

ABSTRACT
Herein, we evaluated whether Placental Mesenchymal Stromal Cells (PDMSCs) derived from normal and Preeclamptic (PE) placentae presented differences in the expression of G1/S-phase regulators p16INK4A, p18INK4C, CDK4 and CDK6. Finally, we investigated normal and PE-PDMSCs paracrine effects on JunB, Cyclin D1, p16INK4A, p18INK4C, CDK4 and CDK6 expressions in physiological term villous explants.
PDMSCs were isolated from physiological (n = 20) and PE (n = 24) placentae. Passage three normal and PE-PDMSC and conditioned media (CM) were collected after 48h. Physiological villous explants (n = 60) were treated for 72h with normal or PE-PDMSCs CM. Explants viability was assessed by Lactate Dehydrogenase Cytotoxicity assay. Cyclin D1 localization was evaluated by Immuofluorescence (IF) while JunB, Cyclin-D1 p16INK4A, p18INK4C, CDK4 and CDK6 levels were assessed by Real Time PCR and Western Blot assay.
We reported significantly increased p16INK4A and p18INK4C expression in PE- relative to normal PDMSCs while no differences in CDK4 and CDK6 levels were detected. Explants viability was not affected by normal or PE-PDMSCs CM. Normal PDMSCs CM increased JunB, p16INK4 and p18INK4C and decreased Cyclin-D1 in placental tissues. In contrast, PE-PDMSCs CM induced JunB downregulation and Cyclin D1 increase in placental explants. Cyclin D1 IF staining showed that CM treatment targeted mainly the syncytiotrophoblast.
We showed Cyclin D1-p16INK4A/p18INK4C altered pathway in PE-PDMSCs demonstrating an aberrant G1/S phase transition in these pathological cells. The abnormal Cyclin D1-p16INK4A/p18INK4C expression in explants conditioned by PE-PDMSCs media suggest a key contribution of mesenchymal cells to the altered trophoblast cell cycle regulation typical of PE pregnancies with fetal-placental compromise.
KEYWORDS: placenta, mesenchymal stromal cells, preeclampsia, cell cycle, pregnancy
Introduction
Eukaryotic cell cycle progression is dependent on a tightly regulated activity of Cyclins (Cyclins A, B, D, E) and Cyclin-Dependent Kinases (CDK1, CDK2, CDK4, CDK6). Upon receiving external stimuli to divide, cells up-regulate CDKs and their activating Cyclins to orchestrate the complex proliferation processes. CDKs activities are in turn regulated by the abundance of Cyclins and by the interaction with CDK-inhibitory proteins (CKIs).1-3 A finely tuned cell cycle progression is pivotal for proper human placental development and functionality, in order to guarantee embryo growth and pregnancy success. Human placentation involves high trophoblast proliferation rate and trophoblast invasion of the uterine wall.4,5 The balance between trophoblast proliferative/invasive phenotypes is controlled by complex interactions among cell cycle promoters and inhibitors6 that are seriously compromised in case of severe placenta-related disorders as Preeclampsia.7 PE represents the main cause of feto-maternal mortality and morbidity worldwide. affecting about 5–8% of all pregnancies. It is characterized by immature hyper-proliferative trophoblast phenotype, shallow trophoblast invasion of maternal spiral arteries8 and aberrant cell cycle progression of placental mesenchymal cells (Placenta-derived Mesenchymal Stromal Cells - PDMSCs)9,10 a unique population stem cells-like features and key immune-regulatory properties.9-12
During early placentation, mesenchymal cells represent the structural support for the forming primary villi and drive placental capillary network establishment.13 Our recent findings on JunB-mediated inhibition of Cyclin D1 in preeclamptic PDMSCs10 lead to the hypothesis that PE-PDMSCs, as previously demonstrated for the trophoblast, may present defects that could cause/contribute to the aberrant placenta development of preeclamptic placentae.7 The Activating Protein 1 (AP-1) family member JunB is well known as a cell division inhibitor14,15 and senescence inducer.15 JunB is able to inhibit the G1/S phase transition by inducing Cyclin-D1 downregulation10 and promoting the expression of the cell-cycle kinase inhibitor p16INK4A.15 Importantly, the G1/S phase is cooperatively regulated by CDK 4 and 6 whose activities are in turn constrained by CKIs p16INK4A and p18INK4C, that specifically inhibit the formation of CDKs-Cyclin D1 complexes.3 In quiescent cells, p16INK4A and p18INK4C are in excess to Cyclin D1–CDK4/6 complexes thus maintaining cells in a non-proliferating state.15 Mitogen stimulation leads to Cyclin D1 synthesis and, as soon as the CKI inhibitory threshold is overcome, cyclin–CDK kinase activity promotes cell cycle progression into S phase.15 CKIs are therefore critical mediators of antiproliferative signals that arrest the cell cycle and allow DNA repair, terminal differentiation and senescence.15 Moreover, Cyclin D1 controls CDK4 and CDK6 activities acting as a positive coactivator16 and when its signal is not strong enough to enhance CDK4 and CDK6 activities, cells cannot enter into S phase.17
In the present study, we characterized the expression of p16INK4A, p18INK4C, CDK4 and CDK6 in normal and PE-PDMSCs. Since mesenchymal stromal cells could influence proliferation and apoptosis in neighboring cells,9,18,19 we investigated the paracrine effect of normal and PE-PDMSCs on JunB, CyclinD1, p16INK4A, p18INK4C, CDK4 and CDK6 expressions in physiological term placental villi. Thus, we increased our knowledge on PDMSCs cell cycle regulation and on their contribution to both physiological and pathological placentation.
Results
Study population
Clinical features of the study population were reported in Table 1. Normal (n = 20) and PE (n = 24) pregnancies were comparable for maternal age, while gestational age (p < 0.01, 0.85 Fold Decrease), neonatal and placental weights at delivery were, as expected, significantly lower in PE group vs controls (p ≤ 0.01). All pregnancies belonging to the PE group presented abnormal umbilical (59%) and/or uterine arteries (76%) Doppler velocimetry and 66% of them presented FGR.
Table 1.
Clinical features of the study population. Comparison s of normal (N, n = 20) and PE (PE, n = 24) pregnancies clinical features. Statistical significance (*) has been considered as p < 0.05.
| Normal (n = 20) | Preeclampsia (n = 24) | |
|---|---|---|
| Nulliparae (%) | 30 | 41.6 |
| Gestational age at delivery (weeks) | 39.5 ± 1.08 (37–41) | 33.6 ± 3.30 (28–41)* |
| Maternal age at delivery (years) | 33.1 ± 4.54 (24–41) | 33.4 ± 5.63 |
| Caucasian ethnicity (%) | 100 | 100 |
| Smokers (%) | 10 | — |
| Alcohol (%) | 15 | — |
| Previous prenatal admission (%) | 10 | 33.3 |
| Systolic blood pressure (mm Hg) | 114.7 ± 14.18 | 150 ± 19.41* |
| Diastolic blood pressure (mm Hg) | 73 ± 10 | 95 ± 12.18* |
| Proteinuria (g/24h) | Absent | 2.92 ± 4.57* |
| A/REDF (%) | 0 | 58.3* |
| Pathological uterine doppler (%) | 0 | 75* |
| Labor (%) | 60 | 25* |
| Caesarian section (%) | 55 | 87.5* |
| Maternal oxygen given at delivery (%) | — | 25 |
| Birth weight (g) | 3530.5 ± 379.54 | AGA (n = 8): 2490 ± 845.3* |
| FGR (n = 16): 1245 ± 476.9* | ||
| Fetal sex (%) | ||
| Male | 65 | 45.8 |
| Female | 35 | 54.2 |
Characterization of placenta-derived MSCs
All PDMSCs cell lines (n = 44) presented proper mesenchymal stromal phenotype as assessed by flow cytometry. Cells were positive for CD105, CD166, CD90, CD73 and negative for HLA-II, for haematopoietic markers CD34 and CD45 and endothelial progenitor markers CD133 and CD31. Normal and PE PDMSCs were also negative for B cells, neutrophils and macrophages markers CD20 and CD14 and for trophoblast and epithelial marker CD326, thus excluding any type of contamination (data not shown). RT-PCR detected the expression of typical stemness markers Oct-4 and Nanog, in all PDMSCs cell lines (n = 44) (data not shown).
P16INK4A, p18INK4C, CDK4 and CDK6 expressions in normal and PE-PDMSCs
We previously demonstrated that JunB overexpression in PE-PDMSCs affects cell cycle progression by inducing Cyclin-D1 downregulation.10 We next investigated the expression of p16INK4A, p18INK4C, CDK4 and CDK6 in normal and PE-PDMSCs in order to complete the characterization of the G1/S regulatory pathway headed by Cyclin D1. We showed that Cyclin D1 post-translational inhibitors p16INK4A (p = 0.01, 1.73 Fold Increase) and p18INK4C (p = 0.04, 2.74 Fold Increase) mRNA levels were significantly higher in PE- relative to normal PDMSCs (Fig. 1a and b left panel). overexpression of p16INK4A (p = 0.011, 2.09 Fold Increase) and p18INK4C (p < 0.01, 2.81 Fold Increase) in PE-PDMSCs were confirmed at the protein level (Fig. 1a and b right panel). No differences were found in CDK4 and CDK6 expression between normal and PE-PDMSCs (p > 0.05, Fig. 2a and b).
Figure 1.
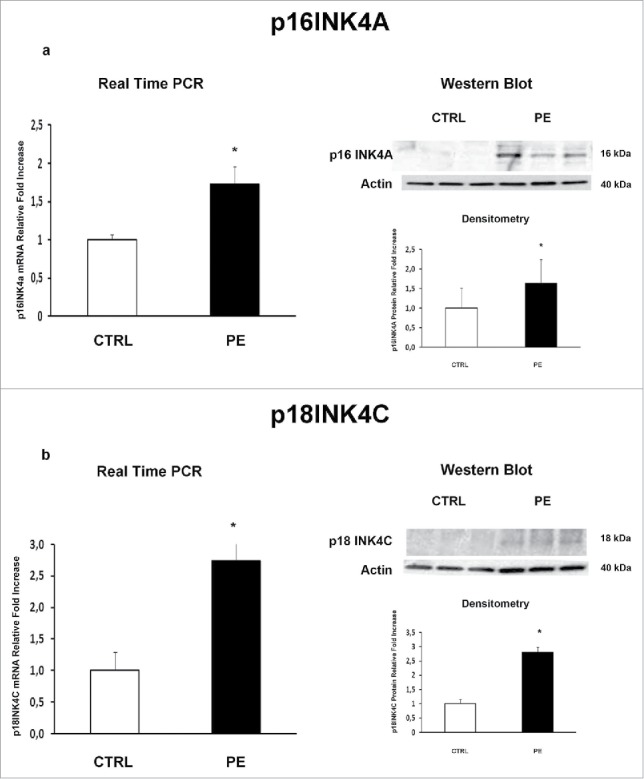
p16INK4A and p18INK4C gene and protein expression in normal vs preeclamptic PDMSCs. (a) mRNA (left panel) and protein (right panel) expression of p16INK4A in normal (N, n = 20) and PE-PDMSCs (PE, n = 24) (b) mRNA (left panel) and protein (right panel) expression of p18INK4C in normal (N, n = 20) and PE-PDMSCs (PE, n = 24). Statistical significance (*) has been considered as p < 0.05.
Figure 2.
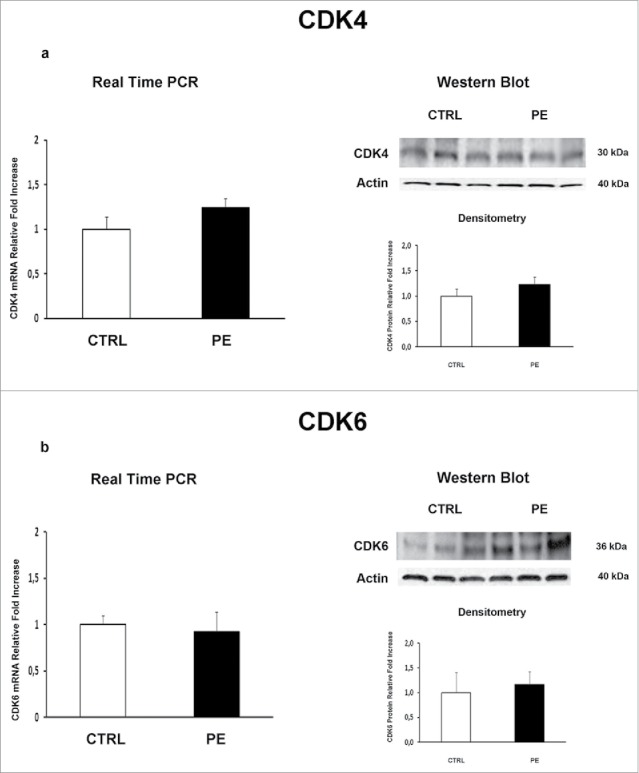
CDK4 and CDK6 gene and protein expression levels in normal vs preeclamptic PDMSCs. (A) mRNA (left panel) and protein (right panel) expression of CDK4 in normal (N, n = 20) and PE-PDMSCs (PE, n = 24) (B) (b) mRNA (left panel) and protein (right panel) expression of CDK6 in normal (N, n = 20) and PE-PDMSCs (PE, n = 24). Statistical significance (*) has been considered as p < 0.05.
PDMSCs conditioned media did not alter physiological term placental villous explants viability
In order to evaluate whether PDMSCs CM affect cell viability of treated physiological term villous explants, the Lactate dehydrogenase (LDH) assay, which assessed the cell membrane integrity by measuring the LDH leakage from cells, was performed. After 72h of treatment, we observed that the relative LDH amount released into the media was comparable among unconditioned media treated explants and those treated by normal or PE-PDMSCs CM (p > 0.05, Fig. 3). Importantly, all CM LDH levels were significantly lower compare with the considered cytotoxicity cut-off of 54% (p < 0.05, Fig. 3).
Figure 3.
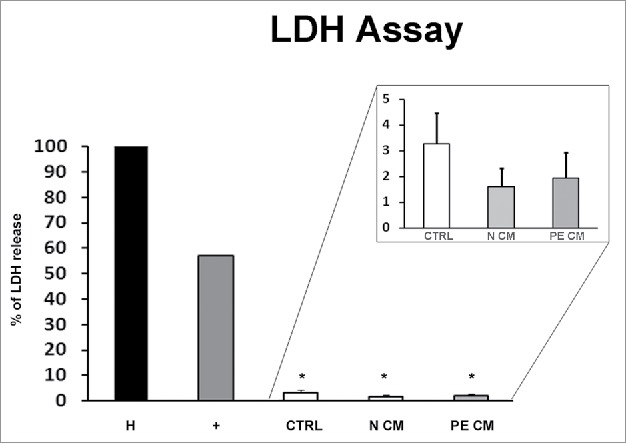
PDMSCs conditioned media effect on physiological term placental villous explants viability. Positive control (+) was provided by the kit. Explants treated by Triton X-100 and by unconditioned culture media for 8 h were used as high (H) and low controls respectively. Cytotoxicity in 72h supernatant of physiological villous explants treated with unconditioned media (CTRL) or media conditioned by normal (N CM) and preeclamptic (PE CM) PDMSCs was assessed by LDH assay. Statistical significance (*) has been considered as p < 0.05.
PDMSCs conditioned media affects JunB, Cyclin D1, p16INK4A, p18INK4C, CDK4, CDK6 and PARP1 expressions in physiological term placental villous explants
PDMSCs exert most of their functions by secreting soluble mediators. Therefore, we cultured physiological term villous explants with CM derived from normal or PE-PDMSCs and we investigated the expression of JunB, Cyclin D1, p16INK4A, p18INK4C, CDK4 and CDK6. Moreover, we evaluated pro-apoptotic PARP1 expression as marker of programmed cell death in treated explants. Normal PDMSCs CM induced overexpression of JunB mRNA (p = 0.01, 1.72 Fold Increase) and protein (p = 0.04, 1.38 Fold Increase) levels (Fig. 4a) and a significant decrease in Cyclin D1 mRNA (p = 0.03, 0.62 Fold Decrease) and protein (p = 0.05, 0.49 Fold Decrease) levels relative to unconditioned media (Fig. 4b). In stark contrast, PE-PDMSCs CM promoted a significant downregulation in JunB expression (p = 0.03, 2.12 Fold Decrease) followed by Cyclin D1 upregulation (mRNA: p = 0.03, 1.52 Fold Increase; protein: p = 0.05, 1.56 Fold Increase) (Fig. 4a and b). Moreover, normal PDMSCs CM promoted p16INK4A and p18INK4C mRNA (p = 0.05, 1.66 Fold Increase; p = 0.04, 1.91 Fold Increase respectively) and protein (p = 0.02, 1.78 Fold Increase; p = 0.04, 1.60 Fold Increase respectively) expression relative to control explants (Fig. 5a and b). There were no significant effects on p16INK4A and p18INK4C expression after PE-PDMSCs CM treatment (Fig. 5a and b). Despite a trend of increase in CDK4 levels in normal PDMSCs CM explants, we showed that neither normal nor PE conditioned medium had significant effects on CDK4 and CDK6 expression (Fig. 6a and b). Finally, we found no significant differences in PARP1 expression levels between untreated controls and normal PDMSCs CM explants while PE-PDMSCs CM induced PARP-1 mRNA (p = 0.024, 2.3 Fold Increase) and protein (2.5 Fold Increase) overexpression relative to unconditioned media (Supplementary Figure S1a and b).
Figure 4.
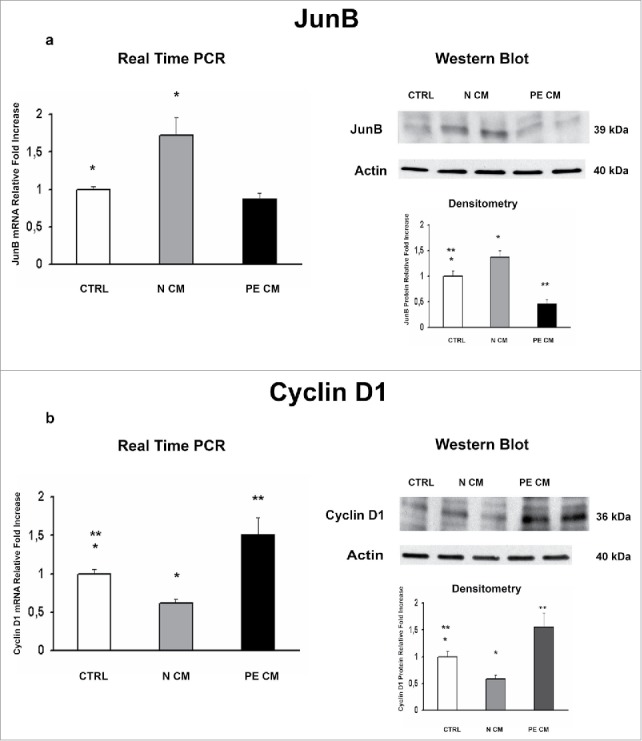
JunB and Cyclin D1 gene and protein expression levels in physiological placental villous explants treated with culture media conditioned by normal or PE-PDMSC. (A)JunB mRNA (left panels) and protein (right panels) expression levels in physiological villous explants treated with unconditioned media (CTRL, n = 16 explants) or media conditioned by normal (N CM, n = 16 explants) and preeclamptic (PE CM, n = 16 explants) PDMSCs as assessed by Real Time PCR and Western Blot analysis. B) Cyclin D1 mRNA (left panels) and protein (right panels) expression levels in physiological villous explants treated with unconditioned media (CTRL, n = 16 explants) or media conditioned by normal (N CM, n = 16 explants) and preeclamptic (PE CM, n = 16 explants) PDMSCs as assessed by Real Time PCR and Western Blot analysis. Statistical significance (*) has been considered as p < 0.05.
Figure 5.
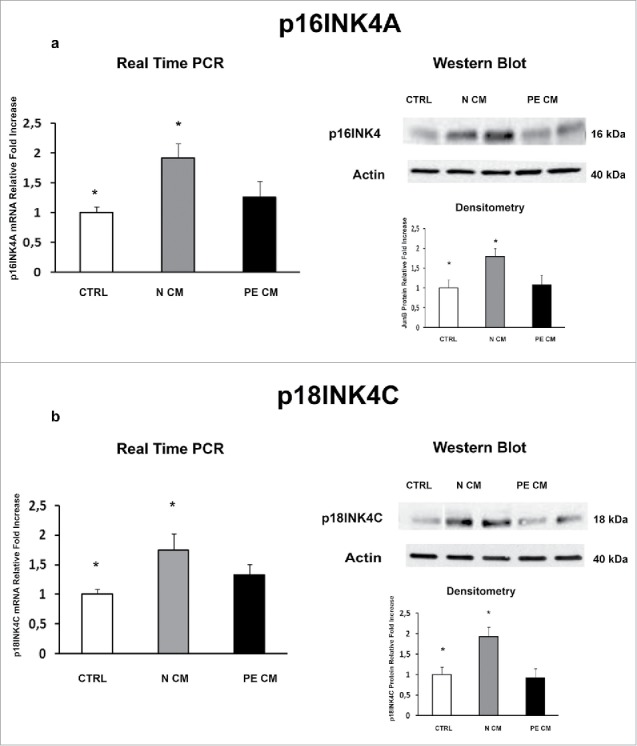
P16INK4A and p18INK4C gene and protein expression levels in physiological placental villous explants treated with culture media conditioned by normal or PE-PDMSC. (A) P16INK4A mRNA (left panels) and protein (right panels) expression levels in physiological villous explants treated with unconditioned media (CTRL, n = 16 explants) or media conditioned by normal (N CM, n = 16 explants) and preeclamptic (PE CM, n = 16 explants) PDMSCs as assessed by Real Time PCR and Western Blot analysis. B) and p18INK4C mRNA (left panels) and protein (right panels) expression levels in physiological villous explants treated with unconditioned media (CTRL, n = 16 explants) or media conditioned by normal (N CM, n = 16 explants) and preeclamptic (PE CM, n = 16 explants) PDMSCs as assessed by Real Time PCR and Western Blot analysis. Statistical significance (*) has been considered as p < 0.05.
Figure 6.
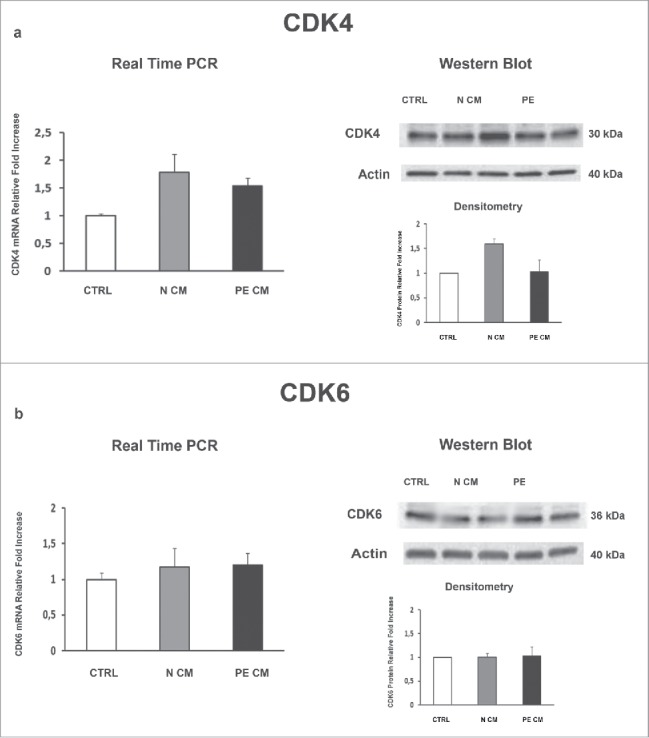
CDK4and CDK6 gene and protein expression levels in physiological placental villous explants treated with culture media conditioned by normal or PE-PDMSC. (A) CDK4 mRNA (left panels) and protein (right panels) expression levels in physiological villous explants treated with unconditioned media (CTRL, n = 16 explants) or media conditioned by normal (N CM, n = 16 explants) and preeclamptic (PE CM, n = 16 explants) PDMSCs as assessed by Real Time PCR and Western Blot analysis. B) CDK6 mRNA (left panels) and protein (right panels) expression levels in physiological villous explants treated with unconditioned media (CTRL, n = 16 explants) or media conditioned by normal (N CM, n = 16 explants) and preeclamptic (PE CM, n = 16 explants) PDMSCs as assessed by Real Time PCR and Western Blot analysis. Statistical significance (*) has been considered as p < 0.05.
In order to clarify which placental cell population was targeted by PDMSC CM, we performed Cyclin D1 immufluorescence (IF) staining on the villous explants treated as explained above. Cyclin D1 staining was mostly localized in the syncytiotrophoblast (TR) layer (Fig. 7), thus identifying TR cells as the main PDMSCs CM target. A weak positivity for Cyclin D1 was found in mesenchymal (M) and perivascular cells (Fig. 7). Importantly, IF confirmed RNA and protein data. Immunoreactivity for Cyclin D1 was markedly decreased in the TR cytoplasm and peri-nuclear areas in normal PDMSCs CM villous explants (Fig. 7c). In stark contrast, Cyclin D1 signal was increased in PE-PDMSCs CM villous explants (Fig. 7d).
Figure 7.
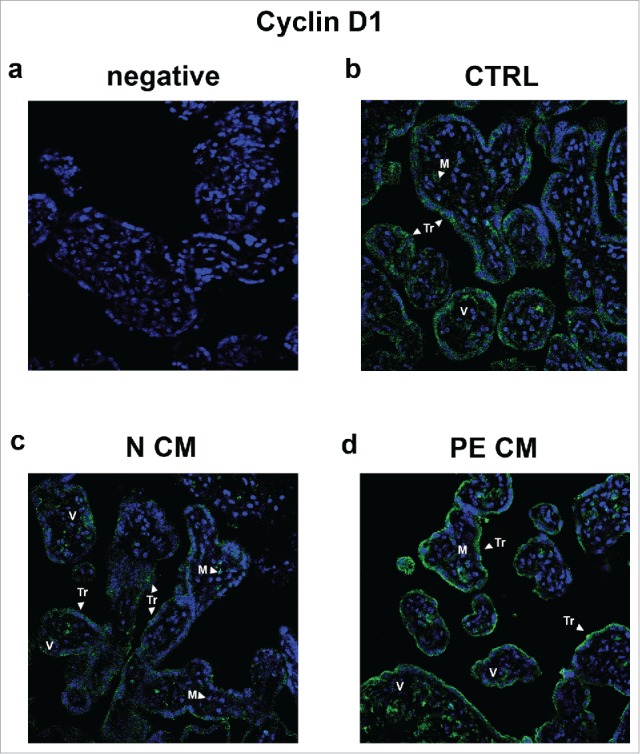
Cyclin D1 spatial localization in physiological placental villous explants treated with culture media conditioned by normal or PE-PDMSC. (A) Absence of positive immunoreactivity for Cyclin D1 in section stained with control IgG. B) Cyclin D1 spatial localization in physiological villous explants treated with unconditioned media (CTRL, n = 4 explants) assessed by immunofluorescent staining. C) Cyclin D1 spatial localization in physiological villous explants treated with media conditioned by normal (N CM, n = 4 explants) assessed by immunofluorescent staining. C) Cyclin D1 spatial localization in physiological villous explants treated media conditioned by with preeclamptic (PE CM, n = 4 explants) assessed by immunofluorescent staining. Cell nuclei are showed in blue by DAPI signal. TR, trophoblast cells; M, mesenchyme; V, vessel. Original magnifications, x40.
Discussion
In the present study, we described, for the first time to our knowledge, the aberrant expressions of G1/S cell cycle transition regulators in preeclamptic chorionic mesenchymal stromal cells. We reported a significant overexpression of Cyclin D1 inhibitors p16INK4A and p18INK4C in PE-PDMSCs, thus emphasizing the presence of major alterations in Cyclin D1 regulatory pathway in this specific placental cell population. Using physiological term villous explants and PDMSCs conditioned media, we demonstrated that normal and preeclamptic mesenchymal cells differentially modulate the expression of G1/S regulators in the placental tissue via paracrine interactions. Normal PDMSCs promoted JunB, p16INK4A and p18INK4C expression and Cyclin D1 downregulation, while preeclamptic PDMSCs induced JunB downregulation accompanied by Cyclin D1 and PARP1 overexpression in physiological placental tissue. Our data strengthen the concept that PDMSCs, along with the well-established trophoblast anomalies, could cause or contribute to the pathological placental alterations typical of preeclampsia.
Cyclin D1 is a cell cycle regulator specific for the G1/S phase that, by binding and activating CDK4 and CDK6, promotes DNA synthesis and cell proliferation. During physiological placentation, cytotrophoblast and extravillous trophoblast cells express Cyclin D1 while it is downregulated in placentae from preeclamptic pregnancies.20,21 Besides the trophoblast, mesenchymal stromal cells resident in both chorionic villi and decidua are actively involved in the impaired placenta development typical of PE.9,10,22-24 In line with PE trophoblast, we previously reported that chorionic PE-PDMSCs are characterized by JunB-mediated Cyclin D1 downregulation,10 stressing the importance of preeclampsia-related G1/S transition anomalies. In the present study, we added another important piece to the intricate puzzle of PE pathogenesis, describing a significant increase of p16INK4α and p18INK4C in preeclamptic PDMSCs. These molecules are G1-specific inhibitors of Cyclin D1-CDK4/6 complex formation that restrain CDKs mitogenic activity.25-27 In accordance with our model, JunB expression induced p16INK4α and p18INK4C accumulation and consequent inhibition of Cyclin D1-associated activities in mouse embryonic fibroblasts, resulting in G1-phase extension and reduced cell proliferation.15, 28
Cell cycle arrest and altered expression of growth regulatory proteins p16INK4α and p18INK4C are key features of senescence, critical cellular response to environmental insults typical of the preeclamptic placental setting as oxidative stress and cytokines stimulation.29,30 We previously reported an increased number of slow proliferating senescent PDMSCs in PE placentae, characterized by enhanced senescence-associated β-galactosidase activity.9 Increased mesenchymal cells senescence was described in the bone marrow of patients affected by systemic lupus erythematosus,31 clinical condition often associated to preeclampsia and characterized as well by hypertension, immunological impairment and exacerbated systemic inflammation. Thus, the dramatic increase of p16INK4α and p18INK4C described in the present study could be interpreted as a cause or consequence of the pathological preeclamptic milieu in which chorionic mesenchymal cells developed.
Importantly, cell cycle is related to placental age and it was described that placental cell proliferation declines throughout the 3 trimesters of pregnancy.32 Therefore, it could be expected a lower expression of cell cycle inhibitors in younger pre-term PE-PDMSCs relative to term cells. Indeed, the over-accumulation of p16INK4A and p18INK4C in pre-term PE-PDMSCs described herein appears an even more significant anomaly of these pathological cells.
As mentioned above, p16INK4α and p18INK4C act by inhibiting Cyclin D1-CDK4/6 complex formation. Herein, we didn't find differences in CDK4 and CDK6 expression between normal and PE-PDMSCs. These data are consistent with previous work by Hara and colleagues that, upon senescence, showed increased p16INK4A and p18INK4C but no effects in CDK4 and CDK6 expression on human cultured fibroblasts.33
In contrast to the early paradigm of cell replacement and differentiation as a therapeutic mechanism of action, increasing evidence suggests that MSCs trophic mediators are responsible for their positive impact on tissue damage.34 They include bioactive soluble factors known to inhibit apoptosis, fibrosis and inflammation, to enhance angiogenesis, to stimulate mitosis and/or differentiation of tissue-intrinsic progenitor cells.35 Moreover, PDMSCs are able to modulate the immune response in an autocrine and/or paracrine manner.36-38 It was previously shown that MSC-mediated immunosuppression is produced via an anti-proliferative action whereby cells of lymphoid and myeloid lineage are arrested at early stages of cell cycle.39 The MSC-mediated cell cycle regulation is also supported by co-culture experiments where mesenchymal cells are able to arrest cancer cell cycle in the G0/G1 phase.19,40 Furthermore, in vitro and in vivo studies on human BV173 acute lymphoblastic leukemia cells and on rat hepatic stellate cells demonstrated that the MSCs-induced cell cycle arrest is mediated by downregulation of Cyclin D1, Cyclin D2, Cyclin H accompanied by expression of negative regulators as p15INK4B, p16INK4A and p18INK4C and p21WAF1/Cip1.19,41 Indeed, PDMSCs could as well modulate the expression of trophoblast cell cycle regulators. We reported a significant Cyclin D1 downregulation and p16INK4A/p18INK4C upregulation in physiological villous explants treated by normal PDMSCs-CM. Our data suggest that normal PDMSCs modulate cell cycle regulators as MSC from other sources. In contrast to the generally accepted idea that the increased senescence is a key contributor in altered placental development, it was described that senescent cells may be important for placental physiological functions during pregnancy. The induction of trophoblast cells senescence contributes to cytokines production that is mandatory for normal placental function. Moreover, trophoblast senescence attracts NK cells pivotal for functional maternal/fetal interface and it maintain cell cycle arrest supporting cell viability.42 The absence of cellular senescence triggers apoptosis and macrophage infiltration to correct the imbalance in cell population.42 Thus, PDMSCs, through the modulation of senescence inducers p16INK4A and p18INK4C, might play a crucial role in physiological placental development maintaining and preserving the normal placental function and homeostasis. Recent preclinical studies on melanoma showed that loss-of-function of proteins as p16INK4A promote the expression and activation of CDK4 and CDK6.43 Since normal PDMSCs-CM induced a significant p16INK4A upregulation, we then examined CDKs expression levels. In contrast to previous data, in our model there was not a p16INK4A-CDK4/6 correlation. In fact, p16INK4A upregulation induced by normal PDMSCs-CM did not affect CDK4 and CDK6 expression suggesting a different control mechanisms mediated by chorionic MSCs. Our results are consistent with reports that demonstrated no detectable changes in the expression of CKDs in B16 melanoma cells treated with media conditioned by adipose mesenchymal stem cells.44 Recently it has been shown that bone marrow MSCs conditioned medium modulates the Activating Protein 1 (AP-1) signaling pathway.45 Even Wharton's jelly-derived MSC conditioned medium regulates cell cycle by triggering the AP-1 pathway in human airway epithelial cells.46 Herein we found that normal PDMSCs-CM induced JunB upregulation and consequent Cyclin D1 downregulation in physiological term villous explants. We recently reported that the AP-1 family member JunB specifically controls Cyclin D1 transcriptional regulation in PDMSCs.10 The modulation of Cyclin D1 as well as Cyclin D1 inhibitors is one of the most common strategies used by MSC to face adverse conditions.41 In the placental context, it could be used by normal PDMSCs to counteract spontaneous apoptosis associated with trophoblast proliferation, thus maintaining tissue homeostasis.47 In line with this hypothesis, we reported that after treatment with normal PDMSCs CM, apoptotic marker PARP-1 expression levels were comparable with those of physiological explants treated with unconditioned media.
MSC's homeostatic functions, exerted through both paracrine and contact-dependent mechanisms, were widely described in other tissues.48,49 In particular, bone marrow MSCs prevented oxidative metabolism thus protecting the neighboring neutrophils from apoptosis.50 Altered expression of G1/S cell-cycle modulators accounts for the aberrant trophoblast proliferation/apoptosis rheostat typical of preeclampsia, leading to trophoblast debries release into the maternal circulation. This disturbance of trophoblast homeostasis produces deleterious effects on placental functionality.51 We next tested the hypothesis that medium conditioned by preeclamptic PDMSCs could disrupt placental homeostasis by altering the expression of trophoblast cell cycle regulators in physiological tissues.
As mentioned above, senescence could function as a protective mechanism that limits trophoblasts proliferation, thus maintaining cell viability and giving cells resistance to apoptosis.52 If this mechanism fails, persisting damaged cells could induce tissue damage.52 Importantly, senescence markers were found in term placentae and they were interpreted as a sign of physiological aging.53 Herein, we reported that term physiological placental explants treated by preeclamptic PDMSCs conditioned medium were characterized by increased proliferation and apoptosis, as indicated by cyclin D1 increase accompanied by JunB downregulation and PARP1 increase. Our data emphasize the ability of PE-PDMSCs to significantly perturb the cell cycle machinery in term explants, thought to be physiologically senescent.53 Moreover, the PE-PDMSCs induced phenotype observed in our model strongly resembles that of severe early onset preeclamptic placentae, characterized by hyperproliferative immature trophoblast cells associated to increased trophoblast cell death.7 In contrast, increased senescence and reduced proliferation levels were described in late onset PE and FGR placentae, suggesting an accelerated aging process in these pathological tissues.30,53 Future investigations should be directed to clarify these key cell cycle regulation differences between early and late onset PE-FGR placentae.
Our data are in line with previous studies that demonstrated the direct involvement of mesenchymal stromal cells in the pathogenesis of several disorders. For example, breast cancer progression and metastasis are promoted by MSCs resident in the tumor niche and we previously described that preeclamptic PDMSCs possess a pro-inflammatory and anti-angiogenic phenotype, thus contributing to the pathogenesis of PE.9 Nevertheless, the described anomalies in PE-PDMSCs could not be a cause but a consequence of preeclampsia. In fact, the adverse stromal microenvironment could alter the properties and trophic actions of the derived MSC.9,54
Next, we focused on Cyclin D1 immunolocalization because it was reported that its subcellular localization regulated entirely p16INK4A/p18INK4C-CDK4/CDK6 pathway.55 Previous data from De Loia and collegues showed that Cyclin D1 protein was present in endothelial cells lining the villous blood vessels.20 In contrast, we reported that Cyclin D1 was mainly expressed in the syncytiotrophoblast layer and less in the perivascular area and mesenchyme. In line with our data, De Falco and colleagues described a strong trophoblast Cyclin D1 positivity.55 In particular, we found a stronger cytoplasmic Cyclin D1 signal. It was shown that Cyclin D1 accumulates in the nucleus throughout G1 phase and relocate in the cytoplasm during the interphase and cell cycle progression.56,57 Cyclin D1 subcellular distribution is a finely tuned equilibrium between nuclear importation and exportation and its overexpression always indicates a cell proliferation advantage.55,58 Thus, PE PDMSCs-CM induced syncytiotrophoblast Cyclin D1 accumulation highlights the ability of preeclamptic mesenchymal cells to induce a PE-like hyper-proliferative phenotype in physiological placentae by the secretion of trophic mediators.
In conclusion, altered Cyclin D1-p16INK4A/p18INK4C pathway in PE-PDMSCs could cause and/or be the consequence of the adverse environment in which mesenchymal cells develop during the early stages of placenta development, thus contributing to the aberrant villous architecture typical of preeclampsia. Due to their ability to interfere cell cycle modulators in neighboring cells, PDMSCs could represent a future therapeutic tool or target for human placenta-related disorders as preeclampsia.
Patients and methods
Ethics statement
The study was approved by the Institutional Ethical Committee of O.I.R.M. S.Anna Hospital and “Ordine Mauriziano di Torino” (n.209; protocol 39226/C.27.1 04/08/09) (Turin, Italy). All patients provided written informed consent.
Study population
The study population included 20 physiological term pregnancies and 24 singleton pregnancies complicated by severe preeclampsia with fetal-placental compromise. We deliberately selected only those PE cases characterized by anomalies at the fetal-placental district since we were interested in specifically investigating the features of those mesenchymal cells resident in the chorionic villi of pathological placentae. The diagnosis of PE was made according to the following criteria: presence of pregnancy-induced hypertension (systolic blood pressure ≥ 140 mmHg or diastolic blood pressure ≥ 90 mmHg) and proteinuria (> 300 mg/24 h) after 20 week of gestational age in previously normotensive women. Fetal-placental compromise was defined as the presence of one or more of the following features: a) Fetal Growth Restriction (FGR), defined as birth weight below the fifth centile according to the Italian growth curves normalized for gestational age and sex59,60; b) pathological umbilical artery Doppler waveforms (absent or reverse end diastolic flow - A/REDF); c) increased resistance to flow in maternal uterine arteries (early diastolic notch or pulsatility index - PtdIns - higher than 0.58).This rigorous classification allowed us to distinguish between maternal and placental PE since placentae are significantly different in these 2 groups When one or more of the above variables are abnormal, preeclampsia is defined of placental origin. If we used the common “early and late PE” classification there would have been a mixed population of maternal and placental preeclampsia in each group.61
Control patients were singleton term physiological normotensive pregnancies with no signs of preeclampsia or FGR. We did not use “age-matched” control pregnancies since no pre-term delivery is physiological. Exclusion criteria were: congenital malformation, chromosomal abnormalities (in number and/or structure), maternal and/or intrauterine infections, cardiovascular diseases and metabolic syndrome.
Placental-derived mesenchymal stromal cells (PDMSCs)
PDMSCs were isolated from normal and PE placental basal plate by enzymatic digestion and gradient as previously described.9 Briefly, after decidua removal, placenta basal plate specimens were cut into pieces of approximately 30 g and washed several times with Hank's Buffered Salt Solution (HBSS, Life Technologies, Cat. No. 14110046). Next, the placental tissues were mechanically minced and processed by enzymatic digestion using 100U/ml collagenase type I (Life Technologies, Cat. No.17100017) plus 5 µg/ml DNAse I (Life Technologies, Cat. No. 18047019) for 3 hours at 37°C, filtered and then processed by density gradient using 1.073 Ficoll Paque Premium (GE Healthcare Europe, Cat. No. geh17544652). PDMSCs were collected from the interphase and washed. Finally, cell counts was performed using an automated cell analyzer (Scepter 2.0 Cell Couter, Merk-Millipore) and PDMSCs were resuspended in Dulbecco's modified Minimum Essential Medium (DMEM, Life Technologies, Cat. No. 11880028) supplemented with 10% Fetal Bovine Serum (FBS Australian origin, Life Technologies, Cat. No. 10099141) in T150 flask maintained at 37 C and 5% CO2. Normal and PE-PDMSCs were characterized at passage 3 by flow cytometry using the following markers panel: HLA-I, HLA-DR, CD34, CD133, CD20, CD326, CD31, CD45 and CD14 (Miltenyi Biotech, Cat. No. 130095198). Moreover, Oct-4 and Nanog expression was evaluated in normal and PE-PDMSCs by semi-quantitative RT-PCR as markers of stemness. Primers were designed as previously described.9
Preparation of PDMSCs conditioned media (CM)
At passage 3, normal and PE-PDMSCs were plated in 6 well plates at a density of 1 × 105 cells/ml in DMEM LG without FBS. PDMSCs conditioned media were then collected at 48 hours of culture and filtered by a 0.22 µm filter (Merck-Millipore, Italy) to remove cellular debris. Cells were harvested and processed for mRNA and protein isolation.
Treatment of physiological villous explants with PDMSCs CM
We next evaluated the paracrine effects of normal and PE PDMSCs-CM on physiological placental villous explants. This model allowed us to determine the sequence of molecular events in tissues characterized by conserved physiological pathways, thus avoiding biases due to previous existing pathological anomalies. Biopsies from 2 different physiological term placentae (n = 30 explants for each placenta) were separated from the placental basal plate and small portions of chorionic villi were excised and cultured in 24-well plates in HAM F-12 Nutrient Medium (Gibco, Invitrogen Life Techonologies, Cat. No. 21765029) at 37°C - 5% CO2 - 20% O2 overnight. Explants, composed by trophoblast, mesenchymal and endothelial cells, were used as model of organized placental tissue. Next, the medium was removed and explants were treated for 72 h with 500 µl of CM by 4 different normal PDMSCs lines (n = 20 explants) and by 4 different PE-PDMSCs lines (n = 20 explants). Explants treated by unconditioned culture media were used as controls (n = 20 explants). Explants were finally immediately fixed for histological analyses (4 for each experimental condition) or frozen for RNA and protein isolation (n = 48).
Lactate dehydrogenase (LDH) cytotoxicity assay
Cytotoxicity of CMs treatment on villous explants was determined by LDH-Cytotoxicity colorimetric assay (Biovision Inc., Cat. No. K726–500) performed using 72h explants supernatant according to the manufacturer's instructions. Explants treated by Triton X-100 and by unconditioned culture media for 8 h were used as high (H, 100%) and low controls (0%) respectively. The positive control (+) was provided by the manufacturer to test whether all reagents were working properly responding to active LDH enzyme. The absorbance at 450 nm was measured using a plate reader spectrophotomether. Percent cytotoxicity values were determined based on the amount of LDH released from 30-min readings, as follows: (test sample – low control) / (high control-low control) × 100. The cut-off values for the LDH assay was determined by the 2 × SD rule where the threshold is defined as 2 × SD beyond the mean of the screened samples. Values equal or exceeding the threshold were considered as sign of cytotoxicity.
RNA isolation and real time PCR
Total RNA was isolated from normal and PE-PDMSCs and villous explants using TRIzol reagent (Life Technologies, Invitrogen, Cat. No. t9424) according to manufacturer instructions. Genomic DNA contamination was removed by DNase I digestion before RT-PCR. cDNA was generated from 5 μg of total RNA using a random hexamers approach and RevertAid H Minus First Strand cDNA Synthesis kit (Fermentas, Cat.No k1632).
Gene expressions levels of JunB, CyclinD1, p16INK4A, p18INK4C, CDK4, CDK6 and PARP1 were determined by Real Time PCR using specific TaqMan primers and probes (Life Technologies, Cat. No 4331182). mRNA levels were normalized using endogenous 18s as internal reference (Life Technologies, Cat. No 4333760F). Relative expression and fold change were calculated according to Livak and Schmittgen.62
Western blot analyses
Total proteins were isolated from PDMSCs and chorionic villous explants using 1X Radio Immuno-precipitation Assay (RIPA) buffer. Thirty μg of total protein from PDMSCs and villous explants were processed by SDS-page on 4–20% and 7.5% polyacrylamide pre-cast gradient gels (Bio-Rad Laboratories S.r.l., Cat. No. 456–1099 and 4561034). Next, proteins were transferred on PVDF membranes and probed at room temperature with primary antibodies using the SnapID system (Merk-Millipore, Italy) following manufacturer instructions. Primary antibodies were: rabbit polyclonal anti-human JunB (1:2000 dilution, Merk-Millipore, Cat. No. 07–1333), mouse monoclonal anti-human Cyclin D1 (1:1500 dilution, Cell Signaling, Cat. No. 2978), rabbit monoclonal anti-human p16INK4A (1:250 dilution, Cell Signaling, Cat.No 4824), mouse monoclonal anti-human p18INK4C (1:250 dilution, Cell Signaling, Cat.No 2896), mouse monoclonal anti-human CDK4 (1:500 dilution, Cell Signaling, Cat.No 12790), mouse monoclonal anti-human CDK6 (1:500 dilution, Cell Signaling, Cat. No. 3136), mouse monoclonal anti-human PARP1 (1:500 diluition, Abcam, Cat. No. ab110915) and mouse monoclonal anti-human β-actin (1:1000, Sigma-Aldrich, Cat.No. A5316). Biotinylated secondary antibodies were goat anti-mouse for Cyclin D1, p18INK4C, CDK4, CDK6, PARP1 and β-actin (1:1000 dilution, Vector Laboratories, Cat.No. sc-2005), donkey anti-rabbit for JunB and p16INK4A (1:1000 dilution, Vector Laboratories, Cat.No. sc-2004). Protein signals were detected by chemoluminescence using LuminataTM Crescendo Western HRP reagent (Millipore, Cat.No wbluc0100).
Cyclin D1 immunofluorescence staining
Villous explants treated by unconditioned medium (n = 4 explants) and with normal (n = 4 explants) or PE PDMSCs CM (n = 4 explants) were fixed in 4% paraphormaldehyde and embedded in paraffin. Sodium citrate antigen retrieval was performed, followed by Sudan Black treatment (0.1% Sudan Black in 70% EtOH) to quench endogenous fluorescence. Sections were pre-incubated in 5% horse serum in PBS (contained 0.04% sodium azide and 0.008% gelatin) to block nonspecific binding and incubated with primary antibodies overnight at 4 °C. Mouse polyclonal antibodies anti-human CyclinD1 (1:100 dilution; Cell Signaling, Cat. No. 2978) was used. Control IgG were used as negative controls. Slides were treated with 0.4% DAPI (4′,6-diamidino-2-phenylindole) for nuclear detection. Fluorescence images were viewed and captured using a confocal microscope (Nikon D-Eclipse C1, Nikon, Chiyoda, Tokyo, Japan).
Statistical analysis
All data are represented as mean ± standard error (SE). For comparison of data between multiple groups, used one-way analysis of variance (ANOVA) with posthoc Dunnett's test was used. For comparison between 2 groups paired and unpaired Student's t-test were used as appropriate. Categorical variables are presented as frequencies (percentages) and the comparison between different groups was done with χ2 by means of a 2 × 2 or 2 × 3 contingency table; Fisher's exact test was used for small sample sizes. Statistical test were performed using SPSS Version 18 statistical software and significance was accepted at P < 0.05.
Supplementary Material
Abbreviations
- A/REDF
absent or reverse end diastolic flow
- AP-1
Activating Protein 1
- CDK
Cyclin-Dependent Kinases
- CKI
CDK-Inhibitory
- CM
Conditioned Media
- FBS
Fetal Bovine Serum
- FGR
Fetal Growth Restriction
- HBSS
Hank's Buffered Salt Solution
- IF
Immuofluorescence
- LDH
Lactate dehydrogenase
- PDMSCs
Placental derived Mesenchymal Stromal Cells
- PE
Preeclamptic
- PI
Pulsatility Index
- PARP1
Poly (ADP-ribose) polymerase 1
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Dr. Elena Olearo, Dr. Annalisa Piazzese and Dr Silvia Renaudi (O.I.R.M. Sant'Anna Hospital, Turin – Italy) for their support in collecting placental tissues.
Funding
This study was supported by Cariplo Foundation, Grant ID: 2011-0495. Alessandro Rolfo is supported by Carlo Denegri Foundation.
References
- [1].Clarke B, Chetty R. Cell cycle aberrations in the pathogenesis of squamous cell carcinoma of the uterine cervix. Gynecologic Oncology 2001; 82:238-46; PMID:11531273; http://dx.doi.org/ 10.1006/gyno.2001.6306 [DOI] [PubMed] [Google Scholar]
- [2].Milde-Langosch K, Riethdorf S. Role of cell-cycle regulatory proteins in gynecological cancer. Journal of Cellular Physiology 2003; 196:224-44; PMID:12811815; http://dx.doi.org/ 10.1002/jcp.10286 [DOI] [PubMed] [Google Scholar]
- [3].Funk JO. Cancer cell cycle control. Anticancer Research 1999; 19:4772-80; PMID:10697591 [PubMed] [Google Scholar]
- [4].Rahat B, Hamid A, Ahmad Najar R, Bagga R, Kaur J. Epigenetic mechanisms regulate placental c-myc and hTERT in normal and pathological pregnancies; c-myc as a novel fetal DNA epigenetic marker for pre-eclampsia. Molecular human reproduction 2014; 20:1026-40; PMID:25024139; http://dx.doi.org/ 10.1093/molehr/gau053 [DOI] [PubMed] [Google Scholar]
- [5].Strickland S, Richards WG. INVASION OF THE TROPHOBLASTS. Cell 1992; 71:355-7; PMID:1423599; http://dx.doi.org/ 10.1016/0092-8674(92)90503-5 [DOI] [PubMed] [Google Scholar]
- [6].Ferretti C, Bruni L, Dangles-Marie V, Pecking AP, Bellet D. Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts. Human Reproduction Update 2007; 13:121-41; PMID:17068222; http://dx.doi.org/ 10.1093/humupd/dml048 [DOI] [PubMed] [Google Scholar]
- [7].Ray JE, Garcia J, Jurisicova A, Caniggia I. Mtd/Bok takes a swing: proapoptotic Mtd/Bok regulates trophoblast cell proliferation during human placental development and in preeclampsia. Cell Death Differ 2010; 17:846-59; PMID:19942931; http://dx.doi.org/ 10.1038/cdd.2009.167 [DOI] [PubMed] [Google Scholar]
- [8].Pijnenborg R, Vercruysse L, Verbist L, Van Assche FA. Interaction of interstitial trophoblast with placental bed capillaries and venules of normotensive and pre-eclamptic pregnancies. Placenta 1998; 19:569-75; PMID:9859859; http://dx.doi.org/ 10.1016/S0143-4004(98)90016-9 [DOI] [PubMed] [Google Scholar]
- [9].Rolfo A, Giuffrida D, Nuzzo AM, Pierobon D, Cardaropoli S, Piccoli E, Piccoli E, Giovarelli M, Todros T. Pro-inflammatory profile of preeclamptic placental mesenchymal stromal cells: new insights into the etiopathogenesis of preeclampsia. PLoS One 2013; 8:e59403; PMID:23527185; http://dx.doi.org/ 10.1371/journal.pone.0059403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nuzzo AM, Giuffrida D, Zenerino C, Piazzese A, Olearo E, Todros T, et al.. JunB/Cyclin-D1 imbalance in placental mesenchymal stromal cells derived from preeclamptic pregnancies with fetal-placental compromise. Placenta 2014; 35:483-90; PMID:24780198; http://dx.doi.org/ 10.1016/j.placenta.2014.04.001 [DOI] [PubMed] [Google Scholar]
- [11].Fukuchi Y, Nakajima H, Sugiyama D, Hirose I, Kitamura T, Tsuji K. Human placenta-derived cells have mesenchymal stem/progenitor cell potential. Stem Cells 2004; 22:649-58; PMID:15342929; http://dx.doi.org/ 10.1634/stemcells.22-5-649 [DOI] [PubMed] [Google Scholar]
- [12].Li C, Zhang W, Jiang X, Mao N. Human-placenta-derived mesenchymal stem cells inhibit proliferation and function of allogeneic immune cells. Cell and Tissue Research 2007; 330:437-46; PMID:17899199; http://dx.doi.org/ 10.1007/s00441-007-0504-5 [DOI] [PubMed] [Google Scholar]
- [13].Demir R, Kaufmann P, Castellucci M, Erbengi T, Kotowski A. FETAL VASCULOGENESIS AND ANGIOGENESIS IN HUMAN PLACENTAL VILLI. Acta Anatomica 1989; 136:190-203; PMID:2481376; http://dx.doi.org/ 10.1159/000146886 [DOI] [PubMed] [Google Scholar]
- [14].Bakiri L, Lallemand D, Bossy-Wetzel E, Yaniv M. Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. EMBO J 2000; 19:2056-68; PMID:10790372; http://dx.doi.org/ 10.1093/emboj/19.9.2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Passegue E, Wagner EF. JunB suppresses cell proliferation by transcriptional activation of p16(INK4a) expression. EMBO J 2000; 19:2969-79; PMID:10856241; http://dx.doi.org/ 10.1093/emboj/19.12.2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Andrecht S, Kolbus A, Hartenstein B, Angel P, Schorpp-Kistner M. Cell cycle promoting activity of JunB through cyclin A activation. J Biol Chem 2002; 277:35961-8; PMID:12121977; http://dx.doi.org/ 10.1074/jbc.M202847200 [DOI] [PubMed] [Google Scholar]
- [17].Berthet C, Kaldis P. Cell-specific responses to loss of cyclin-dependent kinases. Oncogene 2007; 26:4469-77; PMID:17297466; http://dx.doi.org/ 10.1038/sj.onc.1210243 [DOI] [PubMed] [Google Scholar]
- [18].Parolini O, Alviano F, Bergwerf I, Boraschi D, De Bari C, De Waele P, et al.. Toward Cell Therapy Using Placenta-Derived Cells: Disease Mechanisms, Cell Biology, Preclinical Studies, and Regulatory Aspects at the Round Table. Stem Cells and Development 2010; 19:143-54; PMID:19947828; http://dx.doi.org/ 10.1089/scd.2009.0404 [DOI] [PubMed] [Google Scholar]
- [19].Ramasamy R, Lam EW, Soeiro I, Tisato V, Bonnet D, Dazzi F. Mesenchymal stem cells inhibit proliferation and apoptosis of tumor cells: impact on in vivo tumor growth. Leukemia 2007; 21:304-10; PMID:17170725; http://dx.doi.org/ 10.1038/sj.leu.2404489 [DOI] [PubMed] [Google Scholar]
- [20].DeLoia JA, Burlingame JM, Krasnow JS. Differential expression of G1 cyclins during human placentogenesis. Placenta 1997; 18:9-16; PMID:9032805; http://dx.doi.org/ 10.1016/S0143-4004(97)90066-7 [DOI] [PubMed] [Google Scholar]
- [21].Dai Y, Qiu Z, Diao Z, Shen L, Xue P, Sun H, et al.. MicroRNA-155 inhibits proliferation and migration of human extravillous trophoblast derived HTR-8/SVneo cells via down-regulating cyclin D1. Placenta 2012; 33:824-9; PMID:22858023; http://dx.doi.org/ 10.1016/j.placenta.2012.07.012 [DOI] [PubMed] [Google Scholar]
- [22].Mando C, Razini P, Novielli C, Anelli GM, Belicchi M, Erratico S, et al.. Impaired Angiogenic Potential of Human Placental Mesenchymal Stromal Cells in Intrauterine Growth Restriction. Stem Cells Transl Med 2016; 5:451-63; PMID:26956210; http://dx.doi.org/ 10.5966/sctm.2015-0155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen CP, Huang JP, Chu TY, Aplin JD, Chen CY, Wu YH. Human placental multipotent mesenchymal stromal cells modulate trophoblast migration via Rap1 activation. Placenta 2013; 34:913-23; PMID:23896030; http://dx.doi.org/ 10.1016/j.placenta.2013.06.311 [DOI] [PubMed] [Google Scholar]
- [24].Hwang JH, Lee MJ, Seok OS, Paek YC, Cho GJ, Seol HJ, et al.. Cytokine expression in placenta-derived mesenchymal stem cells in patients with pre-eclampsia and normal pregnancies. Cytokine 2010; 49:95-101; PMID:19819721; http://dx.doi.org/ 10.1016/j.cyto.2009.08.013 [DOI] [PubMed] [Google Scholar]
- [25].Neganova I, Lako M. G1 to S phase cell cycle transition in somatic and embryonic stem cells. Journal of Anatomy 2008; 213:30-44; PMID:18638068; http://dx.doi.org/ 10.1111/j.1469-7580.2008.00931.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sherr CJ, Roberts JM. INHIBITORS OF MAMMALIAN G(1) CYCLIN-DEPENDENT KINASES. Genes & Development 1995; 9:1149-63; PMID:7758941; http://dx.doi.org/ 10.1101/gad.9.10.1149 [DOI] [PubMed] [Google Scholar]
- [27].Schwaller J, Pabst T, Koeffler HP, Niklaus G, Loetscher P, Fey MF, et al.. Expression and regulation of G(1) cell-cycle inhibitors (p16(INK4A), p15(INK4B), p18(INK4C), p19(INK4D)) in human acute myeloid leukemia and normal myeloid cells. Leukemia 1997; 11:54-63; PMID:9001419; http://dx.doi.org/ 10.1038/sj.leu.2400522 [DOI] [PubMed] [Google Scholar]
- [28].Piechaczyk M, Farras R. Regulation and function of JunB in cell proliferation. Biochemical Society Transactions 2008; 36:864-7; PMID:18793152; http://dx.doi.org/ 10.1042/BST0360864 [DOI] [PubMed] [Google Scholar]
- [29].Muller M. Cellular senescence: molecular mechanisms, in vivo significance, and redox considerations. Antioxid Redox Signal 2009; 11:59-98; PMID:18976161; http://dx.doi.org/ 10.1089/ars.2008.2104 [DOI] [PubMed] [Google Scholar]
- [30].Biron-Shental T, Sukenik-Halevy R, Sharon Y, Laish I, Fejgin MD, Amiel A. Telomere shortening in intra uterine growth restriction placentas. Early Hum Dev 2014; 90:465-9; PMID:25010904; http://dx.doi.org/ 10.1016/j.earlhumdev.2014.06.003 [DOI] [PubMed] [Google Scholar]
- [31].D'Ippolito S, Di Nicuolo F, Marana R, Castellani R, Stinson J, Tersigni C, et al.. Emerging nonanticoagulant role of low molecular weight heparins on extravillous trophoblast functions and on heparin binding-epidermal growth factor and cystein-rich angiogenic inducer 61 expression. Fertility and Sterility 2012; 98:1028-+; PMID:22818289; http://dx.doi.org/ 10.1016/j.fertnstert.2012.06.042 [DOI] [PubMed] [Google Scholar]
- [32].Smith SC, Price E, Hewitt MJ, Symonds EM, Baker PN. Cellular proliferation in the placenta in normal human pregnancy and pregnancy complicated by intrauterine growth restriction. J Soc Gynecol Investig 1998; 5:317-23; PMID:9824812; http://dx.doi.org/ 10.1016/S1071-5576(98)00035-5 [DOI] [PubMed] [Google Scholar]
- [33].Hara E, Smith R, Parry D, Tahara H, Steven S, Peters G. Regulation of p16(CDKN2) expression and its implications for cell immortalization and senescence. Molecular and Cellular Biology 1996; 16:859-67; PMID:8622687; http://dx.doi.org/ 10.1128/MCB.16.3.859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Caplan AI, Dennis JE. Mesenchymal stem cells as trophic mediators. J Cell Biochem 2006; 98:1076-84; PMID:16619257; http://dx.doi.org/ 10.1002/jcb.20886 [DOI] [PubMed] [Google Scholar]
- [35].Tetta C, Consiglio AL, Bruno S, Tetta E, Gatti E, Dobreva M, et al.. The role of microvesicles derived from mesenchymal stem cells in tissue regeneration; a dream for tendon repair? Muscles Ligaments Tendons J 2012; 2:212-21; PMID:23738299 [PMC free article] [PubMed] [Google Scholar]
- [36].Yagi H, Soto-Gutierrez A, Parekkadan B, Kitagawa Y, Tompkins RG, Kobayashi N, et al.. Mesenchymal stem cells: Mechanisms of immunomodulation and homing. Cell Transplant 2010; 19:667-79; PMID:20525442; http://dx.doi.org/ 10.3727/096368910X508762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Abumaree MH, Al Jumah MA, Kalionis B, Jawdat D, Al Khaldi A, Abomaray FM, et al.. Human placental mesenchymal stem cells (pMSCs) play a role as immune suppressive cells by shifting macrophage differentiation from inflammatory M1 to anti-inflammatory M2 macrophages. Stem Cell Rev 2013; 9:620-41; PMID:23812784; http://dx.doi.org/ 10.1007/s12015-013-9455-2 [DOI] [PubMed] [Google Scholar]
- [38].Pianta S, Magatti M, Vertua E, Bonassi Signoroni P, Muradore I, Nuzzo AM, et al.. Amniotic mesenchymal cells from pre-eclamptic placentae maintain immunomodulatory features as healthy controls. J Cell Mol Med 2016; 20(1):157-69; PMID:26515425; http://dx.doi.org/ 10.1111/jcmm.12715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Glennie S, Soeiro I, Dyson PJ, Lam EW, Dazzi F. Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood 2005; 105:2821-7; PMID:15591115; http://dx.doi.org/ 10.1182/blood-2004-09-3696 [DOI] [PubMed] [Google Scholar]
- [40].Rolfo A, Giuffrida D, Giuffrida MC, Todros T, Calogero AE. New perspectives for prostate cancer treatment: in vitro inhibition of LNCaP and PC3 cell proliferation by amnion-derived mesenchymal stromal cells conditioned media. Aging Male 2014; 17:94-101; PMID:24597941; http://dx.doi.org/ 10.3109/13685538.2014.896894 [DOI] [PubMed] [Google Scholar]
- [41].Qin S, Jiang H, Su S, Wang D, Liang Z, Zhang J, et al.. Inhibition of hepatic stellate cell proliferation by bone marrow mesenchymal stem cells via regulation of the cell cycle in rat. Exp Ther Med 2012; 4:375-80; PMID:23181102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, et al.. Programmed cell senescence during mammalian embryonic development. Cell 2013; 155:1104-18; PMID:24238962; http://dx.doi.org/ 10.1016/j.cell.2013.10.019 [DOI] [PubMed] [Google Scholar]
- [43].Sheppard KE, McArthur GA. The cell-cycle regulator CDK4: an emerging therapeutic target in melanoma. Clin Cancer Res 2013; 19:5320-8; PMID:24089445; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-0259 [DOI] [PubMed] [Google Scholar]
- [44].Lee JH, Park CH, Chun KH, Hong SS. Effect of adipose-derived stem cell-conditioned medium on the proliferation and migration of B16 melanoma cells. Oncol Lett 2015; 10:730-6; PMID:26622561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ooi YY, Dheen ST, Tay SS. Paracrine effects of mesenchymal stem cells-conditioned medium on microglial cytokines expression and nitric oxide production. Neuroimmunomodulation 2015; 22:233-42; PMID:25341618; http://dx.doi.org/ 10.1159/000365483 [DOI] [PubMed] [Google Scholar]
- [46].Chen J, Li Y, Hao H, Li C, Du Y, Hu Y, et al.. Mesenchymal Stem Cell Conditioned Medium Promotes Proliferation and Migration of Alveolar Epithelial Cells under Septic Conditions In Vitro via the JNK-P38 Signaling Pathway. Cell Physiol Biochem 2015; 37:1830-46; PMID:26584283; http://dx.doi.org/ 10.1159/000438545 [DOI] [PubMed] [Google Scholar]
- [47].Wu F, Tian FJ, Lin Y. Oxidative Stress in Placenta: Health and Diseases. Biomed Res Int 2015; 2015:293271; PMID:26693479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bianco P, Gehron Robey P. Marrow stromal stem cells. J Clin Invest 2000; 105:1663-8; PMID:10862779; http://dx.doi.org/ 10.1172/JCI10413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fukada S, Ma Y, Uezumi A. Adult stem cell and mesenchymal progenitor theories of aging. Front Cell Dev Biol 2014; 2:10; PMID:25364718; http://dx.doi.org/ 10.3389/fcell.2014.00010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Raffaghello L, Bianchi G, Bertolotto M, Montecucco F, Busca A, Dallegri F, et al.. Human mesenchymal stem cells inhibit neutrophil apoptosis: a model for neutrophil preservation in the bone marrow niche. Stem Cells 2008; 26:151-62; PMID:17932421; http://dx.doi.org/ 10.1634/stemcells.2007-0416 [DOI] [PubMed] [Google Scholar]
- [51].Roland CS, Hu J, Ren CE, Chen H, Li J, Varvoutis MS, et al.. Morphological changes of placental syncytium and their implications for the pathogenesis of preeclampsia. Cell Mol Life Sci 2016; 73(2):365-76; PMID:26496726; http://dx.doi.org/ 10.1007/s00018-015-2069-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, et al.. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev 2013; 27:2356-66; PMID:24186980; http://dx.doi.org/ 10.1101/gad.227512.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Goldman-Wohl D, Yagel S. United we stand not dividing: the syncytiotrophoblast and cell senescence. Placenta 2014; 35:341-4; PMID:24709558; http://dx.doi.org/ 10.1016/j.placenta.2014.03.012 [DOI] [PubMed] [Google Scholar]
- [54].Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al.. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007; 449:557-63; PMID:17914389; http://dx.doi.org/ 10.1038/nature06188 [DOI] [PubMed] [Google Scholar]
- [55].De Falco M, Fedele V, Cobellis L, Mastrogiacomo A, Giraldi D, Leone S, et al.. Pattern of expression of cyclin D1/CDK4 complex in human placenta during gestation. Cell Tissue Res 2004; 317:187-94; PMID:15221443; http://dx.doi.org/ 10.1007/s00441-004-0880-z [DOI] [PubMed] [Google Scholar]
- [56].Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev 1993; 7:812-21; PMID:8491378; http://dx.doi.org/ 10.1101/gad.7.5.812 [DOI] [PubMed] [Google Scholar]
- [57].Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 1998; 12:3499-511; PMID:9832503; http://dx.doi.org/ 10.1101/gad.12.22.3499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Alt JR, Cleveland JL, Hannink M, Diehl JA. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev 2000; 14:3102-14; PMID: 11124803; http://dx.doi.org/ 10.1101/gad.854900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bertino E, Di Battista E, Bossi A, Pagliano M, Fabris C, Aicardi G, et al.. Fetal growth velocity: kinetic, clinical, and biological aspects. Arch Dis Child Fetal Neonatal Ed 1996; 74:F10-5; PMID:8653429; http://dx.doi.org/ 10.1136/fn.74.1.F10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Parazzini F, Cortinovis I, Bortolus R, Fedele L. [Standards of birth weight in Italy]. Ann Ostet Ginecol Med Perinat 1991; 112:203-46; PMID:1807187 [PubMed] [Google Scholar]
- [61].Ferrazzi E, Zullino S, Stampalija T, Vener C, Cavoretto P, Gervasi MT, et al.. Bedside diagnosis of two major clinical phenotypes of hypertensive disorders of pregnancy. Ultrasound Obstet Gynecol 48:224-31; PMID:26350023; http://dx.doi.org/ 10.1002/uog.15741 [DOI] [PubMed] [Google Scholar]
- [62].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402-8; PMID:11846609; http://dx.doi.org/ 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.