

Abstract
Proline is an amino acid with a unique cyclic structure that facilitates the folding of many proteins, but also impedes the rate of peptide bond formation by the ribosome. As a ribosome substrate, proline reacts markedly slower when compared with other amino acids both as a donor and as an acceptor of the nascent peptide. Furthermore, synthesis of peptides with consecutive proline residues triggers ribosome stalling. Here, we report crystal structures of the eukaryotic ribosome bound to analogs of mono‐ and diprolyl‐tRNAs. These structures provide a high‐resolution insight into unique properties of proline as a ribosome substrate. They show that the cyclic structure of proline residue prevents proline positioning in the amino acid binding pocket and affects the nascent peptide chain position in the ribosomal peptide exit tunnel. These observations extend current knowledge of the protein synthesis mechanism. They also revise an old dogma that amino acids bind the ribosomal active site in a uniform way by showing that proline has a binding mode distinct from other amino acids.
Keywords: hydrolysis‐resistant aminoacyl‐tRNA analogs, peptide bond formation, proline, protein synthesis, ribosome
Subject Categories: Protein Biosynthesis & Quality Control, Structural Biology
Introduction
Among the ~20 amino acids that comprise all proteins in a living cell, proline stands out as the only residue in which the side chain is covalently attached to the α‐amine, forming a rigid cyclic structure. This proline structure facilitates folding of many proteins by introducing rigid turns into the peptide chain and by setting borders of β‐sheets and α‐helices 1, 2. Also, peptides with consecutive proline residues fold into a characteristic polyproline helix (PII‐helix), which constitutes a common protein–protein interaction motif and also endows proteins with unique mechanical properties 3, 4.
However, numerous studies show that, although the unique chemical structure of proline facilitates protein folding, it also markedly impedes the rate of peptide bond formation by the ribosome. When proline is used for protein synthesis—either as a peptidyl donor in ribosomal P‐site 5, 6, 7 or as a peptidyl acceptor in ribosomal A‐site 7, 8, 9—it reacts markedly slower than other amino acids. Most interestingly, the inhibitory effect of proline on protein synthesis becomes progressively stronger when peptides with consecutive proline residues must be produced by the ribosome 10, 11, 12. Thus, synthesis of peptides with three or more consecutive prolines provokes ribosome stalling 10, 11, 12.
In a living cell, ribosome stalling by polyproline sequences is resolved by a universally conserved translation factor, known as eIF5A in eukaryotes and EF‐P in bacteria 10, 11, 12. In eukaryotes, eIF5A alleviates ribosome stalling by contacting the acceptor stem of the P‐site tRNA, using a mechanism that is not yet fully understood 13, 14. The presence of eIF5A in eukaryotic cells enables synthesis of proteins containing polyproline motifs. This factor is essential because polyproline motifs are highly abundant in eukaryotic proteomes. Human proteome, for example, contains ~10,000 motifs with three or more consecutive proline residues, with some proteins having up to 27 consecutive prolines 1, 2.
Extensive kinetic studies of peptide bond formation with proline suggested that proline impedes the rate of protein synthesis by increasing entropy of peptide bond formation 7. Furthermore, cryoelectron microscopy analysis of ribosome complexes with stalling peptides revealed the position of proline residues in the ribosomal P‐site during translational stalling 15, 16, 17. These studies profoundly extended our understanding of protein synthesis chemistry with proline. However, the conformation of proline and its reactive groups in the peptidyl transferase center is still unknown and, therefore, it is unclear how proline slows down the rate of protein synthesis.
Seeking to address this question, we determined crystal structures of the yeast 80S ribosome, bound with prolyl‐ and diprolyl‐tRNA analogs. These initial structures provide high‐resolution snapshots of proline positioning in the active centers of the ribosome, such as the ribosomal A‐site and the nascent peptide tunnel. Our data illustrate that the unique chemical structure of proline cycle has a dramatic impact on the proline position in the ribosomal active centers, preventing proline from adopting an optimal position required for rapid protein synthesis.
Results and Discussion
Synthesis of aminoacyl‐ and peptidyl‐tRNA analogs and ribosome structure determination
To approach structural studies of protein synthesis with proline residues, we chemically synthesized aminoacyl‐ and dipeptidyl‐tRNA analogs (Fig EV1). These analogs comprise the A73C74C75A76 tRNA acceptor stem with a 3′‐amido (instead of the natural ester) linkage between A76 and the C‐terminus of the corresponding amino acid or dipeptide moiety to impede hydrolytic cleavage. Based on a novel 3′‐prolylamino‐3′‐deoxyadenosine solid support (Fig EV1), we produced the ribosome substrates ACCA‐Pro and ACCA‐Pro‐Pro. For reasons of comparison, we additionally produced the ribosome substrates ACC‐puromycin and ACCA‐Leu‐Phe using previously described strategies 18, 19.
Figure EV1. Chemical synthesis of hydrolysis‐resistant ACCA‐Pro and ACCA‐Pro‐Pro conjugates.

- Synthetic scheme overview. Letters indicate conditions: a) 1.25 equivalent of Fmoc‐Pro, 1.25 equivalent of O‐(benzotriazol‐1‐yl)‐N,N,N',N'‐tetramethyluronium hexafluorophosphate, 1.25 equivalent of 1‐hydroxybenzotriazole hydrate, 1.5 equivalent of N,N‐diisopropylethylamine in DMF, room temperature, 12 h, 76% for compound 2; b) 3 equivalent of adipic acid bis(pentafluorophenyl)ester, 1 equivalent of 4‐(N,N‐dimethyl‐amino)pyridine in N,N‐dimethylformamide/pyridine (1/1, v/v), room temperature, 1 h, 54% for compound 3; c) ˜1 equivalent of amino‐functionalized polystyrene support (GE Healthcare, Custom Primer SupportTM 200 Amino), pyridine, N,N‐dimethylformamide, room temperature, 1 day, loading: 45 μmol/g for solid support 4. DMT, 4,4′‐dimethoxytrityl; Fmoc, N‐(9‐fluorenyl)methoxycarbonyl; SPS, solid‐phase synthesis; AE HPLC, anion‐exchange high‐pressure liquid chromatography.
- Anion‐exchange HPLC profiles of crude (top) and purified (middle) ACCA‐Pro‐Pro conjugate and LC‐ESI mass spectra of purified products (bottom). Anion‐exchange chromatography conditions: Dionex DNAPac PA‐100 (4 × 250 mm) column; temperature: 60°C; flow rate: 1 ml/min; eluant A: 25 mM Tris–HCl (pH 8.0), 6 M urea; eluant B: 25 mM Tris–HCl (pH 8.0), 6 M urea, 500 mM NaClO4; gradient: 0–40% B in A within 25 min; UV detection at 260 nm.
Our choice of amido variants of aminoacyl‐tRNA analogs was based not only on the fact that these analogs are hydrolysis resistant and therefore, compatible with crystallization conditions, but also because they were shown to functionally mimic natural ribosome substrates. In particular, it was previously shown that amido derivatives of aminoacyl‐tRNA mimics adopt the same conformation in the peptidyl transferase center as natural aminoacyl‐tRNAs 20.
Having synthesized the ribosome substrates, we determined the crystal structures of these substrates bound to the Saccharomyces cerevisiae 80S ribosome. The ribosome structures bound with the A‐site ACCA‐proline (I/σ = 1 at 3.3 Å) or with ACC‐puromycin (ACCm2A‐methyl tyrosine) (I/σ = 1 at 3.25 Å) (Table EV1, Fig 1A and B) were determined after rapid soaking of preformed ribosome crystals in a 100 μM substrate solution (Materials and Methods). Similarly, the ribosome structures bound with P‐site ACCA‐proline‐proline (I/σ = 1 at 3.1 Å) and ACCA‐leucine‐phenylalanine (I/σ = 1 at 3.5 Å) were determined after soaking of ribosome crystals in 100 μM substrate solution supplemented with 300 μM the antibiotic sparsomycin. Sparsomycin was used to stabilize binding of the peptidyl‐tRNA analogs in the ribosomal P‐site 20, 21, 22 (Table EV1, Fig 1C and D).
Figure 1. Structures and electron density maps of the ribosome‐bound aminoacyl‐ and dipeptidyl‐tRNA analogs.

-
A, BA‐site‐bound substrates ACC‐puromycin (A) and ACCA‐Pro (B).
-
C, DP‐site‐bound substrates ACCA‐Leu‐Phe (C) and ACCA‐Pro‐Pro (D).
Our attempts to simultaneously bind aminoacyl‐ and peptidyl‐tRNA analogs to the A‐site and P‐site, respectively, resulted in datasets in which only aminoacyl‐tRNA analogs were observed in the A‐site, whereas the P‐site remained vacant. These results were similar to the previous studies of the ribosomal complexes with partial tRNA analogs 20, 21, 22. This possibly reflects a relatively weak affinity of the CCA moiety to the ribosomal P‐site 20, 21, 22.
Collectively, we obtained four ribosome crystal structures in which either the A‐site or the P‐site was occupied by a model aminoacyl‐ or dipeptidyl‐tRNA analog (Table EV1).
Proline in the ribosomal A‐site
Ribosome structures with the aminoacyl‐tRNA analogs bound to the A‐site have a similar resolution of ~3.3 Å (Table EV1). The electron density maps reveal the CCA fragment bound with the A‐loop of 25S rRNA, as well as aminoacyl moieties bound with the amino acid binding pocket of the ribosome (Fig 1A and B).
Although the ribosomal P‐site remains vacant in these crystal structures, previous studies showed that the P‐site tRNA binding has a relatively minor effect on the aminoacyl moiety conformation in the ribosomal A‐site 20, 22. Therefore, we used these structures to gain insight into a possible position of proline residue in the ribosomal A‐site during protein synthesis.
In the ribosome structure with the ACC‐puromycin (ACCm2A‐methyl tyrosine), the puromycin moiety has a nearly identical conformation to those observed in prokaryotic ribosomes in the pre‐attack state of the peptide bond formation 21, 23 (Figs 2A and EV2). In this conformation, the α‐amine is directed toward the peptidyl‐tRNA, whereas the amino acid chain flips away from the P‐site and remains bound to the conserved hydrophobic cavity of the ribosome, the A‐site cleft (Figs 2A and EV2). While bound to the A‐site cleft, the amino acid side chain remains physically separated from the actual site of the peptide bond formation by the conserved nucleotide A2820 of the yeast 25S rRNA (A2451 of the E. coli 23S rRNA).
Figure 2. Proline has an atypical side chain position in the ribosomal A‐site.
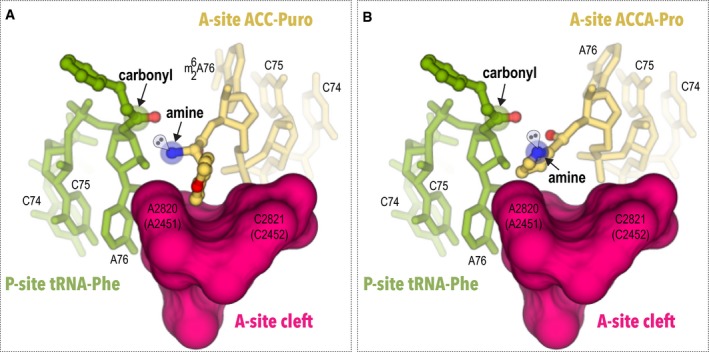
-
A, BThis figure compares conformation of aminoacyl residues of the two A‐site substrates: ACC‐puromycin (A) and ACCA‐Pro (B). To show position of the A‐site substrates relative to the P‐site, the model P‐site substrate tRNA‐Phe is shown as in the pre‐attack complex of the ribosome (PDB ID 1vy4). In both illustrations, the A‐site is viewed as through the peptide exit tunnel. In addition to S. cerevisiae residues numbering, E. coli numbering is shown in parentheses. Comparison of the two structures shows that the side chain of the methyl tyrosine is bound to the conserved hydrophobic A‐site cleft, whereas the side chain of the proline residue penetrates the actual site of peptide bond formation where it may interfere with alignment of the reacting groups and proper positioning of the attacking electron pair of the α‐amine.
Figure EV2. Evolution of the amino acid binding pocket in the ribosomal A‐site.

-
A–DWhen aminoacyl‐tRNAs bind the ribosomal A‐site, the amino acid side chain is accommodated by the A‐site cleft—a hydrophobic cavity formed by universally conserved nucleotides A2820 and C2821 in the 25S rRNA in S. cerevisiae (A2451 and C2452 in E. coli 23S rRNA). The views compare the A‐site cleft of prokaryotic (A, C) and eukaryotic (B, D) ribosomes. In the vacant eukaryotic ribosome (A), the cleft is smaller compared to those in prokaryotes (B), which likely results from a A‐to‐U mutation of the adjacent rRNA nucleotide (U2822 in S. cerevisiae 25S rRNA). In the archaeal ribosome, the corresponding residue is represented by A2488 in H. marismortiu 23S rRNA (PDB ID 3cc2). In bacteria, it is represented by A2452 in E. coli 23S rRNA (PDB ID 4y4o). However, when the aminoacyl‐tRNA or its analog puromycin occupies the A‐site (C, D), the cleft adopts similar conformations in pro‐ and eukaryotic ribosomes. Consequently, the aminoacyl moiety has a similar position in the active site of prokaryotic and eukaryotic ribosome. Therefore, the structural organization of the A‐site cleft is highly evolutionary conserved, despite rRNA sequence variations between prokaryotes and eukaryotes.
In the ribosome structure with the ACCA‐Pro, the proline moiety has a strikingly different and until now unknown conformation of an amino acid in the ribosomal A‐site compared to the typically uniform binding of other amino acids. Unlike methyl tyrosine and other amino acids observed in the A‐site 24, the proline side chain does not occupy the A‐site cleft, but instead flips toward the ribosomal P‐site (Fig 2B). In this conformation, the side chain of the proline residue penetrates the actual site of the peptide bond formation (Fig 2B). It is likely that in this position, the side chain may prevent proper alignment of the A‐site and P‐site substrates in the active site of the ribosome.
Additionally, compared to other amino acids, the α‐amino group of the proline residue is displaced by ~1 Å from the ribosomal P‐site (Fig EV3). In this conformation, proline may have an unusual orientation of the reactive electron pairs in the α‐amine group: The tetrahedral electron pair geometry and limited flexibility of the proline cycle suggest that proline's electron pair should deviate from the favorable position required for optimal nucleophilic attack (Fig 2B).
Figure EV3. Position of the aminoacyl‐tRNA analogs in the active site of the ribosome relative to the peptidyl‐tRNA.

- Position of ACC‐puromycin relative to the P‐site substrate.
- Position of ACCA‐Pro‐Pro relative to the P‐site substrate.
Thus, proline conformation in the ribosomal A‐site suggests that the poor reactivity of proline as a peptidyl acceptor originates from two factors: the unfavorable orientation of the α‐amine and its reactive electron pair as well as the binding of the proline side chain to the outside of the A‐site cleft of the ribosome. Both of these factors appear to prevent the optimal alignment of substrates in the peptidyl transferase center of the ribosome. Notably, despite proline's side chain not entering the A‐site cleft of the ribosome and adopting a highly unusual conformation, the α‐amine of proline remains accessible for the peptide bond formation and has a position in the peptidyl transferase center comparable to the ones observed for other amino acids, illustrating why proline remains reactive, although at an order of magnitude slower rate when it is compared to other proteinogenic amino acids 7, 8, 9.
Diprolyl peptide in the ribosomal nascent peptide exit tunnel
Ribosome structures with dipeptidyl‐tRNA analogs bound to the P‐site have a comparable resolution of 3.1–3.5 Å (Table EV1). The electron density maps reveal the CCA fragment bound to the P‐loop of 25S rRNA, as well as the dipeptidyl moieties at the entrance to the ribosomal nascent peptide exit tunnel (Fig 1C and D).
The antibiotic sparsomycin, which we used in this study to stabilize P‐site substrates on the ribosome, was described to deform the A76 sugar pocket and the carbonyl group in the P‐site peptidyl‐tRNA 20. Therefore, the crystal structures did not reveal the functional state of these critical groups in the P‐site ligands. However, sparsomycin does not affect the overall peptide chain conformation of the model dipeptides 20. Therefore, we used our structures to gain insights into how dipeptides Pro‐Pro and Leu‐Phe are positioned in the nascent peptide‐conducting tunnel.
In the ribosome structure with the ACCA‐Leu‐Phe ligand, the Leu‐Phe peptide enters the tunnel in a way similar to the short model peptides observed earlier 20, 22. The Phe residue appears to be partially disordered, reflecting conformational flexibility of this moiety in the peptide exit tunnel (Fig 3A).
Figure 3. Diprolyl peptide has a bent conformation in the nascent peptide exit tunnel of the ribosome.
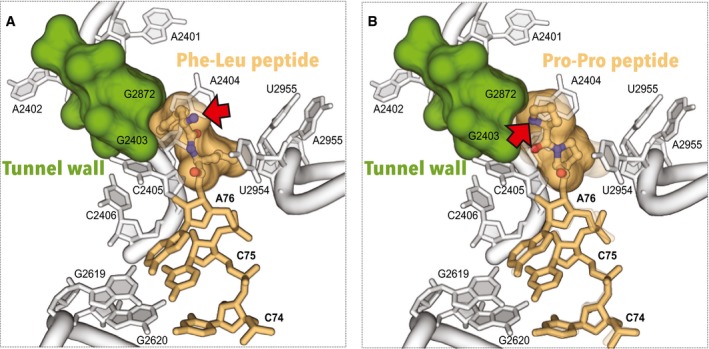
-
A, BThis figure compares conformation of two dipeptides in the ribosome nascent peptide tunnel: Phe‐Leu peptide (A), and Pro‐Pro peptide (B). In both illustrations, the arrow points to the α‐amine of the N‐terminal residue. Comparison of the two structures reveals that the N‐terminus of the Phe‐Leu peptide is directed into the tunnel, whereas the N‐terminus of the Pro‐Pro peptide is directed toward the wall of the tunnel.
In the ribosome structure with ACCA‐Pro‐Pro ligand, the observed diprolyl peptide structure is bent: The N‐terminus of the diprolyl peptide is oriented toward the nascent tunnel wall instead of being directed directly into the tunnel (Fig 3B). It is important to note that this bent conformation reflects unique stereochemical constraints for the pyrrolidine ring of a proline residue (Fig EV4). Consistently with these constraints, the diprolyl peptide has parameters of the trans‐polyproline helix (PII‐helix) (Fig EV4).
Figure EV4. The diprolyl peptide has a PII‐helix geometry.

- A Ramachandran plot, calculated for the diprolyl peptide, illustrates that the peptide has parameters of PII‐helix. Red spot indicates torsion angles in the diprolyl peptide, labels show average values for the three most common elements of protein secondary structure—α‐helix, β structure, and PII‐helix.
- The diprolyl peptide structure shows positions and values of the major torsion angles. Semitransparent spheres represent van der Waals radii of the diprolyl peptide atoms.
- Side‐by‐side structures of the diprolyl peptide, observed in this study, and an ideal PII‐helix. Residues are numbered from the C‐ to N‐terminus.
The diprolyl peptide position in the ribosomal P‐site suggests that bending of a nascent peptide by consecutive proline residues may compromise the peptide passage along the tunnel, because proline‐proline bonds lack the rotatory freedom and do not allow the peptide chain to deviate from the polyproline helix fold (Fig EV5). If correct, this model may explain how residues, which are not located directly in the peptidyl transferase center (e.g., N‐terminal residues in the XPP stalling motifs), may dramatically influence the rate of peptide bond formation 25. Also, if this assumption is correct, then synthesis of polyproline peptides may originate in a way similar way to ribosome stalling by regulatory peptides 15, 16, 17, 26. Although future studies are required to understand this complex problem, our initial data indicate that ribosome stalling by polyproline‐containing peptides may originate in part from interactions between the nascent peptide and the tunnel.
Figure EV5. Structure of the diprolyl peptide in the peptide exit tunnel and a possible mechanism of ribosome stalling.

- Unbiased Fo‐Fc electron density map, contoured at 3 σ, reveals the diprolyl‐tRNA analog in the ribosomal P‐site. In the peptide exit tunnel, the diprolyl peptide has a conformation in which the N‐terminal residue (Pro −1) is directed toward the wall of the tunnel (residues G2403 and A2872). In this conformation, further attachment of proline residues to the nascent peptide would result in a steric clash between the nascent peptide and the ribosomal peptide exit tunnel.
- To illustrate the steric clash, a hypothetical proline residue Pro −2 (in magenta) is attached to the N‐terminus of the diprolyl peptide. The resulting triprolyl peptide illustrates how a characteristic and rigid fold of polyproline peptides—PII‐helix—may potentially create an obstacle for the nascent peptide passage through the ribosomal tunnel.
The observation that diprolyl peptide has parameters of the PII‐helix may have important implications for studies of co‐translational protein folding. The diprolyl peptide structure suggests that PII‐helices are formed simultaneously with the formation of peptide bonds between consecutive proline residues. Because PII‐helix is the third most common secondary structure element after β‐sheets and α‐helices 27, it will be interesting to study whether PII‐helices can indeed be formed in the ribosomal tunnel and how it passes along the tunnel during protein synthesis.
Perspectives
Beyond insights into protein synthesis with proline, our data revise old dogmas suggesting that amino acids bind the ribosomal active site in a uniform way 24. Proline's position in the ribosomal A‐site illustrates that side chain structure may have a tremendous impact on amino acid positioning in the ribosomal active site. Proline position also demonstrates that amino acids have more than a single mode by which to bind the peptidyl transferase center.
In summary, our study provides ribosome structures which describe proline binding within the functional centers of the ribosome: the ribosomal A‐site and the peptide tunnel. In the future, this study may impact not only the field of protein synthesis, but also the emerging field of synthetic biology in which synthetic amino acids are used for protein synthesis to expand the chemistry of living systems 28, 29, 30. The knowledge of how amino acid's structure affects its binding to the ribosome may enable rational design of unnatural ribosome substrates and remodeling of the ribosome active sites to enable rapid protein synthesis with unnatural substrates.
Materials and Methods
Synthesis of hydrolysis‐resistant aminoacyl‐ and peptidyl‐tRNA analogs
As the presence of STM1 protein in the P‐site of our yeast ribosome crystals blocks access of full‐length tRNAs 31, we produced partial aminoacyl‐tRNA and dipeptidyl‐tRNA analogs, using A73C74C75A76 RNA moiety and introduced a 3′‐amido linkage between A76 and the C‐terminus of the diprolyl moiety to prevent hydrolysis of this analog during crystallization. While eukaryotic tRNAPro typically contains C rather than A at residue 73, this residue does not interact with the ribosome and, consistently, does not affect the tRNA position on the ribosome.
The ACC‐puromycin conjugate was produced as previously described 32. The ACCA‐Leu‐Phe conjugate was produced according to the following references 33, 34. The ACCA‐Pro‐Pro and ACCA‐Pro conjugates were produced as outlined in Fig EV1 and as described below:
Compound 2: N6‐[(Di‐n‐butylamino)methylene]‐3′‐[N‐(9‐fluorenyl)methoxycarbonyl‐L‐prolyl] amino‐3′‐deoxy‐5′‐O‐(4,4′‐dimethoxytrityl)‐D‐adenosine. Fmoc‐protected proline (84 mg, 0.25 mmol) was dissolved in 3 ml DMF followed by addition of O‐(benzotriazol‐1‐yl)‐N,N,N',N'‐tetramethyluronium hexafluorophosphate (HBTU, 94 mg, 0.25 mmol), 1‐hydroxybenzotriazole hydrate (HOBt, 38 mg, 0.25 mmol), and N,N‐diisopropylethylamine (DIPEA, 50 μl, 0.29 mmol). After 3 min of activation, 3′‐amino‐N6‐[(di‐n‐butylamino)methylene]‐3′‐deoxy‐5′‐O‐(4,4′‐dimethoxytrityl)‐D‐adenosine 1 35 (135 mg, 0.19 mmol, in 1 ml DMF) was added and the mixture was stirred overnight at room temperature. Then, the solvent was evaporated, the residue dissolved in CH2Cl2 and washed consecutively with half‐saturated aqueous NaHCO3 solution, 5% citric acid solution, and saturated NaCl solution. The organic layer was dried (Na2SO4) and evaporated, and the crude product was purified via SiO2 chromatography yielding 150 mg of compound 2 as white foam (76%). TLC (6% MeOH in CH2Cl2) Rf = 0.4. 1H NMR (300 MHz, CDCl3) δ 9.02 (s, 1H, HC = N(6)), 8.44 (s, 1H, HC(2)), 8.09 (s, 1H, HC(8)), 7.73 (m, 2H, HC(ar)), 7.54 (d, 1H, J = 7.3, HC(ar)), 7.33–7.16 (m, 14H, HC(ar)), 6.98 (s, 1H, HN(3′)), 6.75 (d, 4H, J = 7.2), 6.00 (s, 1H, HC(1′)), 4.83 (s, 1H, HC(2′)), 4.70 (m, 1H, HC(3′)), 4.37 (m, 3H, HC(4′) and OCH2(Fmoc)), 4.22 (m, 2H, HC(9, Fmoc) and HC(α, Pro)), 3.75 (s, 6H, 2xOCH3(DMT)), 3.71 (m, 2H, N(CH2CH2CH2CH3)2), 3.57 (m, 2H, CH2(Pro)), 3.43 (m, 4H, H2C(5′) and N(CH2CH2CH2CH3)2), 2.16–1.91 (m, 4H, 2xCH2(Pro)), 1.66 (m, 4H, N(CH2CH2CH2CH3)2), 1.35 (m, 4H, N(CH2CH2CH2CH3)2), 0.94 (m, 6H, N(CH2CH2CH2CH3)2). 13C NMR (75 MHz, CDCl3) δ 158.6 (HC = N(6)), 152.7 (C(2)), 140.0 (C(8)), 130.2 (C(ar)), 128.3–125.2 (C(ar)), 120.1 (C(ar)), 113.2 (C(ar)), 91.2 (C(1′)), 83.2 (C(4′)), 74.7 (C(2′)), 68.0 (OCH2(Fmoc)), 63.8 (C(5′)), 60.8 (C(α, Pro)), 55.3 (2xOCH3), 53.5 (C(3′)), 52.0 (N(CH2CH2CH2CH3)2), 47.3 (CH(Fmoc)), 46.5 (HC(γ, Pro)), 45.3 (N(CH2CH2CH2CH3)2), 31.1 (N(CH2CH2CH2CH3)2), 29.4 (CH2, Pro), 25.0 (CH2(Pro)), 20.3 and 19.9 (N(CH2CH2CH2CH3)2), 14.0 and 13.8 (N(CH2CH2CH2CH3)2). ESI‐MS (m/z): [M+H]+ calcd for C60H67N8O8, 1027.51; found 1,027.50.
Compound 3: N6‐[(Di‐n‐butylamino)methylene]‐3′‐[N‐(9‐fluorenyl)methoxycarbonyl‐L‐prolyl] amino‐3′‐deoxy‐5′‐O‐(4,4′‐dimethoxytrityl)‐2′‐O‐[1,6‐dioxo‐6‐(pentafluorophenyloxy)hexyl]‐D‐adenosine. To a solution of compound 2 (200 mg, 0.19 mmol) in DMF and pyridine (2.0 ml each) was added DMAP (24 mg, 0.19 mmol) and bis(pentafluorophenyl) adipate (298 mg, 0.61 mmol). The mixture was stirred for one hour followed by evaporation of the solvents. The crude product was purified via SiO2 chromatography yielding 139 mg of compound 3 as white foam (54%). TLC (20% acetone in CH2Cl2) Rf = 0.4. 1H NMR (300 MHz, CDCl3) δ 8.98 (s, 1H, HC = N(6)), 8.47 (s, 1H, HC(2)), 8.01 (HC(8)), 7.74 (d, 2H, J = 7.4, HC(ar)), 7.55 (d, 2H, J = 7.2, HC(ar)), 7.40–7.14 (m, 15H, HN(3′) and HC(ar) and CDCl3), 6.75 (d, 4H, J = 7.7, HC(ar)), 6.12 (d, 1H, J = 2.8, HC(1′)), 5.80 (m, 1H, HC(2′)), 5.14 (q, 1H, HC(3′)), 4.48 (b, 1H, HC(9, Fmoc)), 4.23 (m, 4H, HC(4′) and HC(α, Pro) and OCH2(Fmoc)), 3.75 (s, 6H, 2xOCH3(DMT)), 3.70 (m, 2H, N(CH2CH2CH2CH3)2), 3.52–3.36 (m, 6H, H2C(5′) and CH2(Pro) and N(CH2CH2CH2CH3)2), 2.51 and 2.36 (s, 2H, OOCH2CH2CH2CH2COO), 2.02–1.80 (m, 4H, 2xCH2(Pro)), 1.62 (m, 8H, OOCH2CH2CH2CH2COO and N(CH2CH2CH2CH3)2), 1.35 (m, 4H, N(CH2CH2CH2CH3)2), 0.93 (m, 6H, N(CH2CH2CH2CH3)2). 13C NMR (75 MHz, CDCl3) δ 158.6 (HC = N(6)), 153.0 (C(2)), 141.4 (C(8)), 130.3 (C(ar)), 128.4‐125.0 (C(ar)), 120.2 (C(ar)), 113.3 (C(ar)), 86.8 (C(1′)), 82.7 (C(4′)), 75.2 (C(2′)), 68.0 (OCH3(Fmoc) and CH(Fmoc)), 63.5 (C(5′)), 60.1 (C(α, Pro)), 55.3 (2xOCH3)), 52.0 (N(CH2CH2CH2CH3)2), 50.8 (N(CH2CH2CH2CH3)2), 47.2 (HC(γ, Pro)), 45.3 (N(CH2CH2CH2CH3)2), 33.2 (OOCH2CH2CH2CH2COO), 32.9 (OOCH2CH2CH2CH2COO), 31.5 (N(CH2CH2CH2CH3)2), 31.1 (N(CH2CH2CH2CH3)2), 27.4 (H2C(Pro)), 24.9 (H2C(Pro)), 20.3 (N(CH2CH2CH2CH3)2), 19.9 (N(CH2CH2CH2CH3)2), 14.0 (N(CH2CH2CH2CH3)2), 13.8 (N(CH2CH2CH2CH3)2). ESI‐MS (m/z): [M+H]+ calcd for C72H74F5N8O11, 1,321.54; found 1,321.41.
Compound 4: DMTO‐rA3′‐NH‐Pro‐NHFmoc solid support. Compound 3 (130 mg, 0.09 mmol) was dissolved in dry DMF (5.0 ml), and pyridine (20 μl) was added. To this solution, amino‐functionalized support (GE Healthcare, Custom Primer Support™ 200 Amino, 500 mg) was added, and the suspension was agitated for 20 h at room temperature. Subsequently, the beads were collected on a Büchner funnel and washed with DMF, methanol, and CH2Cl2. For capping of unreacted amino groups, the beads were treated with a mixture of solution A (0.2 M phenoxy acetic anhydride in THF, 10 ml) and solution B (0.2 M N‐methyl imidazole, 0.2 M sym‐collidine in THF, 10 ml) and agitated for 10 min at room temperature. The suspension was filtrated again, the beads were washed with THF, methanol, and CH2Cl2 and dried under vacuum. Loading of the support was 45 μmol/g.
Solid‐phase peptide synthesis on the solid support 4
In a frit‐fitted syringe, the solid support 4 (80 mg) was soaked with dry DMF (2 ml, 30 min). For deprotection of the Nα‐Fmoc group, the solid support was treated two times with piperidine solution (20% in DMF, 1.5 ml, 8, 12 min) and subsequently washed with DMF (3 × 2 ml). Coupling was performed by treating the solid support for one hour with a mixture of Fmoc‐proline solution (0.4 M in DMF, 500 μl), activator solution (HBTU and HOBt, 0.6 M in DMF, 750 μl), and DIPEA (140 μl). This step was performed twice. Then, the solid support was washed with DMF (3 × 2 ml) and acetonitrile (3 × 2 ml) and vacuum‐dried.
RNA synthesis
The ACCA moiety was assembled on an ABI 392 Nucleic Acid Synthesizer following standard synthesis protocols. Detritylation (120 s): dichloroacetic acid/1,2‐dichloroethane (4/96); coupling (120 s): phosphoramidites (0.1 M in acetonitrile, 130 ml) were activated with benzylthiotetrazole (0.3 M in acetonitrile, 180 μl); capping (2 × 10 s, Cap A/Cap B = 1/1): Cap A: phenoxyacetic anhydride (0.2 M in THF), Cap B: N‐methyl imidazole (0.2 M), sym‐collidine (0.2 M) in THF; oxidation (20 s): I2 (0.2 M) in THF/pyridine/H2O (35/10/5). Amidites, benzylthiotetrazole, and capping solutions were dried over activated molecular sieves (4 Å) overnight.
Final product
Deprotection of the 3′‐diprolyl‐ACCA conjugate. A) Fmoc deprotection. In the ABI synthesis column, the solid support was treated with a solution of 20% piperidine in acetonitrile (10 ml, 10 min), washed with acetonitrile, and dried. B) Acyl deprotection and cleavage from the solid support. For the conjugates synthesized on solid support 4, the beads were transferred into an Eppendorf tube and equal volumes of methylamine in ethanol (8 M, 0.5 ml) and methylamine in H2O (40%, 0.5 ml) were added. After 6 h shaking at room temperature, the supernatant was filtered and evaporated to dryness. C) 2′‐O‐TOM deprotection. The obtained residue was treated with TBAF·3 H2O in THF (1 M, 1 ml) overnight at room temperature. The reaction was quenched by the addition of triethylammonium acetate (TEAA) (1 M, pH 7.4, 1 ml). After reducing the volume of the solution, it was applied on a size‐exclusion chromatography column (GE Healthcare, HiPre 26/10 desalting, 2.6 × 10 cm, Sephadex G25). By eluating with H2O, the conjugate‐containing fractions were collected and evaporated to dryness, and the residue was dissolved in H2O (1 ml). Analysis of the crude products was performed by anion‐exchange chromatography on a Dionex DNAPac PA‐100 column (4 × 250 mm) at 60°C. Flow rate: 1 ml/min; eluent A: 25 mm Tris–HCl (pH 8.0), 6 M urea; eluent B: 25 mM Tris–HCl (pH 8.0), 0.5 M NaClO4, 6 M urea; gradient: 0–60% B in A within 45 min or 0–40% B in A within 30 min, UV detection at λ = 260 nm.
Purification of the 3′‐diprolyl‐ACCA conjugate
The crude conjugate was purified on a semipreparative Dionex DNAPac PA‐100 column (9 × 250 mm) at 60°C with flow rate of 2 ml/min (for eluents, see above). Fractions containing the conjugate were loaded on a C18 SepPak Plus cartridge (Waters/Millipore), washed with 0.1–0.15 M (Et3NH)+ , H2O, and eluted with H2O/CH3CN (1:1). Conjugate‐containing fractions were evaporated to dryness and dissolved in H2O (1 ml). The quality of the purified conjugate was analyzed by analytical anion‐exchange chromatography (for conditions, see above). The molecular weight of the synthesized conjugate was confirmed by LC‐ESI mass spectrometry (see Fig EV1B). Yields were determined by UV photometrical analysis of conjugate solutions. The final compound was dissolved in water to achieve ~50 mM concentration for stock solutions and later used for soaking.
Ribosome purification, crystallization, crystal treatments
80S ribosomes from the yeast S. cerevisiae were purified, crystallized, and treated essentially as previously described 14, 31. Ribosome complexes, containing ACCA analogs, were formed by soaking with 100 μM of the corresponding compound and 300 μM of sparsomycin for ~2 h at 4°C in a buffer containing 80 mM Tris‐acetate pH 7.0, 70 mM KSCN, 10 mM Mg(OAc)2, 20% v/v glycerol, 15% w/v PEG 20,000, 6.5 mM spermidine, 7.5 mM NH4OAc, 1.4 mM deoxy‐big‐chap, 2 mM DTT before the transfer into a cryoprotecting buffer containing 80 mM Tris‐acetate pH 7.0, 70 mM KSCN, 10 mM Mg(OAc)2, 18% v/v glycerol, 20% w/v PEG 20,000, 6.5 mM spermidine, 7.5 mM NH4OAc, 20% w/v PEG 6,000, 2 mM Os(NH2)6Cl3.
Data collection and processing
Diffraction data were collected from crystals cooled to 90°K using 0.1o oscillation range and the beam‐line Proxima 1 at the Synchrotron Soleil (France). We used a data collection strategy developed at Swiss Light Source Synchrotron (Switzerland), which exploits the unique features of the single photon counting pixel detector PILATUS 6M 31, 36, 37. During data collection, the beam was attenuated to 3–10% of its maximum flux. This was done to markedly reduce radiation damage and collect a highly redundant dataset from multiple spots of each crystal. Then, data were processed and reduced by the XDS suite 38, yielding the statistics displayed in Table EV1.
Because of highly attenuated beam, this data collection strategy results in unconventionally high Rmeas values when it is compared to CCD‐detector type data collection strategies (no beam attenuation, no fine ϕ‐slicing, low data redundancy). However, it provides more accurate values for reflections' intensities and anomalous signal when single photon counting pixel detectors are used for data collection 37, 39.
Structure determination, refinement, validation, and analyses
The structures were determined by molecular replacement using the vacant yeast 80S ribosome structure (pdb 4v88) as a search model and then subjected to refinement using Phenix.refine 40. Restraints for ACCA analogs and sparsomycin were generated with JLigand 41 and ReadySet from the Phenix suite 40.
Ligands building, fitting, remodeling of ribosomal binding sites and analysis of Ramachandran plots were performed using Coot 42. Ribosome structure and ligand geometry (torsion angles, bond lengths, and bond angles) were refined using Phenix.refine 40. Crystallographic statistics are reported in Table EV1. Ligands geometry was validated with the software Mogul from the CCDC package 43. Compared to the original model of S. cerevisiae ribosome (pdb 4v88), conformation of several rRNA nucleotides was corrected in both monomers for 25S rRNA residues U766, A806, U922, G1152, U2211, C2278, G2283, C2726, A2401, A2872 and A2971 and 16S rRNA residues G163, G337, A542, and A811 in 18S rRNA, and metal ions were modeled de novo. The Ramachandran plot was calculated using Molprobity 44. Figures were prepared using PyMOL (Schrodinger LLC).
Author contributions
SM, MY, TED, and RM devised experiments; LR and SN produced tRNA conjugates, JM crystallized ribosomal complexes and performed the soaking; JM and SM performed crystallographic data collection and processing, SM and JM analyzed crystallographic data; SM, JM, LR, SN, B‐SS, GY, TD, RN and MY wrote and commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Acknowledgements
We are grateful to Petr Sergiev (Lomonosov Moscow State University) for providing us with sparsomycin; to the staff of Proxima 1 beamline at the synchrotron SOLEIL (France), and in particular to Andrew Thompson and Pierre Legrand for providing rapid access to the beam and for assisting with data collection, and to members of T.D., R.M., and M.Y. teams for insightful and inspiring discussions! We also thank Corwin Miller and members of Tom Steitz and Dieter Söll laboratories (Yale University) for critically reading and commenting on the manuscript. We also thank Catherine Dunlavey (University of St. Andrews, UK) for editorial support during the manuscript preparation. This work was supported by Foundation pour la Recherche Médicale en France SPF20111223404 (to S.M.), the Austrian Science Fund FWF (P21641 and I1040 to R.M.), the French National Research Agency ANR‐15‐CE11‐0021‐01 (to G.Y.), the European Research Council advanced grant 294312 and the Human Frontier Science Program grant RGP0062/2012 (both to M.Y.), the Russian Government Program of Competitive Growth of Kazan Federal University (both to M.Y. and G.Y.), and in part by the Intramural Research Program of the National Institutes of Health, NICHD (T.D.). The structures were deposited to the protein data bank with accession codes 5dtv, 5lyb, 5tga, and 5tgm.
EMBO Reports (2016) 17: 1776–1784
Contributor Information
Thomas E Dever, Email: thomas.dever@nih.gov.
Ronald Micura, Email: ronald.micura@uibk.ac.at.
Marat Yusupov, Email: marat@igbmc.fr.
References
- 1. Levitt M (1981) Effect of proline residues on protein folding. J Mol Biol 145: 251–263 [DOI] [PubMed] [Google Scholar]
- 2. Morgan AA, Rubenstein E (2013) Proline: the distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PLoS One 8: e53785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bochicchio B, Tamburro AM (2002) Polyproline II structure in proteins: identification by chiroptical spectroscopies, stability, and functions. Chirality 14: 782–792 [DOI] [PubMed] [Google Scholar]
- 4. Adzhubei AA, Sternberg MJ, Makarov AA (2013) Polyproline‐II helix in proteins: structure and function. J Mol Biol 425: 2100–2132 [DOI] [PubMed] [Google Scholar]
- 5. Wohlgemuth I, Brenner S, Beringer M, Rodnina MV (2008) Modulation of the rate of peptidyl transfer on the ribosome by the nature of substrates. J Biol Chem 283: 32229–32235 [DOI] [PubMed] [Google Scholar]
- 6. Muto H, Ito K (2008) Peptidyl‐prolyl‐tRNA at the ribosomal P‐site reacts poorly with puromycin. Biochem Biophys Res Commun 366: 1043–1047 [DOI] [PubMed] [Google Scholar]
- 7. Doerfel LK, Wohlgemuth I, Kubyshkin V, Starosta AL, Wilson DN, Budisa N, Rodnina MV (2015) Entropic Contribution of Elongation Factor P to Proline Positioning at the Catalytic Center of the Ribosome. J Am Chem Soc 137: 12997–13006 [DOI] [PubMed] [Google Scholar]
- 8. Pavlov MY, Watts RE, Tan Z, Cornish VW, Ehrenberg M, Forster AC (2009) Slow peptide bond formation by proline and other N‐alkylamino acids in translation. Proc Natl Acad Sci USA 106: 50–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Johansson M, Ieong KW, Trobro S, Strazewski P, Aqvist J, Pavlov MY, Ehrenberg M (2011) pH‐sensitivity of the ribosomal peptidyl transfer reaction dependent on the identity of the A‐site aminoacyl‐tRNA. Proc Natl Acad Sci USA 108: 79–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Doerfel LK, Wohlgemuth I, Kothe C, Peske F, Urlaub H, Rodnina MV (2013) EF‐P is essential for rapid synthesis of proteins containing consecutive proline residues. Science 339: 85–88 [DOI] [PubMed] [Google Scholar]
- 11. Ude S, Lassak J, Starosta AL, Kraxenberger T, Wilson DN, Jung K (2013) Translation elongation factor EF‐P alleviates ribosome stalling at polyproline stretches. Science 339: 82–85 [DOI] [PubMed] [Google Scholar]
- 12. Gutierrez E, Shin BS, Woolstenhulme CJ, Kim JR, Saini P, Buskirk AR, Dever TE (2013) eIF5A promotes translation of polyproline motifs. Mol Cell 51: 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schmidt C, Becker T, Heuer A, Braunger K, Shanmuganathan V, Pech M, Berninghausen O, Wilson DN, Beckmann R (2016) Structure of the hypusinylated eukaryotic translation factor eIF‐5A bound to the ribosome. Nucleic Acids Res 44: 1944–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Melnikov S, Mailliot J, Shin BS, Rigger L, Yusupova G, Micura R, Dever TE, Yusupov M (2016) Crystal Structure of Hypusine‐Containing Translation Factor eIF5A Bound to a Rotated Eukaryotic Ribosome. J Mol Biol 428: 3570–3576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang J, Pan X, Yan K, Sun S, Gao N, Sui SF (2015) Mechanisms of ribosome stalling by SecM at multiple elongation steps. elife 4: e09684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bischoff L, Berninghausen O, Beckmann R (2014) Molecular basis for the ribosome functioning as an L‐tryptophan sensor. Cell Rep 9: 469–475 [DOI] [PubMed] [Google Scholar]
- 17. Matheisl S, Berninghausen O, Becker T, Beckmann R (2015) Structure of a human translation termination complex. Nucleic Acids Res 43: 8615–8626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ikeda S, Saito I, Sugiyama H (1998) Facile synthesis of puromycin‐tethered oligonucleotides at the 3′‐end. Tetrahedron Lett 39: 5975–5978 [Google Scholar]
- 19. Moroder H, Steger J, Graber D, Fauster K, Trappl K, Marquez V, Polacek N, Wilson DN, Micura R (2009) Non‐hydrolyzable RNA‐peptide conjugates: a powerful advance in the synthesis of mimics for 3′‐peptidyl tRNA termini. Angew Chem 48: 4056–4060 [DOI] [PubMed] [Google Scholar]
- 20. Schmeing TM, Huang KS, Kitchen DE, Strobel SA, Steitz TA (2005) Structural insights into the roles of water and the 2′ hydroxyl of the P site tRNA in the peptidyl transferase reaction. Mol Cell 20: 437–448 [DOI] [PubMed] [Google Scholar]
- 21. Schmeing TM, Huang KS, Strobel SA, Steitz TA (2005) An induced‐fit mechanism to promote peptide bond formation and exclude hydrolysis of peptidyl‐tRNA. Nature 438: 520–524 [DOI] [PubMed] [Google Scholar]
- 22. Hansen JL, Schmeing TM, Moore PB, Steitz TA (2002) Structural insights into peptide bond formation. Proc Natl Acad Sci USA 99: 11670–11675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Polikanov YS, Steitz TA, Innis CA (2014) A proton wire to couple aminoacyl‐tRNA accommodation and peptide‐bond formation on the ribosome. Nat Struct Mol Biol 21: 787–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Steitz TA (2008) A structural understanding of the dynamic ribosome machine. Nat Rev Mol Cell Biol 9: 242–253 [DOI] [PubMed] [Google Scholar]
- 25. Elgamal S, Katz A, Hersch SJ, Newsom D, White P, Navarre WW, Ibba M (2014) EF‐P dependent pauses integrate proximal and distal signals during translation. PLoS Genet 10: e1004553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ito K, Chiba S (2013) Arrest peptides: cis‐acting modulators of translation. Annu Rev Biochem 82: 171–202 [DOI] [PubMed] [Google Scholar]
- 27. Chebrek R, Leonard S, de Brevern AG, Gelly JC (2014) PolyprOnline: polyproline helix II and secondary structure assignment database. Database 2014: bau102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chin JW (2014) Expanding and reprogramming the genetic code of cells and animals. Annu Rev Biochem 83: 379–408 [DOI] [PubMed] [Google Scholar]
- 29. Neumann H, Wang K, Davis L, Garcia‐Alai M, Chin JW (2010) Encoding multiple unnatural amino acids via evolution of a quadruplet‐decoding ribosome. Nature 464: 441–444 [DOI] [PubMed] [Google Scholar]
- 30. O'Donoghue P, Ling J, Wang YS, Soll D (2013) Upgrading protein synthesis for synthetic biology. Nat Chem Biol 9: 594–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ben‐Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M (2011) The structure of the eukaryotic ribosome at 3.0 A resolution. Science 334: 1524–1529 [DOI] [PubMed] [Google Scholar]
- 32. Neuner S, Micura R (2014) Synthesis of aminoacylated N(6), N(6)‐dimethyladenosine solid support for efficient access to hydrolysis‐resistant 3′‐charged tRNA mimics. Bioorg Med Chem 22: 6989–6995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Steger J, Micura R (2011) Functionalized polystyrene supports for solid‐phase synthesis of glycyl‐, alanyl‐, and isoleucyl‐RNA conjugates as hydrolysis‐resistant mimics of peptidyl‐tRNAs. Bioorg Med Chem 19: 5167–5174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Steger J, Graber D, Moroder H, Geiermann AS, Aigner M, Micura R (2010) Efficient access to nonhydrolyzable initiator tRNA based on the synthesis of 3′‐azido‐3′‐deoxyadenosine RNA. Angew Chem 49: 7470–7472 [DOI] [PubMed] [Google Scholar]
- 35. Geiermann AS, Polacek N, Micura R (2011) Native chemical ligation of hydrolysis‐resistant 3′‐peptidyl‐tRNA mimics. J Am Chem Soc 133: 19068–19071 [DOI] [PubMed] [Google Scholar]
- 36. Ben‐Shem A, Jenner L, Yusupova G, Yusupov M (2010) Crystal structure of the eukaryotic ribosome. Science 330: 1203–1209 [DOI] [PubMed] [Google Scholar]
- 37. Mueller M, Wang M, Schulze‐Briese C (2012) Optimal fine phi‐slicing for single‐photon‐counting pixel detectors. Acta crystallogr D Biol Crystallogr 68: 42–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kabsch W (2010) Xds. Acta crystallogr D Biol Crystallogr 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Melnikov S (2013) Structure of the Eukaryotic Ribosome: Tips and Tricks. Adv Methods Biomol Crystallogr 1: 313–320 [Google Scholar]
- 40. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW et al (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta crystallogr D Biol crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lebedev AA, Young P, Isupov MN, Moroz OV, Vagin AA, Murshudov GN (2012) JLigand: a graphical tool for the CCP4 template‐restraint library. Acta crystallogr D Biol Crystallogr 68: 431–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta crystallog D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bruno IJ, Cole JC, Kessler M, Luo J, Motherwell WD, Purkis LH, Smith BR, Taylor R, Cooper RI, Harris SE et al (2004) Retrieval of crystallographically‐derived molecular geometry information. J Chem Inf Comput Sci 44: 2133–2144 [DOI] [PubMed] [Google Scholar]
- 44. Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66: 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File