

ABSTRACT
Programmed Death 1 (PD-1) and T cell Ig and mucin domain-3 protein (Tim-3) are immune checkpoint receptors that are expressed on tumor-infiltrating lymphocytes (TIL) in tumor-bearing mice and humans. As anti-PD-1 single agent response rates are only <20% in head and neck squamous cell carcinoma (HNSCC) patients, it is important to understand how multiple inhibitory checkpoint receptors maintain suppressed cellular immunity. One such receptor, Tim-3, activates downstream proliferative pathways through Akt/S6, and is highly expressed in dysfunctional TIL. We observed that PD-1 and Tim-3 co-expression was associated with a more exhausted phenotype, with the highest PD-1 levels on TIL co-expressing Tim-3. Dampened Akt/S6 phosphorylation in these PD-1+Tim-3+ TIL, when the PD-1 pathway was ligated, suggested that signaling cross-talk could lead to escape through Tim-3 expression. Indeed, PD-1 blockade of human HNSCC TIL led to further Tim-3 upregulation, supporting a circuit of compensatory signaling and potentially permitting escape from anti-PD-1 blockade in the tumor microenvironment. Also, in a murine HNC tumor model that is partially responsive to anti-PD-1 therapy, Tim-3 was upregulated in TIL from persistently growing tumors. Significant antitumor activity was observed after sequential addition of anti-Tim-3 mAb to overcome adaptive resistance to anti-PD-1 mAb. This increased Tim-3-mediated escape of exhausted TIL from PD-1 inhibition that was mediated by phospho-inositol-3 kinase (PI3K)/Akt complex downstream of TCR signaling but not cytokine-mediated pathways. Taken together, we conclude that during PD-1 blockade, TIL upregulate Tim-3 in a PI3K/Akt-dependent manner, providing further support for dual targeting of these molecules for more effective cancer immunotherapy.
KEYWORDS: Head and neck cancer, immunotherapy, monoclonal antibody, PD-1, Tim-3
Introduction
Tumor-infiltrating lymphocytes (TIL) can recognize and eliminate tumor cells in the microenvironment. Upon activation, these T cells express immune checkpoint receptors such as programmed death (PD)-1 to maintain tolerance to self-antigens. During chronic activation, T cells in the tumor microenvironment acquire a dysfunctional status, leading to lack of responsiveness to antigen stimulation.1-3 These “exhausted” T cells share several important features: loss of effector function, an altered transcriptional state and sustained expression of multiple inhibitory receptors.1 Among the inhibitory receptors expressed by exhausted T cells, PD-1-blocking mAbs are effective in treatment of multiple cancer types,4,5 including head and neck squamous cell carcinoma (HNSCC). In HNSCC cells, membrane and/or intracytoplasmic PD-L1 expression is frequent (40–70%),6,7 coinciding with PD-1 upregulation on the majority of CD8+ TIL.6 This heightened PD-1/PD-L1 axis implies that anti-PD-1 therapy should be more effective at boosting antitumor immunity in HNSCC. However, recent clinical trial data revealed only a modest response rate to anti-PD-1 therapy of 15–20%.8,9 Thus, it is important to understand dynamic changes in the tumor microenvironment after anti-PD-1 therapy and elucidate potential mechanism(s) of escape to design rational combination therapies.
T cell immunoglobulin mucin (Tim)-3 is a type 1 membrane receptor that is highly expressed in Th1/Tc1 T cells.10,11 Several lines of evidence suggest that Tim-3 acts as an inhibitory receptor in the setting of chronic activation,12-14 whereas other data suggest that it may enhance T cell receptor (TCR) signaling.14 In cancer patients, Tim-3 expression on PD-1+ TIL marks the most dysfunctional CD8+ T cells,15-18 and concurrent Tim-3 blockade appears to enhance the effects of anti-PD-1 mAbs in restoring T cell functionality.19 However, dynamic changes in Tim-3 expression and signaling during the response to anti-PD-1 treatment, including potential signaling cross-talk in exhausted TIL, have not been investigated in detail.
In both freshly isolated tumor specimens and a murine HNSCC model, we found that 30–50% of CD8+ TIL are Tim-3+, and this expression correlates with the highest level of PD-1 expression and greater exhaustion. In this study, we show that Tim-3 expression is upregulated in response to PD-1 blockade, both in vitro and in vivo. We also show that the PI3K/Akt/mTOR pathway mediates cross-talk between PD-1 and Tim-3, providing a potential mechanism underlying Tim-3 upregulation in the setting of PD-1 blockade. Our findings support the concept of rationally combining blockade of Tim-3 with anti-PD- mAb therapy. Our data also suggest that targeted signaling pathway interference may help to prevent adaptive Tim-3-mediated escape from anti-PD-1 therapy.
Material and methods
Patients and specimens
Peripheral blood samples and fresh tumor specimens were obtained from previously untreated, stage III/IV HNSCC patients (n = 36). All patients were treated in the Department of Otolaryngology at the University of Pittsburgh Medical Center, and all subjects signed a written informed consent approved by the Institutional Review Board of the University of Pittsburgh (UPCI# 99–069). The clinical-pathological features of the HNSCC patients in this study are shown in Table 1. The patient cohort included 11 females and 25 males with a mean age of 59 y (range: 24–80 y).
Table 1.
Clinicopathological features of the HNSCC patients in this study.
| HN# | Gender | Age (at diagnosis) | Tumor site | T-stage | N-stage |
|---|---|---|---|---|---|
| HN15–8172 | MALE | 52 | Oropharynx | T2 | N2 |
| HN15–8088 | MALE | 51 | Oropharynx | T1 | N2 |
| HN14–7943 | MALE | 43 | Larynx | T4 | N2 |
| HN14–7928 | MALE | 54 | Larynx | T3 | N0 |
| HN14–7800 | FEMALE | 77 | Oral cavity | T4 | N0 |
| HN15–8068 | FEMALE | 57 | Oral cavity | T2 | N2 |
| HN14–7900 | MALE | 49 | Oropharynx | TX | N2 |
| HN14–7893 | MALE | 75 | Oral cavity | T4 | N0 |
| HN14–7927 | MALE | 66 | Larynx | TX | NX |
| HN15–8010 | FEMALE | 80 | Oral cavity | T4 | N0 |
| HN15–8021 | MALE | 24 | Oral cavity | T3 | N0 |
| HN15–8110 | MALE | 74 | Oropharynx | T4 | N2 |
| HN15–8144 | FEMALE | 67 | Oral cavity | T3 | N0 |
| HN15–8173 | MALE | 63 | Larynx | T4 | N0 |
| HN15–8202 | FEMALE | 69 | Oral cavity | T4 | N2 |
| HN15–8228 | MALE | 60 | Oropharynx | T3 | N2 |
| HN15–8253 | MALE | 60 | Oropharynx | T3 | N2 |
| HN15–8290 | MALE | 27 | Oral cavity | T2 | N2 |
| HN15–8296 | MALE | 67 | Larynx | T3 | N2 |
| HN15–8294 | MALE | 64 | Oropharynx | T4 | N2 |
| HN15–8180 | MALE | 50 | Oral cavity | T4 | N1 |
| HN14–7881 | MALE | 54 | Oropharynx | T1 | N2 |
| HN14–7909 | FEMALE | 58 | Oral cavity | T2 | N1 |
| HN14–7949 | MALE | 72 | Larynx | T3 | N2 |
| HN14–7960 | MALE | 61 | Larynx | T4 | N2 |
| HN14–7961 | MALE | 57 | Oral cavity | T2 | N0 |
Collection of peripheral blood mononuclear cells (PBMC) and tumor-infiltrating lymphocytes (TIL)
Venous blood from HNSCC patients was drawn into heparinized tubes and centrifuged on Ficoll-Hypaque gradients (GE Healthcare Life Sciences, Piscataway, NJ). PBMC were recovered, washed in RPMI-1640 medium (Sigma, St. Louis, MO), and either used immediately for experiments or re-suspended in freezing media containing 10% DMSO, transferred to Mr. Frosty containers (Thermo Scientific, Waltham, MA), and stored at −80°C until flow cytometry analysis. For TIL isolation, fresh tumors from HNSCC patients were minced into small pieces manually or using a gentle MACS Dissociator (Miltenyi Biotec, Auburn, CA), then transferred to 70-μm cell strainers (BD) and mechanically separated using the plunger of a 5-mL syringe. The cells passing through the cell strainer were collected and subjected to Ficoll-Hypaque gradient centrifugation. After centrifugation, mononuclear cells were recovered and stored at −80°C until flow cytometry analysis or immediately used for experiments.
Antibodies and flow cytometry
The following anti-human antibodies were used for staining: CD3-Alexa Fluor 700, CD8+-PE, CD4+-PercP/Cy5.5, CD25-APC-Cy7, FOXP3-PerCP/Cy5.5, purchased from BD Biosciences (San Jose, CA), Tim-3-BV421, CD25-PE-Cy7, IFN-γ-APC-Cy7 and TNF-α-FITC purchased from Biolegend (San Diego, CA), CD4-PE-TR purchased from Life Technologies (Carlsbad, CA), phospho-AKT(Ser473)-APC, TIGIT-APC purchased from eBioscience (San Diego, CA), phospho-S6 (Ser235/236)-Alexa Fluor 488 (Cell Signaling Technology, Danvers, MA), CTLA4-FITC purchased from Ancell (Stillwater, MN). The following anti-mouse antibodies were used for staining: PD-1-BV421, CD25-PE, CD4+-APC-Cy7, CD8+-PE-Cy7 purchased from Biolegend (San Diego, CA), Tim-3-APC purchased from eBioscience (San Diego, CA), MHC pentamer (H-2Db RAHYNIVTF)-PE purchased from Proimmune (Oxford OX4 4GA, United Kingdom).
Intracellular staining of p-Akt, p-S6, IFNγ and TNF-α was performed as follows: PBMC or TIL were stained with surface marker antibodies, fixed with fixation/permeabilization buffer (eBioscience), washed, and stained for intracellular antigens in 1X permeabilization buffer. Cells were analyzed on an LSR Fortessa (BD Biosciences) flow cytometer, and data analyzed using Flow Jo (Treestar, Ashland, OR). The acquisition and analysis gates were restricted to the lymphocyte gate based on characteristic properties of the cells in the forward and side scatter. Dead cells were excluded based on viability dye staining (Zombie Aqua Fixable Viability Dye, Biolegend, San Diego, CA).
Sorting of TIL subsets
TIL were isolated from tumor specimens as described above and T cells were purified using EasySep™ Human T Cell Enrichment Kit (Stemcell technologies, Vancouver, Canada). Then, the T cells were stained with CD4+-PercP-Cy5.5, CD8+-PE, CD25-PE-Cy7, PD-1-APC and Tim-3-BV421 antibodies. PD-1+Tim-3−, PD-1−Tim-3+, PD-1+Tim-3+ and PD-1−Tim-3− CD8+ and CD4+CD25lo/− cells were sorted using Beckman Coulter MoFlo Astrios.
Re-stimulation of TIL using anti-CD3/CD28 beads
T lymphocyte subsets were sorted based on PD-1 and Tim-3 expression from freshly isolated TIL from HNSCC patients, rested in 30% Human AB serum overnight and then subjected to re-stimulation experiments. Sorted TIL were cultured with Dynabeads® Human T-Activator CD3/CD28 beads at a fixed cell: bead ratio of 1:5. The cultures were incubated at 37°C with 5% CO2 for indicated time periods. Magnetic beads were removed before analysis.
Freshly isolated HNSCC TIL were cultured with Dynabeads® Human T-Activator CD3/CD28 beads at a fixed cell: bead ratio of 1:1 in the presence of protein transport inhibitor (BD GolgiPlug™) from BD Biosciences (San Jose, CA) at 37°C with 5% CO2 for indicated time period, flow cytometry analysis is used for cytokine detections.
Murine in vivo tumor studies
Female C57BL/6 mice were purchased at 6 to 8 weeks from Jackson laboratories (Bar Harbor, Maine). On arrival, animals were acclimated for 48 h prior to MEER tumor cells (1 × 106 cell per mice at the back of neck) injections, after ketamine (90 mg/kg body weight), xylazine (4.5 mg/kg body weight) mediated anesthesia. All animals were maintained in a pathogen-free facility and cared for and used in accordance with protocol approved by the Institutional Animal Care and Use Committee (IACUC) University of Pittsburgh, which follows the US Public health Service's guide for the care and use of animals. Anti-PD-1 mAb (clone: 4H2) were given at 3 mg/kg starting from day 12 for five doses, anti-Tim-3 mAb (clone: F38–2E2) (eBioscience; San Diego, CA) were given at 5 mg/kg for four doses upon anti-PD-1 treatment completion. Tumor measurement was performed with Vernier Caliper (tumor volume = (Longest diameter2 × smallest diameter)/2), and animals were killed when tumor size was larger than 20 mm in its greatest dimension, the animal was substantially emaciated, or pain or functional impairment was apparent.
Pharmacologic inhibition studies
TIL were cultured for 48 h in medium with Dynabeads® Human T-Activator CD3/CD28 beads at a fixed cell: bead ratio of 1:1 in the presence of either DMSO control or one of the five inhibitory drugs, which were dissolved in DMSO: LY294002 (Cell Signaling) a pan-PI3K inhibitor, Akt 1/2 (EMD Bioscience) an isozyme selective Akt inhibitor VIII, CAY10626 (Abcam) a PI3K α/mTOR inhibitor, GS-1101 (Abcam) PI3K p110δ inhibitor, AS-041164 PI3K p110γ inhibitor (Abcam) together with anti-PD-1 mAb (nivolumab, Bristol Myers-Squibb).
Results
PD-1+Tim-3+ marks exhausted TIL subsets indicated by dampened IFNγ and TNF-α production upon TCR stimulation
In order to define the functional exhaustion status of TIL based on PD-1 and Tim-3 expression,1 we sorted freshly isolated TIL from HNSCC patients (n = 3) into four subsets: PD-1+ Tim-3− (28% ± 6%), PD-1−Tim-3+ (23% ± 10%), PD-1+Tim-3+ (33% ± 14%), and PD-1−Tim-3− (21% ± 14%) in CD8+ T cells. Upon stimulation with anti-CD3/CD28 coated beads for 6 h, PD-1+Tim-3+ and PD-1−Tim-3+ TILs showed the least IFNγ and TNF-α production in both CD8+ T cells (Fig. 1). Thus, PD-1+Tim-3+ T cells appeared to be the most dysfunctional HNSCC TIL subset,18,20 and the expression of Tim-3 correlates with more prominent defects in functionality to single positive PD-1+ cells. These results raised the question of whether, and how, co-expression of Tim-3 might be modulated in response to blockade of the PD-1 pathway, which suffers from a relatively low single-agent response rate.6,9,21
Figure 2.

Tim-3 is upregulated after PD-1 blockade in vitro in human TIL. Freshly isolated tumor-infiltrating lymphocytes from HNSCC patient were treated with anti-PD-1 mAb—Nivolumab, or anti-CTLA-4 mAb—Ipilumumab, and IgG4/IgG1 as isotype control. All the treatments were given at the concentration of 10 µg/mL in vitro for 24 h. Tim-3, CTLA-4, TIGIT expressions were assessed by flow cytometry. (A) Representative flow plots showing Tim-3 expression in CD8+ and CD4+CD25lo/− cells. (B) Summary data showing MFI fold change of Tim-3, CTLA-4 and TIGIT. MFI of isotype control group is normalized to 1. (n = 6) Significance was calculated with multiple t test, *p < 0.05. (C) Summary data showing Tim-3 MFI fold change in isotype, anti-PD-1(nivolumab) or anti-CTLA-4 (ipilumumab) treatment. (n = 5) Significance was calculated with RM-one-way ANOVA, *p < 0.05. All data represent average ± SEM.
Tim-3 is upregulated after PD-1 blockade in vitro in human TIL
Although anti-PD-1 therapy shows promising therapeutic results in patients with HNSCC, only a minority of patients experience therapeutic benefit.8 Thus, we hypothesized that during PD-1 blockade, PD-1 and Tim-3 co-expression might maintain the exhaustion status of TIL, rendering the cells less responsive to PD-1 blockade. We first investigated expression of Tim-3 before and after in vitro PD-1 blockade. Total TIL extracted from freshly excised HNSCC tumors were incubated with anti-PD-1 mAb at 10 μg/mL for 24 h. As shown in Fig. 2A, cell surface expression of Tim-3 was increased by approximately 50% in CD8+ T cells (p = 0.0009) and by 40% in CD4+CD25lo/− effector T cells (p = 0.003). %Tim-3+ T cells were also increased in both CD8+ and CD4+CD25lo/− effector TIL T cells (p = 0.0006) (Fig. S2B). Meanwhile, this effect was relatively selective for Tim-3, since upregulation of other checkpoint receptors such as CTLA-4 and TIGIT was not observed under these conditions (Fig. 2B and Fig. S2A). Furthermore, this upregulation of Tim-3 after anti-PD-1 mAb treatment was not observed in response to CTLA-4 blockade using ipilimumab at 10 ug/mL (Fig. 2C), despite baseline expression of CTLA-4 on the majority of these TIL.22 This finding suggests that compensatory upregulation of Tim-3 might represent a specific adaptive response that functions to sustain the exhaustion status of TIL in response to PD-1 blockade.
Figure 1.
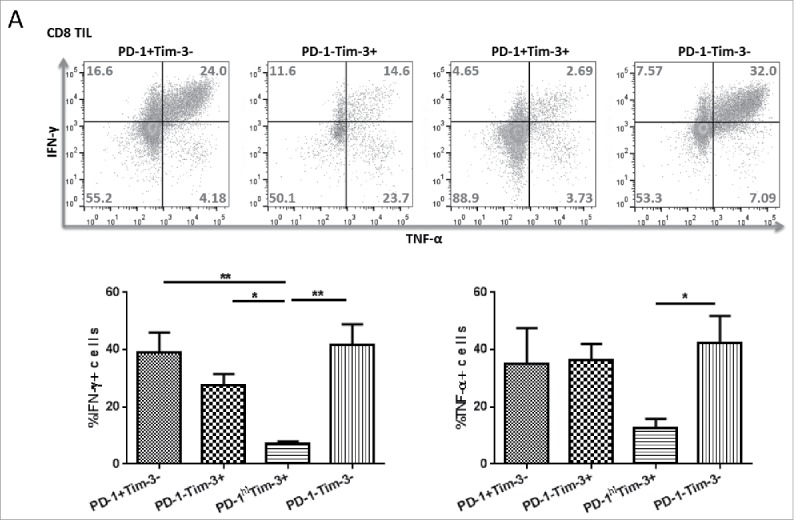
PD-1+Tim-3+ TIL subsets are dampened in IFNγ and TNF-α secretion upon TCR stimulation. Freshly isolated TIL from HNSCC patients were sorted based on PD-1, Tim-3 expression. Sorted cells were stimulated in vitro with anti-CD3/CD28 beads and protein transport inhibitor (BD GolgiPlug™) for 6 h. Cells were then stained intracellularly for cytokines. (A) Intracellular staining results of TNFα and IFNγ in sorted CD8+ T cells are shown. (n = 3) Significance were calculated by RM-one-way ANOVA, *p < 0.05.
Figure 4.
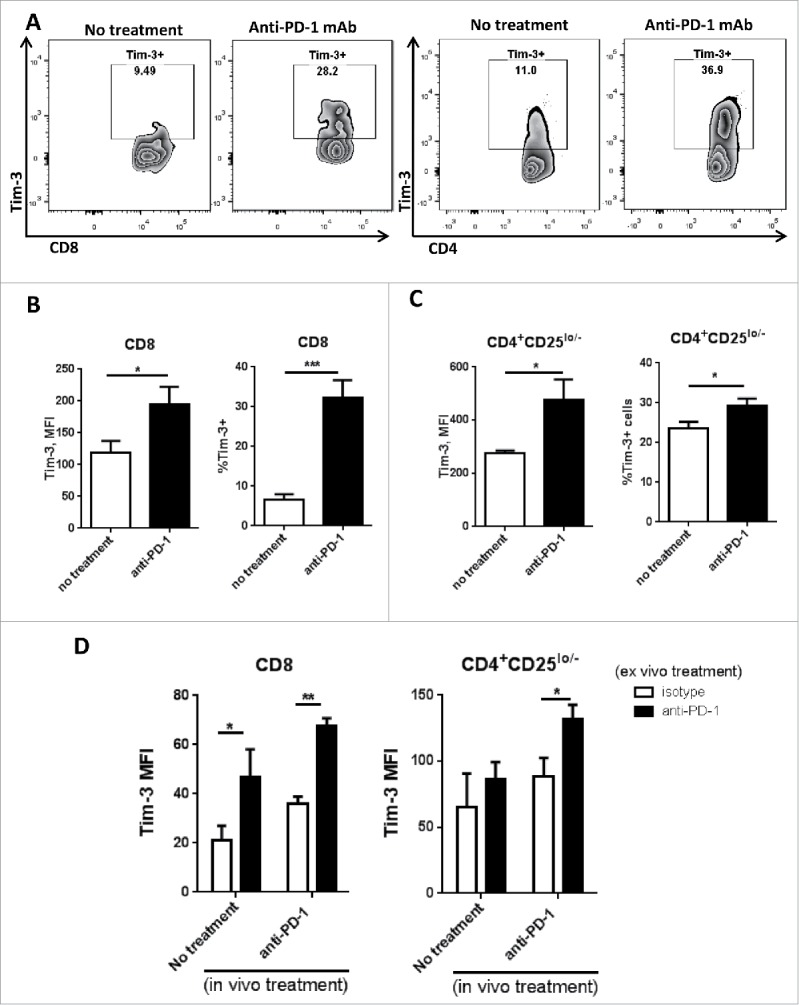
Tim-3 is upregulated in TIL after in vivo anti-PD-1 treatment, and is further upregulated upon in vitro PD-1 blockade in murine HNC model. C57BL/6 mouse were injected 1 × 106 MEER cells in the neck, anti-mouse PD-1 mAb was given as treatment; all the mouse were scarified around day 36 after injection of tumor cells. Freshly isolated tumor-infiltrating lymphocytes from murine HNC model were analyzed for Tim-3 expression by flow cytometry. (A) Representative flow plots of Tim-3 expression pattern in no treatment group and anti-PD-1 treated group were shown in CD8+ and CD4+CD25lo/− T cells. (B, C) Summary data of Tim-3 MFI and %Tim-3+ cells in tumor-infiltrating CD8+ and CD4+CD25lo/− T cells. (n = 6) Significance were calculated by unpaired t test, *p < 0.05, **p < 0.001. (D) Freshly isolated splenocytes were incubated with anti-PD-1 mAb or IgG4 for 48 h and Tim-3 expression was assessed by flow cytometry. Summary data of Tim-3 MFI in CD8+ and CD4+ T cells from splenocytes.(n = 6) Significance were calculated by two-way ANOVA, *p < 0.05, **p < 0.001.
PD-1 blockade upregulates IFNγ and TNF-α expression primarily in Tim-3- cells
In line with previous reports that blockade of the PD-1:PD-L1 pathway can significantly enhance antitumor immune responses,7 we observed significant overall upregulation of IFNγ and TNF-α expression after in vitro PD-1 blockade, by both CD8+ and CD4+CD25lo/− effector T cells.20 Based on our observation that PD-1 blockade induces Tim-3 upregulation, we investigated whether this upregulation of Tim-3 attenuates the Th1/Tc1 skewing promoted by anti-PD-1 treatment. As shown in Fig. 3A, Tim-3+ cells displayed significantly higher baseline PD-1 expression compare with Tim-3- cells in both CD8+ (80% vs. 57%, p = 0.01) and CD4+CD25lo/− (70% vs. 48%, p = 0.03) T cells. Despite the higher level of PD-1 expression in Tim-3+ cells, greater IFNγ (p = 0.0003) and TNF-α (p = 0.0015) production was observed during PD-1 blockade in Tim-3- cells. This finding suggests that augmented induction of Tim-3 expression after PD-1 blockade provides an escape mechanism to enforce dysfunction and exhaustion, since the T cells that manifested higher Tim-3 expression after PD-1 inhibition were more defective in Th1/Tc1 cytokine expression.
Figure 3.

PD-1 blockade induced upregulation of IFNγ and TNF-α expression are mostly seen in Tim-3- cells. Freshly isolated tumor-infiltrating lymphocytes from HNSCC patient were treated with anti-PD-1 mAb—Nivolumab and IgG4 as an isotype control. All treatments were given at the concentration of 10 µg/mL in vitro for 48 h. Anti-CD3/CD28 beads and protein transport inhibitor (BD GolgiPlug™) were added according to manufacture's instruction at least 6 h of cell culture for cytokine detection. Cytokine expressions were assessed by flow cytometry. (A) Summary data of baseline PD-1 expression in Tim-3+ and Tim-3- subsets.(n = 5) (B) Flow plot of IFNγ, TNFα expression in Tim-3+ and Tim-3− subsets. (C) Summary data of IFNγ, TNFα expression in Tim-3− and Tim-3+ subsets. (n = 5) All data represent average ± SEM. Significance were calculated by two-way ANOVA, *p < 0.05, **p < 0.001.
Anti-PD-1 mAb therapy upregulates Tim-3 expression in murine HNC model
HNSCC is highly associated with HPV16 infection,23,24 and recent data indicate that HPV+ HNC are more immunogenic and anti-PD-1 responsive.7,9 In order to identify mechanisms of anti-PD-1 resistance and strategies to enhance this partial anti-PD-1 response, we used an immunocompetent, orthotopic murine HPV+ HNC model. MEER (E6-E7-H-ras) transformed murine tonsillar carcinoma25 syngeneic tumors were injected into C57/BL6 mice. Since we had observed that Tim-3 is upregulated in human TIL after in vitro PD-1 blockade, we investigated whether this upregulation also occurred in vivo. After tumor progression, mice were randomized to receive anti-PD-1 mAb or isotype mAb therapy (3 mg/kg), and tumor growth was measured every 3 d. Within 36 d, anti-PD-1 mAb treatment modestly decreased tumor growth (p = 0.1, isotype mAb vs. anti-PD-1 mAb). Analysis of the TIL from these mice showed that Tim-3 was significantly upregulated in both CD8+ (average: 5% vs. 30%, p = 0.00024) and CD4+CD25lo/− (average: 22% vs. 30%, p = 0.04) T cells from the anti-PD-1 mAb treated mice (Figs. 4A–C). Additionally, ex vivo anti-PD-1 treatment of splenocytes from these mice led to further induction of Tim-3 expression in both CD8+ and CD4+CD25lo/− T cells from both untreated and anti-PD-1 mAb treated mice (Fig. 4D). Since anti-PD-1 treatment failed to show significant therapeutic benefit in this murine HNSCC model, we hypothesized that tumor-infiltrating T cells may develop adaptive resistance to PD-1 blockade in part through compensatory upregulation of Tim-3 expression.
Sequential anti-Tim-3 treatment enhances therapeutic benefit of anti-PD-1 immunotherapy in a murine HNC model
Anti-PD-1 treatment alone showed only modest therapeutic effects in the murine HNC model (Fig. 5A). Based on our observation that Tim-3 was upregulated as a potential compensatory mechanism in response to PD-1 blockade, we administered anti-Tim-3 mAb treatment in mice whose tumors progressed during initial anti-PD-1 therapy. After tumor establishment on day 10, anti-PD-1 mAb was given every 2 d at 3 mg/kg for four doses, and a second group of mice received subsequent anti-Tim-3 treatment after completion of anti-PD-1 treatment. As shown in Fig. 5A, anti-PD-1 treatment alone showed a modest benefit (p = 0.13), whereas sequential anti-Tim-3 treatment significantly suppressed tumor growth, beginning at day 29 (p = 0.02) compare with untreated mice. We then wished to determine whether tumor-specific immunity was stronger in the setting of combination PD-1/Tim-3 blockade. Analyzing TIL from combination vs. single Ab-treated mice, we found that the HPV E749–57-tetramer-specific CD8+ T cells were significantly upregulated in the sequential anti-Tim-3 treated group, suggesting that the delayed blockade of Tim-3 increased T cell activation against tumor antigens expressed in the tumor microenvironment. By analyzing immune phenotypes in CD8+ TIL from all the treatment groups, we found that the proportion of PD-1hi (exhausted) T cells was significantly reduced in both anti-PD-1 treated and anti-PD-1/anti-Tim-3-treated TIL17(Fig. S3A). Thus, tumor-infiltrating T cells in combination Ab-treated mice possessed stronger antitumor activity, and were more likely to restore functionality in response to anti-PD-1 therapy, compare with single anti-PD-1 mAb treated or untreated mice. We conclude that additional inhibitory immune checkpoint receptors (such as Tim-3) may be upregulated upon blockade of PD-1, supporting dual targeting of immune checkpoint receptors in order to maximize tumor-specific immunity.
Figure 5.

Sequential anti-Tim-3 treatment suppresses tumor growth and show better antitumor immune response. C57BL/6 mouse were injected 1 × 106 MEER cells in the neck, after tumor progression (day 10) anti-mouse PD-1 mAb was given at five doses every 2 d, sequential anti-Tim-3 mAb treatment were given to one group of mice starting from day 22 for four doses every 2 d. Tumor volume were measured every 2 d, Mice TIL were harvested at day 31, and HPV E7-tetramer and PD-1 expression were assessed by flow cytometry. (A) Tumor growth curve up to day 31 was shown, (n = 5) significance was calculated by two-way ANOVA, *p < 0.05. (B) Representative flow plots and summary data showing %E7-tetramer+ cells in CD8+ T cells. (n = 5) Significance was calculated by ordinary one-way ANOVA, *p < 0.05.
Tim-3 upregulation upon PD-1 blockade requires activation of PI3K/Akt
Phosphoinositide3-kinases (PI3Ks) plays an important role in inflammatory response in both innate and adaptive immune cells by catalyzing the phosphorylation of inositol phospholipids in the 3-position of the inositol ring. In mammalian cells, there are four class I isoforms of PI3Ks (PI3Kα, β, γ, δ), they differ in catalytic subunit that synthesize the phospholipid “PIP3” which regulates cell movement, growth, survival and differentiation as a “second messenger.” These four isoforms are further divided into two groups: class IA and class IB based on their structure and general mode of regulation. Class I PI3Ks catalyze same reaction with different kinetic parameters, the main distinction between the four isoforms is their adaptation to upstream regulation by receptor transduction pathways. Importantly, class IA PI3K isoforms (PI3Kα, β, δ) are adapted to regulation by receptors signal through protein tyrosin kinases, whereas class IB PI3K (PI3Kγ) is adapted to regulation by GPCRs via direct binding to Gβγ subunits.26-29 Tim-3 expression, in response to certain cytokines, has been reported to be induced by activation of the PI3K pathway.26 We previously showed that pS6 is decreased in TIL after PD-1 stimulation,30 potentially linking PD-1 to the PI3K-Akt-mTOR pathway. Since TCR/CD28 co-stimulation leads to PI3K/Akt activation, and PD-1 ligation causes inhibition of TCR proximal signaling through SHP-227, leading to decreased PI3K activity, we hypothesized that Tim-3 upregulation after PD-1 blockade might be caused by increased PI3K activity due to de-repression by PD-1 blockade.
To test this hypothesis, we used PI3K p110 subunit-selective inhibitors to determine if the upregulation of Tim-3 upon PD-1 is due to enhanced TCR proximal signaling. Among the four isoforms of PI3Ks, p110α, p110γ and p110δ are reported to have important roles in mediating immune cell activation.28 Specifically, p110γ and p110δ are critical for T cell activation, where p110δ mediates T cell activation through TCR signaling, and p110γ is more likely to be mediating signaling downstream of chemokine receptors.29 We used a broad-spectrum PI3K inhibitor (LY294002), class IA PI3K inhibitors: PI3K p110δ subunit inhibitor (GS-1101), PI3K p110α/mTOR inhibitor (CAY10626), and a class IB PI3K inhibitor: selective p110γ inhibitor (AS-041164). TIL were incubated with each inhibitor in the presence of anti-CD3/CD28 bead stimulation for 48 h, and Tim-3 expression was assessed using flow cytometry. As shown in Fig. 6A, TCR stimulation increased baseline Tim-3 expression, and addition of anti-PD-1 mAb further induced Tim-3 upregulation. All the class IA PI3K subunit inhibitors and Akt inhibitor had similar effects on abrogating upregulation of Tim-3 expression as the broad-spectrum PI3K inhibitor upon TCR stimulation. This result is in line with previous findings26 that activation of the PI3K/Akt pathway mediates upregulation of Tim-3 in response to cytokines.
Figure 6.

Tim-3 upregulation after PD-1 blockade is PI3K/Akt pathway dependent. TIL from HNSCC patient were treated with anti-CD3/CD28 beads in combination with DMSO, broad PI3K inhibitor (LY294002), PI3K p110α/mTOR inhibitor (CAY10626), Akt inhibitor, PI3K p110δ inhibitor (GS-1101) and PI3K p110γ inhibitor (AS041164) with or without anti-PD-1 mAbs (Nivolumab) for 48 h. Cells are collected and Tim-3, p-S6 expressions were assessed by flow cytometry. (A) Summary data showing Tim-3 and p-S6 MFI, (n = 4), significance were calculated with ordinary one-way ANOVA, *p < 0.05, **p < 0.001. (B) Summary data showing Tim-3 MFI fold change in the presence of selective PI3K inhibitors with anti-PD-1 or IgG4. (n = 4) Significance was calculated with unpaired t test, *p < 0.05.
Next, we tested whether the upregulation of Tim-3 upon PD-1 blockade is PI3K/Akt dependent. As shown in Fig. 6B, addition of anti-PD-1 mAb further upregulated baseline Tim-3 expression in the presence of TCR activation (p = 0.003). This effect was abrogated in the presence of the broad PI3K inhibitor (LY294002), class IA PI3K inhibitors: PI3K p110α/mTOR inhibitor (CAY10626), PI3K p110δ inhibitor (GS-1101); and Akt inhibitor, while the upregulation effect is still seen in the presence of the class IB PI3K p110γ (AS-041164) (p = 0.04) inhibitor. We conclude that upon PD-1 blockade, upregulation of Tim-3 is mediated by enhanced TCR-proximal signaling to the PI3K/IAkt/mTOR pathway. Based on a previous report that selective PI3K or Akt inhibition results in enhanced antitumor immune response in a HPV+ TC-1 mouse model in a Treg-dependent manner without attenuating conventional T cell activity,31 our work raises a potential therapeutic strategy of combining anti-PD-1 mAb with selective PI3K/Akt inhibition to promote antitumor immunity by overcoming compensatory Tim-3 upregulation.
Discussion
The PD-1:PD-L1 pathway is upregulated in HNSCC and anti-PD-1 therapy has shown some therapeutic benefit in approximately 20% of patients.8 However, the majority of patients still fail to benefit from anti-PD-1 treatment, and the underlying mechanism of resistance to PD-1 blockade is not fully understood. In this study, we show that Tim-3 expression is upregulated in response to PD-1 blockade on both murine and human T cells, an effect that appears to be mediated through enhanced PI3K/Akt/mTOR signaling. We observed that PD-1hiTim-3+ T cells are the most dysfunctional subset in HNSCC TIL, which is in line with previous reports in melanoma15 and leukemia.17 In addition, blockade of PD-1 led to Tim-3 upregulation on HNSCC TIL both in vitro and in vivo. In order to clarify if the upregulation of Tim-3 expression is a sign of T cells dysfunction, we investigated Th1 cytokine production after PD-1 blockade in Tim-3+/− subsets. Anti-PD-1 treatment induced upregulation of IFNγ and TNF-α in Tim-3-cells. Despite the expansion of Tim-3+ T cells, these inflammatory cytokines were not further induced upon PD-1 blockade. At this point, we cannot specifically define the cells which initially are responsible for IFNγ production. Indeed, Tim-3+ cells after anti-PD-1 blockade may originate from Tim-3 negative cells at the start of the activation conditions (Fig. 3).
HPV+ HNSCC has a high incidence rate and HPV+ cancers are generally immunogenic.23,25 And it has been reported that PD-1 and Tim-3 expressions are enriched in HPV+ HNSCC.32,33 We used a murine orthotopic HNSCC model expressing HPV16 E6 and E7 oncogenes25 to investigate the effect of anti-PD-1and anti-Tim-3 therapy on antitumor immunity. Tim-3 upregulation was observed in TIL of the in vivo anti-PD-1-treated mice, and ex vivo PD-1 blockade on splenocytes further induced Tim-3 expression. Sequential anti-Tim-3 treatment following PD-1 blockade significantly suppressed tumor growth. The observation of enriched antigen-specific CD8+ cytotoxic T cells with expansion of PD-1int CD8+ TIL, together with a reduction of PD-1hi cells, indicates that sequential anti-PD-1/anti-Tim-3 treatment induced a more robust antitumor immune response in the orthotopic murine HNC model.
Our findings corroborate a recent report that Tim-3 is significantly upregulated in response to PD-1 blockade in lung cancer.34 Also, other groups have shown that genetic PD-1 depletion led to a more exhausted T cell phenotype, marked by upregulation of multiple inhibitory receptors, including Tim-3.35 Our work extends these findings, revealing a possible mechanism underlying Tim-3 upregulation in the setting of PD-1 blockade. Multiple studies have shown that upon ligation, PD-1 downstream signaling pathway suppresses TCR proximal signaling through SHP-2 phosphorylation36 and increased PTEN phosphatase activity.37 Interestingly, Tim-3 expression has been shown to be induced through Akt and T-bet,38 downstream of cytokine receptor signaling. Thus, we hypothesized that the Tim-3 upregulation induced upon PD-1 blockade may be caused by reduced suppression of the PI3K/Akt/mTOR pathway. By using selective PI3K subunit inhibitors, we find that Tim-3 upregulation can still be observed in the presence of class Ib PI3K inhibitor: p110γ inhibitor, where the inhibition of class Ia PI3K-mediated Akt/mTOR pathway abrogated the upregulation effect of Tim-3 in the setting of PD-1 blockade. Our study is in line with a previous report linking Tim-3 expression to activation of PI3K pathway. However, we studied this mechanism in short-term in vitro settings. Further investigation is needed to elucidate the in vivo mechanism(s) underlying the Tim-3 upregulation observed in response to PD-1 blockade, and the clinical significance of this upregulation in anti-PD-1 treated patients.
We conclude that upon PD-1 blockade in HNSCC, Tim-3 expression is upregulated more significantly, compare with other inhibitory ICRs such as CTLA-4 and TIGIT, and that this upregulation is dependent, at least in part, on activation of PI3K/Akt pathway. However, further observations and experiments will be necessary to document that PD-1 blockade induced Tim-3 upregulation is responsible for adaptive resistance, as our in vivo data suggest, preferably using treated cancer patients. Overall, our study provides a rational combination therapeutic strategy in the treatment of HNSCC, supporting dual or multiple inhibitory receptors targeting, possibly including transient small molecular inhibition of the PI3K pathway.
Supplementary Material
Disclosure of potential conflicts of interest
Robert L. Ferris: consulting or advisory role: AstraZeneca, Bristol- Myers Squibb, Merck, ONO Pharmaceutical and Celgene. Research funding: Amgen (Inst), Bristol-Myers Squibb (Inst.) AstraZeneca (Inst.) and VentiRx (Inst.)
Funding
This work was supported by National Institute of Health grants R01 CA206517, DE019727, P50 CA097190, T32 CA060397 and the University of Pittsburgh Cancer Institute award P30 CA047904. This project used the UPCI Flow Cytometry Facility that is supported in part by award P30CA047904. Gulidanna Shayan and Jing Li were supported by the China Scholarship Council.
References
- 1.Wherry EJ. T cell exhaustion. Nat Immunol 2011; 12:492-9; PMID:21739672; http://dx.doi.org/ 10.1038/ni.2035 [DOI] [PubMed] [Google Scholar]
- 2.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol 2009; 10:29-37; PMID:19043418; http://dx.doi.org/ 10.1038/ni.1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007; 27:670-84; PMID:17950003; http://dx.doi.org/ 10.1016/j.immuni.2007.09.006 [DOI] [PubMed] [Google Scholar]
- 4.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252-64; PMID:22437870; http://dx.doi.org/ 10.1038/nrc3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al.. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Eng J Med 2012; 366:2443-54; PMID:22658127; http://dx.doi.org/ 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferris RL. Immunology and Immunotherapy of Head and Neck Cancer. J Clin Oncol 2015; 33:3293-304; PMID:26351330; http://dx.doi.org/24819861 10.1200/JCO.2015.61.1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zandberg DP, Strome SE. The role of the PD-L1:PD-1 pathway in squamous cell carcinoma of the head and neck. Oral Oncol 2014; 50:627-32; PMID:24819861; http://dx.doi.org/ 10.1016/j.oraloncology.2014.04.003 [DOI] [PubMed] [Google Scholar]
- 8.Laban S, Doescher J, Schuler PJ, Bullinger L, Brunner C, Veit JA et al.. Immunotherapy of head and neck tumors: Highlights of the ASCO Meeting 2015. Hno 2015; 63:612-9; PMID:26319429; http://dx.doi.org/ 10.1007/s00106-015-0054-1 [DOI] [PubMed] [Google Scholar]
- 9.Ferris RL, Blumenschein, Jr. G, Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, Worden F, Saba NF, Docampo LC, Haddad R, Rordorf T, Kiyota N, Tahara M, Monga M, Lynch M, Geese WJ, Kopit J, Shaw JW, Gillison ML. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016 Nov 10;375(19):1856-1867; PMID:27718784; http://dx.doi.org/21697013 10.1056/NEJMoa1602252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakuishi K, Jayaraman P, Behar SM, Anderson AC, Kuchroo VK. Emerging Tim-3 functions in antimicrobial and tumor immunity. Trends Immunol 2011; 32:345-9; PMID:21697013; http://dx.doi.org/ 10.1016/j.it.2011.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu C, Anderson AC, Kuchroo VK. TIM-3 and its regulatory role in immune responses. Curr Topics Microbiol Immunol 2011; 350:1-15; PMID:20700701; http://dx.doi.org/ 10.1007/82_2010_84 [DOI] [PubMed] [Google Scholar]
- 12.Gorman JV, Colgan JD. Regulation of T cell responses by the receptor molecule Tim-3. Immunologic Res 2014; 59:56-65; PMID:24825777; http://dx.doi.org/ 10.1007/s12026-014-8524-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res 2011; 71:3540-51; PMID:21430066; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-0096 [DOI] [PubMed] [Google Scholar]
- 14.Ferris RL, Lu B, Kane LP. Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion 2014; 193:1525-30; PMID:25086175; http://dx.doi.org/20819923 10.4049/jimmunol.1400557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C et al.. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med 2010; 207:2175-86; PMID:20819923; http://dx.doi.org/ 10.1084/jem.20100637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, Freeman GJ, Kuchroo VK, Ahmed R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A 2010; 107:14733-8; PMID:20679213; http://dx.doi.org/ 10.1073/pnas.1009731107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kong Y, Zhang J, Claxton DF, Ehmann WC, Rybka WB, Zhu L et al.. PD-1(hi)TIM-3(+) T cells associate with and predict leukemia relapse in AML patients post allogeneic stem cell transplantation. Blood Cancer J 2015; 5:e330; PMID:26230954; http://dx.doi.org/ 10.1038/bcj.2015.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Shayan G, Avery L, Jie H-B, Gildener-Leapman N, Schmitt N, Lu B, Kane LP, Ferris RL. Tumor-infiltrating Tim-3+ T cells proliferate avidly except when PD-1 is co-expressed: evidence for intracellular cross talk. Oncoimmunology. 2016 Sep 22;5(10):e1200778; PMID:27853635; http://dx.doi.org/20819927 10.1080/2162402X.2016.1200778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med 2010; 207:2187-94; PMID:20819927; http://dx.doi.org/ 10.1084/jem.20100643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Jie HB, Lei Y, Gildener-Leapman N, Trivedi S, Green T et al.. PD-1/SHP-2 inhibits Tc1/Th1 phenotypic responses and the activation of T cells in the tumor microenvironment. Cancer Res 2015; 75:508-18; PMID:25480946; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferris RL, Lu B, Kane LP. Too Much of a Good Thing? Tim-3 and TCR signaling in T cell exhaustion. J Immunol 2014; 193:1525-30; PMID:25086175; http://dx.doi.org/24169351 10.4049/jimmunol.1400557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jie HB, Gildener-Leapman N, Li J, Srivastava RM, Gibson SP, Whiteside TL et al.. Intratumoral regulatory T cells upregulate immunosuppressive molecules in head and neck cancer patients. Br J Cancer 2013; 109:2629-35; PMID:24169351; http://dx.doi.org/ 10.1038/bjc.2013.645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith EM, Ritchie JM, Summersgill KF, Klussmann JP, Lee JH, Wang D et al.. Age, sexual behavior and human papillomavirus infection in oral cavity and oropharyngeal cancers. Int J Cancer 2004; 108:766-72; PMID:14696105; http://dx.doi.org/ 10.1002/ijc.11633 [DOI] [PubMed] [Google Scholar]
- 24.Kreimer AR, Clifford GM, Boyle P, Franceschi S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: a systematic review. Cancer Epidemiol Biomarkers Prev 2005; 14:467-75; PMID:15734974; http://dx.doi.org/17515506 10.1158/1055-9965.EPI-04-0551 [DOI] [PubMed] [Google Scholar]
- 25.Hoover AC, Spanos WC, Harris GF, Anderson ME, Klingelhutz AJ, Lee JH. The role of human papillomavirus 16 E6 in anchorage-independent and invasive growth of mouse tonsil epithelium. Archives Otolaryngol Head Neck Surg 2007; 133:495-502; PMID:17515506; http://dx.doi.org/ 10.1001/archotol.133.5.495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stark AK, Sriskantharajah S, Hessel EM, Okkenhaug K. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr Opin Pharmacol 2015; 23:82-91; PMID:26093105; http://dx.doi.org/ 10.1016/j.coph.2015.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hawkins PT, Stephens LR. PI3K signalling in inflammation. Biochim Biophys Acta 2015; 1851:882-97; PMID:25514767; http://dx.doi.org/ 10.1016/j.bbalip.2014.12.006 [DOI] [PubMed] [Google Scholar]
- 28.Koyasu S. The role of PI3K in immune cells. Nat Immunol 2003; 4:313-9; PMID:12660731; http://dx.doi.org/ 10.1038/ni0403-313 [DOI] [PubMed] [Google Scholar]
- 29.Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annual Rev Immunol 2013; 31:675-704; PMID:23330955; http://dx.doi.org/ 10.1146/annurev-immunol-032712-095946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J, Srivastava RM, Ettyreddy A, Ferris RL. Cetuximab ameliorates suppressive phenotypes of myeloid antigen presenting cells in head and neck cancer patients. J Immunother Cancer 2015; 3:54; PMID:26579227; http://dx.doi.org/ 10.1186/s40425-015-0097-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abu-Eid R, Samara RN, Ozbun L, Abdalla MY, Berzofsky JA, Friedman KM, Mkrtichyan M, Khleif SN. Selective inhibition of regulatory T cells by targeting the PI3K-Akt pathway. Cancer Immunol Res 2014; 2:1080-9; PMID:25080445; http://dx.doi.org/ 10.1158/2326-6066.CIR-14-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Partlova S, Boucek J, Kloudova K, Lukesova E, Zabrodsky M, Grega M, Fučíková J, Truxová I, Tachezy R, Špíšek R et al.. Distinct patterns of intratumoral immune cell infiltrates in patients with HPV-associated compared to non-virally induced head and neck squamous cell carcinoma. Oncoimmunology 2015; 4:e965570; PMID:25949860; http://dx.doi.org/ 10.4161/21624011.2014.965570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Badoual C, Hans S, Merillon N, Van Ryswick C, Ravel P, Benhamouda N, Levionnois E, Nizard M, Si-Mohamed A, Besnier N et al.. PD-1-expressing tumor-infiltrating T cells are a favorable prognostic biomarker in HPV-associated head and neck cancer. Cancer Res 2013; 73:128-38; PMID:23135914; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-2606 [DOI] [PubMed] [Google Scholar]
- 34.Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H et al.. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 2016; 7:10501; PMID:26883990; http://dx.doi.org/ 10.1038/ncomms10501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med 2015; 212:1125-37; PMID:26034050; http://dx.doi.org/ 10.1084/jem.20142237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med 2012; 209:1201-17; PMID:22641383; http://dx.doi.org/ 10.1084/jem.20112741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol 2013; 33:3091-8; PMID:23732914; http://dx.doi.org/ 10.1128/MCB.00319-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mujib S, Jones RB, Lo C, Aidarus N, Clayton K, Sakhdari A, Benko E, Kovacs C, Ostrowski MA. Antigen-independent induction of Tim-3 expression on human T cells by the common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 is associated with proliferation and is dependent on the phosphoinositide 3-kinase pathway. J Immunol 2012; 188:3745-56; PMID:22422881; http://dx.doi.org/ 10.4049/jimmunol.1102609 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.