ribosomes (55S, mitoribosomes). We show that in the absence of CLPP, ERAL1, a putative 12S rRNA chaperone, strongly associates with the small ribosomal subunit (28S), therefore preventing formation of functional mitoribosomes.
Results
Generation of Clpp knockout mice
To determine the in vivo function of CLPP in mammals, we generated a conditional knockout allele of the mouse Clpp gene (Fig 1A and B). CLPP‐deficient mice (Clpp −/− ) were obtained from intercrossing of Clpp +/− animals (Fig 1C), and the loss of CLPP was confirmed on both transcript and protein level (Fig 1D and E). Although we regularly acquired Clpp −/− mice, they were not born in Mendelian ratios (Table EV1), likely dying in utero, since no postnatal lethality prior to the age of weaning was observed. Currently, we follow an aging cohort of CLPP‐deficient mice that show the same survival as respective wild‐type littermates at least up to 2 years of age. This is in contrast to findings on a similar mouse model reported to have increased postnatal mortality and possibly shorter life span, although we also observed general sterility of both CLPP‐deficient genders and shorter stature of the mice, as describe earlier (Gispert et al, 2013). Besides differences in the genetic approach used to generate CLPP‐deficient mice (conditional knockout targeting versus gene‐trap technology), also different hygienic level in animal facilities might contribute to these disparities. Overall, our results suggest that CLPP function is essential in some critical period during development and that in the postnatal life mice can tolerate a CLPP deficiency.
Figure 1. Creation of Clpp knockout mice.

- Targeting strategy for the conditional disruption of the Clpp gene. Map of the genomic locus region of the wild‐type Clpp gene (wild type), the targeting vector, the Clpp locus after homologous recombination (Targeted Allele) and targeted allele after Flp‐recombinase‐mediated excision of the PuroR cassette (Conditional KO Allele Clpp loxP), and the knocked‐out allele after Cre‐recombinase‐mediated excision of the exons 3–5 (Constitutive KO Allele Clpp − ). The position of primers used for genotyping is indicated by red arrows.
- Southern blot analysis of targeted ES cells. Positions of wild‐type (WT, 14.7 kb) and targeted (loxP, 12.8 kb) alleles are indicated.
- Genotyping analysis of wild‐type (Clpp +/+), heterozygous (Clpp +/−), and homozygous (Clpp −/−) Clpp knockout animals. DNA fragments corresponding to the wild‐type (WT, 199 bp) and knockout (KO, 273 bp) Clpp loci are indicated.
- Analysis of Clpp transcript levels in heart (n = 4). Bars represent mean ± standard deviation (SD). Student's t‐test was used to determine the level of statistical difference.
- Western blot analysis of CLPP protein levels in heart (He), quadriceps (Quad), liver (Li), kidney (Ki), lung (Lu), brain (Br), spleen (Sp), and pancreas (Pa). Tubulin and actin are used as loading controls. The arrowhead indicates position of CLPP, and the star indicates position of cross‐reacting bands.
Source data are available online for this figure.
Loss of CLPP causes moderate respiratory deficiency
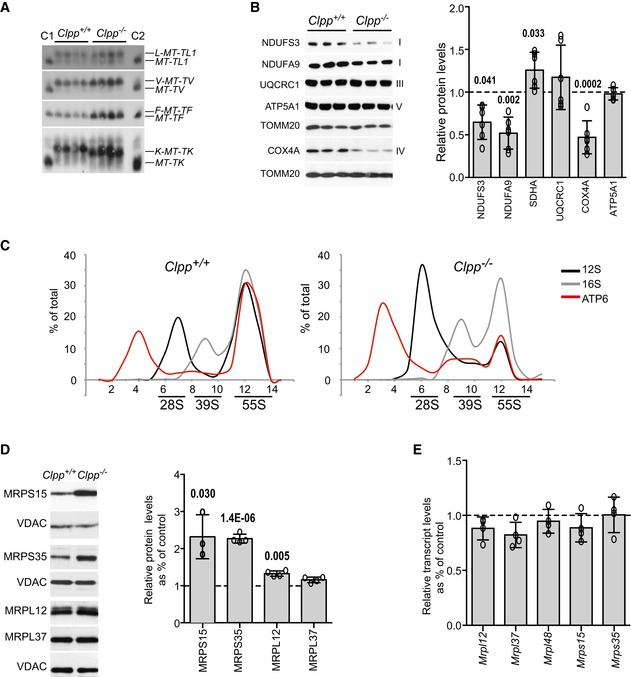
To understand the role of CLPP in the organelle physiology, we analyzed the global protein changes in the presence and absence of CLPP. Proteomes of sucrose‐cushion‐purified heart mitochondria were analyzed by liquid chromatography–tandem mass spectrometry (LC‐MS/MS). With this approach, we were able to identify 736 proteins, of which 595 (81%) were mitochondrial (Table EV2), with high reproducibility as determined by Pearson correlation coefficient for biological replicates R > 0.97 (Fig EV1A). In addition, we detected similar numbers of significantly upregulated and downregulated proteins (51 and 62, respectively, at a FDR < 0.05) upon CLPP depletion (Table EV2).
Figure EV1. Mitochondrial RC complex levels in Clpp knockout mice.

-
ACorrelation matrix of log2‐transformed label‐free quantification (LFQ) intensities for each biological replicate of Clpp knockout (KO) and wild‐type (WT) heart mitochondria. Reported values represent Pearson correlation coefficient.
-
BWestern blot analysis of respiratory chain complexes in heart, skeletal muscle, and liver mitochondria separated by BN‐PAGE, of control (Clpp +/+) and Clpp knockout (Clpp −/−) mice. Immunoblotting was performed to detect subunits of C I (NDUFA9), C II (SDHA), C III (UQCRC1), C IV (COXI), and C V (ATP5A1). Presence of subcomplex of complex V that is likely the F1 part of the ATP synthase is indicated with an arrow.
-
C, DIn‐gel activity of complex (C) I and (D) IV in heart and skeletal muscle mitochondria performed after BN‐PAGE. Position of supercomplexes (SC) and individual complexes (C I and C IV) is indicated.
Source data are available online for this figure.
GO term analysis (Eden et al, 2009) was used to obtain a more systematic evaluation of specific changes in our dataset related to CLPP deficiency (Fig 2B). Proteins that are annotated as mitochondrial respiratory chain (RC), in particular complex I (NADH:ubiquinone oxidoreductase, EC 1.6.5.3) subunits, showed a clear downregulation in CLPP‐deficient mitochondria (Fig 2A and B, and Table EV2). In contrast, mitochondrial translation machinery was highly upregulated, as the levels of multiple mitoribosome subunits (mainly part of 28S) and proteins involved in translation initiation, elongation, and ribosome recycling were increased (Fig 2A and Table EV2). Taken together, these results demonstrate that loss of CLPP causes downregulation of RC subunits and alteration in mitochondrial gene expression, indicating respiratory chain deficiency.
Figure 2. Mitochondrial function in wild‐type (Clpp +/+) and Clpp knockout (Clpp −/−) heart mitochondria.

- Volcano plot of proteomic data reveals significant changes in proteins of the respiratory chain and factors involved in mitochondrial protein synthesis (FDR < 0.05).
- Bar chart representing enriched Gene Ontology (GO) terms. FDR was calculated by Benjamini–Hochberg correction (n = 4).
- Respiratory chain enzyme activities. Relative enzyme activities of respiratory chain enzyme C I (NADH coenzyme Q reductase), C I/III (NADH cytochrome c reductase), C II (succinate dehydrogenase), C II/III (succinate cytochrome c reductase), and C IV (cytochrome c oxidase) in mitochondrial extracts from wild‐type and Clpp knockout hearts (n = 5). Bars present mean levels ± SD. Statistical difference was calculated by Student's t‐test, and changes are presented in the figure (n = 5).
- Substrate‐dependent oxygen consumption rates in isolated heart mitochondria. Complex I substrates (pyruvate, malate, glutamate); complex II substrate (succinate). State 3: ADP stimulated oxygen consumption rate; State 4: respiration rate after addition of oligomycin; Uncoupled: oxygen consumption rate in the presence of FCCP. Bars present mean levels ± SD. Student's t‐test was used to determine the level of statistical difference (n = 7).
- Decrease in steady‐state levels of respiratory chain subunits in Clpp knockout hearts relative to wild type (set to 1). VDAC or TOMM20 was used as loading controls. Bars present mean levels ± SD. Student's t‐test was used to determine the level of statistical difference (n = 4).
- Blue Native PAGE (BN‐PAGE) analysis of the respiratory chain supercomplexes followed by Western blot analysis using antibodies against C I (anti‐NDUFA9), C III (anti‐UQCRC1), and C IV (anti‐COX4A).
Source data are available online for this figure.
Thus, we next measured RC enzyme activities in CLPP‐deficient heart mitochondria (Fig 2C). An isolated complex I (C I) deficiency was detected at 15 weeks of age and was followed by complex IV deficiency (C IV) at 35 weeks (Fig 2C). Additional experiments allowed us to further characterize the nature of the RC deficiency in Clpp knockout mice. We measured the oxidation of complex I or complex I + II substrates in freshly isolated heart mitochondria and found a general reduction in all inducible respiratory states resulting from the loss of CLPP (Fig 2D). The decrease in the RC complex activities was accompanied by a significant reduction in the steady‐state levels of RC subunits encoded either by mitochondrial or nuclear DNA (Fig 2E). Likewise, a clear reduction in the level of assembled complexes I and IV was observed in CLPP‐deficient heart and skeletal muscle mitochondria, while liver mitochondria showed a milder decrease in both RC complexes (Fig EV1B). Reduction in complex I and IV levels in heart and skeletal muscle mitochondria was mirrored by a reduction in the level of supercomplexes (Fig 2F) and a decrease in in‐gel activity (Fig EV1C and D). An additional sign of existing respiratory chain dysfunction was the presence of a subcomplex of complex V (Fig EV1B) that is likely the F1 part of the ATP synthase and was found in other models defective in mitochondrial gene expression like Mterf3 (Park et al, 2007), Mterf4 (Camara et al, 2011), and Dars2 knockout mice (Dogan et al, 2014).
CLPP deficiency increases mitochondrial transcription rate
The respiratory dysfunction in Clpp knockout mice prompted us to inspect mtDNA levels and mitochondrial expression patterns. Southern blot analysis showed no increase in the level of mtDNA in Clpp knockout hearts (Fig 3A), in contrast to a previous report on a similar model (Gispert et al, 2013). The difference might stem from methods applied to examine mtDNA levels (Southern blot analysis versus RT–PCR) or from the genetic approach used to generate CLPP‐deficient mice (conditional knockout targeting versus gene‐trap technology) (Gispert et al, 2013). Remarkably, this study also reports that the marked rise in the amount of mtDNA is not accompanied by an increase in steady‐state levels of mitochondrial transcription factor A (TFAM) (Gispert et al, 2013), a protein that packages mtDNA and is absolutely necessary for mtDNA maintenance (Freyer et al, 2010), making this result hard to interpret.
Figure 3. MtDNA and mtRNA levels in wild‐type (Clpp +/+) and Clpp knockout (Clpp −/−) hearts.

-
AQuantification of mtDNA levels after Southern blot analysis. Bars present mean levels ± SD (n = 4).
-
B, CNorthern blot analysis and corresponding quantification of mitochondrial (B) rRNAs and mRNAs or (C) tRNAs. 18S rRNA is used as a loading control. Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (12S n = 5 and the rest of samples n = 4).
-
DWestern blot analysis of LRPPRC levels. VDAC was used as a loading control.
-
EIn organello transcription analysis upon 60 min labeling with α‐32P‐UTP.
Source data are available online for this figure.
We next examined the levels of mitochondrial RNAs and detected a substantial increase in most (Fig 3B and C). Matching the increase in steady‐state levels of mitochondrial transcripts, we found higher levels of LRPPRC, an RNA binding protein essential for the stability and polyadenylation of mitochondrial mRNAs (Fig 3D) (Ruzzenente et al, 2012). Remarkably, a twofold increase in 12S rRNA was observed, while the levels of 16S rRNA were not significantly different (Fig 3B). De novo transcription rates in CLPP‐deficient mitochondria were also increased, matching the observed higher levels of mtRNA species (Fig 3E). An enhanced transcription is often detected in mouse models with defects in mitochondrial protein synthesis, as a likely compensatory mechanism for respiratory chain deficiency (Camara et al, 2011; Dogan et al, 2014).
Loss of CLPP decreases the amount of fully formed mitoribosomes and diminishes protein synthesis
To assess mitochondrial translation, we measured the rate of de novo protein synthesis in CLPP‐deficient heart mitochondria and detected a general decrease in mitochondrial translation rates (Fig 4A). The absence of a major protease might prevent the generation of peptides and possibly decreases the pool of free amino acids in the mitochondrial matrix. However, the analysis of different tRNAs showed that aminoacylation levels of mt‐tRNAs is not diminished by CLPP deficiency and might be even increased, corresponding to higher steady‐state levels of mt‐tRNAs (Fig EV2A).
Figure 4. Analysis of mitochondrial protein synthesis and mitoribosome assembly in wild‐type (Clpp +/+) and Clpp knockout (Clpp −/−) heart mitochondria.

- De novo synthesized mitochondrial proteins were isolated after labeling with 35S‐methionine for 1 h. Positions of individual mitochondrial encoded proteins are indicated.
- Western blot analysis of RC subunits in MEFs upon 48‐h treatment with actinonin, doxycycline, and chloramphenicol (+ antibiotic), followed by a 48‐h recovery period (− antibiotic). During both periods, proteins were isolated at 12, 24, and 48 h. Antibodies against subunits of C I (NDUFB6), C III (UQCRC1), C IV (COXI), MRPS35, and MRPL37 were used as indicated.
- Quantitative analysis of the COXI recovery upon treatment with antibiotics (actinonin, doxycycline (DOXY), and chloramphenicol (CAP)) as in (B). The values represent ratios of recovered (48 h, − antibiotics) versus untreated (0 h) COXI levels. Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (actinonin and CAP n = 3, DOXY n = 4).
- Sucrose density gradient sedimentation profile of ribosomal proteins and mtRNAs from heart mitochondria. The migration of ribosomal subunits (28S and 39S) and mitoribosome (55S) was detected by immunoblotting with antibodies against MRPS35 and MRPL12. The profile of a given mt‐RNA is calculated after qRT–PCR analysis of individual fractions and corrected for its abundance in the cell according to results from Northern blot analysis. Results of individual experiments are provided for 12S and 16S rRNA, and for mt‐mRNAs, results of distribution from two independent experiments using probes against ATP6, COXIII, and ND5 were combined. Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (n = 3).
- Relative association of 12S rRNA, 16S rRNA, and mt‐mRNAs with mitoribosomes (55S) in Clpp knockout mitochondria comparing to wild type. Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (12S and 16S n = 3; mRNA n = 6).
Source data are available online for this figure.
Figure EV2. Mitochondrial tRNA aminoacylation grade and steady‐state levels of ribosomal subunits.
- Aminoacylation assay for mitochondrial tRNA Leu1 , tRNA Val , tRNA Phe, and tRNA Lys in heart of wild‐type (Clpp +/+) and Clpp knockout (Clpp −/−) mice. Positions of charged (AA‐MT‐tRNA‐AA) and uncharged (MT‐tRNA‐AA) tRNAs are indicated. Deacylated tRNAs from wild‐type and Clpp knockout mice are used as controls (C1 and C2, respectively).
- Western blot analysis and respective quantification of steady‐state levels of respiratory chain subunits from Clpp knockout MEFs (Clpp −/−) relative to wild type (Clpp +/+). Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (ATP5A1 n = 5; the rest n = 6).
- Sucrose density gradient sedimentation profile of ribosomal transcripts from heart mitochondria. Individual mt‐RNAs were analyzed by qRT–PCR, and the abundance of RNA in each fraction is represented as percentage of the sum of the total abundance in all fractions (set to 100%).
- Western blot analysis and respective quantification of steady‐state levels of mitochondrial 28S and 39S ribosomal subunits from Clpp knockout heart mitochondria relative to wild type (set to 1). Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (MRPS15 n = 3; the rest n = 4).
- Quantitative PCR analysis of the transcript levels encoding mitochondrial 28S and 39S ribosomal subunits in Clpp knockout heart mitochondria relative to wild type (set to 1). Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (n = 4).
Source data are available online for this figure.
To further understand the defect in mitochondrial protein synthesis caused by the loss of CLPP, we treated immortalized mouse embryonic fibroblasts (MEFs) with antibiotics that block mitochondrial translation at different steps. Clpp knockout MEFs recapitulated a decrease in the steady‐state level of individual OXPHOS subunits observed in CLPP‐deficient hearts (Fig EV2B). We monitored the level of mtDNA‐encoded COXI/MT‐CO1 subunit upon 48 h incubation in the presence of the antibiotic, followed by a recovery period of additional 2 days. Treatment of MEFs with actinonin, a peptidomimetic antibiotic, which inhibits peptide deformylase that catalyzes the removal of the formyl group from the starter methionine (Richter et al, 2013), demonstrated that Clpp knockout cells have a lower recovery of COXI levels, in agreement with defective mitochondrial synthesis and possibly diminished translation initiation (Fig 4B and C). Doxycycline and chloramphenicol are known inhibitors of mitochondrial translation elongation, occupying the ribosomal A‐site, the former binding to the small (28S), and the latter to the large (39S) ribosomal subunit (LaPolla & Lambowitz, 1977; Kearsey & Craig, 1981). Chloramphenicol had an identical effect on both cell lines, resulting in an almost full recovery of the COXI levels (Fig 4B and C). In contrast, doxycycline‐treated Clpp knockout cells were not able to recover COXI levels, even though this antibiotic had an overall milder effect on wild‐type MEFs (Fig 4B and C). This result suggests that loss of CLPP specifically affects the function of the 28S small ribosome.
To assess the state of mitoribosome assembly, we performed sucrose gradient sedimentation analyses of mitochondrial extracts. Following the distribution of MRPS35, we observed high accumulation of the 28S small ribosome in Clpp knockout mitochondria, while levels of fully assembled mitoribosomes (55S) were diminished (Fig 4D). The distribution of MRPL12, a subunit of the 39S large ribosome, further confirmed these results (Fig 4D). In agreement with previous results, we detected a high accumulation of 12S rRNA in the 28S fraction, while the distribution of 16S rRNA was less affected by CLPP deficiency (Fig 4D and EV2C). Both 12S and 16S rRNAs showed decreased association with the fully assembled mitoribosome, matching the decrease observed on the protein level (Fig 4D and E). Different mt‐mRNAs presented an abnormal pattern of distribution resulting in a smaller proportion of mt‐mRNAs bound to the mitoribosome (Figs 4D and E, and EV2C). Correspondingly, the steady‐state levels of individual 28S ribosomal proteins were highly increased, while the subunits of 39S ribosome showed milder changes (Fig EV2D). The upregulation was likely the result of increased stability, rather than synthesis, as transcript levels of these ribosomal subunits were normal (Fig EV2E).
CLPP‐substrate screen reveals proteins involved in mitochondrial translation
To identify mammalian ClpXP substrates, we took advantage of the ability of inactivated CLPP to accept and retain proteins translocated into its chamber. We constructed a FLAG‐tagged variant of murine CLPP in which the conserved Ser‐149 residue of the consensus active site is replaced with alanine (S149A) (Fig EV3A). The homologous active‐site mutation in Escherichia coli, Staphylococcus aureus, and Caulobacter crescentus was previously successfully used as a CLPP‐substrate trap (Flynn et al, 2003; Bhat et al, 2013; Feng et al, 2013). Immortalized Clpp knockout MEFs were transfected with either empty plasmid or plasmids carrying genes encoding wild‐type (CLPPWT‐FLAG) or the mutant variant (CLPPS149A‐FLAG) of CLPP (Fig EV3A). The ability of FLAG‐tagged CLPP to form a functional chamber was confirmed on BN‐PAGE, and we show that CLPPS149A produced stable tetradecamers and steadily interacted with CLPX likely due to a failure to release substrates (Fig EV3B). After isolation of mitochondria and subsequent co‐immunoprecipitation with anti‐FLAG antibodies, CLPP‐bound proteins were analyzed by LC‐MS/MS. The list of 66 possible substrates and interactors of ClpXP contains proteins highly enriched in CLPPS149A compared to CLPP−/− and CLPPWT fractions (Table EV3). In order to decipher the interactome obtained by our LC‐MS/MS analysis, we used the string webtool (http://www.ncbi.nlm.nih.gov/pubmed/10982861) (Fig 5A). Here, we observed a strong association of several potential CLPP substrates and we found 15 proteins which have bacterial orthologues known to be ClpXP substrates (Table EV3) indicating that our approach is suitable for detection of potential substrates. The STRING analysis revealed a network of proteins involved in mitochondrial translation, but not replication or transcription, in agreement with our results. We also identified MRC protein subunits and enzymes involved in multiple metabolic processes in mitochondria (Table EV3).
Figure EV3. Analysis of potential substrates and interactors of CLPP .

- Strategy for identification of CLPP substrates and interactors in mammalian Clpp knockout (Clpp −/−) MEFs. Schemes for an empty expression plasmid or plasmids containing genes encoding the murine wild‐type (CLPPwt) and proteolytically inactive CLPP (CLPPS149A) are represented.
- Assembly of the CLPP/CLPXP protease complex with the ectopically expressed wild‐type (CLPPwt) and catalytically inactive (CLPPS149A) CLPP in Clpp knockout (−/−) MEFs. Wild‐type (Clpp +/+) and Clpp knockout (Clpp −/−) MEFs were used as a control. Crude mitochondria lysed with digitonin (left panel) or whole cells lysed with Triton X‐100 (right panel) were subjected to BN‐PAGE followed by Western blot analysis.
- Western blot analysis and subsequent quantification of protein levels in Clpp knockout MEFs (Clpp −/−) relative to wild‐type (Clpp +/+) cells. Tubulin is used as a loading control. Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (n = 6).
- Western blot analysis and subsequent quantification of P32/C1QBP, PNPT1, and MRPP1 levels in Clpp knockout heart mitochondria (Clpp −/−) relative to wild type (Clpp +/+). Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (P32 n = 4; MRPP1 and PNPT1 n = 3). Arrowhead indicates position of the PNPT1 protein.
- Cycloheximide (CHX) chase assay to determine the stability of P32/C1QBP, MRPP1, and PNPT1. Proteins are visualized by Western blot with respective antibodies. Proteins are isolated after 2, 4, 6, 8, 10, and 12 h after beginning of chase (0).
Source data are available online for this figure.
Figure 5. Analysis of potential CLPP substrates involved in mitochondrial translation.

- Network analysis of 66 potential CLPP substrates using only high confidence interactions (> 0.7). Known ClpXP substrates are shown in boxes with a green frame. Respiratory chain proteins and ribonucleoproteins are indicated in dark gray and orange, respectively.
- Western blot analysis and subsequent quantification of ERAL1 and EFG1 levels from Clpp knockout (Clpp −/−) heart mitochondria relative to wild type (Clpp +/+). Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (n = 4). The arrowhead indicates positions of EFG1.
- Expression levels of potential CLPP substrates from Clpp knockout (Clpp −/−) heart mitochondria relative to wild type (set to 1). Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (n = 4).
- Cycloheximide (CHX) chase assay to determine the stability of ERAL1 and EFG1. Proteins are visualized by Western blot with respective antibodies. Proteins are isolated after 2, 4, 6, 8, 10, and 12 h after beginning of chase (0). The arrowhead indicates positions of ERAL1.
- Western blot analysis and subsequent quantification of protein levels in wild‐type (C1, C2) and Clpp knockout (KO1, KO2) MEFs upon Clpx siRNA (Clpx) relative to untreated (Ctrl) wild type. Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (n = 6). The arrowhead indicates positions of either CLPX or ERAL1, and stars indicate position of cross‐reacting bands.
Source data are available online for this figure.
We then proceeded to further analyze selected candidates with a role in mitochondrial translation (EFG1, ERAL1, and P32/C1QBP) and also tested proteins involved in post‐transcriptional processing of mtRNAs (MRPP1 and PNPT1). As expected from protease substrates, the steady‐state levels of EFG1, one of mitochondrial translation elongation factors that catalyzes the movement of mRNA and tRNA within the 28S subunit (Sharma et al, 2003) and ERAL1, a 12S rRNA chaperone needed for the formation of functional small ribosomal subunits (Dennerlein et al, 2010; Uchiumi et al, 2010), were highly upregulated in Clpp knockout heart mitochondria and MEFs (Fig 5B and EV3C). The upregulation is a result of increased stability and no changes in transcript levels (Fig 5C). Conversely, we did not observe a change in the steady‐state transcript or protein level of P32/C1QBP, proposed to be an essential RNA‐binding factor in mitochondrial translation (Yagi et al, 2012), in cardiomyocytes or MEFs, suggesting that this protein might be an interactor of the ClpXP protease (Fig EV3C–E), in agreement with a previous report (Lowth et al, 2012).
Further evidence that EFG1 and ERAL1 are likely ClpXP substrates came from the analysis in which we have blocked cytoplasmic protein synthesis by cycloheximide treatment and followed the turnover of these proteins. While in wild‐type cells levels of both EFG1 and ERAL1 decreased over time, they were clearly stabilized in Clpp knockout MEFs (Fig 5D). Correspondingly, a knockdown of Clpx unfoldase, the interacting partner of CLPP, in wild‐type MEFs also caused an increase in EFG1 and ERAL1 levels (Fig 5E). The same treatment of CLPP‐deficient cells caused no additional effect on the levels of these two proteins, confirming that the observed phenotype is a result of general ClpXP deficiency (Fig 5E).
Increased steady‐state levels of MRPP1 and PNPT1 were detected in Clpp knockout hearts, with no change in transcript levels, suggesting that these two proteins could be bona fide ClpXP substrates (Figs 5C and EV3D). Nevertheless, no upregulation was detected in CLPP‐deficient MEFs and we could not observe a clear difference in the turnover rate of these proteins upon cycloheximide chases, owing to their high stability even in wild‐type mitochondria (Fig EV3E). Together, these data leave the open question if these proteins are indeed the substrates of ClpXP protease.
As we have detected a strong defect in complex I activity and levels, we also analyzed NDUFV2 and NDUFS2, two highly scored C I subunits from our screen as potential CLPP substrates (Table EV3). Our results demonstrate that both, NDUFV2 and NDUFS2, are not stable in the absence of CLPP (Fig EV4A). In contrast, the C II subunit SDHA is highly stable under the same condition of CLPP deficiency (Fig EV4A). Thus, it is likely a ClpXP substrate (Fig 5A), as others and we have suggested (Cole et al, 2015; Seiferling et al, 2016). Transcripts of both C I subunits are present in normal quantities (Fig EV4B), while steady‐state protein levels are either normal (NDUFV2) or even downregulated (NDUFS2) in CLPP‐deficient cardiomyocytes (Fig EV4C). Taken together, these results suggest that NDUFV2 and NDUFS2 are likely not substrates of CLPP, and hence, we have not investigated them further.
Figure EV4. Analysis of putative CLPP substrates that are components of mitochondrial complex I.

- Cycloheximide (CHX) chase assay to determine the stability of NDUFV2 and NDUFS2. Proteins were visualized by Western blot with respective antibodies. Proteins were isolated after 2, 4, 6, 8, 10, and 12 h after beginning of chase (0). SDHA was used as a positive control for a putative ClpXP substrate.
- Expression levels of different subunits of mitochondrial respiratory chain complexes from Clpp knockout (Clpp −/−) heart mitochondria relative to wild type (Clpp +/+, set to 1). Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (n = 4).
- Western blot analysis and subsequent quantification of levels of putative CLPP substrates belonging to complex I (NDUFV2 and NDUFS2) or mitochondrial translation machinery (ERAL1 and EFG1) in Clpp knockout heart mitochondria (Clpp −/−) relative to wild type (Clpp +/+). Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (n = 4).
Source data are available online for this figure.
ERAL1 strongly binds to the 28S subunit and affects formation of the mitoribosome
To unravel how increased levels of EFG1 and ERAL1 could affect the formation of the mitoribosome, we performed gradient sedimentation analyses of mitochondrial ribosomes. To this end, we used shorter gradients that have a lower separation power, but allowed us to collect more material needed for subsequent studies. We confirmed our previous results, showing accumulation of 12S rRNA in the 28S small ribosomal subunit fraction, but decreased levels of fully assembled mitoribosomes (Fig 6A). Similarly, we detected a massive increase (fivefold) in the level of unassembled (“free”) 39S ribosome subunits (MRPL12) in the absence of CLPP, accompanied by a milder rise in the level of unassembled 28S ribosome subunits (MRPS35) (Fig 6A and EV5A).
Figure 6. Association of potential candidates with small ribosomal subunits.

- Sucrose density gradient sedimentation profile (“short gradients”) of ribosomal proteins and transcripts from control (Clpp +/+) and Clpp knockout (Clpp −/−) heart mitochondria. The migration of ribosomal subunits (28S and 39S) and mitoribosome (55S) was detected by immunoblotting with antibodies against MRPS35 and MRPL12. 12S rRNA was analyzed by Northern blot.
- Sedimentation profiles of ERAL1, MRPS28, EFG1, CLPP, CLPX, EFTU, and MTIF2 in heart mitochondria as in (A).
- Relative abundance of proteins bound to the 28S ribosomal subunit (fractions 3 and up). Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (ERAL1 n = 4; EFG1, CLPX, MTIF2 n = 3; EFTU n = 2).
- Sucrose density gradient sedimentation profile (“long gradients” as in Fig 4D) of ERAL1, MRPS35, MRPL37, CLPX, and EFG1 from control (Clpp +/+) and Clpp knockout (Clpp −/−) heart mitochondria. The same amount of protein lysates was separated by the gradient, and the amount of each individual protein is estimated after Western blot analysis (Input). Only fractions 4–11 containing the 28S, 39S, and 55S ribosomal fractions were blotted to avoid high signals arising in free fractions (1–3).
Source data are available online for this figure.
Figure EV5. Association of potential CLPP substrates with the mitoribosomes.

- Quantification of distribution of MRPS35 and MRPL12 on sucrose density gradient sedimentation profile of wild‐type (Clpp +/+) and Clpp knockout (Clpp −/−) heart mitochondria as in Fig 6A, presented as percentage of total. Fractions are divided into: unbound (“free”, 1–2); small ribosomal subunit (“28S”, 3–4); large ribosomal subunit (“39S”, 5–6); and assembled mitoribosomes (“55S”, 7–10). Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (n = 3). Statistical difference between specific fractions in wild‐type and Clpp knockout heart mitochondria is presented.
- Sucrose density gradient sedimentation of MRPS35 and ERAL1 in heart mitochondria from wild‐type (Clpp +/+) and Clpp knockout (Clpp −/−) mice. In a parallel experiment, lysed mitochondria were treated with RNase A to disintegrate mitochondrial ribosomes.
- Sucrose density gradient sedimentation of ERAL1 and mitoribosomal proteins upon Clpx siRNA in wild‐type (Clpp +/+) and Clpp knockout (Clpp −/−) MEFs. TOMM20 was used as a mitochondrial loading control.
- Quantification of the Western blot determined steady‐state levels of respiratory chain subunits upon Clpx siRNA in a Clpp knockout (Clpp −/−) MEFs. Actin was used as loading control. Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (n = 3).
- Western blot analysis and subsequent quantification of steady‐state levels of translational factors EFTU, MTIF2, MTIF3 in Clpp knockout heart mitochondria (Clpp −/−) relative to wild type (Clpp +/+; set to 1). Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (n = 4).
Source data are available online for this figure.
Next we investigated the migration of EFG1 and ERAL1 with the small (28S) and large (39S) ribosomal subunit or complete mitoribosomes (55S). Both of these proteins primarily interact with the 28S small ribosomal subunit (Coenen et al, 2004; Dennerlein et al, 2010). In wild‐type mitochondria, EFG1 loosely associates with 28S ribosome and we detected it mainly as a free protein (Fig 6B and D). In the absence of CLPP, EFG1 is present in higher levels in the mitochondria, but the association with the small ribosomal subunit is only mildly increased, suggesting that ClpXP is likely involved in turnover of this protein in its free form (Fig 6B–D).
In the absence of CLPP, ERAL1 association with the 28S ribosomal subunit was highly increased and spans over many fractions, suggesting that ERAL1 is not sufficiently removed from the 28S subunit and might still be present in the complete mitoribosome (Fig 6B and C). This is even more obvious on long gradients, where we have corrected for the amount of the protein present in the sample (input) (Fig 6D). Pretreatment of mitochondrial lysates with RNase that disrupts formation of mitoribosomes also leads to the loss of ERAL1 in the high molecular‐mass fractions, suggesting that it indeed binds the ribosomal subunits (Fig EV5B). Collectively, these data suggest that this abnormal association of ERAL1 where approximately 20% of the protein present in the mitochondria is bound to multiple ribosomal fractions, likely has a major effect on mitoribosomal function and might be the main cause for the mitochondrial translation defect.
We also detected lower levels of MRPS28 associated with the 28S ribosomal subunit (Fig 6B). MRPS28 is predicted to be a functional and structural homolog of bacterial bS1 (Amunts et al, 2015) that shares the same binding site as Era, a bacterial homolog of ERAL1, on the bacterial 30S small ribosomal subunit. bS1 cannot be recruited to finish the maturation of the 30S subunit before Era is removed (Sharma et al, 2005). Our results demonstrate that MRPS28 is less recruited to the 28S ribosome in the CLPP‐deficient mitochondria, likely because ERAL1 is still strongly bound to it (Fig 6B). Furthermore, we provide first evidence that MRPS28 behaves more as a maturation factor, rather than a structural component of the 28S ribosome, as shown for its bacterial homolog (Komarova et al, 2002; Sharma et al, 2005).
The binding of CLPX to the 28S ribosome was also increased in the absence of CLPP protease, possibly through interactions with its substrate ERAL1 (Fig 6B–D). However, this interaction seems to be transient as it varies in different isolation conditions, for example, in ribosomal gradients from isolated heart mitochondria CLPX seems to bind more to the ribosomal fractions (Fig 6B–D), while in gradients from whole cell lysates, we did not observe this interaction (Fig EV5C). Knockdown of Clpx failed to normalize the levels of 28S and 55S ribosomes or decrease the high association of ERAL1 with different ribosomal intermediates (Fig EV5C). More importantly, knockdown of Clpx did not have an impact on the levels of OXPHOS subunits in CLPP‐deficient cells (Fig EV5D).
In contrast, a lower level of binding was observed for EFTU, the other mitochondrial translation elongation factor and MTIF2, the initiation factor of mitochondrial protein synthesis (Fig 6B and C). This was not the result of decreased steady‐state levels of both proteins, as they were unaffected by the loss of CLPP (Fig EV5E).
Normalization of ERAL1 levels partially rescues respiratory chain deficiency in Clpp knockout mitochondria
As the primary defect observed in CLPP‐deficient mitochondria seems to be in the maturation of 28S ribosomal subunits, we have turned our attention to ERAL1. First, we wanted to establish whether ERAL1 interacts with the ClpXP protease. To this end, we performed co‐immunoprecipitation experiments with antibodies raised against endogenous ERAL1 (Fig 7A). Our results show that, although ERAL1 interacts poorly with either CLPX or CLPP in wild‐type cells, the interaction with CLPX is increased in the absence of the protease subunit (Fig 7A). This was confirmed in the reciprocal experiment where we have precipitated much higher levels of ERAL1 with an antibody raised against CLPX in the Clpp knockout background (Fig 7A). These data verify the direct interaction of the proteins within mitochondria and suggest that in wild‐type cells this interaction is transient, while in the absence of CLPP, ERAL1 stays more permanently bound to CLPX. Remarkably, ectopic expression of ERAL1 in wild‐type cells did not result in increased binding to mitochondrial ribosomal subunits, likely due to intact ClpXP activity that prevented increased and/or more permanent binding that might be disadvantageous (Fig EV6A). Consistently, further overexpression of ERAL1 in CLPP‐deficient cells proved to be deleterious for cell growth and/or survival.
Figure 7. Effect of ERAL1 knockdown on wild type (Clpp +/+) and Clpp knockout (Clpp −/−) MEFs.

- Coimmunoprecipitation analysis using antibodies raised against ERAL1 and CLPX proteins. Western blot analysis of input (I), control (C), and eluate (E) is shown. Asterisk indicates the cross‐reacting band.
- Sucrose density gradient migration profile: association of ERAL1 with the 28S ribosomal subunit (MRPS35) upon Eral1 siRNA. TOMM20 was used as a loading control.
- De novo synthesis of mitochondrial proteins in wild‐type (Clpp +/+) and Clpp knockout (Clpp −/−) MEFs upon Eral1 siRNA. Proteins were isolated after labeling with 35S‐methionine for 1 h from cells on control siRNA (Ctrl) or cells on Eral1 siRNA (Eral1). Positions of individual mitochondrial encoded proteins are indicated. Coomassie blue staining was used to ensure equal loading.
- Quantification of Western blot analysis of respiratory chain subunits upon knockdown of Eral1. Levels relative to wild‐type cells treated with control siRNA (set to 1) are presented. Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (COXI n = 4; ATP5A1 n = 2; the rest n = 6). Combined results from 2 to 3 blots were used for quantification as only two samples of each condition are used in a single experiment. Numbers within bars represent statistical difference to control wild‐type cells, and numbers above bars represent difference in Clpp knockout cells upon Eral1 siRNA.
- Western blot analysis of RC subunits upon Eral1 knockdown in MEFs. Cells (0) were treated with doxycycline for 48 h (T) and upon doxycycline removal allowed to recover for 24 h (R).
- Quantification of the relative recovery of COXI and NDUFB6 upon Eral1 siRNA. The values are calculated as ratio between recovered (R) to doxycycline‐untreated (0) protein levels in Eral1 knockdown cells (Eral1) and compared to the same results obtained in control cells (Ctrl). Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (COXI n = 4; NDUFB6 n = 3). Numbers within bars represent statistical difference between control/Eral1 siRNA‐treated cells, and numbers above bars represent difference between +/+; Eral1 siRNA and −/−; Eral1 siRNA.
Source data are available online for this figure.
Figure EV6. Analysis of respiratory chain subunits and supercomplexes upon Eral1 and Clpx knockdown.

- Sucrose density gradient sedimentation of ERAL1 and mitoribosomal proteins upon CAG::Eral1 overexpression (Eral1 OE) in wild‐type (Clpp +/+) MEFs compared to Clpp knockout (Clpp −/−) MEFs. TOMM20 was used as a mitochondrial loading control.
- Western blot analysis of steady‐state levels of respiratory chain subunits in wild‐type (Clpp +/+; C1, C2) and Clpp knockout (Clpp −/−; KO1, KO2) MEFs upon Eral1 siRNA. TOMM20 was used as loading control.
- Quantification of the steady‐state levels of ERAL1 upon Eral1 siRNA relative to wild‐type (Clpp +/+) MEFs treated with control siRNA. Bars represent mean ± SD. Student's t‐test was used to determine the level of statistical difference (n = 6).
- BN‐PAGE analysis of the assembled respiratory chain super complexes in MEFs upon Eral1 siRNA. Western blot analysis was used to visualize C I (NDUFA9), C III (UQCRC1), and C V (ATP5A1).
- Sucrose density gradient sedimentation of mitoribosomal proteins upon expression of wild‐type (Clpp WT) or catalytically inactive (Clpp S149A) CLPP protein in wild‐type (Clpp +/+) and Clpp knockout (Clpp −/−) MEFs. The position of ERAL1 is indicated by arrows and position of MRPS35 by arrowheads.
Source data are available online for this figure.
If ERAL1 upregulation due to the absence of CLPP leads to development of the observed phenotypes, then normalization of ERAL1 levels should, at least in part, rescue the observed phenotypes. To test this, we have moderately downregulated ERAL1 levels in CLPP‐deficient mitochondria to match the amount present in wild‐type cells (Fig EV6B and C). Regulation of ERAL1 levels resulted in a decrease in the amount of 28S subunit in CLPP‐deficient mitochondria and a weaker binding of ERAL1 to the higher fractions (Fig 7B). Normalization of ERAL1 levels in CLPP‐deficient MEFs also salvaged previously observed translation defect (Fig 7C), resulting in a partial rescue of the steady‐state levels of complex I and IV subunits (Fig 7D and EV6B). The increase in the amount of individual subunits resulted in higher level of assembled complex I, but did not affect the level of complex III (Fig EV6D). In contrast, ERAL1 knockdown in wild‐type cells led to a decrease in the levels of the same RC subunits (Fig 7D and EV6B), in agreement with previous results showing that ERAL1 is needed for mitochondrial protein synthesis (Uchiumi et al, 2010).
We proceeded to test whether normalization of ERAL1 levels in CLPP‐deficient cells can rescue the observed defect in the recovery from doxycycline treatment. We monitored the recovery of mtDNA‐encoded COXI subunit and nDNA‐encoded NDUFB6 upon 48 h of doxycycline treatment followed by 24 h recovery (Fig 7E and F). In line with previous results, we detected higher survival of cells and the recovery of COXI was increased in CLPP‐deficient cells (Fig 7F). As shown for other nDNA‐encoded RC subunits (Fig 7D and EV6B), NDUFB6 levels were also increased by the ERAL1 knockdown, although the recovery was not affected by the treatment (Fig 7F). This result suggests that ERAL1 knockdown in CLPP‐deficient cells rescues the nDNA‐encoded subunits by increasing the amount of available mtDNA‐encoded polypeptides needed for the assembly of functional RC complexes, therefore decreasing the turnover of unassembled subunits. The interdependence of nDNA‐encoded RC subunits on functional mitochondrial protein synthesis is well established in multiple models and across different phyla (Park et al, 2007; Kwasniak et al, 2013; Dogan et al, 2014). In wild‐type cells, knockdown of ERAL1 led to a significant decrease in the recovery of COXI levels (Fig 7F).
Remarkably, overexpression of wild‐type CLPP reduced the levels of ERAL1, bound to the 28S ribosome, and restored the mitoribosomes (55S) (Fig EV6E). In contrast, ectopic expression of catalytically inactive CLPP (CLPPS149A) reduced the overall levels of ribosomal subunits, without restoring the mitoribosome assembly (Fig EV6E). Moreover, catalytically inactive CLPP protein strongly binds multiple ribosomal fractions and in its presence CLPX behaves in the same way, suggesting an active role for ClpXP on the mitochondrial ribosomes. These data also strengthen the role of ClpXP in the turnover of ERAL1 during ribosomal assembly.
Taken together, these results show that normalization of ERAL1 levels in CLPP‐deficient mitochondria leads to increased mitochondrial protein synthesis, likely by allowing efficient maturation of mitoribosomes. At the same time, increased availability of mtDNA‐encoded subunits allows more nDNA‐encoded polypeptides to be present in mitochondria and incorporated in RC complexes.
Discussion
Our results demonstrate that the CLPP protease has an unforeseen role in the regulation of ribosomal biogenesis and mitochondrial translation. Mitochondrial translation is impaired in Clpp knockout mitochondria because the two ribosomal subunits are not assembled into a functional 55S mitoribosome. Consequently, respiratory chain dysfunction seems to be the major effect of CLPP deficiency in mammalian mitochondria, with complex I downregulation as a foremost consequence.
The respiratory deficiency caused by CLPP depletion is moderate, and Clpp knockout mice represent a faithful model of Perrault syndrome caused by CLPP deficiency in humans, primarily characterized by sensorineural hearing loss and a premature ovarian failure (Gispert et al, 2013; Jenkinson et al, 2013). A spectrum of additional clinical features, including cerebellar ataxia, peripheral neuropathy, and muscle atrophy, has been described in some affected individuals (Jenkinson et al, 2012; Morino et al, 2014). Here we provide the first evidence that loss of CLPP leads to a defect in mitochondrial translation that may be the primary cause of the disease. In agreement, mutations in HARS2 (Pierce et al, 2011), LARS2 (Pierce et al, 2013), and TWINKLE (Morino et al, 2014), three out of four other genes causing Perrault syndrome also have a direct effect on mitochondrial protein synthesis.
Our study identified many potential CLPP substrates involved in various pathways including mitochondrial translation and transcript processing, respiratory chain function, and multiple metabolic pathways. Some of the identified and partially characterized candidate proteins, like EFG1 and PNPT1, have been already detected as CLPP substrates in other organisms (Zybailov et al, 2009; Bhat et al, 2013; Feng et al, 2013). Remarkably, a recent study, using a BioID‐mass spectrometry method to identify ClpP‐interacting partners, provides a list of 49 possible ClpXP substrates and adaptors in human cells (Cole et al, 2015). The list of candidates, however, only partially overlaps with the one created by us and contains only nine identical hits, most of them involved in either TCA cycle or fatty acid metabolism (Cole et al, 2015). In addition, the list included a number of proteins involved in mtDNA maintenance (TFAM, MTSS1, POLG2), but not in mitochondrial translation (Cole et al, 2015). The obvious discrepancies between these two studies could be attributed to differences in the experimental approach, as the previous study selected proteins that equally interact with wild‐type and mutant CLPP, thus excluding many of the CLPP substrates, as they would be instantly degraded in the wild‐type background. Further differences could be explained by the cell‐type‐specific interactions (HEK293 T‐Rex cells were used) and by the fact that cells were treated with tetracycline/doxycycline that has a profound effect on mitochondrial function, specifically on the maturation of 28S ribosomal subunit (LaPolla & Lambowitz, 1977; Moullan et al, 2015), and we showed here that this is especially detrimental in the Clpp knockout background.
Our results strongly suggest that the role for CLPP in ribosomal biogenesis could be explained by its regulation of ERAL1 levels. We believe that the upregulation of other proteins involved in mitochondrial translation, like translation elongation factor EFG1 or mitochondrial tRNA synthetases (VARS2 and IARS2) do not contribute significantly to the translation defect. Further support for this comes from a study in A. thaliana CLPP‐deficient chloroplasts showing that over‐accumulation of both translation elongation factors and aminoacyl‐tRNA synthetases does not result in a significant loss of translation (Zybailov et al, 2009).
We show that accumulation of ERAL1 results in much higher binding to the small ribosomal subunit that prevents full maturation and assembly of 28S and 39S subunits into a functional mitoribosome. This is reminiscent of the situation in prokaryotes where binding of Era to the 30S small ribosomal subunit results in a conformation change that is less favorable for association with the 50S large subunit (Sharma et al, 2005). Furthermore, in an in vitro assay, 2.5‐fold increase in the amount of Era leads to a 50% decreased formation of mature bacterial ribosomes, while a fivefold increase, almost completely prevents it (Sharma et al, 2005). Our results argue that the strong binding of ERAL1 also prevents binding of other translation initiation and elongation factors that are not substrates of ClpXP protease. It is currently not known how ERAL1 is extracted from the 28S subunit during the last steps of small subunit maturation, and our results propose that ClpXP might play an active role in this process. This is supported by our data showing that not only ERAL1 levels were increased, but also the ratio of ribosome‐bound to free ERAL1 is much higher in Clpp knockout mitochondria, while MRPS28 association with the 28S ribosome is diminished. This goes hand in hand with the observation that CLPX is also much more present on the small ribosomal subunit and possibly bound to ERAL1 in the CLPP‐deficient mitochondria, likely because protease activity and/or conformation changes mediated by ClpXP are needed to release this interaction. Interestingly, strong downregulation of ERAL1 levels also has detrimental effects on cell survival and mitoribosome assembly, even though two published studies do not agree on which effect is more pronounced (Dennerlein et al, 2010; Uchiumi et al, 2010). Taken together, these data suggest that ERAL1 is essential for the 28S ribosome assembly, but needs to be timely removed to allow translation initiation to proceed and 28S to interact with 39S to form functional mitoribosome. Therefore, both increased and decreased levels of ERAL1 would be unfavorable for the assembly of the mitoribosome and the mitochondrial translation rate. It is possible to envision that the changes of ClpXP levels under stress condition might provide a way to rapidly downregulate mitochondrial translation, by regulating ERAL1 and hence 28S levels, thus preventing further proteotoxic burden to mitochondria.
In conclusion, our results provide clear evidence for the novel role of CLPP in regulation of mitochondrial ribosome biogenesis through its target‐substrate ERAL1. The level and binding of ERAL1 to the 28S ribosomal subunit needs to be tightly regulated to allow efficient formation of a mature mitoribosome, and we show that ClpXP has an active role in this process. Our study also produced an extensive list of other potential ClpXP substrates that need to be evaluated. Further analyses are clearly necessary to determine their role in mitochondrial physiology.
Materials and Methods
Generation and genotyping of Clpp knockout mice
Clpp gene targeting was carried out at Taconic Artemis, Germany, in Art B6/3.5 embryonic stem cell line on a genetic background of C57BL/6 NTac. LoxP sites flanked exons 3–5 of the Clpp gene. A puromycin resistance selection marker was inserted into intron 5 and flanked by F3 sites. The targeted locus was transmitted through germline that resulted in heterozygous Clpp +/PuroR−loxP mice. Animals were bred with B6 Flp_deleter transgenic (TG) mice to remove the puromycin selection marker. Finally, Clpp +/loxP mice were mated with mice expressing Cre recombinase under the promoter of beta‐actin resulting in deletion of exons 3–5. This deletion resulted in loss of function of the Clpp gene, by removing a part of the protease domain and generated a frameshift from exon 2–6. Clpp +/− heterozygous mice were intercrossed to obtain the Clpp −/− homozygous knockout mice. The genotyping primers used are:
forward (5′‐TGTGCATTCTTACCATAGTCTGC‐3′);
reversewt (5′‐CCCAGACATGATTCCTAGCAC‐3′);
reverseko (5′‐CCCAGACATGATTCCTAGCAC‐3′).
Animal experiments were in accordance with guidelines for humane treatment of animals and were reviewed and approved by the Animal Ethics Committee of North‐Rheine Westphalia, Germany.
Generation of MEF lines
Primary MEFs were isolated from E13.5 embryos obtained as a result of intercrossed Clpp +/− animals as described previously (Trifunovic et al, 2005). Immortalized cell lines were generated upon transformation with the SV40 T antigen (Richter‐Dennerlein et al, 2014).
Protein turnover in MEFs
Cells were grown to 80–90% confluency in DMEM supplemented with galactose (10 mM), pyruvate (100 μg/ml), and uridine (50 μg/ml). After blockage of cytoplasmic protein synthesis by cycloheximide treatment (100 μg/ml), cells were collected at indicated time points, counted, and lysed with RIPA buffer, proportional to the number of living cells. Western blot was used to analyze the obtained results.
Trapping of CLPP substrates in mouse embryonic fibroblasts
Plasmids containing genes encoding the murine wild‐type (Clpp wt) and proteolytically inactive CLPP (Clpp S149A) fused to the double FLAG tag were transiently overexpressed for 48 h under the control of CMV promoter in Clpp knockout cells. As a negative control, cells transfected with the empty plasmid (pcDNA3.1, Invitrogene) were used. Mitochondria were isolated as described below, lysed in a buffer containing 25 mM Tris, 150 M NaCl, 1 mM EDTA, 1% NP‐40, 5% glycerol, protease inhibitor cocktail, pH 7.4, and cleared by 1‐h centrifugation at 20,000 × g. Anti‐FLAG M2 antibody (Sigma) was immobilized on the AminoLink Plus resin (Thermo) according to the manufacturer's instructions. Lysates were precleared with the Control Agarose Resin (Thermo) and incubated with immobilized beads overnight at 4°C. Beads were washed five times with the lysis buffer followed by the elution of the immobilized protein complexes with the low pH elution buffer (100 mM glycine; pH 2.8). Immunoprecipitated proteins were subjected to SDS–PAGE and stained by Coomassie colloidal blue staining. Protein containing gel slices were trypsinized, separated by the HPLC, and subjected to LC‐ESI‐MS/MS analysis. In total, we identified 286 proteins assigned to the mitochondria and a fraction of 143 proteins were equally detected in samples from CLPP‐deficient cells containing either empty vector or FLAG‐tagged CLPPWT or CLPPS149A protein.
Assembly of the CLPPWT/CLPPS149A‐2xFLAG containing protease complexes for a substrate‐trapping experiment was monitored by BN‐PAGE on crude mitochondria solubilized in the 4% digitonin PBS buffer or using whole cells lysed in the 1% Triton X‐100 PBS buffer. BN‐PAGE resolved protein complexes were blotted on a PVDF membrane and immunodecorated with indicated antibodies. For ectopic CLPPWT/CLPPS149A expression, cells were transfected with Nucleofector™ electroporation technology (Lonza) according to the manufacturer's instructions.
Antibiotics treatment
The recovery of RC subunits from defects caused by treatments with antibiotics that inhibit mitochondrial translation were followed in wild‐type and Clpp knockout MEFs incubated in full DMEM with high glucose content (4.5 g/l). Cells in the exponential growth phase (50–70% confluence) were supplemented with the inhibitors of mitochondrial translation (doxycycline 15 μg/ml; chloramphenicol 200 μg/ml; actinonin 150 μM) and incubated up to 48 h. After that, cells were transferred to the antibiotics‐free medium for the next 48 h. At indicated time points, cells were collected and lysed with a RIPA buffer and subsequent Western blotting was used to monitor the recovery of respiratory chain subunits. For the analysis of recovery of RC subunits upon ERAL1 depletion, cells were transfected with Eral1 siRNA 48 h prior to the start of the assay and retransfected 24 h into the assay.
Southern, Northern, and Western blot analysis
DNA isolation and Southern blot analysis were performed as described previously (Hance et al, 2005). RNA was extracted from tissues with Trizol reagent (Invitrogen), and Northern blot analysis was performed with α‐32P‐dCTP‐labeled DNA probes or α‐32P‐dATP‐labeled oligonucleotide probes for mt‐mRNAs and mt‐tRNAs, respectively (Park et al, 2007).
Protein lysates were obtained from either homogenized tissue or isolated mitochondria and subsequently subjected to Western blot analysis as described previously (Hance et al, 2005). MEFs were lysed with the RIPA buffer followed by sonication and lysate clearance. Western blot analysis was performed using the following primary antibodies at indicated concentrations: MTIF2 and MTIF3, 1:1,000 (a kind gift from Prof. A. Filipovska, University of Western Australia); MRPS15, 1:300; LRPPRC, 1:1,000 (a kind gift from Prof. N.‐G. Larsson, MPI for Biology of Aging Cologne); EFTU, 1:1,000 (a kind gift from Assistant Prof. Nono Tomita‐Takeuchi); MTCO2, 1:1,000 (a kind gift from Prof. T. Langer, CECAD Cologne). All other antibodies were used in dilutions recommended by the manufacturers: CLPP, CLPX, MRPL37, β‐actin, α‐tubulin (Sigma); ERAL1, PNPT1, MRPS35, NDUFV2 (Proteintech); EFG1, MRPL12, NDUFS2 (Abcam); MRPP1 (Aviva Systems Biology); P32/C1QBP (Millipore); SDHA, NDUFA9, UQCRC1, MTCO1, COX4A (Invitrogen), VDAC (Cell Signaling), NDUFS3, NDUFB6, ATP5A1, UQCRFS1 (MitoSciences); TOMM20, CYTB (Santa Cruz Biotechnology).
Real‐time quantitative PCR
Isolated heart RNA was treated with DNase (DNA‐free Kit, Ambion) and subsequently reverse transcribed with the High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Gene expression levels were determined with qPCR technique using the TaqMan Assay Kit (Applied Biosystems) or Brilliant III Ultra‐Fast SYBR Green QPCR Master Mix (Agilent Technologies). Samples were adjusted for total RNA content by TATA box binding protein (Tbp) or hypoxanthine‐guanine phosphoribosyltransferase (Hprt). Clpp (Mm00489940_m1), Clpx (Mm00488586_m1), Eftu (Mm01136248_m1), Hrpt (Mm00446968_m1), and Tbp1 (Mm00446973_m1) probes were obtained from TaqMan Assay‐on‐Demand kits (Applied Biosystems). Primers were as follows:
| FWD 5′–3′ | REV 5′–3′ | |
|---|---|---|
| Eral1 | GGACCGTATCCTTGGATTTTCTC | GAGGACCCGTGGATTCTCAG |
| P32/C1qbp | AAGATCCAGAAACACAAGTCCCT | CCTCCTCACCATCAAATGTTGG |
| Pnpt1 | AATCGGGCACTCAGCTATTTG | CAGGTCTACAGTCACCGCTC |
| Mrpp1 | TGTCCTCCAAAGCACCTTCTT | TGAATGCTCGACTTCATTGTAGC |
| Mrpl12 | GCCACCGAAGGAGTGAAGC | GGAGCGTAAGGCTGGCAAT |
| Mrpl37 | AACCCTGCTACATCTTCCACC | TGGTCTTAGTAAGCCAGAGAGC |
| Mrpl48 | TGGGTGTGTGGACCAATACTG | CTTTGTCTTGTAGTGCCGACT |
| Mrps15 | AAGTAGCGCCAGTCTGCTC | GAGGGTGGTTCATCGTCCT |
| Mrps35 | GGCCCAGTGTTTACCCCAC | GTGCCTTTTAATTGCCACAGG |
| Ndufv2 | GGCTACCTATCTCCGCTATGA | TCCCAACTGGCTTTCGATTATAC |
| Ndufs2 | TCGTGCTGGAACTGAGTGGA | GGCCTGTTCATTACACATCATGG |
| Ndufa9 | GGCCAGCTTACCTTTCTGGAA | GCCCAATAAGATTGATGACCACG |
| Ndufb6 | TGGAAGAACATGGTCTTTAAGGC | TTCGAGCTAACAATGGTGTATGG |
| Ndufs3 | CTGTGGCAGCACGTAAGAAG | GCTTGTGGGTCACATCACTCC |
| Sdha | TCCTACCCGATCACATACTGTT | GCTCTGTCATGTAATGGATGGCA |
| Ndufab1 | ATGGCGTCTCGTGTCCTCT | GCGGCACAAATGTGTGACT |
| Uqcrc1 | ACGCAAGTGCTACTTCGCA | CAGCGTCAATCCACACTCCC |
| Hprt | GCCCCAAAATGGTTAAGGTT | TTGCGCTCATCTTAGGCTTT |
| Tbp1 | GGGAGAATCATGGACCAGAA | TTGCTGCTGCTGTCTTTGTT |
Analysis of de novo transcription and translation in isolated mitochondria
In organello transcription assay and translation assays were performed as previously described (Dogan et al, 2014).
tRNA aminoacylation assay
For analysis of the tRNA aminoacylation, total RNA was isolated with TRIzol reagent (Invitrogen) according to the manufacturer's protocol. Total RNA was resuspended in 0.3 M NaOAc (pH 5.0) and 1 mM EDTA. Aminoacylation status was determined by acid–urea PAGE, using 6.5% (19:1) polyacrylamide, 8 M urea gels in 0.1 M NaOAc (pH 5.0) buffer. Gels were run for 48 h at 4°C with regular buffer changes. tRNAs were detected using specific γ‐32P‐dATP‐labeled oligonucleotide probes:
MT‐TL1: 5′‐AAG TCT TAC GCA ATT TCC TGG CTC TG‐3′
MT‐TF: 5′‐CAT TTT CAG TGC TTT GCT TTG TTA TTA‐3′
MT‐TV: 5′‐TGG TCA TGA AAT CTT CTG GGT GTA GG‐3′
MT‐TK: 5′‐TCA CTA TGG AGA TTT TAA GGT C‐3′
Isolation of mitochondria
For majority of experiments, including all Western blot and BN‐PAGE analysis, mitochondria were isolated from the heart, liver, or skeletal muscle as previously reported (Edgar et al, 2009). For isokinetic sucrose gradient and co‐immunoprecipitation (coIP) experiments, mitochondria were isolated as follows: Organs were homogenized in a mitochondria isolation buffer (320 mM sucrose, 10 mM Tris–HCl pH 7.5, protease inhibitor cocktail w/o EDTA) with a rotating Teflon potter (Potter S, Sartorius) at 1,200 rpm followed by differential centrifugation. To obtain mitochondria from MEFs, cells were grown at standard conditions, collected, washed with PBS, and resuspended in a homogenization buffer [20 mM HEPES pH 7.6, 220 mM mannitol, 70 mM sucrose, 1 mM EDTA, protease inhibitor cocktail (Sigma)]. Cell suspension was homogenized with a rotating Teflon potter (Potter S, Sartorius) at 1,000 rpm followed by differential centrifugation.
BN‐PAGE and in‐gel activity
BN‐PAGE was carried out using the NativePAGE Novex Bis‐Tris Mini Gel system (Invitrogen) according to the manufacturer's specifications. Proteins were transferred onto a PVDF (polyvinylidene fluoride) membrane, and immunodetection of mitochondrial OXPHOS complexes was performed (CI: NDUFA9; CII: SDHA; CIII: UQCRC1; CIV: COXI; CV: ATP5A1). In‐gel activity assays for complex I and complex IV were performed as previously described (Dogan et al, 2014).
Sucrose gradient fractionation of mitochondrial ribosomes
Protein and RNA analysis with long isokinetic sucrose gradient fractionation of the mitoribosomes was performed as described previously (Ruzzenente et al, 2012). In the control experiments, mitoribosomes were cleared out with RNase treatment (4,800 U/ml, AppliChem) following the lysis of mitochondria.
Short isokinetic sucrose gradients: Mitochondria obtained from single organs or whole cultured cells were resuspended in a lysis buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X‐100, 10 mM MgCl2, protease inhibitor cocktail, RNAse inhibitor), lysed for 30 min, and clarified by centrifugation at 12,000 × g at 4°C. 0.6–0.9 mg of supernatant was loaded onto a linear sucrose gradient (10–30% [vol/vol], 1 ml) in a gradient forming buffer (50 mM Tris–HCl, pH 7.2, 10 mM MgAc, 40 mM NH4Cl, 100 mM KCl, 50 μg/ml chloramphenicol, protease inhibitor cocktail, RNase inhibitor) and centrifuged for 2 h 15 min at 100,000 × g at 4°C. Fractions of 100 μl were collected and analyzed by Western blotting or subjected to the Northern blot analysis or qRT–PCR estimation of the RNA loading. The RNA profiles obtained from isokinetic sucrose gradient fractionation were determined with qRT–PCR technique using the TaqMan probes (Applied Biosystems): 16S rRNA (Mm03975671_s1), Atp6 (Mm03649417_g1), Cox3 (Mm04225261_g1), Cytb (Mm04225271_g1), Nd6/Nd5 (Mm04225315_s1). The 12S rRNA probe was kindly provided by Prof. N.‐G. Larsson, MPI for Biology of Aging Cologne.
Eral1 and Clpp overexpression
Transfections for sucrose density gradient sedimentations were carried out using the 4D‐Nulceofector System (Lonza) according to the manufacturer protocol. Plasmids used for Clpp overexpression correspond to the plasmids used in the substrate‐trapping screen. M. musculus Eral1 was expressed for 3 days from the pRT[Exp] plasmid under the CAG promoter, which was obtained from VectorBuilder (Cyagen).
Oxygen consumption measurements and respiratory chain enzyme activities
The measurements of respiratory chain enzyme complex activities and citrate synthase activity were performed as previously described (Wibom et al, 2002). Oxygen consumption rates were determined with Oxygraph‐2k (Oroboros Instruments) following the substrate–uncoupler–inhibitor–titration (SUIT) protocol. Briefly, mitochondria were isolated as described above and the state 3 was determined in the presence of CI substrates (pyruvate, malate, glutamate). Then, the state 3 for CI + CII was estimated after addition of CII substrate, succinate. Next, to evaluate mitochondrial coupling, oligomycin‐mediated inhibition of ATP synthase was performed (state 4) followed by a multiple‐step FCCP titration in order to determine the maximum respiration through the CI + CII (uncoupled).
Eral1 and Clpx siRNA‐mediated knockdown
Mouse embryonic fibroblast lines were transfected with siRNA oligonucleotide duplexes (ERAL1 siRNA: CCCAAGUGAUUCUGCUUGA; CLPX siRNA: GCUUCGCUGUCCUAAAUGU) (Eurogentec), using the lipofectamin reagent RNAi Max (Invitrogen) following the reverse transfection protocol provided by the manufacturer. The efficiency of downregulation was assessed by Western blot analysis.
Co‐immunoprecipitation analysis
For ERAL1 co‐immunoprecipitation, 1 mg of mitochondria was lysed in a solubilization buffer (1.5% w/v digitonin, 150 mM KAc, 30 mM Tris–HCl pH 7.4, complete protease inhibitor cocktail) and cleared by centrifugation at 16,000 × g. Lysates were incubated with 2 μg of respective antibodies (ERAL1 (Proteintech) or anti‐rabbit IgG (Sigma)) overnight at 4°C followed by 2 h incubation with Protein‐A Sepharose CL‐4b beads. Beads were washed five times with a buffer containing 0.5% digitonin, 150 mM KAc, 30 mM Tris–HCl pH 7.4, and protease inhibitor cocktail. Protein complexes were eluted with Laemmli buffer and subjected to SDS–PAGE followed by Western blot analysis.
For CLPX co‐immunoprecipitation experiments, 15 μg of CLPX antibody (Sigma) was immobilized on the AminoLink Plus resin (Thermo Fisher Scientific) according to the manufacturer's instructions. 1 mg of mitochondria was lysed in a solubilization buffer (1.5% w/v digitonin, 150 mM KAc, 30 mM Tris–HCl pH 7.4, complete protease inhibitor cocktail) and cleared by centrifugation at 16,000 × g. Lysates were incubated overnight with immobilized beads at 4°C followed by five times washing in a buffer containing 0.5% digitonin, 150 mM KAc, 30 mM Tris–HCl pH 7.4, and complete protease inhibitor cocktail. Protein complexes were eluted with low pH buffer (100 mM glycine pH 2.8) and subjected to SDS–PAGE followed by Western blotting.
Label‐free quantification of mitochondrial proteome
Crude mitochondria isolated from heart as described above were resuspended in 0.6 M sucrose, layered on a 1–1.5 M sucrose gradient, and spun at 71,000 g for 30 min at 4°C. After ultracentrifugation, mitochondria were collected from the interface between two sucrose phases and washed with 10 mM Tris–HCl, pH 7.5; 5 mM EDTA. Mitochondrial pellets were subjected to FASP digestion followed by LC‐MS/MS analysis.
FASP digestion
Mitochondrial pellets were lysed with 8 M Urea and applied to the 10 kDA Amplicon Filter. FASP digestion was performed as described previously (Wisniewski et al, 2010). In brief, proteins were washed several times with 8 M Urea, reduced using 10 mM DTT (Dithiothreitol) for 30 min, and alkylated with 55 mM IAA (Iodoacetamide) for 40 min in the dark. Then, conditions were set for digestion using 10 mM Tris–HCl pH = 7.5 for several washing steps. Trypsin was added in 1:100 enzyme to protein ratio and incubated at 37°C overnight. Peptides were eluted from the filter with water and acidified using TFA (Trifluoroacetic acid) to a final concentration of 1%.
Liquid chromatography and mass spectrometry
Generated peptides were desalted prior to liquid chromatography–tandem mass spectrometry (LC‐MS/MS) analysis using STAGE tip technique2 (Rappsilber et al, 2003). The eluate was concentrated in a speed vac to complete dryness and resuspended in 10 μl acidified water (Buffer A: 0.1% acetic acid). LC‐MS/MS instrumentation consisted out of an EASY nano‐LC (Proxeon, now Thermo Scientific) coupled via a nano‐electrospray ionization source (Thermo Scientific) to an ion‐trap‐based LTQ Discovery instrument (Thermo Scientific). 4 μl of peptide mixture was loaded onto an in‐house packed 15‐cm column (3‐μm C18 beads, Dr. Maisch). Peptides were separated by a binary buffer system: (A) 0.1% acetic acid and (B) 0.1% acetic acid in acetonitrile and the following gradient was applied: within 220 min, buffer B content was raised in a linear shape from 7% to 20% and further increased within 60 min to 40%. Then, 95% B was used to wash the column and held there for 20 min. The flow rate was constant at 200 nl/min over the complete gradient. After each run, a blank run (5% B = const.) was performed to re‐equilibrate the column.
Eluting peptides from the column were ionized by an applied voltage of 1.8 kV. The capillary voltage was set to 44 V, and Multipole RF Amplifier (Vp‐p) was set to 400. For MS1 scans, a resolution of 30,000 (400 m/z), a maximal injection time of 500 ms, and an AGC (Automatic gain control) target of 2e5 were used. In a data‐dependent Top10 mode, the 10 most intense peaks were selected for MS2 level scans in the ion trap. A resolution of 7,500 (400 m/z), an AGC target of 1E54, an isolation window of 3.0 Th, and a maximal injection time of 200 ms were used. The normalized collision energy for CID scans was 35, and the activation time for MS2 scans was 30 ms. For both scan types, the injection waveform setting was enabled.
All raw files were processed by MaxQuant 1.5.1.0 3, and the implemented Andromeda search engine 4. MS/MS spectra were correlated against the mouse Uniprot database (downloaded Feb. 2015) including a list of common contaminants. Search parameters were set as following: minimal peptide length: 7 amino acids; mass tolerance for MS/MS ITMS spectra: 0.5 Dalton. The FDR was estimated by the implemented decoy algorithm utilizing the revert database on the peptide–spectrum match (PSM) and protein level to 1%. The label‐free quantification, match between runs, and requantification of algorithms were enabled using default settings. Acetylation at the protein N‐term and oxidation of methionine residues were defined as variable modification, and carbamidomethylation at cysteine residues was set as a fixed modification. LFQ intensities were log2 transformed and tested for differential expression using a two‐sided t‐test. To correct against multiple testing errors, the FDR was calculated by a permutation‐based algorithm using a FDR cutoff of 5% and fudge factor s0 of 0.1. To identify significant enriched GO terms, we utilized the 1D enrichment tool in Perseus 5. Data visualization was done in the statistical environment R.
String network
Protein Uniprot IDs from Table EV3 were entered in String webtool (Snel et al, 2000), and the confidence level was set to 0.7 (“high confidence”). Active predictive models were as follows: Co‐expression, Experiments, and Database. This network was imported into Cytoscape for visualization (Shannon et al, 2003). Proteins not showing any connectivity were added to the network separately.
Data accessibility
Proteomics raw data have been uploaded to the PRIDE repository under the Accession Code:
Project Name: Mouse heart mitochondria LC‐MS/MS
Project accession: PXD002935
Project DOI: Not applicable
Statistical analysis
Student's t‐test was used to determine statistical significance. Error bars represent standard deviation (SD). Unless otherwise indicated, all experiments were performed on six biological replicates.
Author contributions
AT conceived the project and together with PM, KS, AK, and MK designed the experiments, analyzed the data, and wrote the manuscript. PM, KS, AK, EH, KS, HN, CB, BR, H‐TH‐D, RW, and RJW performed the experiments, interpreted, and analyzed data.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
The work was supported by grants of the European Research Council (ERC‐StG‐2012‐310700), and German Research Council (DFG). P.M and C.B. received scholarships from CECAD graduate school.
The EMBO Journal (2016) 35: 2566–2583
References
- Amunts A, Brown A, Toots J, Scheres SH, Ramakrishnan V (2015) Ribosome. The structure of the human mitochondrial ribosome. Science 348: 95–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat NH, Vass RH, Stoddard PR, Shin DK, Chien P (2013) Identification of ClpP substrates in Caulobacter crescentus reveals a role for regulated proteolysis in bacterial development. Mol Microbiol 88: 1083–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camara Y, Asin‐Cayuela J, Park CB, Metodiev MD, Shi Y, Ruzzenente B, Kukat C, Habermann B, Wibom R, Hultenby K, Franz T, Erdjument‐Bromage H, Tempst P, Hallberg BM, Gustafsson CM, Larsson NG (2011) MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab 13: 527–539 [DOI] [PubMed] [Google Scholar]
- Coenen MJ, Antonicka H, Ugalde C, Sasarman F, Rossi R, Heister JG, Newbold RF, Trijbels FJ, van den Heuvel LP, Shoubridge EA, Smeitink JA (2004) Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N Engl J Med 351: 2080–2086 [DOI] [PubMed] [Google Scholar]
- Cole A, Wang Z, Coyaud E, Voisin V, Gronda M, Jitkova Y, Mattson R, Hurren R, Babovic S, Maclean N, Restall I, Wang X, Jeyaraju DV, Sukhai MA, Prabha S, Bashir S, Ramakrishnan A, Leung E, Qia YH, Zhang N et al (2015) Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 27: 864–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damerau K, St John AC (1993) Role of Clp protease subunits in degradation of carbon starvation proteins in Escherichia coli . J Bacteriol 175: 53–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennerlein S, Rozanska A, Wydro M, Chrzanowska‐Lightowlers ZM, Lightowlers RN (2010) Human ERAL1 is a mitochondrial RNA chaperone involved in the assembly of the 28S small mitochondrial ribosomal subunit. Biochem J 430: 551–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan SA, Pujol C, Maiti P, Kukat A, Wang S, Hermans S, Senft K, Wibom R, Rugarli EI, Trifunovic A (2014) Tissue‐specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab 19: 458–469 [DOI] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar D, Shabalina I, Camara Y, Wredenberg A, Calvaruso MA, Nijtmans L, Nedergaard J, Cannon B, Larsson NG, Trifunovic A (2009) Random point mutations with major effects on protein‐coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metab 10: 131–138 [DOI] [PubMed] [Google Scholar]
- Feng J, Michalik S, Varming AN, Andersen JH, Albrecht D, Jelsbak L, Krieger S, Ohlsen K, Hecker M, Gerth U, Ingmer H, Frees D (2013) Trapping and proteomic identification of cellular substrates of the ClpP protease in Staphylococcus aureus. J Proteome Res 12: 547–558 [DOI] [PubMed] [Google Scholar]
- Fischer F, Weil A, Hamann A, Osiewacz HD (2013) Human CLPP reverts the longevity phenotype of a fungal ClpP deletion strain. Nat Commun 4: 1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA (2003) Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX‐recognition signals. Mol Cell 11: 671–683 [DOI] [PubMed] [Google Scholar]
- Freyer C, Park CB, Ekstrand MI, Shi Y, Khvorostova J, Wibom R, Falkenberg M, Gustafsson CM, Larsson NG (2010) Maintenance of respiratory chain function in mouse hearts with severely impaired mtDNA transcription. Nucleic Acids Res 38: 6577–6588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gispert S, Parganlija D, Klinkenberg M, Drose S, Wittig I, Mittelbronn M, Grzmil P, Koob S, Hamann A, Walter M, Buchel F, Adler T, Hrabe de Angelis M, Busch DH, Zell A, Reichert AS, Brandt U, Osiewacz HD, Jendrach M, Auburger G (2013) Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum Mol Genet 22: 4871–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hance N, Ekstrand MI, Trifunovic A (2005) Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. Hum Mol Genet 14: 1775–1783 [DOI] [PubMed] [Google Scholar]
- Jenal U, Fuchs T (1998) An essential protease involved in bacterial cell‐cycle control. EMBO J 17: 5658–5669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson EM, Clayton‐Smith J, Mehta S, Bennett C, Reardon W, Green A, Pearce SH, De Michele G, Conway GS, Cilliers D, Moreton N, Davis JR, Trump D, Newman WG (2012) Perrault syndrome: further evidence for genetic heterogeneity. J Neurol 259: 974–976 [DOI] [PubMed] [Google Scholar]
- Jenkinson EM, Rehman AU, Walsh T, Clayton‐Smith J, Lee K, Morell RJ, Drummond MC, Khan SN, Naeem MA, Rauf B, Billington N, Schultz JM, Urquhart JE, Lee MK, Berry A, Hanley NA, Mehta S, Cilliers D, Clayton PE, Kingston H et al (2013) Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP‐dependent chambered protease. Am J Hum Genet 92: 605–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SG, Ortega J, Singh SK, Wang N, Huang NN, Steven AC, Maurizi MR (2002) Functional proteolytic complexes of the human mitochondrial ATP‐dependent protease, hClpXP. J Biol Chem 277: 21095–21102 [DOI] [PubMed] [Google Scholar]
- Kardon JR, Yien YY, Huston NC, Branco DS, Hildick‐Smith GJ, Rhee KY, Paw BH, Baker TA (2015) Mitochondrial ClpX activates a key enzyme for heme biosynthesis and erythropoiesis. Cell 161: 858–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearsey SE, Craig IW (1981) Altered ribosomal RNA genes in mitochondria from mammalian cells with chloramphenicol resistance. Nature 290: 607–608 [DOI] [PubMed] [Google Scholar]
- Komarova AV, Tchufistova LS, Supina EV, Boni IV (2002) Protein S1 counteracts the inhibitory effect of the extended Shine‐Dalgarno sequence on translation. RNA 8: 1137–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwasniak M, Majewski P, Skibior R, Adamowicz A, Czarna M, Sliwinska E, Janska H (2013) Silencing of the nuclear RPS10 gene encoding mitochondrial ribosomal protein alters translation in Arabidopsis mitochondria. Plant Cell 25: 1855–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPolla RJ, Lambowitz AM (1977) Mitochondrial ribosome assembly in Neurospora crassa. Chloramphenicol inhibits the maturation of small ribosomal subunits. J Mol Biol 116: 189–205 [DOI] [PubMed] [Google Scholar]
- Lowth BR, Kirstein‐Miles J, Saiyed T, Brotz‐Oesterhelt H, Morimoto RI, Truscott KN, Dougan DA (2012) Substrate recognition and processing by a Walker B mutant of the human mitochondrial AAA+ protein CLPX. J Struct Biol 179: 193–201 [DOI] [PubMed] [Google Scholar]
- Morino H, Pierce SB, Matsuda Y, Walsh T, Ohsawa R, Newby M, Hiraki‐Kamon K, Kuramochi M, Lee MK, Klevit RE, Martin A, Maruyama H, King MC, Kawakami H (2014) Mutations in Twinkle primase‐helicase cause Perrault syndrome with neurologic features. Neurology 83: 2054–2061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moullan N, Mouchiroud L, Wang X, Ryu D, Williams EG, Mottis A, Jovaisaite V, Frochaux MV, Quiros PM, Deplancke B, Houtkooper RH, Auwerx J (2015) Tetracyclines disturb mitochondrial function across eukaryotic models: a call for caution in biomedical research. Cell Rep 10: 1681–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CB, Asin‐Cayuela J, Camara Y, Shi Y, Pellegrini M, Gaspari M, Wibom R, Hultenby K, Erdjument‐Bromage H, Tempst P, Falkenberg M, Gustafsson CM, Larsson NG (2007) MTERF3 is a negative regulator of mammalian mtDNA transcription. Cell 130: 273–285 [DOI] [PubMed] [Google Scholar]
- Pierce SB, Chisholm KM, Lynch ED, Lee MK, Walsh T, Opitz JM, Li W, Klevit RE, King MC (2011) Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc Natl Acad Sci USA 108: 6543–6548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce SB, Gersak K, Michaelson‐Cohen R, Walsh T, Lee MK, Malach D, Klevit RE, King MC, Levy‐Lahad E (2013) Mutations in LARS2, encoding mitochondrial leucyl‐tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am J Hum Genet 92: 614–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, Mann M (2003) Stop and go extraction tips for matrix‐assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75: 663–670 [DOI] [PubMed] [Google Scholar]
- Richter U, Lahtinen T, Marttinen P, Myohanen M, Greco D, Cannino G, Jacobs HT, Lietzen N, Nyman TA, Battersby BJ (2013) A mitochondrial ribosomal and RNA decay pathway blocks cell proliferation. Curr Biol 23: 535–541 [DOI] [PubMed] [Google Scholar]
- Richter‐Dennerlein R, Korwitz A, Haag M, Tatsuta T, Dargazanli S, Baker M, Decker T, Lamkemeyer T, Rugarli EI, Langer T (2014) DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab 20: 158–171 [DOI] [PubMed] [Google Scholar]
- Ruzzenente B, Metodiev MD, Wredenberg A, Bratic A, Park CB, Camara Y, Milenkovic D, Zickermann V, Wibom R, Hultenby K, Erdjument‐Bromage H, Tempst P, Brandt U, Stewart JB, Gustafsson CM, Larsson NG (2012) LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J 31: 443–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer RT, Baker TA (2011) AAA+ proteases: ATP‐fueled machines of protein destruction. Annu Rev Biochem 80: 587–612 [DOI] [PubMed] [Google Scholar]
- Seiferling D, Szczepanowska K, Becker C, Senft K, Hermans S, Maiti P, Konig T, Kukat A, Trifunovic A (2016) Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. EMBO Rep 17: 953–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma MR, Barat C, Wilson DN, Booth TM, Kawazoe M, Hori‐Takemoto C, Shirouzu M, Yokoyama S, Fucini P, Agrawal RK (2005) Interaction of Era with the 30S ribosomal subunit implications for 30S subunit assembly. Mol Cell 18: 319–329 [DOI] [PubMed] [Google Scholar]
- Sharma MR, Koc EC, Datta PP, Booth TM, Spremulli LL, Agrawal RK (2003) Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins. Cell 115: 97–108 [DOI] [PubMed] [Google Scholar]
- Snel B, Lehmann G, Bork P, Huynen MA (2000) STRING: a web‐server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res 28: 3442–3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson NG (2005) Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci USA 102: 17993–17998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiumi T, Ohgaki K, Yagi M, Aoki Y, Sakai A, Matsumoto S, Kang D (2010) ERAL1 is associated with mitochondrial ribosome and elimination of ERAL1 leads to mitochondrial dysfunction and growth retardation. Nucleic Acids Res 38: 5554–5568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wibom R, Hagenfeldt L, von Döbeln U (2002) Measurement of ATP production and respiratory chain enzyme activities in mitochondria isolated from small muscle biopsy samples. Anal Biochem 311: 139–151 [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Nagaraj N, Zougman A, Gnad F, Mann M (2010) Brain phosphoproteome obtained by a FASP‐based method reveals plasma membrane protein topology. J Proteome Res 9: 3280–3289 [DOI] [PubMed] [Google Scholar]
- Yagi M, Uchiumi T, Takazaki S, Okuno B, Nomura M, Yoshida S, Kanki T, Kang D (2012) p32/gC1qR is indispensable for fetal development and mitochondrial translation: importance of its RNA‐binding ability. Nucleic Acids Res 40: 9717–9737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zybailov B, Friso G, Kim J, Rudella A, Rodriguez VR, Asakura Y, Sun Q, van Wijk KJ (2009) Large scale comparative proteomics of a chloroplast Clp protease mutant reveals folding stress, altered protein homeostasis, and feedback regulation of metabolism. Mol Cell Proteomics 8: 1789–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
Proteomics raw data have been uploaded to the PRIDE repository under the Accession Code:
Project Name: Mouse heart mitochondria LC‐MS/MS
Project accession: PXD002935
Project DOI: Not applicable