

ABSTRACT
Previously, we have identified a new epitope in EMMPRIN, a multifunctional protein that mediates tumor cell–macrophage interactions and induces both MMP-9 and VEGF. Here, we synthesized this epitope as an octa-branched multiple antigenic peptide (MAP) to vaccinate mice implanted with subcutaneous syngeneic colon (CT26), prostate (TRAMP-C2) or renal (RENCA) cell line carcinomas. Vaccination inhibited, and sometimes regressed, tumor growth in a dose-dependent manner, reaching 94%, 71% and 72% inhibition, respectively, at a 50 μg dose (p < 0.01). Mice with regressed tumors demonstrated immune memory, preventing tumor recurrence upon re-implantation (p < 0.001). When tumor cells were administered through the tail vein to generate lung metastases, vaccination reduced the number of metastatic foci (by 15- and 23-folds, p < 0.001), and increased the median survival time by 25% and 53% in RENCA and CT26 metastases, respectively (p < 0.01) relative to scrambled-MAP controls. No significant adverse responses were observed in all experiments. We show that the tumor microenvironment was immune modulated, as vaccination induced production of EMMPRIN-specific antibodies, increased CD8+ T cells infiltration and cytotoxicity, alleviated immune suppression by decreasing TGFβ concentrations, reduced angiogenesis and cell proliferation, and enhanced apoptosis. Thus, our successful active peptide vaccination strategy differs from previous, unsuccessful attempts, both in the selected target (the EMMPRIN epitope) and in the use of a modified, MAP configuration, and demonstrates that this may be an efficient approach for the treatment and prevention of some types of cancer.
KEYWORDS: Active peptide vaccination, angiogenesis, EMMPRIN/CD147, multiple antigenic peptide (MAP), tumor cell–macrophage interactions, tumor microenvironment
Abbreviations
- DEG
differentially expressed genes
- EMMPRIN
extracellular matrix metalloproteinase inducer
- MAP
multiple antigenic peptide
- MDSCs
myeloid-derived suppressor cells
- MMP
matrix metalloproteinase
- TAA
tumor-associated antigen
- TME
tumor microenvironment
- TRA
tumor rejection antigen
- VEGF
vascular endothelial growth factor
Introduction
Cancer peptide vaccination has drawn a lot of attention, especially in pre-clinical studies. However, to date, peptide-based antitumor vaccines show only limited efficacy and insufficient clinical benefits. Several explanations may exist.1,2 First, to escape immune recognition, tumor cells tend to suppress the expression of the targeted antigen (antigen loss).3,4 Second, some epitopes chosen for vaccination are differentiation antigens or overexpressed tumor-associated antigens (TAAs) that are self-antigens, and subject to central and peripheral tolerance. Therefore, only a limited repertoire of T cell clones remain to recognize them, thus limiting the efficiency of the immune response toward them.5 Moreover, the few T cells that may escape tolerance and arrive at the tumor site encounter a local immunosuppressive tumor microenvironment (TME) triggered by regulatory cells (Tregs, MDSCs) and anti-inflammatory cytokines (e.g., IL-10 and TGFβ) that strongly inhibit their effector functions.6 Lastly, peptide vaccinations are usually administered to patients with advanced-stage disease, thus, minimizing the time allowed for the immune response to exert a productive, efficient response.
Nonetheless, peptide vaccinations still represent an interesting treatment modality,1,2,7 as they exhibit important advantages such as high specificity, low adverse effects with minimal toxicity, no oncogenic potential, and easy and inexpensive synthetic production mode free of pathogenic contamination. So, improvements in vaccination protocols, choice of target, and delivery systems are required. The ideal antigens are conserved epitopes that are derived from tumor-rejection antigens (TRAs), indispensable for tumor cell survival and proliferation8,9 and the ideal adjuvant and route of vaccination (e.g., i.d. vs. s.c.) should enhance the immune response, break tolerance and overcome the immunosuppressive TME.
Short peptides are considered weak immunogens, unstable and rapidly degraded by plasma proteases. The synthesis of long, linear peptides with one or multiple epitopes facilitates their presentation on both MHC class I and II and enhances their immunogenicity, but still demonstrates insufficient efficacy and no improvement in overall survival,1,10 although specific subsets of patients may benefit from them.7
Multiple antigenic peptides (MAPs) contain a core matrix of lysine residues that form a scaffold, on which short peptides are attached in parallel using standard solid phase chemistry. This forms a three-dimensional protein structure with four or eight repeats of the peptide.11 The advantages of using MAPs over monomeric peptides for vaccination include: (a) increased peptide stability with retention of biological activity.12,13; (b) improved immunogenicity and increased TCR affinity due to generation of a neo-antigen by the conjugation of lysine residues and the three-dimensional structure changes; (c) increased concentrations due to repetitions of the peptide sequence.14 Together, these may break tolerance and allow immune reactivity toward the antigen. However, MAP vaccinations for the treatment of cancer are still very rare, focusing on in vitro or ex vivo generation of antibodies or cytotoxic effector functions.15,16
Extracellular matrix metalloproteinase inducer (EMMPRIN/CD147) is a member of the Ig superfamily, with two heavily glycosylated extracellular domains that are important to its activity.17 This protein is weakly or moderately expressed on several cell types (including monocytes, T cells and glandular epithelial cells), but its expression is markedly increased on many types of malignant cells, correlating with higher grade and stage of tumors, and with increased invasiveness and poor prognosis.18,19 EMMPRIN is best known for its pro-angiogenic and pro-invasive activities, as it can induce several types of matrix metalloproteinases (MMPs), as well as VEGF.20-22 Additionally, EMMPRIN is a multifunctional protein with many activities, including cell metabolism through its interaction with the lactate transporters MCT-1 and MCT-4, leukocyte chemotaxis via binding to extracellular cyclophilins, and more.18,23,24 We have recently identified a novel epitope in the first of the two highly glycosylated extracellular domains (EC-I), located at position 52–63, which is responsible for the induction of both VEGF and MMPs, especially MMP-9.25 Targeting this epitope with a polyclonal antibody, we demonstrated marked reduction of tumor growth in several subcutaneous and orthotopic mouse models.
Synthesizing this epitope sequence as a MAP (designated 161-MAP), we hypothesized that we can efficiently inhibit tumor growth and metastases by actively vaccinating mice against EMMPRIN.
Results
161-MAP reduces tumor growth and prevents recurrence through immune memory
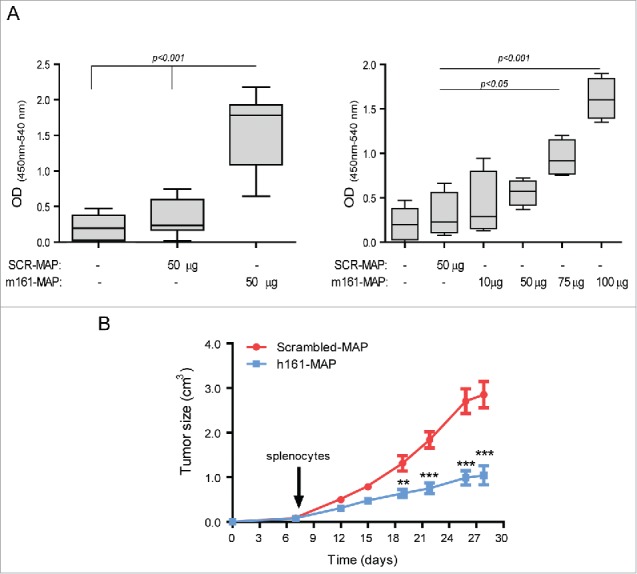
We synthesized the EMMPRIN epitope as an octa-branched peptide, emulsified it in Freund's adjuvant and injected it to mice that were previously subcutaneously implanted with one of three types of tumorigenic cell lines (renal cell carcinoma RENCA, colon carcinoma CT26 or prostate carcinoma TRAMP-C2). As negative control we synthesized the octa-branched scrambled-MAP (Scr-MAP), which has the same amino acid composition in a scrambled order. In all experiments, we used complete Freund's adjuvant (CFA) for the first injection, and incomplete Freud's adjuvant (IFA) for the following boost injections. The 161-MAP inhibited the growth of the RENCA (Fig. 1A) and CT26 tumors (Fig. 1B), in a dose-dependent manner, by 72% (p < 0.01) and 94% (p < 0.001), respectively, relative to the scramble-MAP controls. In the TRAMP-C2 model (Fig. 1C) we used only the 50μg dose for each vaccine injection, and the 161-MAP, which was synthesized according to the mouse (m161-MAP) or human (h161-MAP) epitope sequences with a three amino acids difference between them, equally inhibited tumor growth by about 51% (p < 0.001) on average, relative to the scrambled control (Scr-MAP).
Figure 1.
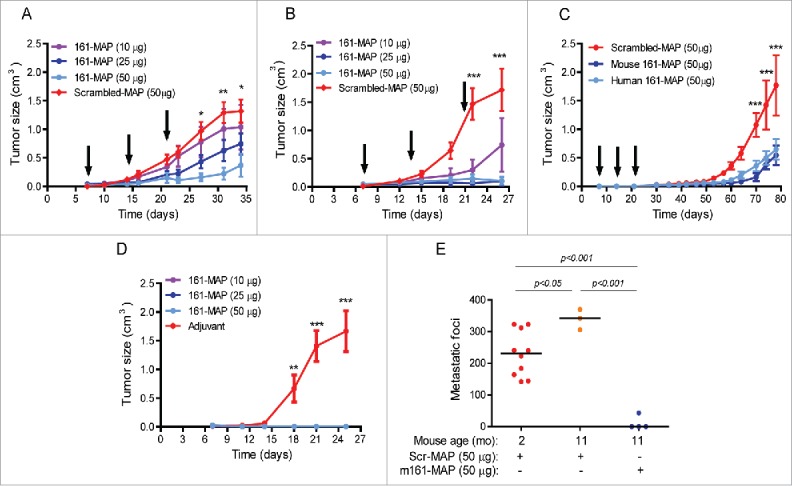
161-MAP inhibits growth of subcutaneous tumors and prevents recurrence. Tumor cells (2 × 106) were injected to the flank of mice (day 0), and then vaccinated every 7 d (black arrows) with different amounts of mouse or human 161-MAP emulsified in CFA (first injection) or IFA (next boost injections). (A) RENCA renal cell carcinoma cells (two independent experiments, n = 5 in each group) injected to BALB/c mice; (B) CT26 colon carcinoma cells injected to BALB/c mice (three independent experiments, n = 5–6 in the 10 and 25 μg groups, and n = 12 in the 50 μg groups); (C) TRAMP-C2 prostate cell carcinoma cells injected to C57BL/6 mice (two independent experiments, n = 5–7 in each group); (D) all BALB/c mice where CT26 tumors fully regressed in the previous experiment (panel B) were re-challenged with another s.c. inoculation of 2 × 106 CT26 cells, 1.5 mo after the first injection of cells. As control, a new batch of mice was injected with CT26 cells and vaccinated with the control Scr-MAP; (E) The same mice whose tumors were regressed (panel B) and were re-challenged with the tumor cells twice (panel D) were re-challenged 11 mo after first implantation with i.v. inoculation of 106 CT26 cells injected to the tail vein. Two-way ANOVA was used to compare the groups. *p < 0.05, **p < 0.01, ***p < 0.001 relative to the group treated with 50 μg 161-MAP.
In the CT26 model, which responded best to the vaccination, we observed that all mice that received the highest dose (50μg) and some of the mice that received the lower doses exhibited full regression of tumors that were previously small but palpable. We then collected the 14 mice that demonstrated regression, waited 6 weeks to make sure that tumors did not reappear, and then re-challenged them with another dose of the CT26 tumor cells (2 × 106 cells) injected to the opposite flank, without repeating the vaccination procedure. As control, we injected a new batch of mice with the tumor cells and vaccinated them with Scr-MAP, as before. The control group demonstrated normal rate of tumor growth (Fig. 1D), indicating that the tumor cells implanted the second time were viable, but no tumors grew in any of the mice that had exhibited full regression of their previous tumors. To make sure that immune memory was initiated and could protect the mice for a long period of time, we waited 6 more months and repeated this experiment, with the same results (one mouse of the 14 died of un-related causes). To verify that the route of tumor cell implantation did not have an effect, we waited 3 more months (total of 11 mo after the first tumor cells were implanted), randomly selected 4four mice and injected to their tail vein 106 CT26 tumor cells, in order to generate lung metastases. 16 d later the mice were sacrificed and the metastatic foci were counted. In three of these mice no metastatic foci were observed at all, and in the remaining mouse we counted only 43 metastatic foci (Fig. 1E), relative to an average of 340 metastatic foci of age-matched mice, and 230 metastatic foci of young, 8-weeks-old mice, that received the Scr-MAP vaccination prior to the injection of CT26 tumor cells.
161-MAP reduces the number of metastatic foci and extends survival
To generate a severe model with high tumor cell burden that simulates the later stages of the metastatic process, we used the accepted model of injecting tumor cells directly to the circulation, where they disseminate and colonize organs, specifically the lung.26 We injected 106 of the RENCA or CT26 tumor cells to the tail vein. Because of the short survival time in these models, especially in the CT26 model, we initially attempted to vaccinate the mice before the injection of the tumor cells, at days −21, −14 and −7, using the same doses, adjuvant and control as before (Fig. 2A). In the CT26 model, the number of metastatic foci was reduced in a dose-dependent manner (15-folds at the high dose, p < 0.001 relative to control), and the weight of the wet lung, that reflects tumor burden, was similarly reduced (Fig. 2I–K). In the RENCA model, even the low dose of 10 μg was sufficient to inhibit generation of metastases (12-folds, p < 0.001 relative to the Scr-MAP control), and the weight of the lungs in all 161-MAP vaccinated was equivalent to that of healthy, untreated and tumor-free mice (Fig. 2C–E). In these two models, the effects of the m161-MAP and h161-MAP were equivalent, and both markedly reduced the number of metastatic foci (p < 0.001, Fig. 2G and M).
Figure 2.
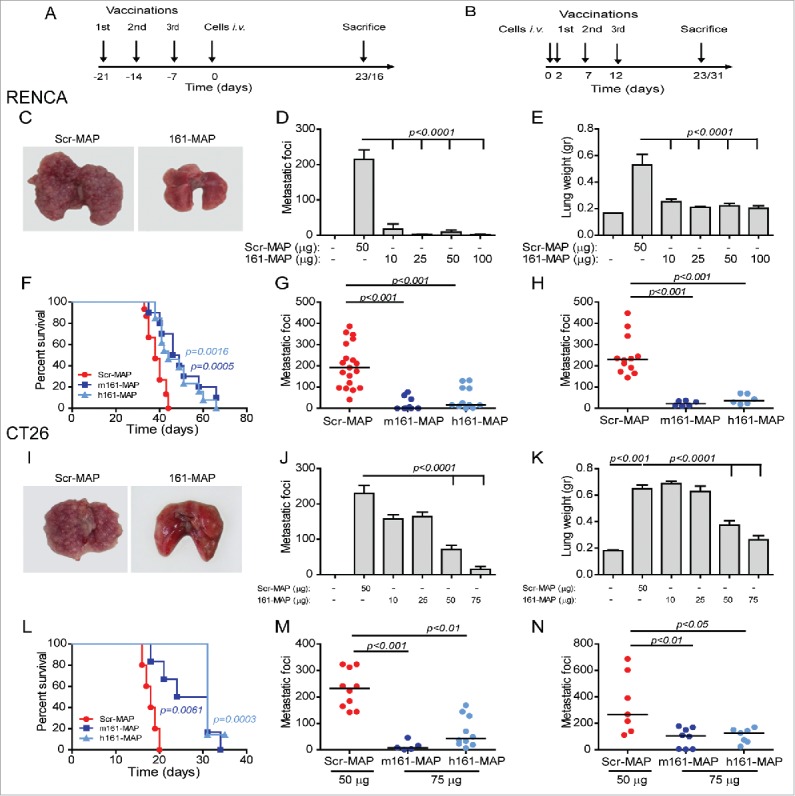
161-MAP inhibits metastatic foci and extends survival. (A, B) Timeline of experimental design where mice were sacrificed at day 23 (RENCA model) or 16 (CT26 model). BALB/c mice were vaccinated with the control Scr-MAP or 161-MAP prior to (at days −21, −14 and −7, panels C–G and I–M) or after (at days 2, 7, 12, panels H and N) tail vein injection (day 0) of tumor cells (106). Vaccination was administered at 50 μg/boost injection, unless indicated otherwise. (C, I) Representative images of lungs, and (D, J) number of metastatic foci grossly counted in the lungs (RENCA metastases: two independent experiments, n = 12 for the control, 25 and 50 μg groups, n = 6 for the 10 and 100 μg groups; CT26 metastases, three independent experiments, n = 5 for all groups); (E, K) wet lung weight, as a measure of metastatic spread; (G, M) comparison of the number of metastatic foci in mice vaccinated with the mouse or human 161-MAPs at the optimal dosage (RENCA two independent experiments, n = 11 in each group, CT26 three independent experiments, n = 10 in the control and human groups and n = 5 in the mouse group). (H, N) number of metastatic foci after therapeutic vaccination, with optimal dose of MAPs used (50 μg for 106 RENCA cells and 75 μg for 0.25 × 106 CT26 cells injected. Mice injected with CT26 cells were sacrificed at day 31); (F, L) survival curves for RENCA (three independent experiments, n = 11 in each group) and CT26 (two independent experiments, n = 6 in each group) after i.v. tumor cell injection.
Both the m161-MAP and the h161-MAP significantly extended the median survival time of mice injected i.v. with the RENCA cells by 25% and 15%, respectively (Fig. 2F), and with the CT26 cells by 53% and 72% respectively, relative to the Scr-MAP controls (Fig. 2L).
To examine the therapeutic manner of vaccination, we next administered the vaccine injections after i.v. injections of the cells, and in a narrow window of time, on days 2, 7 and 12 (Fig. 2B). In the RENCA model, m161-MAP and the h161-MAP successfully reduced the number of metastatic foci by 10-folds and 6-folds, respectively (Fig. 2H, p < 0.001), whereas in the CT26 model we had to extend the duration of the model by reducing the number of cells injected (0.25 × 106 cells), to allow enough time for the immune system to mount a productive response, and only then vaccination significantly reduced the number of metastatic foci (Fig. 2N, 3.7-folds p < 0.01 and 3.2-folds p < 0.05 for the mouse and human 161-MAP, respectively).
161-MAP structure, and not the monomeric sequence, is immunogenic
To demonstrate that the MAP structure was critical for the inhibitory effects of the vaccine, we compared tumor sizes of mice s.c. implanted with the CT26 cells and vaccinated with the monomeric sequence to those vaccinated with the 161-MAP. No difference was observed between the group vaccinated with the scrambled negative control and the group vaccinated with the monomeric epitope (Fig. S1A), whereas the group vaccinated with 161-MAP exhibited reduced tumor sizes. Likewise, when the tumorigenic CT26 cells were injected i.v. to the tail vein, the human monomeric sequence of the epitope resulted in a number of metastatic foci that was not significantly different than that of the Scr-MAP control (Fig. S1B, 127 ± 7.5 vs. 197 ± 20 metastatic foci, respectively). Similar results were obtained for the group receiving the mouse monomeric peptide vaccination (data not shown). These data suggest that the monomeric peptide demonstrated low antigenicity or was very unstable.
161-MAP recruits cytotoxic CD8+ T cells to the tumor
To explore the mechanisms of action for 161-MAP vaccination, we used tumor sections obtained from the 50 μg dose groups, or if the tumors were too small to analyze, from the 25 μg dose group. Metastatic foci from the CT26 model were analyzed from the 75 μg dose group that yielded optimal results.
Infiltration of CD8+ T cells into tumors is usually inhibited by the immunosuppressive TME, leading to their accumulation mostly at the rims of the tumor. We immunohistochemically (IHC) stained sections for CD8+ and evaluated the change in CD8+ T cell infiltration by quantifying the stained area. Due to the high background, we considered only the densely stained areas that resulted in dark spots, consistent with the small size of lymphocytes. We observed a significant increase in the CD8+ positive area, ranging between 2- and 3-folds (Fig. 3A), in all 3 s.c. models (RENCA, CT26 and TRAMP-C2) and in both metastatic models (RENCA and CT26), in the tumors obtained from 161-MAP relative to Scr-MAP vaccinated mice.
Figure 3.
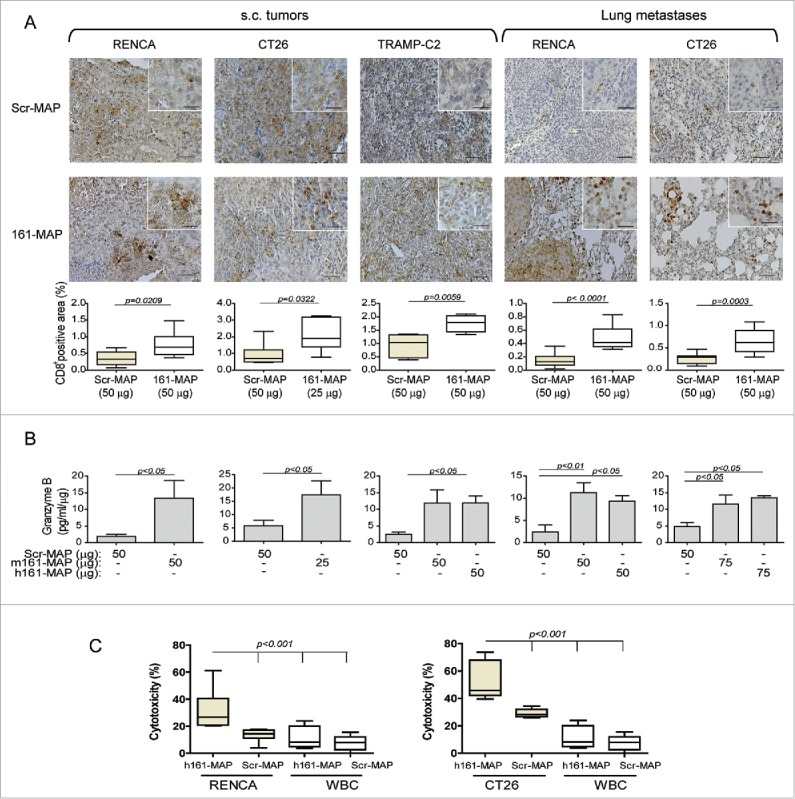
161-MAP vaccination enhances CD8+ (T)cells infiltration into the tumor and lung metastases, and increases their cytotoxic activity. (A) Representative images of IHC staining for CD8+ and their quantification in RENCA, CT26 and TRAMP-C2 s.c. tumor sections, and in RENCA and CT26 metastatic lung section (n = 4–5). Bar size for all images is 50 μm and for all insets is 25 μm. (B) Granzyme B concentrations determined in s.c. tumor and metastatic lung lysates and normalized to the amount of total protein (n = 3–5). (C) Cytotoxic activity of positively selected CD8+ T cells, isolated from tumors of Scr-MAP or 161-MAP vaccinated mice, was determined by incubating them in vitro with fluorescently-labeled RENCA or CT26 tumor cells for 24 h in 40:1 effector (CD8+ T cells) to target (CT26 or RENCA cells) ratio (E:T). As negative control, CD8+ T cells were incubated with labeled autologous leukocytes (three independent experiments, n = 5–6). The two-tailed unpaired t-test was used to compare two groups, and the one-way ANOVA was used to compare the groups in the cytotoxicity assay.
To assess the activity of these CD8+ T cells, we measured the concentrations in tumor lysates of granzyme B, the serine protease that initiates apoptosis in target cells. In all 161-MAP vaccinated groups, granzyme B was significantly elevated by 3–5-folds relative to the control groups, suggesting immune activation of CD8+ T cells (Fig. 3B). Furthermore, to demonstrate the ability of CD8+ T cells to kill their tumor target cells, we positively isolated them directly from RENCA or CT26 s.c. tumors, and incubated them in vitro with previously labeled RENCA or CT26 cells, as described in the methods. The ability of these cells to kill autologous leukocytes (WBC), which express low amounts of EMMPRIN, served as negative control, and was minimal in both vaccinated groups (Fig. 3C). In contrast, CD8+ T cells that were obtained from h161-MAP vaccinated tumors exhibited enhanced ability to kill their respective tumor cells in comparison to these cells obtained from Scr-MAP vaccinated tumors (2–2.5-folds, p < 0.001).
161-MAP alters the TME and modulates macrophage activity
IHC staining for F4/80, the pan-macrophage marker, demonstrated that macrophages were present in in all tumor implants and metastatic foci, infiltrating deep into the necrotic regions. Upon vaccination their amount was significantly reduced (p < 0.05, Fig. S2A). As macrophages are main producers, albeit not sole producers, of both pro- and anti-inflammatory mediators, (e.g., TNFα, IL-1β, IL-10 and TGFβ), and their role as immune suppressors may be changed depending on the local TME, we next measured the local concentrations of such mediators in tumor and metastatic lysates, rather than in serum samples. In Scr-MAP vaccinated mice, concentrations of TNFα, IL-1β and IL-10 were very low in both RENCA and CT26 s.c. tumors and lung metastases, whereas TGFβ levels were 1,000–2,500-folds higher than those, suggesting that TGFβ, a major immunosuppressive cytokine, dominates the TME in the tumors (Fig. 4A and B). 161-MAP vaccination markedly reduced the concentrations of all cytokines, but because of the initial high concentrations of TGFβ, its reduction was most pronounced, and suggests alleviation of immune suppression. Furthermore, the levels of nitrites, the stable product of nitric oxide (NO), were significantly increased in the TME of both s.c. tumors and lung metastases after 161-MAP vaccination (about 2–3-folds, p < 0.05). Since iNOS is not expressed in RENCA and CT26 cells due to miR-146a-induced translational inhibition,27 nitrite accumulation must originate from the infiltrating macrophages, suggesting that activation of the remaining macrophages was skewed toward M1 activation.
Figure 4.
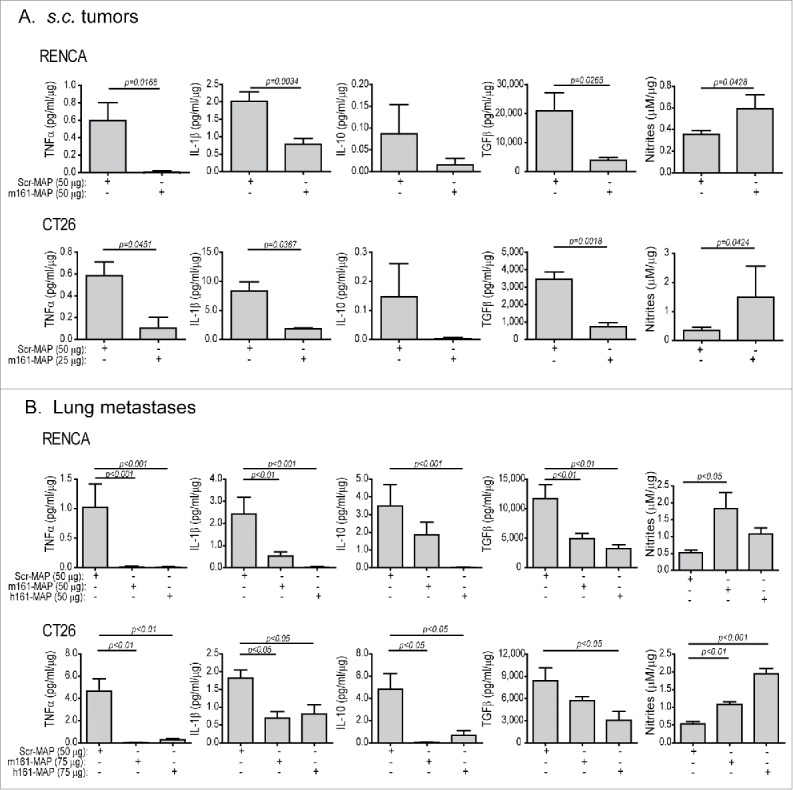
161-MAP vaccination changes the tumor microenvironment (TME). Accumulation of nitrites and cytokine concentrations of TNFα, IL-1β, IL-10 and TGFβ in lysates extracted from (A) s.c. tumors and (B) metastatic lungs were determined by ELISA, and normalized to the amount of total protein in each sample (n = 4–6). The two-tailed unpaired t test was used to compare two groups.
To have a broader sense of the change in the TME, we compared the transcriptome of s.c. CT26 tumors that were vaccinated with either Scr-MAP or 161-MAP using RNAseq, as described in the methods. Pathway and network analyses using the FGNet and IPA tools were used to determine the differentially expressed genes (DEGs). The list of DEGs was uploaded into IPA and analyzed with stringent parameters of log ratio of 1 (2-fold change) or above at adjusted p-value of 0.05 or lower and using only data from primary cells within the ingenuity knowledge base. The results (Fig. 5A) clearly show four metagroups of genes that were significantly and differentially expressed. The most significant canonical pathway was EIF2 signaling (z score of 3.4) with many ribosomal proteins inhibited (Fig. 5A, the translational inhibition cluster). The most significant network had IFNγ as its central hub with 33 focus molecules from the gene list out of possible 35. Together these suggest an upregulation of an antitumoral response induced by the peptide vaccination. Among these DEGs we noticed that many genes associated with M1/Th1 mode of activation (marked in red, Fig. 5B) were elevated by more than 2-folds at the mRNA level, whereas M2/Th2 genes (marked in blue, Fig. 5B) had a more mixed response, where some were elevated at similar rates, and others, such as CD206 and VEGFB, were inhibited. Notably, many of the significantly increased M1/Th1 genes (e.g., NOS2, Indoleamine (2,3)-dioxygenase (IDO1 and IDO2), TNFsf10/TRAIL) can be induced by the Th1 cytokine IFNγ, and mediate its antitumoral response.28 This suggests an alleviation of immunosuppression and a shift toward an antitumoral mode.
Figure 5.
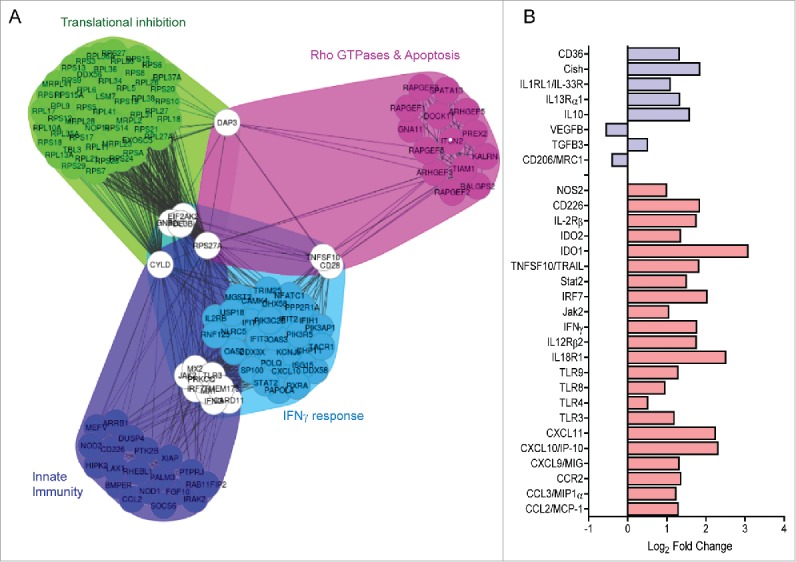
161-MAP vaccination alters the transcriptome. (A) Total RNA was extracted from CT26 s.c. tumors of Scr-MAP and 161-MAP vaccinated mice at day 26 (n = 3 each), and RNAseq data were obtained as described in the methods. Functional gene networks were derived from significantly differentially expressed genes (DEG, p-adj < 0.05) and resulted in 4 distinct metagroups of genes. (B) The change in the expression of specific DEGs (p < 0.05) that are associated with M1/Th1 (red bars) or M2/Th2 (blue bars) activation of macrophages and lymphocytes is presented.
161-MAP vaccination induces both adaptive humoral and cellular immunity
Activation of the humoral adaptive immune response can be demonstrated by presence of EMMPRIN-specific antibodies in the serum of vaccinated mice. Using a direct ELISA assay with recombinant mouse EMMPRIN to coat the wells, we demonstrated a significant (p < 0.001) elevated antibody recognition of this antigen in RENCA tumor-bearing mice that were vaccinated with 161-MAP in comparison to the Scr-MAP. Healthy mice that were not vaccinated and the Scr-MAP control group demonstrated similar low levels of EMMPRIN recognition (Fig. 6A, left panel). In CT26 tumor-bearing mice, this could be repeated in a dose-response manner (Fig. 6A, right panel). Similar results were obtained when we used the m161-MAP as the antigen coating the plate (data not shown), verifying the specific recognition of the epitope.
Figure 6.
161-MAP vaccination triggers both humoral and cellular immune responses. (A) Serum samples obtained from mice that were i.v. injected with RENCA (n = 8–12 in each group, left panel) or CT26 cells (n = 4 in each group, right panel) and vaccinated with Scr-MAP or 161-MAP were collected. Presence of EMMPRIN-specific antibodies was determined using direct ELISA. Healthy mice (without tumors or vaccinations) were used as negative controls (n = 12). (B) Splenocytes (30 × 106 cells) from BALB/c mice that were vaccinated with either Scr-MAP or h161-MAP (50 μg each) were pooled from four mice, and adoptively transferred by tail vein injections to CT26 s.c. tumor-bearing BALB/c naïve mice, and tumor growth was monitored (two independent experiments, n = 5–6 in each group). **p < 0.01, ***p < 0.001 relative to the same time point. The two-way ANOVA test was used to compare groups in different time points.
When we adoptively transferred splenocytes from naïve mice vaccinated with either h161-MAP or Scr-MAP into CT26 tumor-bearing mice (Fig. 6B), the receiving mice exhibited inhibition of tumor growth (p < 0.001), although the transfer occurred only once. In contrast, similar adoptive transfer of serum, rather than spleen cells, had no effect on tumor growth (data not shown), although the serum was injected in three boost injections.
Thus, both humoral and cellular adaptive responses were triggered, and the cellular response was sufficient to mediate tumor inhibition, but antibodies present in the serum were not, suggesting that the dominant and effective adaptive arm is the cellular response. It is likely that EMMPRIN-specific B and T lymphocytes continued to clonally expand in the recipient mice, thus establishing an adequate, long-lasting response.
161-MAP vaccination reduces angiogenesis and proliferation, enhances apoptosis
Since EMMPRIN can induce the expression of both VEGF and MMP-9 to regulate angiogenesis, we examined the effects of vaccination on tumor angiogenesis by IHC staining for the endothelial cell marker CD31. Mean vessel density was significantly (p < 0.01) reduced by 161-MAP vaccination in both s.c. tumors and lung metastases (Fig. 7A) by about 1.6–2.7-folds (p < 0.001). Concentrations of both MMP-9 (Fig. 7B) and VEGF (Fig. 7C) in tumor lysates were reduced by 2–3-folds (p < 0.05) in lysates obtained from vaccinated mice in comparison to the control.
Figure 7.
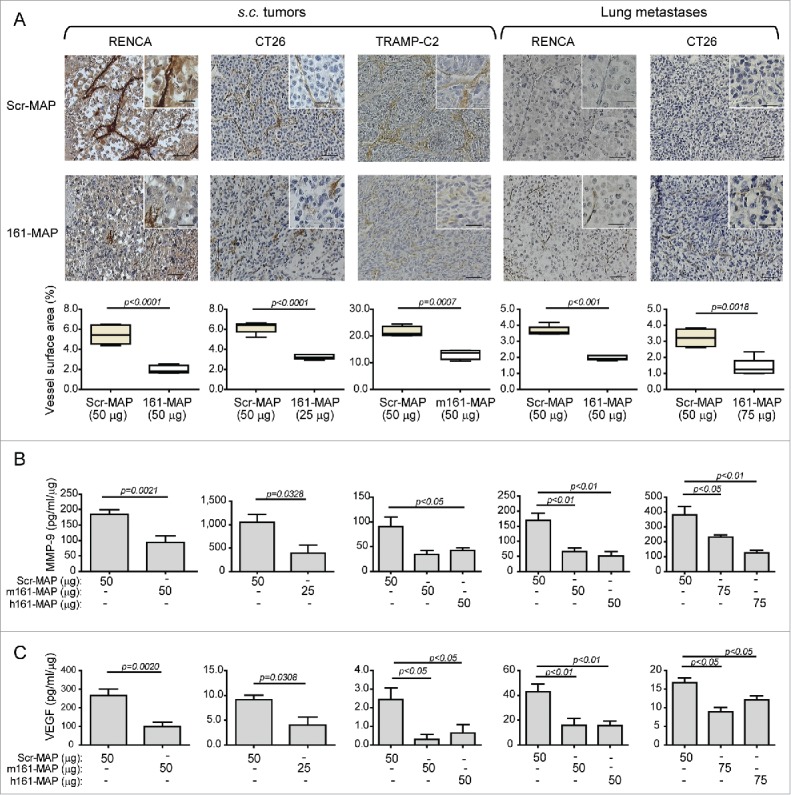
161-MAP reduces tumor angiogenesis. (A) Representative images of IHC staining for CD31, the marker of endothelial cells and their quantification, in RENCA, CT26 and TRAMP-C2 s.c. tumor sections and in sections of RENCA and CT26 metastatic lungs (n = 4–5). Bar size for all images is 50 μm and 25 μm in the insets. Vessel density was calculated using the Weibel grid applied to at least three different fields in each slide. Concentrations of (B) MMP-9 and (C) VEGF in lysates extracted from s.c. tumors and metastatic lungs were determined by ELISA, and normalized to the amount of total protein in each sample (n = 4–6). The two-tailed unpaired t test was used to compare between two groups.
Proliferation rate, estimated by the IHC staining for the Ki-67, was reduced in 161-MAP vaccinated mice by about 1.6–3.4-folds (p < 0.05) in both s.c. tumors and lung metastases (Fig. S3) relative to the Scr-MAP vaccinated mice. In addition, the rate of apoptosis, as assayed by TUNEL staining was increased by about 2–4-folds (p < 0.05, Fig. S4A), and the concentrations of activated caspase-3, that served as a marker for increased activation of programmed cell death, were likewise elevated by at least 3–4-folds (p < 0.05, Fig S4B) in tumor lysates derived from vaccinated mice relative to control.
Discussion
We demonstrate that peptide vaccination using our newly found EMMPRIN-specific epitope sequence,25 modified as octa-branched MAP, can effectively inhibit tumor growth, prevent tumor recurrence, and reduce metastasis. This demonstrates that: (a) the peptide must be modified to elicit a productive immune response; (b) vaccination can be administered directly, without the ex vivo mediation of dendritic cells; and (c) EMMPRIN is a good target for vaccination.
Our previously defined epitope is short and consists of only 11 amino acids. As haptens, monomeric peptides are unstable and poorly immunogenic, requiring their conjugation to a carrier protein that triggers its own unproductive immune response.1,2 Our octa-branched MAP modified peptide has the molecular weight of a small protein (∼12 kDa), and demonstrated higher stability and immunogenicity even without conjugation to a carrier protein.13,29 Similar to other studies,30,31 we show that the monomeric form of the epitope sequence had no impact on tumor size or on the number of metastatic foci, suggesting that the modification was necessary to elicit a productive immune response against EMMPRIN.
We have previously passively immunized tumor-bearing mice against EMMPRIN with a polyclonal antibody directed against this epitope, and successfully inhibited tumor progression and metastasis in three subcutaneous and orthotopic in vivo tumor models.25 Although some results are similar and the same epitope was targeted, several differences exist between that study and our current one. First, only active vaccination triggers long-term memory that protects the host from additional cancerous challenges, without the need for any additional boost injections. This implies that 161-MAP vaccination could be used not only therapeutically, but also to prevent tumor recurrence. This is a crucial point, as current immunotherapies that are based on antibody administration such as anti-VEGF (bevacizumab) or anti-CD20 (rituximab), often succeed in temporarily reducing tumor burden, only to demonstrate a “rebound effect” with rapid recurrence of the tumor upon therapy withdrawal.32 Second, when we used passive immunization, the antibody was the driving force that recruited macrophages to the tumor and enhanced their cytotoxicity. In contrast, here all arms of the adaptive immune system were activated: recruiting EMMPRIN-specific CD8+ T cells into the tumor and increasing their cytotoxicity, producing EMMPRIN-specific antibodies, and reducing macrophages in the tumor while shifting them toward M1-activation, as suggested by the increased production of nitrites. These immune modulations cumulatively overcame immune suppression, increased tumor cell death, and reduced angiogenesis. Although it is clear that the anti-EMMPRIN specific antibodies that we injected in the passive vaccination play a critical role in initiating changes in the TME and in macrophage activation mode, their role in the active vaccination is not yet fully elucidated. The antibodies may recognize slightly different epitopes, as rabbit antibodies that recognize the monomeric epitope were used for the passive vaccination, whereas the antibodies generated in the mouse after 161-MAP injections could be directed partially against the MAP scaffold. Furthermore, the facts that only spleen lymphocytes were sufficient to induce tumor inhibition upon adoptive transfer, but serum antibodies were not, and that CD8+ T cells exhibited increased cytotoxicity when extracted from 161-MAP vaccinated mice, may cumulatively suggest that the cellular arm of the immune response is more dominant in the active vaccination. However, the role that antibodies may play during active vaccination deserves further investigation.
The RNAseq data generally supports the cytokine data that indicates a shift toward M1/Th1 activation, and reveals the induction of an anti-tumoral IFNγ response. Similar IFNγ-induced antitumoral responses, that involve signaling proteins and transcription factors (Jak2, Stat2, IRF-7, SOCS6), cytotoxic genes (e.g., TNFsf10/TRAIL, iNOS, IDO1/2) and pro-apoptotic genes (NOD1, NOD2, OAS2, OAS3, CD226), all differentially elevated, were also observed in other studies utilizing immune therapies.28,33-35 Of note, some discrepancies between the RNA and protein levels could be observed. For example, although TGFβ1 mRNA levels were unchanged, the protein was greatly reduced by the vaccination, suggesting a post-transcriptional regulatory level that should be explored further.
MAPs have been used for different purposes, including with antimicrobial peptides, inhibitors of protein-protein interactions,29 and in vaccines against pathogens.36,37 Previous attempts to use MAP vaccination to reduce tumor growth used four-branched MAP epitopes of heparanse or human telomerase reverse transcriptase (hTRET) that were loaded onto dendritic cells ex vivo and then injected to tumor-bearing mice.16,31 Alternatively, naïve rabbits were vaccinated with an octa-branched B cell epitope of heparanse, and the polyclonal antibodies were used to passively immunize nude mice bearing human tumors.38 Both of these indirect methods require elaborate and intensive work that is not cost effective. In contrast, we show that MAP vaccination can be induced directly, without the ex vivo mediation of dendritic cells or B cells, to trigger all components of the immune response. A study that directly vaccinated against a MAP-synthesized mutated EGFR epitope, also demonstrated activation of both CD4+ and CD8+ T cells, as well as antibody production.14 We did not test for the presence of CD4+ T cells, which are associated with providing help for CD8+ T cells,39 and focused on the effector cells themselves.
EMMPRIN may be viewed as a stimulatory immune checkpoint, as it mediates interactions between immune cells and endothelial or epithelial cells, including tumor cells. In fact, the overexpression of EMMPRIN in tumors of higher grade and stage and in metastases in many types of cancer,40 and its important role as a regulator of angiogenesis, suggests that it may be a universal TAA, much like heparanse or hTRET that have been targeted with MAPs with considerable success.30,31 Moreover, the additional roles of EMMPRIN, as a facilitator of lactate efflux and as a leukocytes recruiter through interactions with different protein partners,18,24,40,41 are also compromised by initiating a full-scale immune response against the protein. As these functions are all crucial to tumor survival, EMMPRIN may be a TRA, whose expression the tumor cannot afford to lose, and targeting it may prevent this route of tumor escape from immune recognition.
Despite the fact that we vaccinate against a self-protein, our mice did not show any adverse responses, their weight and behavior were normal, and only in some cases a limited hair loss, especially in the forehead and mustache areas, was observed. However, EMMPRIN is also expressed in non-cancerous cells, including normal colon and kidney epithelial cells and in leukocytes. Once activated the immune system could potentially recognize EMMPRIN in all cells that express it, but the lack of any adverse responses suggests otherwise. In fact, the inability of CD8+ T cells isolated from vaccinated tumors to kill autologous leukocytes that express EMMPRIN, may suggest that the immune system is activated to kill only above a certain threshold of expression, but this premise must be further investigated. It is relevant to note that the 13 mice injected with the CT26 and vaccinated with 61-MAP that were used to demonstrate immune memory, survived for a year from the initial implantation until they were sacrificed, despite two repeated assaults, and with no adverse health effects from the cancer or the vaccination.
In conclusion, we have shown that by modifying the peptide sequence using MAP synthesis, we can elicit an effective immune response that overcomes the immunosuppressive TME and re-activates immune mechanisms to combat the tumor. Our results lay the basis for EMMPRIN therapeutic vaccination in patients with malignant tumors. Further research should explore the applicability of these results to humans. That would include targeting the equivalent human epitope instead of the mouse one, applying an adjuvant approved for use in humans, injecting the tumor cell lines orthotopically rather than relying on experimental metastasis, or delaying the vaccination until after tumor-resection surgery. As we could observe that different tumor models exhibited variations in the host response, we expect that human patients could also behave differently. This may depend on the level of EMMPRIN expression on different tumor cells, and this point must also be further examined in animal models.
Materials and methods
Cells
The tumorigenic mouse renal (RENCA, ATCC CRL-2947), and colon (CT26, ATCC CRL-2638) carcinoma cell lines were cultured in RPMI-1640 medium, 10% fetal calf serum (FCS), 1% L-Glutamine and antibiotics, with addition of 100 mM HEPES buffer (pH 7.4) for the RENCA cells, or 1% sodium pyruvate for the CT26 cells. The mouse TRAMP-C2 prostate cancer cell line (ATCC CRL-2731) was cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% FCS, 1% L-glutamine and antibiotics, with 5μg/ml Insulin and 10−8 mol/L Dihydrotestosteron (Perkin-Elmer). All cell lines were regularly tested for morphological changes and presence of mycoplasma.
Synthetic peptides
Our epitope was synthesized as monomeric peptides or as octa-branched MAPs (GHRWLKGGVVLC for the mouse sequence designated m161-MAP, or GHRWLKGGVVLC for the human sequence designated m161-MAP). The negative control MAP had the same amino acids in a scrambled order (WCRGGGLKMRVH, designated Scr-MAP). MAPs were synthesized by the standard stepwise solid-phase procedure using Fmoc chemistry on β-Ala-Wang resin, conjugating the peptides onto an octa-branched lysine core (Yuan Yu Bio-Teck). Purity and identity were confirmed by high-performance liquid chromatography (HPLC) and mass spectrographic analysis.
Determination of nitrites
Nitrites, the stable product of NO, were determined in tumor lysates by the Griess reagent system (Promega), and normalized to total protein.
Sandwich ELISA
The concentrations of MMP-9 and VEGF, as well as TNFα, IL-1β, IL-10, and TGFβ in tumor lysates were determined using ELISA kits (R&D systems), and normalized to total protein, determined by Bradford reagent (Bio-Rad).
Direct ELISA
EMMPRIN-specificity of serum antibodies was determined by direct ELISA (reagents from R&D Systems, all incubations for 2 h at room temperature, unless indicated). Wells were coated with mouse recombinant EMMPRIN (50 ng/mL) overnight at 4 °C, followed by blocking (1% BSA in PBS), three washes (0.05% Tween-20 in PBS) and incubation with 1:100 diluted serum samples or mouse anti-EMMPRIN antibody as control. After three washes, 1:5,000 diluted biotinylated goat anti-mouse was added and washed, and 1:200 diluted streptavidin-HRP was added. After three washes, the TMB/E solution was added for 5 min, the reaction stopped and the absorbance of each well was measured at 450 nm and 540 nm.
Experimental mouse models and vaccination
BALB/c female and C57BL/6 male mice (8 weeks old, Harlan Laboratories), were kept with a 12 h light/dark cycle and access to food and water ad libitum. Tumors were generated by subcutaneous injections of 2 × 106 syngeneic tumor cells suspended in Matrigel® in a total volume of 200 μL into the flank. A total of three vaccination injections were administered, one every 7 d. Peptides were emulsified in CFA for the first injection and in IFA for the following boosts, and were administered in the foot pad (in 30 μL) or subcutaneously, with no difference observed. Tumors were measured every 3–4 d and their volume calculated (length × width × 0.5 cm3). At the end of the experiment or when tumors were greater than 1.5 cm3, mice were euthanized and their tumor tissue, serum, lungs and spleens were harvested. To simulate metastases, RENCA and CT26 tumor cells were injected to the tail vein (106 cells/100 μL saline), and the mice were vaccinated, before (21, 14 and 7 d) or after (2, 7 and 12 d) tumor cell injections. In survival experiments, the end point was the beginning of respiratory distress. Parts of the tumor and lung tissues were freshly frozen for evaluation of cytokine concentrations, while other parts were formalin-fixed and paraffin-embedded for later analysis.
Mice were cared for in accordance with the procedures outlined in the NIH Guideline for the Care and Use of laboratory Animals, and all experiments were performed under the approved protocols (IL-1231013 and IL-0670614) issued by the Animal Care and Use Committee of the Technion, Israel Institute of Technology.
Adoptive transfer of spleen cells
Naïve BALB/c mice were vaccinated with 3 injections, one every 7 d, of either Scr-MAP or 161-MAP (50 μg each) as described above, and after 4 weeks were euthanized and their spleens removed. Spleens were meshed to single cell suspensions, pooled together from 4 mice of the same group, and a single injection of splenocytes (30 × 106 cells/mouse) to the tail vein of mice bearing s.c. CT26 tumors was administered 7 d after tumor implantation.
In vitro cytotoxicity assay
Tumor cells were made into single cell suspension and CD8+ T cells (effector cells) were isolated using EasySept beads (STEMCELL Technologies). Target cells (5 × 104 CT26 or RENCA cells) were labeled with 5 μM of Cell Tracker Orange™ (Molecular Probes, Invitrogen) and then incubated for 6 h with the effector CD8+ T cells or with white blood cells obtained from the same mouse as control, at different E:T ratios, as indicated. Release of the fluorescent stain to the supernatants (measured in relative fluorescent units, RFU) indicated cell death, and percent cytotoxicity of target cells was calculated by the formula:
where spontaneous release was measured from target cells incubated alone, and maximal RFU was measured from target cells incubated with 5% Triton X-100.
Immunohistochemistry (IHC)
Four micron thick paraffin embedded tissue sections were deparaffinized on a glass slide with xylene substitute K-Clear Plus (Kaltex) and rehydrated with decreasing ethanol immersions. Antigen retrieval for Ki-67 and F4/80 was performed by microwave heating in citrate buffer pH 6.0, for CD31 by immersing the slides in 42 mg/mL Proteinase XXIV solution (Sigma) for 10 min at 37 °C, or in 20 mg/mL of Proteinase K in Tris buffer, pH 8.0 for the TUNEL kit. Endogenous peroxidase was quenched in 3% H2O2 solution for 10 min, slides were blocked with 5% BSA and incubated overnight at 4 °C with the following primary antibodies: rat monoclonal anti-CD31 (Acris Antibodies) diluted 1:50, rabbit monoclonal anti-Ki67 (Abcam) diluted 1/140, rat monoclonal anti-F4/80 (Abcam) diluted 1:200, and rabbit polyclonal anti-CD8+ (Bioss) diluted 1:400. After washing, the antibodies were detected with HRP-Polymer anti-rabbit (Zytomed) or with the N-Histofine Simple Stain Mouse MAX PO (Rat) (Nichirei Bioscience) for 1 h and the DAB substrate Kit (Zytomed). All sections were counterstained with hematoxylin (Sigma) and coverslips were applied using Pertex mounting medium (Histolab Products AB). TUNEL staining was performed using the in situ death detection kit POD (Roche Life Science) according to manufacturer's instructions. All sections were viewed under the Olympus BX-60 bright field trinocular microscope equipped with a Sony DXC-950P digital camera. Images were acquired using the GrabBee X video grabber (VideoHome Technology Corp.). Vessel densities were assessed by CD31 staining using a Weibel grid and expressed as percent vessel surface area.42 The fraction of Ki-67-positive tumor cells was calculated by the digital image analysis web application ImageJS.43
RNA extraction, library construction and data generation and analysis
Total RNA was extracted using the Qiacube (Qiagen, Hilden, Germany) with the RNeasy kit from 161-MAP and Scr-MAP vaccinated CT26 s.c. tumors, three of each group. Total RNA quality measurements were performed using TapeStation (Agilent), with RINe values ranging from 6.5 to 10. Six RNAseq libraries (NEBNext® Ultra™ RNA Library Prep Kit for Illumina, cat no. E7530) were produced according to manufacturer protocol using 800 ng total RNA. mRNAs pull-up was performed using Magnetic Isolation Module (NEB, cat no. E7490). The six libraries were mixed into a single tube with equal molarity of all samples. The RNAseq data was generated on NextSeq500 mid-output (75 bp paired-end reads) kit v2 (Illumina, FC-404-2005). Quality control was performed using FastQC (v0.11.2) and overrepresented sequences were removed using CUTADAPT tool (v 1.8). The 75 paired-end reads were aligned to mouse reference genome and annotation file (Mus_musculus.GRCm38 downloaded from ENSEMBL) using TopHat (v2.0.13) allowing two mismatches per read with options –very-sensitive and –GTF. The number of reads per gene was counted using Htseq (0.6.1). Principal components analysis (PCA), samples' clustering, and differential expressed genes (DEGs), were calculated using Deseq2 (version 1.8.1). DEGs were further investigated for function and pathway enrichment using Ingenuity Pathway Analysis (IPA). In addition to IPA analysis, we conducted functional gene and term networks from clustering of enrichment analysis using Bioconductor R package FGNet (v3.2.1). FGNet generates gene networks derived from the results of functional enrichment analysis (FEA) and annotation clustering using GeneTerm Linker. This part of the study was conducted by the Genomic Center of the Biomedical Core Facility (BCF), at the Faculty of Medicine, Technion.
Data availability
The RNAseq data discussed here have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE854400.
Statistical analyses
All values are presented as means±SE of two or three independent experiments. Statistical analyses were performed using the two-tailed unpaired t test for comparing two groups, the analysis of variance (ANOVA) and the post-hoc Bonferroni's multiple comparison tests for three groups or more, and the two-way ANOVA following Bonferroni's post-tests for comparing time and groups. For survival analysis the Kaplan–Meier curve and the log-rank test were used. The p values exceeding 0.05 were not considered significant.
Supplementary Material
Disclosure of potential conflict of interest
M.W. and M.A.R. are the inventors of a pending patent application related to the research described in the manuscript. All other authors have declared that no conflict of interest exists.
Acknowledgments
We would like to thank Dr. Liat Linde and Dr.Nili Avidan (The Genomics Center, Technion) for performing RNAseq experiments and analysis, and also Prof. Ze'ev Ronai for his advice and critical reading of the manuscript.
Funding
This work was supported by the KAMIN project from the Office of the Chief Scientist in Israel's Ministry of Economy (Grant Nos. 48440 and 51111), and by the Israel Science Foundation (Grant No. 1392/14).
Author Contributions
E.S., M.M.R., E.D., M.W. and J.S. prepared cell lines and conducted the animal experiments. V.B. performed the IHC analysis, E.S. performed ELISA analyses and evaluated cellular toxicity, and M.A.R. designed the experiments, analyzed the results, performed statistical analysis, drew the conclusions and wrote the manuscript.
References
- 1.Melief CJM, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. J Clin Invest 2015; 125:3401-12; PMID:26214521; http://dx.doi.org/ 10.1172/JCI80009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iero M, Filipazzi P, Castelli C, Belli F, Valdagni R, Parmiani G, Patuzzo R, Santinami M, Rivoltini L. Modified peptides in anti-cancer vaccines: Are we eventually improving anti-tumour immunity? Cancer Immunol Immunother 2009; 58:1159-67; PMID:18998128; http://dx.doi.org/22723522 10.1007/s00262-008-0610-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jensen SM, Twitty CG, Maston LD, Antony PA, Lim M, Hu H-M, Petrausch U, Restifo NP, Fox BA. Increased frequency of suppressive regulatory T cells and T cell-mediated antigen loss results in murine melanoma recurrence. J Immunol 2012; 189:767-76; PMID:22723522; http://dx.doi.org/ 10.4049/jimmunol.1103822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olson BM, McNeel DG. Antigen loss and tumor-mediated immunosuppression facilitate tumor recurrence. Expert Rev Vaccines 2012; 11:1315-7; PMID:23249231; http://dx.doi.org/ 10.1586/erv.12.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pedersen SR, Sørensen MR, Buus S, Christensen JP, Thomsen AR. Comparison of vaccine-induced effector CD8 T cell responses directed against self- and non-self-tumor antigens: implications for cancer immunotherapy. J Immunol 2013; 191:3955-67; PMID:24018273; http://dx.doi.org/ 10.4049/jimmunol.1300555 [DOI] [PubMed] [Google Scholar]
- 6.Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 2013; 14:1014-22; PMID:24048123; http://dx.doi.org/ 10.1038/ni.2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kissick HT, Sanda MG. The role of active vaccination in cancer immunotherapy: lessons from clinical trials. Curr Opin Immunol 2015; 35:15-22; PMID:26050634; http://dx.doi.org/ 10.1016/j.coi.2015.05.004 [DOI] [PubMed] [Google Scholar]
- 8.Ilyas S, Yang JC. Landscape of Tumor Antigens in T Cell Immunotherapy. J Immunol 2015; 195:5117-22; PMID:26589749; http://dx.doi.org/ 10.4049/jimmunol.1501657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pol J, Bloy N, A Buqué, Eggermont A, Cremer I, Sautès-Fridman C, Galon J, Tartour E, Zitvogel L, Kroemer G et al.. Trial Watch: Peptide-based anticancer vaccines. Oncoimmunology 2015; 4:e974411; PMID:26137405; http://dx.doi.org/ 10.4161/2162402X.2014.974411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harao M, Mittendorf EA, Radvanyi LG. Peptide-based vaccination and induction of CD8+ T-cell responses against tumor antigens in breast cancer. BioDrugs 2015; 29:15-30; PMID:25523015; http://dx.doi.org/ 10.1007/s40259-014-0114-1 [DOI] [PubMed] [Google Scholar]
- 11.Tam JP. Synthetic peptide vaccine design: synthesis and properties of a high-density multiple antigenic peptide system. Proc Natl Acad Sci USA 1988; 85:5409-13; PMID:3399498; http://dx.doi.org/ 10.1073/pnas.85.15.5409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bracci L, Falciani C, Lelli B, Lozzi L, Runci Y, Pini A, De Montis MG, Tagliamonte A, Neri P. Synthetic peptides in the form of dendrimers become resistant to protease activity. J Biol Chem 2003; 278:46590-5; PMID:12972419; http://dx.doi.org/ 10.1074/jbc.M308615200 [DOI] [PubMed] [Google Scholar]
- 13.Falciani C, Lozzi L, Pini A, Corti F, Fabbrini M, Bernini A, Lelli B, Niccolai N, Bracci L. Molecular basis of branched peptides resistance to enzyme proteolysis. Chem Biol Drug Des 2007; 69:216-21; PMID:17441908; http://dx.doi.org/ 10.1111/j.1747-0285.2007.00487.x [DOI] [PubMed] [Google Scholar]
- 14.Ciesielski MJ, Kazim AL, Barth RF, Fenstermaker RA. Cellular antitumor immune response to a branched lysine multiple antigenic peptide containing epitopes of a common tumor-specific antigen in a rat glioma model. Cancer Immunol Immunother 2005; 54:107-19; PMID:15340764; http://dx.doi.org/ 10.1007/s00262-004-0576-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J, Yang J, Han X, Zhao Z, Du L, Yu T, Wang H. Overexpression of heparanase multiple antigenic peptide 2 is associated with poor prognosis in gastric cancer: Potential for therapy. Oncol Lett 2012; 4:178-82; PMID:22807984; http://dx.doi.org/ 10.3892/ol.2012.703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang X-D, Wang G-Z, Guo J, Lu M-H, Li C, Li N, Chao Y-L, Li C-Z, Wu Y-Y, Hu C-J et al.. Multiple antigenic peptides based on H-2Kb-restricted CTL epitopes from murine heparanase induce a potent antitumor immune response in vivo. Mol Cancer Ther 2012; 11:1183-92; PMID:22442309; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0607 [DOI] [PubMed] [Google Scholar]
- 17.Bai Y, Huang W, Ma L-T, Jiang J-L, Chen Z-N. Importance of N-glycosylation on CD147 for its biological functions. Int J Mol Sci 2014; 15:6356-77; PMID:24739808; http://dx.doi.org/ 10.3390/ijms15046356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weidle UH, Scheuer W, Eggle D, Klostermann S, Stockinger H. Cancer-related issues of CD147. Cancer Genomics Proteomics 2010; 7:157-69; PMID:20551248 [PubMed] [Google Scholar]
- 19.Kanekura T, Chen X. CD147/basigin promotes progression of malignant melanoma and other cancers. J Dermatol Sci 2010; 57:149-54; PMID:20060267; http://dx.doi.org/ 10.1016/j.jdermsci.2009.12.008 [DOI] [PubMed] [Google Scholar]
- 20.Voigt H, Vetter-Kauczok CS, Schrama D, Hofmann UB, Becker JC, Houben R. CD147 impacts angiogenesis and metastasis formation. Cancer Invest 2009; 27:329-33; PMID:19160100; http://dx.doi.org/ 10.1080/07357900802392675 [DOI] [PubMed] [Google Scholar]
- 21.Zhou J, Zhu P, Jiang JL, Zhang Q, Wu ZB, Yao XY, Tang H, Lu N, Yang Y, Chen ZN. Involvement of CD147 in overexpression of MMP-2 and MMP-9 and enhancement of invasive potential of PMA-differentiated THP-1. BMC Cell Biol 2005; 6:25; PMID:15904490; http://dx.doi.org/ 10.1186/1471-2121-6-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bougatef F, Quemener C, Kellouche S, Naïmi B, Podgorniak MP, Millot G, Gabison EE, Calvo F, Dosquet C, C Lebbé et al.. EMMPRIN promotes angiogenesis through hypoxia-inducible factor-2α-mediated regulation of soluble VEGF isoforms and their receptor VEGFR-2. Blood 2009; 114:5547-56; PMID:19837976; http://dx.doi.org/ 10.1182/blood-2009-04-217380 [DOI] [PubMed] [Google Scholar]
- 23.Zhu X, Song Z, Zhang S, Nanda A, Li G. CD147: a novel modulator of inflammatory and immune disorders. Curr Med Chem 2014; 21:2138-45; PMID:24372217; http://dx.doi.org/ 10.2174/0929867321666131227163352 [DOI] [PubMed] [Google Scholar]
- 24.Yurchenko V, Constant S, Eisenmesser E, Bukrinsky M. Cyclophilin-CD147 interactions: a new target for anti-inflammatory therapeutics. Clin Exp Immunol 2010; 160:305-17; PMID:20345978; http://dx.doi.org/ 10.1111/j.1365-2249.2010.04115.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walter M, Simanovich E, Brod V, Lahat N, Bitterman H, Rahat MA. An epitope-specific novel anti-EMMPRIN polyclonal antibody inhibits tumor progression. Oncoimmunology 2015; 5:e1078056; PMID:27057452; http://dx.doi.org/ 10.1080/2162402X.2015.1078056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saxena M, Christofori G. Rebuilding cancer metastasis in the mouse. Mol Oncol 2013; 7:283-96; PMID:23474222; http://dx.doi.org/ 10.1016/j.molonc.2013.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perske C, Lahat N, Sheffy Levin S, Bitterman H, Hemmerlein B, Rahat MA. Loss of inducible nitric oxide synthase expression in the mouse renal cell carcinoma cell line RENCA is mediated by microRNA miR-146a. Am J Pathol 2010; 177:2046-54; PMID:20709800; http://dx.doi.org/ 10.2353/ajpath.2010.091111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brandacher G, Winkler C, Schroecksnadel K, Margreiter R, Fuchs D. Antitumoral activity of interferon-gamma involved in impaired immune function in cancer patients. Curr Drug Metab 2006; 7:599-612; PMID:16918315; http://dx.doi.org/ 10.2174/138920006778017768 [DOI] [PubMed] [Google Scholar]
- 29.Falciani C, Pini A, Bracci L. Oligo-branched peptides for tumor targeting: from magic bullets to magic forks. Expert Opin Biol Ther 2009; 9:171-8; PMID:19236247; http://dx.doi.org/ 10.1517/14712590802620501 [DOI] [PubMed] [Google Scholar]
- 30.Wang GZ, Tang XD, MH Lü, Gao JH, Liang GP, Li N, Li CZ, Wu YY, Chen L, Cao YL et al.. Multiple antigenic peptides of human heparanase elicit a much more potent immune response against tumors. Cancer Prev Res 2011; 4:1285-95; http://dx.doi.org/ 10.1158/1940-6207.CAPR-11-0083 [DOI] [PubMed] [Google Scholar]
- 31.Liao ZL, Tang XD, MH Lü, Wu YY, Cao YL, Fang DC, Yang SM, Guo H. Antitumor effect of new multiple antigen peptide based on HLA-A0201-restricted CTL epitopes of human telomerase reverse transcriptase (hTERT). Cancer Sci 2012; 103:1920-8; PMID:22909416; http://dx.doi.org/ 10.1111/j.1349-7006.2012.02410.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zuniga RM, Torcuator R, Jain R, Anderson J, Doyle T, Schultz L, Mikkelsen T. Rebound tumour progression after the cessation of bevacizumab therapy in patients with recurrent high-grade glioma. J Neurooncol 2010; 99:237-42; PMID:20151176; http://dx.doi.org/ 10.1007/s11060-010-0121-0 [DOI] [PubMed] [Google Scholar]
- 33.Ulloa-Montoya F, Louahed J, Dizier B, Gruselle O, Spiessens B, Lehmann FF, Suciu S, Kruit WHJ, Eggermont AMM, Vansteenkiste J et al.. Predictive gene signature in MAGE-A3 antigen-specific cancer immunotherapy. J Clin Oncol 2013; 31:2388-95; PMID:23715562; http://dx.doi.org/ 10.1200/JCO.2012.44.3762 [DOI] [PubMed] [Google Scholar]
- 34.Wang E, Bedognetti D, Marincola FM. Prediction of response to anticancer immunotherapy using gene signatures. J Clin Oncol 2013; 31:2369-71; PMID:23715576; http://dx.doi.org/ 10.1200/JCO.2013.49.2157 [DOI] [PubMed] [Google Scholar]
- 35.Zaidi MR, Merlino G. The two faces of interferon-gamma in cancer. Clin Cancer Res 2011; 17:6118-24; PMID:21705455; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-0482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pini A, Falciani C, Mantengoli E, Bindi S, Brunetti J, Iozzi S, Rossolini GM, Bracci L. A novel tetrabranched antimicrobial peptide that neutralizes bacterial lipopolysaccharide and prevents septic shock in vivo. FASEB J 2010; 24:1015-22; PMID:19917670; http://dx.doi.org/ 10.1096/fj.09-145474 [DOI] [PubMed] [Google Scholar]
- 37.Wang XY, Huang ZX, Chen YG, Lu X, Zhu P, Wen K, Fu N, Liu BY. A multiple antigenic peptide mimicking peptidoglycan induced T cell responses to protect mice from systemic infection with Staphylococcus aureus. PLoS One 2015; 10:1-17; PMID:26317210; http://dx.doi.org/25522727 10.1371/journal.pone.0136888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J, Yang J, Cai Y, Jin N, Wang H, Yu T. Multiple antigenic polypeptide composed of heparanase B-cell epitopes shrinks human hepatocellular carcinoma in mice. Oncol Rep 2015; 33:1248-56; PMID:25522727; http://dx.doi.org/ 10.3892/or.2014.3679 [DOI] [PubMed] [Google Scholar]
- 39.Kennedy R, Celis E. Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol Rev 2008; 222:129-44; PMID:18363998; http://dx.doi.org/ 10.1111/j.1600-065X.2008.00616.x [DOI] [PubMed] [Google Scholar]
- 40.Xiong L, Edwards CK, Zhou L. The biological function and clinical utilization of CD147 in human diseases: a review of the current scientific literature. Int J Mol Sci 2014; 15:17411-41; PMID:25268615; http://dx.doi.org/ 10.3390/ijms151017411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grass GD, Toole BP. How, with whom and when: an overview of CD147-mediated regulatory networks influencing matrix metalloproteinase activity. Biosci Rep 2016; 36:e00283; http://dx.doi.org/ 10.1042/BSR20150256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weibel ER, Kistler GS, Scherle WF. Practical stereological methods for morphometric cytology. J Cell Biol 1966; 30:23-38; PMID:5338131; http://dx.doi.org/ 10.1083/jcb.30.1.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Almeida JS, Iriabho EE, Gorrepati VL, Wilkinson SR, Grüneberg A, Robbins DE, Hackney JR. ImageJS: Personalized, participated, pervasive, and reproducible image bioinformatics in the web browser. J Pathol Inform 2012; 3:25; PMID:22934238; http://dx.doi.org/ 10.4103/2153-3539.98813 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNAseq data discussed here have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE854400.