

Graphical abstract
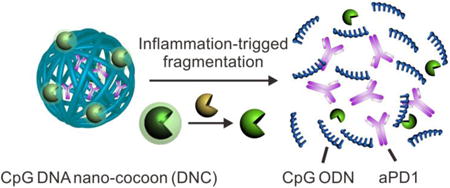
Inflammation-triggered combination delivery of anti-PD-1 antibody (aPD1) and CpG oligodeoxynucleotides (CpG ODNs) has been demonstrated to prevent cancer relapse utilizing post-surgical inflammatory response. The controlled release of anti-PD1 antibodies and CpG ODN by CpG DNA-based “nano-cocoon” can induce considerable immune response, which in turn significantly prolonged the survival time and complete response (CR) rate. This study provides a new strategy to reduce the risk of cancer relapse and metastasis after resection of the primary tumor.
Keywords: anti-PD1, CpG, drug delivery, nanobiotechnology, cancer immunotherapy
Surgical treatment is usually the most effective therapeutic method for solid tumors. However, many patients develop recurrent disease post-surgery, which can lead to significant morbidity as well as mortality for cancer patients.1, 2 There is also suggestion that the inflammatory processes during wound healing following tumor resection may promote cancer progression.3-6 Increasing amounts of evidence indicate that the perioperative inflammation induced by trauma may pose a high risk for the development of tumor reoccurrence, acceleration of local remaining tumor relapse as well as the promotion of tumor invasion and metastasis.7-11 Hence, there has been strong interest in the development of strategies to prevent cancer recurrence after surgery. Current approaches rely on established treatments including systemic chemotherapy and radiotherapy, which carry high toxicity profiles.12 Recent successes of cancer immunotherapy suggest that it can be utilized to prevent cancer recurrence.13-15 Immune regulatory checkpoint inhibitors, including anti-CTLA-4 and anti-PD-1/PD-L1, have shown exciting results in treating various types of cancer, such as melanoma.16-23. Programmed cell death protein 1 (PD-1) is expressed on various immune cells including activated T and B lymphocytes.21 The interaction between the PD-1 and its ligand 1 and 2 (PD-L1/2) is a key pathway hijacked by tumors to suppress immune response. Therefore, inhibition of the interaction between PD-1 and PD-L1 by anti-PD antibodies can boost the immune response against cancer cells.16-23
Despite the impressive clinical data of anti-PD immunotherapy, the objective response rate in metastatic melanoma remains potential to increase.24, 25 Besides, side effects, such as autoimmune disorders from these agents can be significant.20, 26-28 Therefore, how to improve the therapy efficacy with insignificant side effects is a central theme for the anti-PD-based cancer immunotherapy. For example, checkpoint inhibition combined with other immunomodulators has demonstrated a synergistic increase of antitumor activity.29, 30 Moreover, the limited efficacy may also due to the lack of co-stimulation tumor microenvironment in the setting where the antigen-presenting cells (APCs) encounter the tumor cells and T cells.31-33 Current clinical research are investigating whether utilizing cancer immunotherapy earlier in the disease course will be more effective as there is less tumor burden and heterogeneity of the tumor cells.34, 35 Therefore, further efforts are demanded to improve the clinical benefits of these immunomodulatory therapies while avoiding their untoward side effects.36-39
To address these limitations, we propose to develop a new cancer immunotherapy agent for the prevention of post-surgical tumor relapse. Tumor burden is the lowest after cancer surgery. Moreover, the local pro-inflammatory environment is conducive to immunotherapy by converting quiescent precursor lymphocytes into activated lymphocytes required for tumor eradication. To accomplish this, we have developed an innovative delivery carrier for the controlled release of loaded aPD1 and CpG ODNs in response to inflammation conditions (Fig. 1A). CpG ODNs trigger cells that express Toll-like receptor 9, including human plasmacytoid dendritic cells (pDCs), have potent immunostimulatory effects and can enhance the anti-cancer activity of a variety of cancer treatments.40, 41 Through an enzymatic rolling circle amplification (RCA) method42-46 specifically based on a template encoded with the CpG sequence, the carrier (designated as DNA “nano-cocoons”, DNCs) is assembled by a long-chain single-stranded DNA (ssDNA).47 The DNA is repeatedly containing interval CpG sequences and cutting sites of restriction enzyme HhaI, which are capable of digesting DNCs and subsequently generating CpG ODN fragments. To make the release event bio-responsive, HhaI is caged into triglycerol monostearate (TGMS) nanoparticles (TGMS NPs) and attached to DNCs. TGMS is an amphiphile whose ester linkage enables cleavage by esterases and matrix metalloproteinases (MMPs) that are highly expressed at the wound sites for developmental tissue remodeling.48 Triggered by the inflammatory condition occurring in the wound site of the tumor resection incision (Fig. 1B), TGMS can be enzymatically cleaved,48, 49 thereby disassembling the cage and releasing the HhaI, which can further sequentially convert DNCs to CpG ODNs and release aPD1 as well. The combination action of sustained released CpG ODNs and aPD1 (Fig. 1C) can synergistically facilitate induction of durable and specific anti-tumor T-cell responses in a melanoma model, with a potential to avoid the toxic peak level in the body.
Figure 1.

Schematic illustration of delivery of CpG and anti-PD1 antibody (aPD1) by DNA nano-cocoon under an inflammation condition. (A) CpG-sequence containing DNA nano-cocoon (DNC) loaded with anti-PD1 antibody and caged restriction enzyme for inflammation-trigged fragmentation of DNC for releasing both CpG DNA and aPD1. (B) A schematic representation of in vivo tumor immunotherapy after primary tumor resection, local injection and treatment of DNC-based delivery system. (C) Released CpG DNA activates dendritic cells (DCs) to drive T cell response with aPD1 for PD 1 blockade.
As shown in Supplementary Fig. S1, the synthesized ssDNA self-assembled into the three-dimensional “cocoon”-like structure50, 51 with an average particle size of 150 nm, as revealed by the transmission electron microscopy (TEM) and atomic force microscopy (AFM).- As expected, the obtained DNCs can be digested into small homogeneous fragments treated with HhaI (Supplementary Fig. S2). More importantly, these homogeneous fragments can synergistically induce TNF-α and IL6 production in RAW264.7 cells (Supplementary Fig. S2), indicating that the DNCs can be degraded into CpG ODNs after restriction enzyme treatment. Two different control DNCs (cDNC) with a non-specific sequence were synthesized (Table S1) to verify our design. As shown in agarose gels (Supplementary Fig. S3), without HhaI treatment, these cDNCs cannot migrate in the gel as DNCs. While being treated with HhaI, the cDNC (with cutting sites, without CpG sequence) could be readily fragmented. However, the fragments cannot induce TNF-α and IL6 production in RAW264.7 cells (Fig. S2), while the other cDNCs (with CpG, without cutting sites) were resistant to cleavage by HhaI.
Next, we found that the enzyme could be caged into TGMS NPs with a diameter about 30 nm monodispersed in PBS (Supplementary Fig. S4), with a loading efficiency about 0.4%. We validated that the enzyme activity of HhaI was significantly inhibited when caged into TGMS NPs (Supplementary Fig. S5). We further demonstrated that TGMS NPs can efficiently attach to DNA nano-cocoon to form monodispersed nanocomposites TGMS-DNC NPs after mixing, as revealed by TEM and dynamic light scattering (DLS) (Fig. 2A-B). Once attached with TGMS NPs, DNA nano-cocoon displayed an obvious increase in the DLS measured diameter, from 150 nm to 210 nm. The nanocomposites showed excellent stability in physiological solutions as determined by DLS data. The cell viability assay further suggested that the nanocomposites showed insignificant cytotoxicity (Fig. S6). In addition, it is found that the aPD1 could be loaded into TGMS-DNC NPs after ultrasonication and incubation overnight at 4 °C, probably due to the hydrophobic and electrostatic nonspecific-interaction when antibodies penetrated into DNCs. The maximal loading efficiency of aPD1 was about 5.8%, as determined by the ELISA assay (Supplementary Fig. S7).
Figure 2.

Characterization of CpG DNCs loaded with aPD1 and caged enzyme and enzyme-responsive drug release. (A) TEM imaging of HhaI-TGMS-DNCs-aPD1 nanocomposites (Scale bar: 500 nm). Inset: zoom-in image (Scale bar: 200 nm). (B) Dynamic light scattering characterization of HhaI-TGMS-DNCs-aPD1 nanocomposites. (C) Schematic of LPS activation of RAW264.7 macrophages for mimicking inflammatory conditions. (D) Gel electrophoresis of HhaI-TGMS-DNCs-aPD1 nanocomposites incubated with cell culture supernatant from the activated and non-activated macrophages at the difference time points (Lane 0, 0min; Lane 1, 30min; Lane 2, 1h; Lane 3, 2h; Lane 4, 4h; Lane 5, 6h; Lane M, DNA ladder). (E-F) Percentage of DNA and aPD1 released from TGMS-DNC nanocomposites when incubated with cell culture supernatant from activated and non-activated macrophages at different time points. (G-H) AFM images and hydrodynamic size of HhaI-TGMS-DNCs-aPD1 nanocomposites when incubated with cell culture supernatant from non-activated macrophages. (I-J) AFM images and hydrodynamic size of HhaI-TGMS-DNCs-aPD1 nanocomposites when incubated with cell culture supernatant from activated macrophages (Scale bar: 500 nm). The error bars are based on the standard deviations (SD) of triplicated samples.
We then studied their controlled release profiles in response to inflammation conditions in vitro. Proteolytic enzymes, such as MMPs, are significantly up-regulated under inflammatory conditions like perioperative trauma.52, 53 We identified and confirmed that TGMS NPs were quickly dissociated by MMP9 treatment, as visualized by TEM imaging (Supplementary Fig. S8). We next evaluated the ability of TGMS-DNCs to disassemble and release the loaded aPD1 in response to the proteolytic enzymes. TGMS-DNCs were incubated at 37°C with lipase (esterase) or MMP-9 enzyme (500 ng/mL) in PBS. The degradation of DNA nano-cocoon and release profile of aPD1 were quantified by the agarose gel electrophoresis and ELISA assay, respectively (Supplementary Fig. S9). The data suggest that DNA nano-cocoons were degraded into small DNA fragments upon the MMP-9 treatment as time increased. The cumulative release profiles of DNA and aPD1 revealed that lipase and MMPs could trigger fragmentation of DNCs and subsequently release the loaded aPD1. Whereas TGMS-DNCs in the PBS control solution remained stable and release insignificant amount of the aPD1 over time (Supplementary Fig. S9). Furthermore, the DNCs after MMP-9 treatment can effectively induce IL-6 and TNF-α production in murine macrophage (RAW264.7) cells, indicating that DNCs were digested into CpG nucleotides (Supplementary Fig. S10).
To further mimic the conditions of inflammation in vitro, RAW 264.7 cells were activated by treatment with lipopolysaccharide (LPS) (Fig. 2C). After 24 hours, the cell culture medium from the activated macrophages was added to TGMS-DNCs and incubated at 37°C. The release of CpG nucleotides and aPD1 were quantified in medium at different time points (Fig. 2D-F). Plotting cumulative release versus time revealed that the medium from the LPS activated macrophages triggered TGMS-DNCs degradation, leading to the sustained release of the aPD1. Whereas the medium of the nonactivated macrophage cultures did not. Furthermore, the atomic force microscopy (AFM) image and DLS data suggested that the TGMS-DNCs complexes remained stable when treated with the nonactivated macrophage cultures; while a high degradation efficiency of TGMS-DNCs complexes was observed when treated with the medium from the LPS activated macrophages (Fig. 2G-J).
To evaluate anti-tumor relapse of the TGMS-DNCs complexes post-operative, we next used the B16F10 mouse melanoma incomplete tumor resection model (which mimics local and systemic recurrences arising post-surgically) (Supplementary Fig. S11).13 To verify the up-regulated expression of MMP-9 in the operative site, medium from wound tissues at day 1–4 post operation were tested for MMP-9 activity by ELISA (Supplementary Fig. S12). Significantly increased levels of active MMP-9 were observed in the wounds cultured ex vivo, compared to the presurgery and unwounded tissue, thereby indicating that surgery promotes the MMP-9 secretion in the wound site. After incomplete operation to removal of the tumor, the mice were local injected with single dose of varying drug formulations to resection bed, including PBS control (Group 1 (G1)), HhaI-TGMS-DNCs (G2), HhaI-TGMS-cDNCs (including cutting sites of HhaI without CpG sequence)-aPD1 (G3), free aPD1/free CpG nucleotides (G4) and HhaI-TGMS-DNCs-aPD1 (G5) (aPD1, 0.5mg/kg. DNA, 10mg/kg). Tumor growth was monitored by the bioluminescence signals of B16F10 cells and the sizes of the tumor (Fig. 3A-C). We demonstrated that mice receiving HhaI-TGMS-DNCs-aPD1 treatment showed the smallest relapsed tumor volumes. Of note, 40% of mice showed complete responses to our combination therapy. The remaining mice showed substantially delayed tumor growth (Fig. 3B). In other tested groups, free aPD1 and free CpG nucleotides co-administered mice showed a modest delay of tumor growth but did not prevent relapse. Furthermore, the prevention of tumor relapse in mice treated with HhaI-TGMS-DNCs and HhaI-TGMS-cDNCs-aPD1 was not more successful than the controls. It was also observed that these antibodies alone, without CpG DNCs, had insignificant efficacy toward tumor regression, indicating that CpG was critical to enhance the anti-tumor immune response together with aPD1 treatment. In addition to the prevention of tumor relapse, tumor size in mice was correlated with their survival (Supplementary Fig. S13). Complete tumor regression and survival (40%) was observed 60 days after combination therapy. Co-administered free aPD1 and free CpG nucleotides modestly increased average survival times. Also neither HhaI-TGMS-DNCs nor HhaI-TGMS-cDNCs-aPD1 treatment improved survival over PBS controls.
Figure 3.

In vivo tumor therapy to reduce post-surgical tumor relapse via CpG DNC delivery system. (A) In vivo bioluminescence imaging of the B16F10 tumors of the different groups after removal of primary tumor. (Group 1, PBS control; G2, HhaI-TGMS-DNCs; G3, HhaI-TGMS-cDNCs-aPD1; G4, free aPD1/free CpG nucleotides; G5, HhaI-TGMS-DNCs-aPD1) (B) Quantified tumor signals and (C) mean tumor growth of different groups of mice after various treatments indicated. Pie chart shows percent CR rate (orange) (n=10). The black arrow indicates the surgery time. (D) Representative plots of T cells in relapsed tumors analyzed by the flow cytometry. (Gated on CD3+ T cells). (E) Representative plots of activated CD8 T cells (CD44+CD62L-) in relapsed tumors analyzed by the flow cytometry (gated on CD8+ T cells). (F) Immunofluorescence of relapsed tumors showed CD4+ T cells and CD8+ T cells infiltration (Scale bar: 100 μm). (G) Absolute number of the activated CD8 cells present in tumors for the study shown in C&D. (H) Ratios of the tumor-infiltrating CD8+ T cells and effective CD4+ T cells over regulatory T cells in the relapsed tumors upon various treatments. Statistical significance was calculated by 2-way ANOVA using the Tukey post-test. P value: *, P<0.05; **, P<0.01; ***P<0.005.
To investigate the immunologic mechanisms behind the synergistic therapeutic effects of our formulation, tumor-infiltrating lymphocytes (TILs) from relapsed tumors were harvested and analyzed by the flow cytometry and immunofluorescence 10 days after surgery. It was verified that HhaI-TGMS-DNCs-aPD1 combined treatment increased both the percentage and absolute numbers of activated CD8+ T cells (CD8+CD44+CD62L-) in the relapsed tumors (Fig. 3D-E). Immunofluorescence staining also revealed that the relapsed tumors in control group had limited T-cell infiltration (Fig. 3F & Fig. S14). In contrast, tumors from HhaI-TGMS-DNCs-aPD1 treated mice were remarkably infiltrated by both CD8+ and CD4+ T cells. When absolute cell numbers and the mass of the pooled tumors were accounted for in the analysis, the total CD8+ T cells per mg of tumor in HhaI-TGMS-DNCs-aPD1 combination-treated mice increased 2-fold over free aPD1 / free CpG nucleotides and HhaI-TGMS-cDNCs-aPD1 treated mice, and 10-fold over HhaI-TGMS-DNCs treatment and PBS controls (Fig. 3G). Besides, the tumor-infiltrating CD4+ FoxP3+ T cells were also studied (Supplementary Fig. S15). Likewise, the effective CD4+ cells were increased in combination-treated tumors as well compared with control groups. Collectively, the combination treatment not only increased the ratio of CD8+ T cells to Tregs, but also increased effective CD4+ T cells to Tregs (Fig. 3H).
To further demonstrate the potency of this formulation, we tested it in a metastasis tumor model by challenging mice with i.v. injection of luciferase expressing B16F10 cells one day before surgery. After incomplete surgery to remove the most primary tumor, the mice were peritumorally injected with single dose of different formulations as indicated. The systemic antitumor immune response generated by injection of HhaI-TGMS-DNCs-aPD1 after surgery was clearly observed (Fig. 4A-B). Both the primary tumor and the disseminated tumors showed significantly delayed growth, with 40% of mice surviving 40 days. In sharp contrast, the antitumor immune response generated by the control groups had little effect on the distant metastatic in multiple organs, and showed negligible improvement of survival compared to the untreated groups (Fig. 4C). We further examined splenocytes responses by measuring their production of IFN-γ. Splenocytes harvested from mice in the 30th day were treated with irradiated B16F10 cell for 16 h. It was shown that after HhaI-TGMS-DNCs-aPD1 treatment, the proportion of IFN-γ-secreting CD8 T cells in the spleen increased approximately 2-3 fold in mice compared to the control groups (Fig. 4D-E). Moreover, enhanced cytotoxic responses were also observed when incubated the splenocytes to B16F10 cancer cells in vitro (Fig. 4F). Taken together, the systemic antitumor efficacy can be achieved by locally injecting HhaI-TGMS-DNCs-aPD1 into the operative site, eradicating tumors in situ as well as disseminating metastasized tumors.
Figure 4.

Systemic antitumor efficacy could be obtained by the local injection of DNC delivery system at the surgical site. (A) In vivo bioluminescence imaging of the B16F10 metastasis of different groups after removing of primary tumors at different time points. (G1, PBS control; G2, HhaI-TGMS-DNCs; G3, HhaI-TGMS-cDNCs-aPD1; G4, free aPD1 / free CpG nucleotides; G5, HhaI-TGMS-DNCs-aPD1). (B) Quantified tumor signals according to A. Every line represents one animal and each dot shows the whole animal photon count (n=3). (C) Kaplan Meier survival curves for treated and control mice. Shown are ten mice per treatment group. (D) IFN-γ CD8 CTLs T-cell in splenocytes of mice with various treatments indicated in A. (E) Quantified IFN-γ CD8 CTL T-cell in splenocytes from three independent experiments. (F) CTL-mediated immune responses measured by incubating restimulated splenocytes with B16 tumor cells. The error bars are based on the standard error of the mean (SEM) of three mice.
Furthermore, besides the experimental metastasis model, we further tested the aPD1 delivery system in the spontaneous metastasis animal model. After spontaneous metastasis to the lung, the most primary tumor was removed by surgery as described above. The mice were injected with a single dose of different formulations as indicated. We then conducted the systemic administration of aPD1 by intravenous (i.v.) injection as a positive control. On day 22, lungs were harvested for analysis. Compared to the mice treated with aPD1 i.v. injection or co-administrated with free agents peritumorally injected, the mice treated with the CpG DNA-based aPD1 delivery system showed remarkably reduced relapsed tumor as well as lung metastasis, as evidenced by the bioluminescence signals of B16F10 cells, the weight of relapsed tumor and photos of the whole lungs with their Hematoxylin and Eosin (H&E) staining. Next, we studied the survival of mice treated with the CpG DNA-based aPD1 delivery system. The survival time of treated mice was significantly increased compared with the control groups.
In summary, we have developed a local delivery system for cancer immunotherapy that can substantially improve the ability of the anticancer immune response to treat remaining or metastasis tumors after the resection of primary tumors. The novel CpG DNA-based carrier not only served as a therapeutic loading matrix for aPD1, but can also enhance treatment efficacy after fragmentation. The sustained release and synergistic action of aPD1 and CpG ODNs in tumor sites can be activated by the inflammatory microenvironment induced by operation. It was substantiated that the bio-responsive controlled release of CpG and aPD1 was more effective than free CpG nucleotides and aPD1. The controlled release can also prevent the risk of toxic peak dosage of aPD1 in the body. Beyond its potential for cancer immunotherapy, this programmed combination delivery strategy may inspire new treatments that involve the localized and bio-responsive release of other therapeutics.
Supplementary Material
Figure 5.

Antitumor efficacy by the local injection of DNC delivery system in spontaneous metastasis model. (A) In vivo bioluminescence imaging at different time points of the B16F10 metastasis of different groups after removal of primary tumors. (G1: PBS control, G2: aPD1 i.v. Inj., G3: aPD1+CpG local Inj., G4: HhaI-TGMS-DNCs-aPD1) (B) Representative lung photographs and H&E-stained lung slices collected from mice post different treatments indicated (Scale bar: 150 μm). (C) Average weights of relapsed tumors collected from mice in the end of various treatments indicated. (D) Quantification of lung metastasis nodules after different treatments. (E) Quantified tumor signals according to A. (F) Kaplan Meier survival curves of mice in after various treatments indicated. Shown are ten mice per treatment group. P value: *, P<0.05.
Acknowledgments
This work was supported by grants from NC TraCS, the NIH (Clinical and Translational Science Awards (CTSA, NIH grant 1L1TR001111) at UNC-CH to Z.G.), and the pilot grant from the UNC cancer center. AZW is supported by 1R01CA178748, R21CA182322 and U54CA198999 from NIH/NCI. We acknowledge Dr. Leaf Huang for providing the B16F10-Luc cell line.
Footnotes
Supporting Information: Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Chao Wang, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Raleigh, NC 27695, USA; Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
Wujin Sun, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Raleigh, NC 27695, USA; Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Grace Wright, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Raleigh, NC 27695, USA; Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Andrew Wang, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Raleigh, NC 27695, USA; Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; Department of Radiation Oncology, University of North Carolina, Chapel Hill, NC 27599, USA
Zhen Gu, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Raleigh, NC 27695, USA; Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; Department of Medicine University of North Carolina School of Medicine Chapel Hill, NC 27599, USA
References
- 1.Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T. N Engl J Med. 2004;351:2817. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 2.Turajlic S, Swanton C. Science. 2016;352:169. doi: 10.1126/science.aaf2784. [DOI] [PubMed] [Google Scholar]
- 3.Vakkila J, Lotze MT. Nat Rev Immunol. 2004;4:641. doi: 10.1038/nri1415. [DOI] [PubMed] [Google Scholar]
- 4.Coffey JC, Wang J, Smith M, Bouchier-Hayes D, Cotter T, Redmond H. Lancet Oncol. 2003;4:760. doi: 10.1016/s1470-2045(03)01282-8. [DOI] [PubMed] [Google Scholar]
- 5.Demicheli R, Retsky M, Hrushesky W, Baum M, Gukas I. Ann Oncol. 2008:mdn386. doi: 10.1093/annonc/mdn386. [DOI] [PubMed] [Google Scholar]
- 6.Grivennikov SI, Greten FR, Karin M. Cell. 2010;140:883. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ceelen W, Pattyn P, Mareel M. Crit Rev Oncol Hematol. 2014;89:16. doi: 10.1016/j.critrevonc.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 8.Segatto I, Berton S, Sonego M, Massarut S, Perin T, Piccoli E, Colombatti A, Vecchione A, Baldassarre G, Belletti B. Oncotarget. 2014;5:6267. doi: 10.18632/oncotarget.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antonio N, Bonnelykke-Behrndtz ML, Ward LC, Collin J, Christensen IJ, Steiniche T, Schmidt H, Feng Y, Martin P. EMBO J. 2015;34:2219. doi: 10.15252/embj.201490147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker D, Masterson T, Pace R, Constable W, Wanebo H. Surgery. 1989;106:525. [PubMed] [Google Scholar]
- 11.Tohme S, Yazadani HO, Al-Khafaji AB, Chidi AP, Lougharn P, Mowen K, Wang Y, Simmons RL, Huang H, Tsung A. Cancer Res. 2016 doi: 10.1158/0008-5472.CAN-15-1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albain KS, Swann RS, Rusch VW, Turrisi AT, Shepherd FA, Smith C, Chen Y, Livingston RB, Feins RH, Gandara DR. The Lancet. 2009;374:379. doi: 10.1016/S0140-6736(09)60737-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stephan SB, Taber AM, Jileaeva I, Pegues EP, Sentman CL, Stephan MT. Nat Biotechnol. 2015;33:97. doi: 10.1038/nbt.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwon ED, Foster BA, Hurwitz AA, Madias C, Allison JP, Greenberg NM, Burg MB. Proc Natl Acad Sci U S A. 1999;96:15074. doi: 10.1073/pnas.96.26.15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Q, Zhai B, Yang W, Yu LX, Dong W, He YQ, Chen L, Tang L, Lin Y, Huang DD. Mol Ther. 2009;17:2049. doi: 10.1038/mt.2009.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ. Nature. 2014;515:577. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pardoll DM. Nat Rev Cancer. 2012;12:252. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang C, Xu LG, Liang C, Xiang J, Peng R, Liu Z. Adv Mater. 2014;26:8154. doi: 10.1002/adma.201402996. [DOI] [PubMed] [Google Scholar]
- 19.Curran MA, Montalvo W, Yagita H, Allison JP. Proc Natl Acad Sci U S A. 2010;107:4275. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buchbinder EI, Hodi FS. Nat Rev Clin Oncol. 2016;13:77. doi: 10.1038/nrclinonc.2015.237. [DOI] [PubMed] [Google Scholar]
- 21.Zou W, Wolchok JD, Chen L. Sci Transl Med. 2016;8:328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ribas A. N Engl J Med. 2015;373:490. doi: 10.1056/NEJMp1510079. [DOI] [PubMed] [Google Scholar]
- 23.Wang C, Ye Y, Hochu G, Sadeghifar H, Gu Z. Nano Lett. 2016 doi: 10.1021/acs.nanolett.5b05030. [DOI] [PubMed] [Google Scholar]
- 24.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, Mc Neil C, Lotem M, Larkin J, Lorigan P, Neyns B, Blank CU, Hamid O, Mateus C, Shapira-Frommer R, Kosh M, Zhou H, Ibrahim N, Ebbinghaus S, Ribas A K.-. investigators. N Engl J Med. 2015;372:2521. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 25.Sharma P, Allison JP. Science. 2015;348:56. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 26.Naidoo J, Page DB, Li BT, Connell LC, Schindler K, Lacouture ME, Postow MA, Wolchok JD. Ann Oncol. 2015:mdv383. doi: 10.1093/annonc/mdv383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mellati M, Eaton KD, Brooks-Worrell BM, Hagopian WA, Martins R, Palmer JP, Hirsch IB. Diabetes Care. 2015;38:e137. doi: 10.2337/dc15-0889. [DOI] [PubMed] [Google Scholar]
- 28.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD. N Engl J Med. 2015;373:23. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minn AJ, Wherry EJ. Cell. 2016;165:272. doi: 10.1016/j.cell.2016.03.031. [DOI] [PubMed] [Google Scholar]
- 30.Sharma P, Allison JP. Cell. 2015;161:205. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, Spranger S. Curr Opin Immunol. 2013;25:268. doi: 10.1016/j.coi.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 32.Hegde PS, Karanikas V, Evers S. Clin Cancer Res. 2016;22:1865. doi: 10.1158/1078-0432.CCR-15-1507. [DOI] [PubMed] [Google Scholar]
- 33.Sabado RL, Bhardwaj N. Nature. 2015;519:300. doi: 10.1038/nature14211. [DOI] [PubMed] [Google Scholar]
- 34.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, Watkins TB, Shafi S, Murugaesu N, Mitter R, Akarca AU, Linares J, Marafioti T, Henry JY, Van Allen EM, Miao D, Schilling B, Schadendorf D, Garraway LA, Makarov V, Rizvi NA, Snyder A, Hellmann MD, Merghoub T, Wolchok JD, Shukla SA, Wu CJ, Peggs KS, Chan TA, Hadrup SR, Quezada SA, Swanton C. Science. 2016 [Google Scholar]
- 35.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS. Science. 2015;348:124. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goldberg MS. Cell. 2015;161:201. doi: 10.1016/j.cell.2015.03.037. [DOI] [PubMed] [Google Scholar]
- 37.Irvine DJ, Hanson MC, Rakhra K, Tokatlian T. Chem Rev. 2015;115:11109. doi: 10.1021/acs.chemrev.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gu L, Mooney DJ. Nat Rev Cancer. 2015;16:56. doi: 10.1038/nrc.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koshy ST, Mooney DJ. Curr Opin Biotechnol. 2016;40:1. doi: 10.1016/j.copbio.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, Rajapaksa R, Green MR, Torchia J, Brody J. J Clin Invest. 2013;123:2447. doi: 10.1172/JCI64859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krieg AM. Oncogene. 2008;27:161. doi: 10.1038/sj.onc.1210911. [DOI] [PubMed] [Google Scholar]
- 42.Ali MM, Li F, Zhang Z, Zhang K, Kang DK, Ankrum JA, Le XC, Zhao W. Chem Soc Rev. 2014;43:3324. doi: 10.1039/c3cs60439j. [DOI] [PubMed] [Google Scholar]
- 43.Roh YH, Lee JB, Shopsowitz KE, Dreaden EC, Morton SW, Poon Z, Hong J, Yamin I, Bonner DK, Hammond PT. ACS Nano. 2014;8:9767. doi: 10.1021/nn502596b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lv Y, Hu R, Zhu G, Zhang X, Mei L, Liu Q, Qiu L, Wu C, Tan W. Nat Protoc. 2015;10:1508. doi: 10.1038/nprot.2015.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun W, Gu Z. Biomaterials Sci. 2015;3:1018. doi: 10.1039/c4bm00459k. [DOI] [PubMed] [Google Scholar]
- 46.Sun W, Lu Y, Gu Z. Ther Deliv. 2015;6:765. doi: 10.4155/tde.15.27. [DOI] [PubMed] [Google Scholar]
- 47.Jones MR, Seeman NC, Mirkin CA. Science. 2015;347:1260901. doi: 10.1126/science.1260901. [DOI] [PubMed] [Google Scholar]
- 48.Gajanayake T, Olariu R, Leclère FM, Dhayani A, Yang Z, Bongoni AK, Banz Y, Constantinescu MA, Karp JM, Vemula PK. Sci Transl Med. 2014;6:249ra110. doi: 10.1126/scitranslmed.3008778. [DOI] [PubMed] [Google Scholar]
- 49.Karp JM, Vemula PK, Campbell NR, Syed AM, Zhang S, Farokhzad OC, Langer RS. U.S Patent Application No 13/825,486. 2011
- 50.Sun W, Ji W, Hall JM, Hu Q, Wang C, Beisel CL, Gu Z. Angew Chem Int Ed. 2015;54:12029. doi: 10.1002/anie.201506030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun W, Jiang T, Lu Y, Reiff M, Mo R, Gu Z. J Am Chem Soc. 2014;136:14722. doi: 10.1021/ja5088024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ryan TJ. Australas J Dermatol. 1994;35:35. doi: 10.1111/j.1440-0960.1994.tb01799.x. [DOI] [PubMed] [Google Scholar]
- 53.Armstrong DG, Jude EB. J Am Podiatr Med Assoc. 2002;92:12. doi: 10.7547/87507315-92-1-12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.