

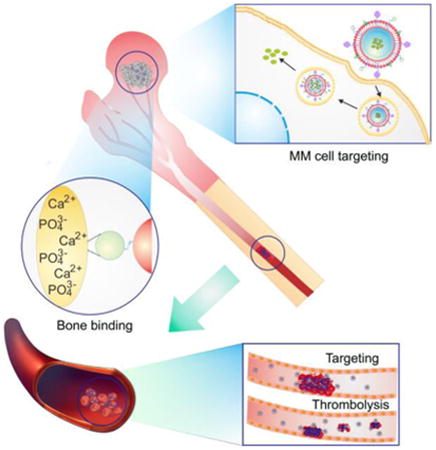
Graphical abstract
A platelet membrane-coated biomimetic nanocarrier, which could sequentially target bone microenvironment and myeloma cells to enhance the drug availability at the myeloma site and decrease the off-target effects, was developed for inhibiting the multiple myeloma growth and simultaneously eradicating the thrombus complication.
Keywords: drug delivery, multiple myeloma, thrombus, platelet, biomimetic
Multiple myeloma (MM), one of the most frequently occurred hematological cancers with an average overall survival of 5 years after diagnosis depending on the tumor types, is often characterized by the clonal proliferation of plasma cells in the bone marrow.[1] The treatment of MM remains a big challenge despite the great advances in radiation therapy, chemotherapy and stem cell implantation.[2] The first line treatment for patients ineligible for transplantation is proteasome inhibitors combination regimens, such as bortezomib and carfilzomib, aiming at targeting the MM cells and modulating the bone marrow microenvironment including alleviating osteolysis.[3] However, the non-specific biodistribution and short circulation time after administration could result in severe side effects, including peripheral neuropathy, and significantly limit their application in clinics.[4] Another contributing factor to the unsatisfactory treatment efficacy of MM is the emergence of thrombus complication,[5] which has been observed in MM patients after treatment with immunomodulatory drugs in combination with proteasome inhibitors.[5-6] Recent clinical studies showed that the risk of death of MM patients had increased by 3-fold after diagnosis of thrombus when compared to MM patients without a thrombus.[7] The mechanism underlying the risk of thrombus formation is multifactorial but mainly relies on the tumor-specific clot-promoting mechanisms, such as increased blood viscosity, the upregulation of procoagulant and fibrinolytic activities.[8] Therefore, the development of a new drug delivery system with the capability of improving the current standard-of-care treatment and eliminating the complications would be greatly beneficial to MM therapy.
Here we describe a platelet membrane-coated nanoparticulate platform (designated PM-NP) for targeted delivery of bortezomib at the myeloma site based on the bone microenvironment and myeloma cell sequential targeting strategy. The tissue plasminogen activator (tPA) is co-delivered for efficient dissolution of the thrombus by taking the advantage of the vital role of platelets in thrombus formation (Figure 1A-C).[9] As a US Food and Drug Administration (FDA) approved proteasome inhibitor,[10] bortezomib facilitates the programmed cell death by preventing the degradation of pro-apoptotic factors and inhibiting the regular functions of proteasome.[11] The clot-lysing drug—tPA is able to dissolve both preexisting and nascent clots by catalyzing the conversion of plasminogen to plasmin, which is the major enzyme responsible for clot dissolution.[12]
Figure 1. Schematic design and characterization of tPA-Ald-PM-NP-bort.
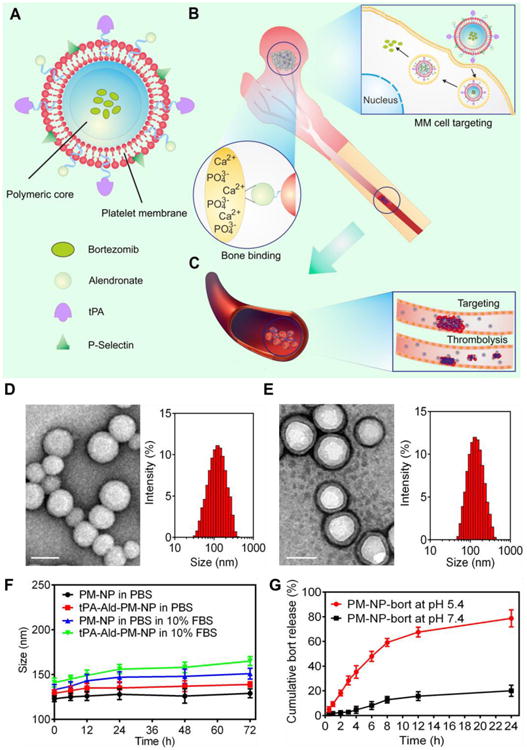
(A) The main components of tPA-Ald-PM-NP-bort: the platelet membrane derived from the platelets; polymeric nanoparticle made of acid-responsive modified dextran. (B) After intravenous injection, tPA-Ald-PM-NP-bort could sequentially target bone microenvironment through efficient binding between Ald and calcium ions and home to MM cells via specific affinity of P-Selectin and overexpressed CD44 receptors. After internalization, the matrix of tPA-Ald-PM-NP-bort could be dissociated by the acidity of lyso-endosome, releasing the encapsulated bortezomib. (C) tPA-Ald-PM-NP-bort could further target the thrombus that happens during the anti-MM treatment and dissolute the thrombus readily and effectively. (D) The TEM image and hydrodynamic size distribution of bare m-dextran NP. Scale bar: 100 nm. (E) The TEM image and hydrodynamic size distribution of PM-NP. Scale bar: 100 nm. (F) In vitro stability of PM-NP and tPA-Ald-PM-NP in PBS and 10% FBS. Error bars indicate s.d. (n=3). (G) Cumulative release of bortezomib from PM-NP in PBS with different pH levels. Error bars indicate s.d. (n=3).
To deliver both bortezomib and tPA to their most active destination efficiently, we construct a core-shell structured nanocarrier, where the platelet membrane is wrapped on the surface of a polymeric nanoparticle. The bortezomib-loaded core nanoparticle is prepared using a nano precipitation method.[13] with the integration of an acid-degradable modality (Figure 1A). tPA is decorated on the platelet membrane via biotin-streptavidin affinity.[12b, 14] To endow the PM-NP with bone targetability, alendronate (Ald) with the capability of chelating calcium ions rich in the bone microenvironment.[15] is adopted as the targeting ligand to enhance the drug accumulation at the bone sites and decrease the off-target effects. In our previous study, we have validated that the platelet membrane-coated nanocarrier could target the tumor cells based on the selective affinity between P-selectin on the platelet membrane and CD44 overexpressed on the tumor cells.[16]
After intravenous (i.v.) injection, tPA-Ald-PM-NPs are expected to accumulate at the myeloma site through a sequential targeting manner by first targeting the bone microenvironment through bone mineral binding capability of functionalized Ald, and subsequently targeting the myeloma cells through the specific affinity between P-selectin and CD44 receptor (Figure 1B). This programmed targeting strategy is expected to enhance the drug concentration at the bone marrow and decrease the side effects to the normal marrow cells. Furthermore, after internalization by the myeloma cells, the acidity in the lyso-endosome will readily cleave the mildly acidic responsive m-dextran-nanoparticle,[17] releasing the encapsulated bortezomib to induce the cell death. Of note, when the thrombus complication happens during the MM treatment, the tPA-Ald-PM-NP is expected to home to the thrombus site due to the intrinsic property of platelets that is indispensable for thrombus formation (Figure 1C). Meanwhile, the conjugated tPA on the PM-NP could readily dissolute the thrombus and reduce the mortality of MM patients. Taken together, this platelet membrane-coated nanoparticle with sequential actions toward the bone microenvironment and myeloma cells targeting capability can enhance the drug availability at the bone marrow, decreasing the side effects and reducing the emergence of the complications for promoting anti-MM treatment efficacy.
The platelet membrane-coated nanocarrier was prepared by wrapping the purified platelet membrane on the surface of acid-responsive nanocarrier.[16, 18] composed of modified dextran that is biocompatible and biodegradable.[19] The average size of the uncoated nanocarrier was determined to be 113 nm by the dynamic light scattering (DLS), which was increased to 127 nm after coating with the platelet membrane (Figure 1D, E). Additionally, the PM-NP possessed a similar surface charge to that of platelets (Figure S1). The transmission electron microscopy (TEM) images demonstrated the existence of a unilamellar membrane on the surface of nanocarrier, where the morphology was distinct from the bare nanocarrier (Figure 1D, E). To investigate the colloidal stability, the size change of tPA-Ald-PM-NP was evaluated over time. Both PM-NP and tPA-Ald-PM-NP were stable in PBS and 10% FBS solution for 72 h, which was attributed to the hydrophilic glycans on the platelet membrane (Figure 1F).18c Additionally, the surface modification of tPA and Ald did not affect the stability of PM-NP. The conjugation of tPA on the surface was validated by the confocal laser scanning microscopy (CLSM). The colocalization of the fluorescence signals from Cy 5.5 (blue) in Cy5.5-labeled tPA, rhodamine (red) in rhodamine-labled PM and coumarin-6 (green) in coumarin-6-loaded nanocarrier substantiated the successful cloaking of PM on the nanoparticle and surface conjugation of tPA (Figure S2). We further investigated the in vitro release profile of bortezomib in different pH levels. The results demonstrated that an accelerated release rate with over 70% cumulative release of bortezomib within 24 h was obtained for PM-NP-bort at pH 5.4, suggesting its acid-responsive behavior (Figure 1G).
NCI-H929 MM cell with overexpression of CD44 receptors.[20] was selected as the model cell line in this study. Additionally, coumarin-6 was decorated as the fluorescence probe to track the intracellular behavior of PM-NP. We first investigated the intracellular delivery efficiency and endosome escape of PM-NP via the confocal imaging. After 1 h of incubation, the majority of internalized PM-NP was entrapped in the endosome, as evidenced by the yellow fluorescence, resulted from colocalized coumarin-6-loaded nanoparticle and texas red-labeled endo-lysosome. In contrast, the green fluorescence signal was found to separate from red ones after 4 h of incubation, suggesting the liberation of encapsulated coumarin-6 from the endocytosed PM-NP (Figure 2A). The enhanced release of the encapsulated cargo to the cytosol indicated the dissociation of the PM-NP, which was attributed to the acid-responsive behavior of the polymeric matrix composed of the modified dextran.
Figure 2. Evaluation of the in vitro myeloma cells targetability.
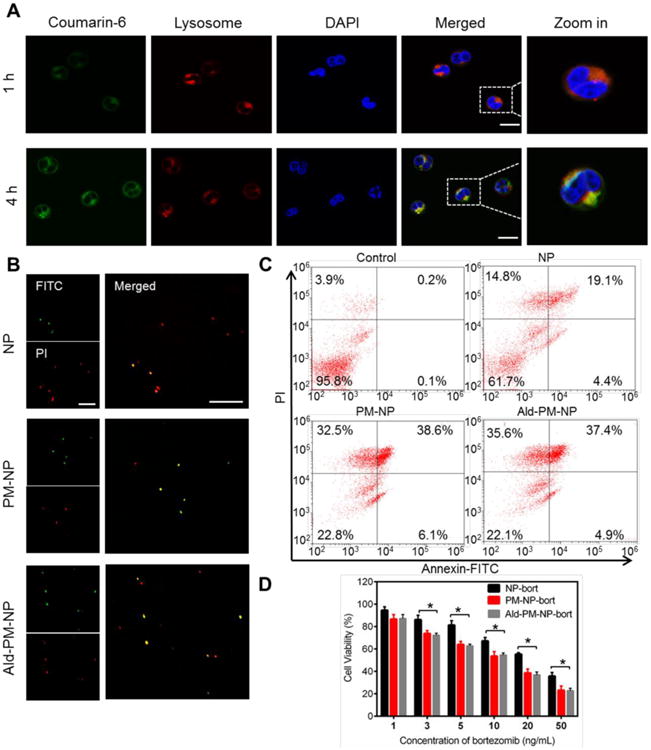
(A) Intracellular delivery of Coumarin-6-PM-NP on NCI-H929 cells at different time intervels observed by CLSM. The late endo-lysosomes were stained by LysoTracker Red, and the nuclei were stained by Hoechst 33342. Scale bar: 20 μm. (B) The APO-BrdU TUNEL assay of the induced apoptosis of NCI-H929 cells treated with NP-bort, PM-NP-bort and Ald-PM-NP-bort for 12 h. Green fluorescence indicates Alexa Fluor 488-stained nick end label DNA fragment, and red fluorescence indicates PI-stained nuclei. Scale bar: 100μm. (C) Flow cytometry analysis of NCI-H929 cells treated with drug free RPMI 1640 medium, NP-bort, PM-NP-bort and Ald-PM-NP-bort at the bort concentration of 10 ng/mL for 12 h. The cells were stained with Annexin V-FITC and PI for analysis. (D) In vitro cytotoxicity of NP-bort, PM-NP-bort, and Ald-NP-bort after incubation for 24 h. Error bars indicate s.d. (n=3).
After confirming the effective internalization of PM-NP, the apoptosis inducing capability of PM-NP was demonstrated via the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay.[21] and the Annexin-V/PI double staining assay.[22] NCI-H929 cells treated with PM-NP-bort and Ald-PM-NP-bort exhibited remarkably higher levels of the apoptotic DNA fragmentation as observed from the green fluorescence signals compared with NP-bort (Figure 2B). The enhanced intracellular accumulation of bortezomib could be ascribed to the selective affinity between platelet membrane and CD44 overexpressed MM cells. Furthermore, the quantitative flow cytometry results substantiated the enhanced apoptosis inducing capability of PM-NP. The late apoptosis ratios were 19.1% for NP-bort. While after coating with PM, the PM-NP-bort and Ald-PM-NP-bort exhibited higher ratios of apoptotic cells with the late apoptosis rate of 38.6% and 37.4%, respectively (Figure 2C). We further evaluated the cytotoxicity of different bortezomib formulations via cell counting kit-8 (CCK8) assay. PM-NP-bort and Ald-PM-NP-bort displayed higher cytotoxicity against NCI-H929 cells with the IC50 values of 13.6 ng/mL and 13.1 ng/mL, respectively, which were significantly lower than that of NP-bort (23.2 ng/mL) (Figure 2D). Collectively, the specific affinity between platelets and myeloma cells contributed to the enhanced internalization of the PM-NP and further increased the apoptosis inducing capability and cytotoxicity against MM cells. Furthermore, the addition of Ald did not affect the efficacy of PM-NP-bort.
The bone targetability of Ald-PM-NP was investigated by studying the binding efficiency between Ald-PM-NP and bone fragment and further evaluated via whole body imaging after in vivo administration. Hydroxyapatite (HAP), the most abundant mineral in the bone, was used to mimic the bone microenvironment.[23] Alendronate could bind to the HAP through chelating the calcium ions available on the structure HAP with a high affinity. As displayed in Figure 3A, over 70% of Ald-PM-NP was observed to bind to HAP within 4 h, while the PM-NP and NP displayed minimized binding efficacy with less than 10% binding efficiency. We next tested the targeting capability of Ald-PM-NP on the bone fragment. After incubation for 1 h, a clearly high fluorescence signal was found on the bone fragment treated with Ald-PM-NP. In contrast, the femurs treated with PM-NP and NP showed a barely observable fluorescence signal (Figure 3B). Taken together, these results demonstrated the superior binding efficacy of Ald-PM-NP to the bone in vitro.
Figure 3. Bone targetablity evaluation.
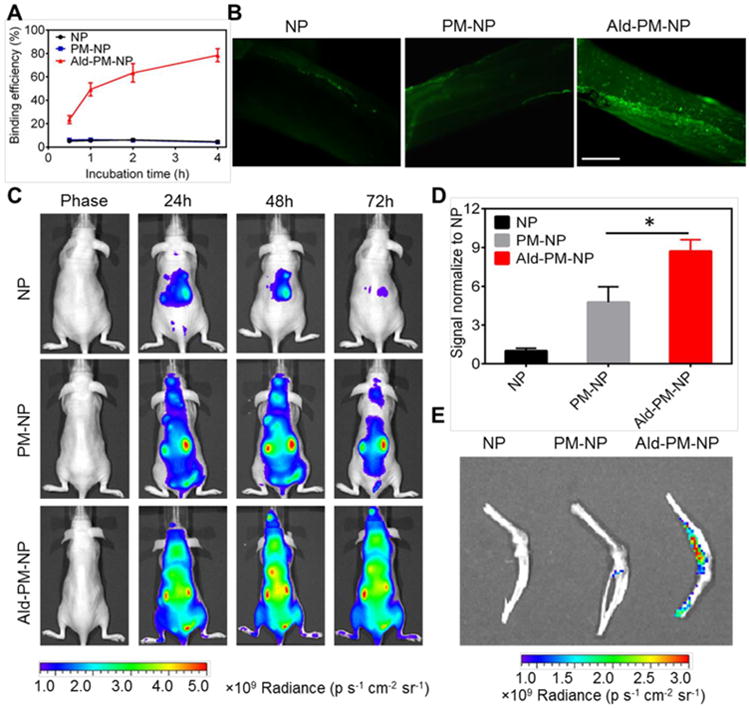
(A) In vitro hydroxyapatite (HAP) binding efficiency. HAP was incubated with coumarin-6-NP, coumarin-6-PM-NP and coumarin-6-Ald-PM-NP for different time intervals. Error bars indicate s.d. (n=3). (B) In vitro bone fragment targeting capability. The bone fragments were treated with coumarin-6-NP, coumarin-6-PM-NP and coumarin-6-Ald-PM-NP at coumarin-6 concentration of 200 ng/mL for 1 h and then observed by fluorescence microscope. Scale bar: 500 μm. (C) In vivo fluorescence imaging of the nude mice at 24, 48 and 72 h after intravenous injection of Cy5.5-loaded NP, Cy5.5-loaded PM-NP and Cy5.5-loaded Ald-PM-NP at Cy5.5 dose of 30 nmol/kg. (D) Region-of-interest analysis of fluorescent intensities from whole body at 72 h. The analysis was based on the whole body fluorescence intensities from multiple mice (three mice for each group). Error bars indicate s.d. (n=3). *P<0.05 (two-tailed Student's t-test). (E) Ex vivo fluorescence imaging of the femur tissues at 72 h post injection.
The in vivo bone-homing ability of Ald-PM-NP was investigated by performing biodistribution studies using Cy5.5-loaded nano-formulations via an in vivo imaging system (IVIS) in the nude mice. All animals were treated in accordance with the Guide for Care and Use of Laboratory Animals, approved by the Institutional Animal Care and Use Committee (IACUC) of University of North Carolina at Chapel Hill and North Carolina State University. The mice were treated with Cy5.5-loaded NP, Cy5.5-loaded PM-NP and Cy5.5-loaded Ald-PM-NP and the whole body imaging was taken during different time intervals. Ald-PM-NP showed a higher fluorescence intensity than PM-NP and NP at every detected time point. After 72 h post-injection, the PM-NP displayed enhanced fluorescence signal than NP that was barely observed, which was mainly attributed to abundant “self-recognized” proteins.[18d] on the platelet membrane, resulting in the long circulation time of PM-NP. In contrast, Ald-PM-NP presented the highest fluorescence intensity among all the treated groups with the main retention of the Ald-PM-NP located at the bone-associated tissue including spine and femur (Figure 3C). The quantitative whole body imaging results demonstrated that the fluorescence intensity of the mice treated with Ald-PM-NP was about 1.5 folds higher than PM-NP and 9 folds higher than NP (Figure 3D). After 72 h, the dissected femurs were taken out and imaged ex vivo. The results were in good accordance with the whole body imaging and further validated the effective bone targetability of Ald-PM-NP (Figure 3E).
To assess the potency of thrombolysis of tPA-PM-NP, we first tested the bioactivity of tPA by hydrolyzing the tripeptide chromogenic substrates after conjugation.[24] The results demonstrated a similar bioactivity between free tPA and tPA-PM-NP (Figure 4A), suggesting a good preservation of the bioactivity of tPA. We further evaluated the in vitro thrombus dissolution capability of various tPA formulations by incubating with fibrinogen, followed by the addition of thrombin.[14] The results displayed insignificant difference between free tPA and tPA-conjugated NP formulations (Figure 4B). Next, the thrombolysis potency of tPA-PM-NP was demonstrated after circulation in the blood. The free tPA showed a lower dissolution capability, which was mainly due to the quick clearance of tPA after administration. The longer circulation of tPA-PM-NP contributed significantly to the superior thrombolysis ability, as evidenced by the higher fibrinolysis ratio compared to tPA-NP (Figure 4C). Taken together, the decoration of the platelet membrane on the nanocarrier elongated the circulation of the tPA-PM-NP, which was beneficial to the tPA-mediated clot dissolution.
Figure 4. Thrombus dissolution activity determination.
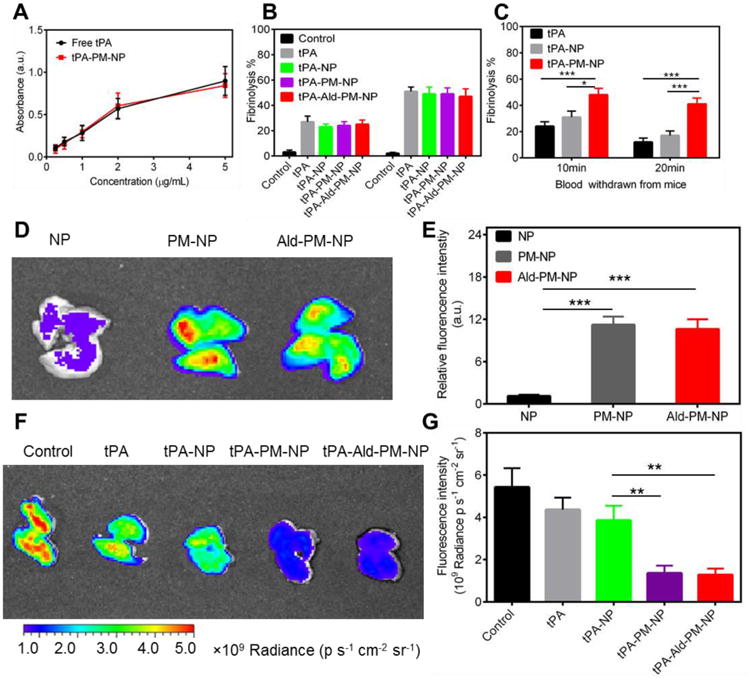
(A) The bioactivity assay of tPA conjugated on the PM-NP. Error bars indicate s.d. (n=3). (B) In vitro fibrinolysis of fibrin clots by saline, free tPA, tPA-NP, tPA-PM-NP and tPA-Ald-PM-NP. Error bars indicate s.d. (n=3). Fibrin clot treated with saline served as the control. (C) In vitro fibrinolysis of fibrin clots by blood drawn from the mice treated with tPA, tPA-NP and tPA-PM-NP. Error bars indicate s.d. (n=3). *P<0.05, ***P<0.001 (two-tailed Student's t-test). (D) In vivo fluorescence imaging of lungs treated with Cy 5.5-loaded NP, Cy5.5-loaded PM-NP and Cy5.5-loaded Ald-PM-NP at Cy5.5 concentration of 30 nmol/kg. (E) Quantification of the fluorescence intensity of the lungs. Error bars indicate s.d. (n=3). ***P<0.001 (two-tailed Student's t-test). (F) In vivo fluorescence imaging of lungs treated with saline, tPA, tPA-NP, tPA-PM-NP and tPA-Ald-PM-NP. The lung treated with saline served as the control. (G) Quantification of the fluorescence intensity of the lungs. Error bars indicate s.d. (n=3). **P<0.01 (two-tailed Student's t-test).
To evaluate whether the platelet membrane coating increased the accumulation of the nanocarriers in the tissues containing thrombus, a mouse thrombosis model was established by intravenously injecting fibrinogen and thromboplastin to trigger the clotting cascade in the lung.[25] As displayed in Figure 4D, the lung treated with Cy5.5-loaded PM-NP and Cy5.5-loaded Ald-PM-NP showed stronger fluorescence intensity compared to Cy5.5-loaded NP, suggesting the excellent targeting capability of PM-NP and Ald-PM-NP endowed by platelet membrane decoration. Quantification showed that the intensity of the lung signal was similar to that in the PM-NP and Ald-PM-NP groups, while over 10-fold higher than the NP group. There was no significant difference in thrombolysis potency between tPA-PM-NP and tPA-Ald-PM-NP (Figure 4E). The homing ability of tPA-PM-NP was mainly due to the intrinsic properties of platelets that are essential for the thrombus formation.
To investigate efficacy of the platelet membrane-coating in enhancing the lung thrombus formation inhibition, saline, free tPA, tPA-NP, tPA-PM-NP and tPA-Ald-PM-NP were tested in the lung thrombosis model after administration of the Cy5.5-labeled fibrinogen. As showed in Figure 4F, the strong fluorescence signal in the saline group validated the successful establishment of lung thrombus model. The tPA-PM-NP and tPA-Ald-PM-NP displayed the most effective dissolution potency, as evidenced by the lowest fluorescence intensity of the lungs. Quantitative data showed over 2-fold lower in the fluorescence intensity of lungs treated with tPA-PM-NP and tPA-Ald-PM-NP compared to the free tPA and tPA-NP (Figure 4G). Collectively, these results demonstrated that the enhanced targeting capability of nanoparticles enabled by platelet membrane coating contributed to the improved accumulation of tPA at the thrombus site, leading to the efficient dissolution of the thrombus formation.
The bone marrow targeting efficiency was investigated by injecting the MM-bearing Nod/SCID mice with different coumarine-6-loaded NP formulations. As observed by the confocal imaging, the bone marrow treated with Ald-PM-NP and tPA-Ald-PM-NP displayed the highest accumulation of the nanoparticles, which were significantly higher than PM-NP and Ald-NP (Figure 5A). This result suggested that the sequential bone and MM cells targeting strategy could facilitate increase of the drug availability at the myeloma site, thereby leading to the superior treatment efficacy. The targetability of PM-NP could be compromised due to the lack of homing capability to the bone, while the binding potency of Ald-NP was unsatisfactory because of anchoring at the bone mineral only.
Figure 5. In vivo anti-MM and thrombolysis efficacy.
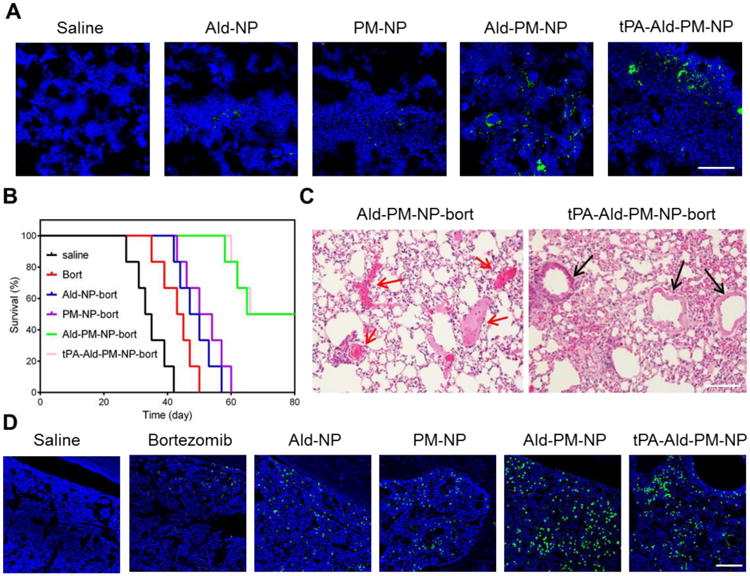
(A) In vivo fluorescence images of the bone marrow of the Nod/SCID mice treated with saline, coumarin-6-loaded Ald-NP, coumarin-6-loaded PM-NP, coumarin-6 loaded Ald-PM-NP and coumarin-6 loaded tPA-Ald-PM-NP for 6 h. Scale bar: 100 μm. (B) Survival curves of MM-bearing Nod/SCID mice (n=6) following the administration of saline, bortezomib, Ald-NP-bort, PM-NP-bort, Ald-PM-NP-bort and tPA-Ald-PM-NP, respectively. (C) Histological observation of the lungs of the mice treated with Ald-PM-NP-bort and tPA-Ald-PM-NP-bort after induction of lung thrombus. Black arrows indicate the blood vessel. Red arrows indicate the blocked sites. Scale bar: 100μm. (D) Detection of apoptosis in the bone marrow tissue after treatment using fluorescein-dUTP (green) for staining apoptotic cells. Scale bar: 100μm.
We next evaluated the pharmacokinetics of PM-NP and tPA-Ald-PM-NP by quantitatively monitoring the bortezomib concentration in the blood plasma. The circulation time of PM-NP and tPA-Ald-PM-NP were significantly higher than that of the NP, suggesting the decrease clearance rate after platelet membrane coating. Furthermore, the decoration of Ald and tPA did not significantly affect the in vivo pharmacokinetics of PM-NP (Figure S3). Additionally, we investigated the anti-MM efficacy in the MM-bearing Nod-SCID mice by administering with various bortezomib formulations. As displayed in Figure 5B, all the mice treated with saline died within 42 days, indicating the successful establishment of MM model. The free bortezomib group resulted in a less than 50 days survival of the mice. Additionally, the survival time of the mice treated with Ald-NP-bort and PM-NP-bort was less than 60 days. In a sharp contrast, the mice treated with Ald-PM-NP-bort and tPA-Ald-PM-NP-bort achieved the longest survival time with half of mice surviving over 80 days. This remarkable anti-MM efficacy was attributed to the programmed targeting manner of Ald-PM-NP by combining the bone microenvironment and MM cells targeting efficacy, leading to the significantly increased drug accumulation at the MM site. The fluorescence images obtained using the in situ TUNEL assay displayed the highest level of cell apoptosis in the mice treated with Ald-PM-NP-bort and tPA-Ald-PM-NP-bort (Figure 5D and Figure S4). No obvious pathological abnormalities were observed in the normal organs (Figure S5). After induction of the lung thrombus, tPA-Ald-PM-NP-bort exhibited a superior dissolution capability over Ald-PM-NP-bort, as evidenced by the presence of the blocked blood vessels in the lung hematoxylin-eosin staining (H&E) staining of Ald-PM-NP-bort (Figure 5C and Figure S6), verifying that the tPA-Ald-PM-NP-bort reduced the thrombus complication during anti-MM treatment.
In conclusion, compared to the traditional active targeting nanoparticle-based anti-MM treatment strategies, our delivery system could precisely bind to the myeloma cells by taking advantage of the programmed targeting of bone microenvironment-specific binding and myeloma cell-selective homing modules. Furthermore, the emergence of the thrombus during anti-MM therapy could be eliminated via tPA conjugated on the surface of PM-NP. This “all-in-one” drug delivery strategy generates the promising anti-MM treatment efficacy, offering new guideline for treating the MM patients and improving prognosis.
Supplementary Material
Acknowledgments
This work was supported by the grants from NC TraCS, NIH's Clinical and Translational Science Awards (CTSA, NIH grant 1UL1TR001111) at UNC-CH and Sloan Research Fellowship. We acknowledge the use of the Analytical Instrumentation Facility (AIF) at NC State, which is supported by the State of North Carolina and the National Science Foundation (NSF). We also acknowledge the use of Cellular and Molecular Imaging Facility (CMIF) at NC State University.
Footnotes
Supporting Information: Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Quanyin Hu, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Raleigh, NC 27695, USA and Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Chenggen Qian, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Raleigh, NC 27695, USA and Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; Department of Polymer Science and Engineering and Key Laboratory of High Performance Polymer Materials and Technology of MOE, School of Chemistry and Chemical Engineering, Nanjing University Nanjing 210023, China.
Wujin Sun, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Raleigh, NC 27695, USA and Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Jinqiang Wang, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Raleigh, NC 27695, USA and Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
Zhaowei Chen, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Raleigh, NC 27695, USA and Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
Hunter N. Bomba, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Raleigh, NC 27695, USA and Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
Prof. Hongliang Xin, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Raleigh, NC 27695, USA and Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; School of Pharmacy, Nanjing Medical University, Nanjing 211166, China
Prof. Qundong Shen, Department of Polymer Science and Engineering and Key Laboratory of High Performance Polymer Materials and Technology of MOE, School of Chemistry and Chemical Engineering, Nanjing University Nanjing 210023, China
Prof. Zhen Gu, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Raleigh, NC 27695, USA and Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; Department of Medicine, University of North Carolina School of Medicine, Chapel Hill, NC 27599, USA.
References
- 1.a) Kyle R, Rajkumar S. Leukemia. 2009;23:3–9. doi: 10.1038/leu.2008.291. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kuehl WM, Bergsagel PL. Nat Rev Cancer. 2002;2:175–187. doi: 10.1038/nrc746. [DOI] [PubMed] [Google Scholar]; c) Barlogie B, Mitchell A, van Rhee F, Epstein J, Morgan GJ, Crowley J. Blood. 2014;124:3043–3051. doi: 10.1182/blood-2014-07-552059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Mahindra A, Laubach J, Raje N, Munshi N, Richardson PG, Anderson K. Nature reviews Clinical oncology. 2012;9:135–143. doi: 10.1038/nrclinonc.2012.15. [DOI] [PubMed] [Google Scholar]; b) Kyle RA, Rajkumar SV. Clin Lymphoma Myeloma. 2009;9:278–288. doi: 10.3816/CLM.2009.n.056. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McCarthy PL, Owzar K, Hofmeister CC, Hurd DD, Hassoun H, Richardson PG, Giralt S, Stadtmauer EA, Weisdorf DJ, Vij R. N Engl J Med. 2012;366:1770–1781. doi: 10.1056/NEJMoa1114083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H. N Engl J Med. 2005;352:2487–2498. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]; b) Richardson PG, Weller E, Lonial S, Jakubowiak AJ, Jagannath S, Raje NS, Avigan DE, Xie W, Ghobrial IM, Schlossman RL. Blood. 2010;116:679–686. doi: 10.1182/blood-2010-02-268862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Argyriou AA, Iconomou G, Kalofonos HP. Blood. 2008;112:1593–1599. doi: 10.1182/blood-2008-04-149385. [DOI] [PubMed] [Google Scholar]
- 5.Palumbo A, Rajkumar S, Dimopoulos M, Richardson PG, San Miguel J, Barlogie B, Harousseau J, Zonder J, Cavo M, Zangari M. Leukemia. 2008;22:414–423. doi: 10.1038/sj.leu.2405062. [DOI] [PubMed] [Google Scholar]
- 6.Zangari M, Anaissie E, Barlogie B, Badros A, Desikan R, Gopal AV, Morris C, Toor A, Siegel E, Fink L. Blood. 2001;98:1614–1615. doi: 10.1182/blood.v98.5.1614. [DOI] [PubMed] [Google Scholar]
- 7.Kristinsson SY, Pfeiffer RM, Björkholm M, Schulman S, Landgren O. Haematologica. 2012;97:1603–1607. doi: 10.3324/haematol.2012.064444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Zangari M, Elice F, Fink L, Tricot G. Expert Rev Anticancer Ther. 2007;7:307–315. doi: 10.1586/14737140.7.3.307. [DOI] [PubMed] [Google Scholar]; b) Kristinsson SY. ASH Education Program Book. 2010;2010:437–444. doi: 10.1182/asheducation-2010.1.437. [DOI] [PubMed] [Google Scholar]
- 9.a) Ruggeri ZM. Nat Med. 2002;8:1227–1234. doi: 10.1038/nm1102-1227. [DOI] [PubMed] [Google Scholar]; b) Nesbitt WS, Westein E, Tovar-Lopez FJ, Tolouei E, Mitchell A, Fu J, Carberry J, Fouras A, Jackson SP. Nat Med. 2009;15:665–673. doi: 10.1038/nm.1955. [DOI] [PubMed] [Google Scholar]; c) Furie B, Furie BC. N Engl J Med. 2008;359:938–949. doi: 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- 10.Kane RC, Bross PF, Farrell AT, Pazdur R. The oncologist. 2003;8:508–513. doi: 10.1634/theoncologist.8-6-508. [DOI] [PubMed] [Google Scholar]
- 11.Kane RC, Dagher R, Farrell A, Ko CW, Sridhara R, Justice R, Pazdur R. Clin Cancer Res. 2007;13:5291–5294. doi: 10.1158/1078-0432.CCR-07-0871. [DOI] [PubMed] [Google Scholar]
- 12.a) Wardlaw JM, Murray V, Berge E, del Zoppo G, Sandercock P, Lindley RL, Cohen G. The Lancet. 2012;379:2364–2372. doi: 10.1016/S0140-6736(12)60738-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Korin N, Kanapathipillai M, Matthews BD, Crescente M, Brill A, Mammoto T, Ghosh K, Jurek S, Bencherif SA, Bhatta D. Science. 2012;337:738–742. doi: 10.1126/science.1217815. [DOI] [PubMed] [Google Scholar]
- 13.Bilati U, Allémann E, Doelker E. Eur J Pharm Sci. 2005;24:67–75. doi: 10.1016/j.ejps.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Murciano JC, Medinilla S, Eslin D, Atochina E, Cines DB, Muzykantov VR. Nat Biotechnol. 2003;21:891–896. doi: 10.1038/nbt846. [DOI] [PubMed] [Google Scholar]
- 15.Swami A, Reagan MR, Basto P, Mishima Y, Kamaly N, Glavey S, Zhang S, Moschetta M, Seevaratnam D, Zhang Y. Proceedings of the National Academy of Sciences. 2014;111:10287–10292. doi: 10.1073/pnas.1401337111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu Q, Sun W, Qian C, Wang C, Bomba HN, Gu Z. Adv Mater. 2015;27:7043–7050. doi: 10.1002/adma.201503323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Gu Z, Aimetti AA, Wang Q, Dang TT, Zhang Y, Veiseh O, Cheng H, Langer RS, Anderson DG. ACS Nano. 2013;7:4194–4201. doi: 10.1021/nn400630x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mo R, Gu Z. Materials Today. 2015 [Google Scholar]
- 18.a) Hu CMJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Proceedings of the National Academy of Sciences. 2011;108:10980–10985. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hu CMJ, Fang RH, Copp J, Luk BT, Zhang L. Nat Nanotechnol. 2013;8:336–340. doi: 10.1038/nnano.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hu CMJ, Fang RH, Wang KC, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV. Nature. 2015;526:118–121. doi: 10.1038/nature15373. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Li J, Ai Y, Wang L, Bu P, Sharkey CC, Wu Q, Wun B, Roy S, Shen X, King MR. Biomaterials. 2016;76:52–65. doi: 10.1016/j.biomaterials.2015.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhou H, Fan Z, Lemons PK, Cheng H. Theranostics. 2016;6:1012. doi: 10.7150/thno.15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Bachelder EM, Beaudette TT, Broaders KE, Dashe J, Fréchet JM. J Am Chem Soc. 2008;130:10494–10495. doi: 10.1021/ja803947s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Broaders KE, Cohen JA, Beaudette TT, Bachelder EM, Fréchet JM. Proceedings of the National Academy of Sciences. 2009;106:5497–5502. doi: 10.1073/pnas.0901592106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Driel M, Gunthert U, Van Kessel A, Joling P, Stauder R, Lokhorst H, Bloem A. Leukemia. 2002;16:135–143. doi: 10.1038/sj.leu.2402336. [DOI] [PubMed] [Google Scholar]
- 21.Hu Q, Sun W, Lu Y, Bomba HN, Ye Y, Jiang T, Isaacson AJ, Gu Z. Nano Lett. 2016 doi: 10.1021/acs.nanolett.5b04343. [DOI] [PubMed] [Google Scholar]
- 22.Mo R, Jiang T, DiSanto R, Tai W, Gu Z. Nat Commun. 2014;5:3364. doi: 10.1038/ncomms4364. [DOI] [PubMed] [Google Scholar]
- 23.De Miguel L, Noiray M, Surpateanu G, Iorga BI, Ponchel G. Int J Pharm. 2014;460:73–82. doi: 10.1016/j.ijpharm.2013.10.048. [DOI] [PubMed] [Google Scholar]
- 24.Higazi AAR, Barghouti II, Abu-Much R. J Biol Chem. 1995;270:9472–9477. doi: 10.1074/jbc.270.16.9472. [DOI] [PubMed] [Google Scholar]
- 25.She ZG, Liu X, Kotamraju VR, Ruoslahti E. ACS Nano. 2014;8:10139–10149. doi: 10.1021/nn502947b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.