

Ischemic conditions as diverse as chronic heart failure (CHF) and frostbite inflict tissue damage via inadequate O2 delivery. Herein we demonstrate that direct application of sodium nitrite enhances the O2 supply-O2 demand relationship, raising microvascular O2 pressure in healthy skeletal muscle. This therapeutic action of nitrite-derived nitric oxide occurred without inducing systemic hypotension and has the potential to relieve focal ischemia and preserve tissue vitality by enhancing O2 delivery.
Keywords: beetroot, nitrate, nitric oxide, microcirculation, oxygen delivery, blood flow, exercise, vascular control
Abstract
Exercise intolerance characteristic of diseases such as chronic heart failure (CHF) and diabetes is associated with reduced nitric oxide (NO) bioavailability from nitric oxide synthase (NOS), resulting in an impaired microvascular O2 driving pressure (Po2mv; O2 delivery/O2 utilization) and metabolic control. Infusions of the potent NO donor sodium nitroprusside augment NO bioavailability yet decrease mean arterial pressure (MAP) thereby reducing its potential efficacy for patient populations. To eliminate or reduce hypotensive sequelae, was superfused onto the spinotrapezius muscle. It was hypothesized that local administration would elevate resting Po2mv and slow Po2mv kinetics [increased time constant (τ) and mean response time (MRT)] following the onset of muscle contractions without decreasing MAP. In 12 anesthetized male Sprague-Dawley rats, Po2mv of the circulation-intact spinotrapezius muscle was measured by phosphorescence quenching during 180 s of electrically induced twitch contractions (1 Hz) before and after superfusion of sodium nitrite (NaNO2 30 mM). superfusion elevated resting Po2mv (control: 28.4 ± 1.1 vs. : 31.6 ± 1.2 mmHg; P ≤ 0.05), τ (control: 12.3 ± 1.2 vs. : 19.7 ± 2.2 s; P ≤ 0.05), and MRT (control: 19.3 ± 1.9 vs. : 25.6 ± 3.3 s; P ≤ 0.05). Importantly, these effects occurred in the absence of any reduction in MAP (103 ± 4 vs. 105 ± 4 mmHg, pre- and postsuperfusion respectively; P > 0.05). These results indicate that supplementation delivered to the muscle directly through superfusion enhances the blood-myocyte oxygen driving pressure without compromising MAP at rest and following the onset of muscle contraction. This strategy has substantial clinical utility for a range of ischemic conditions.
NEW & NOTEWORTHY Ischemic conditions as diverse as chronic heart failure (CHF) and frostbite inflict tissue damage via inadequate O2 delivery. Herein we demonstrate that direct application of sodium nitrite enhances the O2 supply-O2 demand relationship, raising microvascular O2 pressure in healthy skeletal muscle. This therapeutic action of nitrite-derived nitric oxide occurred without inducing systemic hypotension and has the potential to relieve focal ischemia and preserve tissue vitality by enhancing O2 delivery.
sustained muscle contractions require a robust and appropriate hyperemic response that is contingent on arteriolar vasodilation increasing muscle O2 delivery (Q̇o2) in proportion to the elevated O2 demands (V̇o2). It is also important to recognize that the instantaneous Q̇o2/V̇o2 ratio sets microvascular O2 pressures (Po2mv) that are crucial for blood-myocyte O2 flux and also the intracellular Po2, which influences metabolic control (31, 57).
Within the spectrum of vasoactive mediators for the exercise hyperemia such as ADP, cAMP, and K+, nitric oxide (NO) has a key role. Thus blockade of the endogenous endothelial NO synthase (eNOS) and/or neuronal NOS (nNOS) systems reduces exercising muscle(s) blood flow and Q̇o2 (10, 30), lowering Po2mv (24) and impairing function. In diseases such as chronic heart failure (CHF), dysfunction of the NOS system reduces NO bioavailability as well as the capability to reduce vascular tone due to decreased arteriolar compliance. There is also a reduced efficacy of the muscle pump that is induced by venous congestion that reduces the muscle hyperemia following the onset of contractions (53). Thus in CHF, Po2mv is compromised compared with healthy muscle especially in the transient phase immediately following the onset of contractions. This phenomenon slows V̇o2 kinetics and contributes to the muscle dysfunction characteristic of CHF (17, 23, 28, 46).
Given the reduced capacity for endogenous NO production in disease, possibly as a consequence of tissue hypoxia and elevated reactive oxygen species, which constrain NOS function, there has been substantial interest in providing exogenous NO precursors such as sodium nitroprusside (SNP) and inorganic nitrate/nitrite (/) (39, 61). A particularly attractive feature of the / pathway is that the pathological muscle hypoxia found in CHF and other patients promotes reduction to NO and hence enhances Q̇o2 locally with the potential to not compromise systemic blood pressure as might be expected for systemic vasodilators such as SNP or hydralazine, for example (58, 59). Recently, supplementation (i.e., beetroot juice) has been advocated as a / source that reduces mean arterial pressure, enhances muscle Q̇o2 and Po2mv, and improves muscle contractile function and efficiency (21, 22, 35). Unfortunately, after absorption in the gut the - reduction requires salivary gland secretion and the participation of commensal bacteria in the oral cavity before resorption into the blood stream as circulating . These steps typically take hours to raise plasma [] (60) and can be blocked by mouthwash-induced bactericide (41).
Given these limitations the utility of increasing vascular [] more directly has been considered (40). However, the results have been controversial with up to 36 µmol/min infused intra-arterially in the brachial artery supplying the forearm of healthy subjects being unable to produce vasodilation (36). Arguing that hypoxia is crucial for the -NO reduction Maher et al. (40) subsequently demonstrated that direct arterial infusions into the resting forearm of subjects breathing 12% O2 induced a robust arterial vasodilation. These low O2 pressures are precisely the conditions extant in the microvasculature of contracting muscles. Specifically, in the rat during exercise muscle Po2mv falls to ~20 mmHg in muscles comprised predominantly of slow-twitch fibers or ~10 mmHg in fast-twitch muscles (6, 42). Moreover, we have demonstrated that, when NO bioavailability is reduced by CHF (25) or NOS blockade (20), infusions induce a robust increase in muscle Q̇o2 that is especially pronounced in fast-twitch muscles. Yet, the effects of on systemic blood pressure have been inconclusive with adults and CHF patients reporting no change (7, 16, 25) while a modest, transient decrease in MAP has been reported in diabetic patients and during L-NAME induced NOS blockade (20, 26).
Because enhanced NO bioavailability has the potential to both elevate Q̇o2 and reduce V̇o2, direct measurements of Po2mv are necessary to evaluate the efficacy of treatment to enhance blood-myocyte O2 flux. Since changes in systemic blood pressure may influence Po2mv measurements in the spinotrapezius muscle of the rat, we first hypothesized that directly applying sodium nitrite (NaNO2) to the muscle via superfusion, when compared with intra-arterial (IA) infusion, would allow systemic pressure to remain stable. Moreover, because V̇o2 demands change most rapidly within the first minute or so of contractions we argue that assessing the efficacy of to raise Po2mv across this interval with high temporal fidelity is crucial. We also tested the hypothesis that local superfusion under normoxic conditions would elevate Po2mv significantly and, importantly, would do so in the absence of systemic hypotension.
METHODS
Three male Sprague-Dawley rats (body wt = 449 ± 16 g; Charles River Laboratories, Wilmington, MA) were utilized in protocol 1 and 12 male Sprague-Dawley rats (body wt = 421 ± 23 g) were used in protocol 2. Rats were provided food and water ad libitum while housed in a 12/12 h light-dark cycle facility at Kansas State University. All procedures were approved by the Institutional Animal Care and Use Committee of Kansas State University and conducted according to National Institutes of Health Guidelines.
Surgical Preparation
Rats were initially anesthetized with a 5% isoflurane-O2 mixture (isoflurane vaporizer; Ohio Medical Products) and subsequently maintained on 3% isoflurane-O2. An incision was made on the ventral side of the neck and the right carotid artery was isolated and cannulated with PE-10 connected to PE-50 (Intra-Medic polyethylene tubing; Clay Adams Brand, Becton, Dickinson, Sparks, MD) for measurements of mean arterial pressure (MAP) and pulse rate (HR), and infusion of the phosphorescent probe (see below). The Digi-Med Blood Pressure Analyzer (model 400; Micro-Med, Louisville, KY) was utilized to measure MAP and pulse rate, where pulse rate was interpreted and recorded as heart rate (HR). A second catheter was inserted into the caudal artery for the infusion of pentobarbital sodium anesthesia and arterial blood sampling. After the incisions for the carotid and caudal catheters were closed, rats were transitioned to pentobarbital sodium anesthesia (~20 mg/kg body wt) given intra-arterially and concentrations of isoflurane were decreased and subsequently discontinued. The level of anesthesia was regularly monitored via toe pinch and palpebral reflex, with pentobarbital anesthesia supplemented (3.5–7.0 mg/kg) as necessary. Rats were placed in the prone position on a heating pad to maintain a core temperature of ~38°C (measured via rectal probe). Incisions were then made to carefully expose the right spinotrapezius muscle with overlying skin and fascia reflected such that the integrity of the neural and vascular supply was maintained (2). With the use of 6-0 silk sutures, silver wire electrodes were secured to the rostral (cathode) and caudal (anode) regions of the muscle (protocol 2 only). Exposed muscle tissue was superfused with warmed (38°C) Krebs-Henseleit bicarbonate-buffered solution equilibrated with 5% CO2-95% N2 (pH 7.4). Surrounding exposed tissue was covered with Saran wrap (Dow Brands, Indianapolis, IN) to minimize solution contact between nonspinotrapezius tissue and superfused solutions. The spinotrapezius muscle was selected based on its mixed muscle fiber-type composition and citrate synthase activity, which resembles the quadriceps muscle in humans (15, 37).
Experimental Protocol 1: NaNO2 Superfusion vs. Intra-Arterial Infusion
The phosphorescent probe palladium meso-tetra (4 carboxyphenyl)tetrabenzoporphyrin dendrimer (G2: 15–20 mg/kg dissolved in 0.4 ml saline) was infused in bolus form (~10 s) via the carotid artery catheter followed by a saline flush. Following a brief stabilization period (~10 min), the common end of the light guide of a frequency domain phosphorimeter (PMOD 5000; Oxygen Enterprises, Philadelphia, PA) was positioned ~2–4 mm superficial to the dorsal surface of the exposed right spinotrapezius muscle as described previously (2). Fields containing large vessels were avoided to ensure that the measurements obtained were of principally capillaries and associated microvessels.
Po2mv was measured during 180-s continuous superfusion of NaNO2 (30 mM in 3.0 ml Krebs-Henseleit bicarbonate buffered solution) along the exposed muscle and 600 s of postsuperfusion rest via phosphorescence quenching (see below) and recorded at 2-s intervals. This NaNO2 dose and a 180-s continuous method of delivery were chosen due to the curved nature of the spinotrapezius muscle overlying the rib cage. Once the superfused solution was applied, the portion that did not diffuse into the muscle ran down the tissue covered by Saran wrap and onto a waste collection site. A minimal amount (2–5 drops) of Krebs-Henseleit bicarbonate-buffered solution was applied periodically to ensure that the exposed spinotrapezius muscle surface did not become dry. Central hemodynamics (MAP and HR) were continuously measured and recorded at 30-s intervals throughout the 780-s protocol. Following a 30-min stabilization, the aforementioned protocol was used with the administration of a bolus intra-arterial infusion of NaNO2 (~10 s; 7.0 mg/kg body wt in 0.4 ml heparinized saline solution).
Experimental Protocol 2: Muscle Contractions During Control and Following NaNO2 Superfusion
In the second set of experiments, Po2mv was measured at rest and during the 180-s contraction protocol (1 Hz, ~6 V, 2-ms pulse duration; Grass stimulator model S88, Quincy, MA) via phosphorescence quenching (see below) and recorded at 2-s intervals. Following the contraction period, blood samples were taken for analysis and Po2mv was monitored to ensure that microvascular control was preserved and values returned to baseline. After a 30-min stabilization period, NaNO2 (30 mM in 3.0 ml Krebs-Henseleit bicarbonate buffered solution) was continuously superfused for 180 s along the exposed muscle. Approximately 1 min following superfusion, after Po2mv stabilized, the aforementioned contraction protocol was repeated. Rats were then euthanized via pentobarbital sodium overdose (≥50 mg/kg administered into the carotid artery catheter).
Po2mv Measurement and Curve Fitting
Po2mv was calculated using the Stern-Volmer relationship. Direct measurement of phosphorescence lifetime utilized in the following equation yields Po2mv (51);
where kQ is the quenching constant and τ° and τ are the phosphorescence lifetimes in the absence of O2 and at the ambient O2 concentration, respectively. For G2, kQ is 273 mmHg−1·s−1and τ° is 251 μs (18). Since the G2 phosphorescent probe binds to blood proteins, G2 is evenly distributed throughout plasma and compartmentalized to the vascular space (18, 45). Therefore, in vivo, the phosphorescence lifetimes are determined directly by O2 pressure because kQ and τ° do not change appreciably over the physiological ranges of temperature and pH (18, 51).
With the use of computer software (SigmaPlot 11.0; Systat Software, San Jose, CA), Po2mv responses were curve-fitted from the collected Po2mv data points. Monoexponential responses to muscle contractions were fit using a one-component model whereas three control (CON) and two profiles expressed biexponential responses (exponential elevation of Po2mv following the initial contracting nadir) requiring a two-component model to best fit the data. The models used to fit either one-component or two-component models are described below:
where Po2mv(t) represents the Po2mv at any given time t, Po2mvBL corresponds to the pre-contracting resting baseline Po2mv, Δ1 and Δ2 are the amplitudes for the first and second component, respectively, TD1 and TD2 are the time delays for each component, and τ1 and τ2 are the time constants (i.e., time to reach 63% of the final response value) for each component.
Appropriate fits using either one-component or two-component models were determined using the following criteria: 1) the coefficient of determination, 2) sum of the squared residuals, and 3) visual inspection and analysis of the model fits to the data and the residuals. To provide an index of the overall principal kinetics response, the first and second components were used to calculate MRT1 and MRT2, respectively. Mean response time (MRT) was calculated using the following equations:
where TD1, TD2, τ2, and τ2 are as described above.
Statistical Analysis
Data are presented as means ± SE. Superfusion vs. Intra-arterial results were compared using two-way repeated-measures ANOVA (MAP) with Tukey’s post hoc analyses. Muscle contraction results were compared within (pre- vs. postsuperfusion) groups using one-way repeated measures ANOVA (MAP and HR) or paired 1- and 2- tail Student’s t-tests as appropriate for a priori directional hypotheses (blood gases, blood [lactate], arterial pH, and Po2mv kinetics variables). Significance was accepted at P ≤ 0.05.
RESULTS
Experimental Protocol 1: NaNO2 Superfusion vs. Intra-Arterial Infusion
Results in Figs. 1 and 2 indicated that NaNO2 (30 mM) during 180-s superfusion elevated Po2mv without significantly changing MAP. In marked contrast, bolus (~10 s) intra-arterial infusion of NaNO2 (7 mg/kg in 0.4 ml heparinized saline solution) in the caudal artery decreased MAP significantly and transiently decreased Po2mv within the initial 180 s.
Fig. 1.
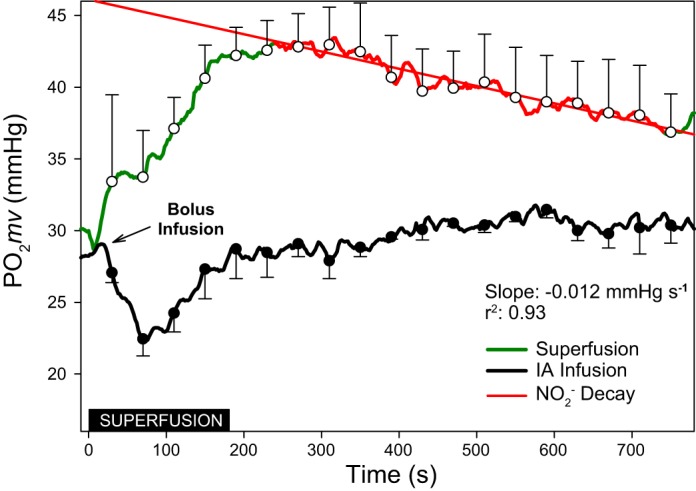
Averaged microvascular O2 driving pressure (Po2mv) from results (n = 3) during 180 s of NaNO2 superfusion (30 mM in Krebs-Henseleit solution) and a bolus (~10 s) intra-arterial infusion of NaNO2 (7 mg/kg in 0.4 ml heparinized saline solution). Inset: black box denotes the superfusion period. Measurements were recorded for an additional 600 s to monitor the effects of direct vs. systemic administration in the absence of muscle contraction. Following the bolus intra-arterial infusion Po2mv declined at a rate of −0.098 mmHg/s (regression not shown) before returning to baseline levels. In the superfusion condition, following the initial increase and plateau in Po2mv, there was a decline in Po2mv at the rate of −0.012 mmHg/s. Data are presented as means ± SE every 30 s throughout each condition.
Fig. 2.
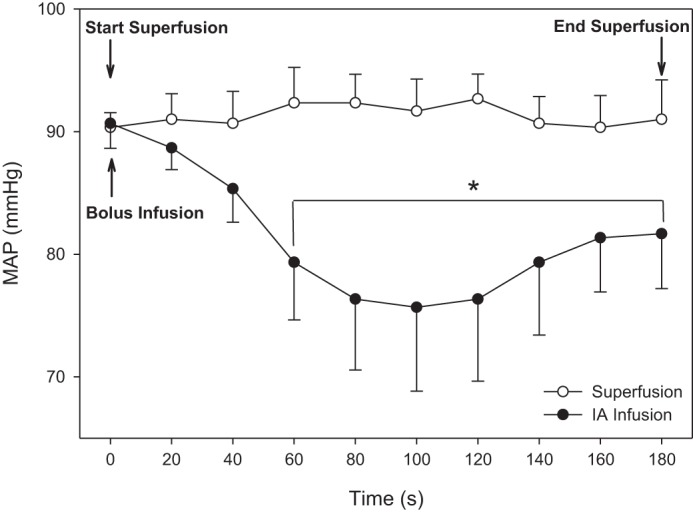
Mean arterial pressure (MAP) during 180 s of NaNO2 superfusion (30 mM in Krebs-Henseleit solution) remained constant whereas a bolus (~10 s) intra-arterial infusion of NaNO2 (7 mg/kg in 0.4 ml heparinized saline solution) produced a sustained reduction in MAP by as much as ~14–15 mmHg on average (n = 3; first experimental group of rats). Data are presented as means ± SE.
Experimental Protocol 2: Muscle Contractions During Control and Following NaNO2 Superfusion
Microvascular oxygen pressures (Po2mv).
Within 180-s superfusion of NaNO2 Po2mv was significantly elevated (Po2mvBL; CON: 28.4 ± 1.1 vs. : 31.6 ± 1.2 mmHg; P ≤ 0.05) (Table 1 and Fig. 3). Following the onset of contractions, exhibited a significantly larger Po2mv amplitude (CON: 10.5 ± 0.9 vs. : 12.7 ± 0.8 mmHg; P ≤ 0.05), slower τ (CON: 12.3 ± 1.2 vs. : 19.7 ± 2.2 s; P ≤ 0.05), and increased MRT (CON: 19.3 ± 1.9 vs. : 25.6 ± 3.3 s; P ≤ 0.05; Fig. 4A). As shown in Fig. 4B, the difference in driving pressure between conditions continued for the first 40 s of electrically stimulated contractions.
Table 1.
Po2mv variables in the spinotrapezius muscle at rest and following the onset of contractions under control and NaNO2 conditions
| CON | ||
|---|---|---|
| Po2mvBL, mmHg | 28.4 ± 1.1 | 31.6 ± 1.2* |
| Δ1Po2mv, mmHg | 10.5 ± 0.9 | 12.7 ± 0.8* |
| Po2mv (steady state), mmHg | 18.0 ± 0.9 | 18.9 ± 1.2 |
| TD1, s | 7.0 ± 1.2 | 6.0 ± 1.6 |
| τ1, s | 12.3 ± 1.2 | 19.7 ± 2.2* |
| MRT1, s | 19.3 ± 1.9 | 25.6 ± 3.3* |
| Δ2Po2mv, mmHg | 3.4 ± 0.5 | 2.9 ± 0.1 |
| TD2, s | 78.5 ± 22.3 | 68.9 ± 9.5 |
| τ2, s | 67.9 ± 7.7 | 77.1 ± 14.2 |
| MRT2, s | 146.4 ± 25.7 | 97.3 ± 48.8 |
Values are means ± SE. Po2mvBL, resting Po2mv; Δ1Po2mv and Δ2Po2mv, amplitude of the first and second components; Po2mv(steady state), contracting steady-state Po2mv; TD1 and TD2, time delay of the first and second component; τ1, and τ2, time constant of the first and second component; MRT1 and MRT2, mean response time of the first and second component. Primary components were calculated for all rats (n = 12). Secondary components were present in 3 control (CON) and 2 rats.
P ≤ 0.05 vs. CON.
Fig. 3.
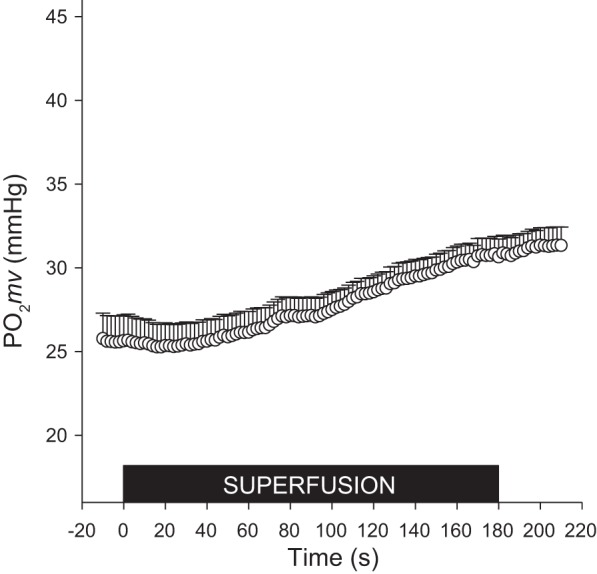
Microvascular oxygen pressure (Po2mv) of the spinotrapezius muscle for the second experimental group of rats (n = 12) was measured every 2 s during the 180-s superfusion of 30 mM NaNO2 in Krebs-Henseleit solution. Inset: black box denotes the superfusion period. Data are presented as means ± SE.
Fig. 4.
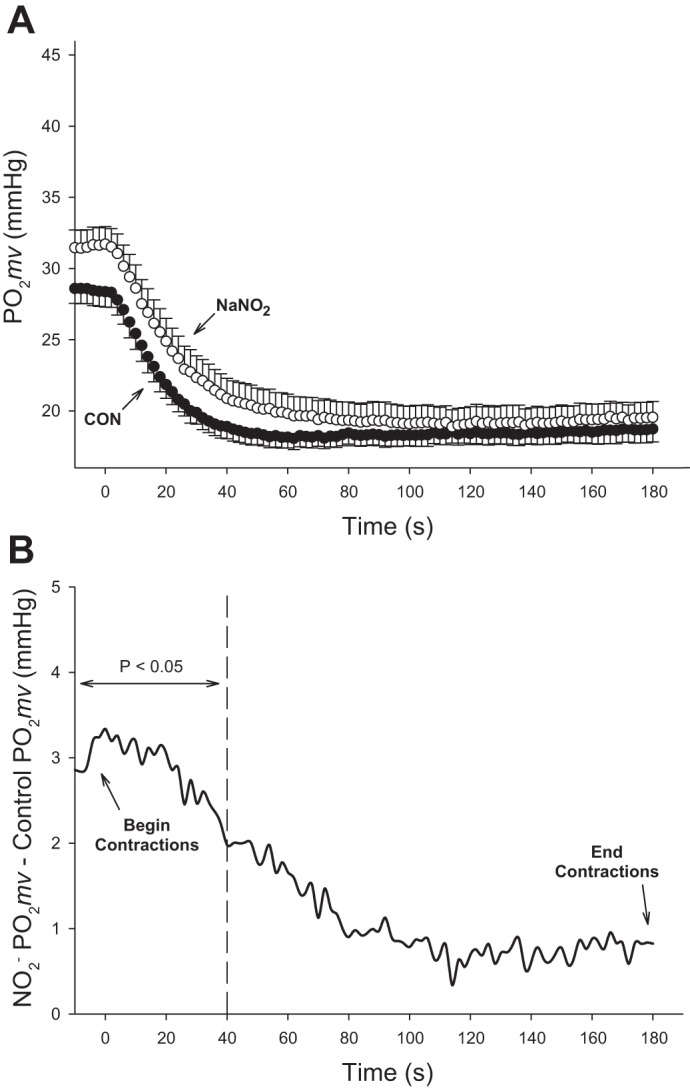
A: average spinotrapezius Po2mv kinetics for the second experimental group of rats (n = 12) during 180 s electrically stimulated contractions in the control condition (closed circles) and following NaNO2 superfusion (open circles). Mean arterial pressure was recorded at the beginning (105 ± 5 mmHg) and end (105 ± 5 mmHg) of the contraction protocol. Data are presented as means ± SE. B: Po2mv differences for each 2 s measurement for the experimental group of rats (n = 12) between NaNO2 and CON conditions during spinotrapezius muscle contractions. Note that before (i.e., left of) the dashed line, NaNO2 is significantly different from CON (P < 0.05).
Central hemodynamics and blood gases.
Superfusion of NaNO2 did not significantly alter MAP or HR from presuperfusion values (MAP: 103 ± 4 vs. 105 ± 4 mmHg, HR: 330 ± 10 vs. 321 ± 11 beats/min, pre and post, respectively; both P > 0.05). Likewise, there was no significant difference in MAP or HR between CON and conditions following the contraction protocol (P > 0.05). Finally, there were no significant differences in %O2 saturation (CON: 91.5 ± 1.5 vs. : 93.1 ± 0.7%), Pco2 (CON: 42 ± 2 vs. : 41 ± 2 mmHg), blood [lactate] (CON: 1.6 ± 0.1 vs. : 1.6 ± 0.2 mM), or arterial pH (CON: 7.36 ± 0.01 vs. : 7.37 ± 0.01) between conditions (P > 0.05).
DISCUSSION
The principal findings of the present investigation demonstrate that NaNO2 superfusion enhances the muscle Po2mv at rest (i.e., elevated Po2mvBL) and during the transition from rest-to-contractions, in part, by slowing the Po2mv fall (i.e., increased τ and MRT). Importantly, these effects occurred concomitant with stable MAP. The ability of to enhance the driving pressure of O2 before and for the 40 s after the onset of muscle contractions (Fig. 4, A and B) would be expected to potentially decrease reliance on immediate, nonoxidative energy pathways and delay the production of fatigue-inducing metabolic products (31, 57). In disease conditions attended by cardiovascular complications, the inability to deliver O2 may induce ischemic damage, impair recovery, and, for active skeletal muscle, constrain exercise performance and perhaps daily activities essential to life quality. The delivery of exogenous is known to improve the central and systemic derangements prevalent in CHF (7, 39, 44) and we now demonstrate that it rapidly enhances peripheral control directly by circumventing the NOS pathway.
Microvascular Oxygen Pressure (Po2mv) Dynamics
In skeletal muscle at rest, NaNO2 has the potential to elevate the driving pressure of O2 from blood to myocyte (Po2mvBL) under normoxic conditions. This NOS-independent effect coheres with previous investigations that have administered NO precursors (22, 24, 27). With respect to the Po2mv elevations, these may be the consequence of increased Q̇o2, decreased V̇o2, or a combination of both. Q̇o2 increases with increasing [] because the one-step reduction to NO occurs in the low Po2 environment of resistance vessels in healthy individuals (20, 21). The advantage of the -NO reduction is that NOS reliance is avoided, with being reduced to NO in environments of low O2/pH via interactions with deoxyhemoglobin, tissue and vascular myoglobin, xanthine oxidoreductase, and aldehyde dehydrogenase. Furthermore, by experimental blockade of endogenous production of NO from eNOS and nNOS pharmacologically [nitro-l-arginine methyl ester (l-NAME; eNOS and nNOS) and S-methyl-l-thiocitrulline (SMTC; nNOS)], blood flow is reduced independent of any increases in sympathetic activity/vasoconstriction (11, 12, 29, 30). The present results clearly demonstrate that NaNO2 increases baseline (resting) Po2mv and suggest strongly that the -NO reduction compliments any NO emanating from the intact NOS system to augment Po2mv. The direct measurement of Po2mv herein extends previous observations of -induced vasorelaxation and increased Q̇o2 in healthy subjects (13, 14, 40).
Potentially, the increased Po2mv could also be explained by mitochondrial inhibition (↓V̇o2) where NO competitively binds to complexes I and IV of the respiratory chain (8, 9). However, if the primary effect of on Po2mv is mitochondrial inhibition, then a continuous increase in steady-state Po2mv during muscle contractions would be expected but clearly this is not found (Fig. 4A). It is pertinent that using a similar protocol with SNP, Hirai et al. (27) demonstrated the integrity of mitochondrial respiration (and thus V̇o2) concomitant with elevated and NO. Thus the elevation in baseline Po2mv seen herein is most likely the consequence of enhanced blood flow and Q̇o2 rather than decreased V̇o2.
The increases in Po2mv presumed to be produced by an increased Q̇o2 with NaNO2 superfusion (13, 30, 52) persists into the onset of contractions (slowed Po2mv kinetics) as demonstrated in Fig. 4, A and B. In Fig. 4B the rate at which Po2mv is falling is slowed by disproportionate changes in Q̇o2 and V̇o2 across the initial 40 s. Enhancing the Q̇o2-to-V̇o2 relationship at this crucial transition would potentially improve energy production via oxidative metabolism, decrease the acidic sequelae of glycolytic pathways, and delay the onset of fatigue processes (31, 57).
Potential Therapeutic Significance
precursors such as containing beetroot juice effectively increase plasma [] and can improve exercise performance in healthy individuals (1, 34, 60) and patient populations where cardiovascular function is impaired such as CHF (61) and peripheral artery disease (PAD) (33). Specifically, CHF patients have an increased cardiac output reserve secondary to the reduction of vascular resistance without any changes in MAP (61). Likewise, PAD patients exhibit improved exercise/walking performance and delayed onset of claudication following beetroot juice supplementation (33). However, elevating circulating [] via dietary means takes 2–3 h, which may constitute too great a delay for an effective therapeutic outcome. A potential alternative is oral administration of NaNO2, which elevates circulating [] within 30 min in older adults (16) and diabetic patients (26). More direct routes of NaNO2 (i.e., intravascular infusion or oral administration) can bypass the need for bacterial breakdown of and absorption in the gut and increase vascular [] rapidly. Thus intravenous infusions have improved cardiac function and exercise performance in CHF patients (7). Additionally, infusion during l-NAME-induced NOS blockade or CHF (20, 25) reverses the consequences of absent endogenous NO bioavailability and provides an avenue for enhanced Q̇o2-to-V̇o2 matching.
Administration of NaNO2 protects against hypoxic damage to liver, cerebral, and myocardial tissue via the reduction of to NO (19, 32, 54, 56) but possibly not the kidney (3, 55). Therefore topical administration of NaNO2, potentially via implantable osmotic micro-pump, may provide tissue protection during acute surgical or other medical conditions by elevating Q̇o2 and Po2mv within 180 s of application (see Fig. 3) without confounding peripheral vascular effects [i.e., reductions in tissue Po2mv (Fig. 1) that are coincident with reductions in MAP (Fig. 2)]. Local administration may open up the avenue for long-term administration at a desired location enhancing Q̇o2 and Po2mv for extended durations. The progressive decay of effects on Po2mv (Figs. 1 and 4B) indicates that setting the required [NaNO2] and timing of application would be crucial.
Experimental Considerations
Although Po2mv was significantly different at rest and the beginning of muscle contractions, the effect of is largely diminished by the time the steady-state Po2mv is reached (Fig. 4, A and B). It remains unclear whether this is a direct effect of wearing off irrespective of muscle contractions or whether contractions and the attendant hypoxia are elevating utilization. Figure 1 demonstrates that in the absence of muscle contractions, Po2mv plateaus and then declines at a rate of ~0.7 mmHg/min after superfusion has ceased. This would indicate, in part, why NaNO2 superfusion does not enhance steady-state Po2mv relative to the control condition. Importantly, the linear decay in Po2mv sheds light on the utilization of in a localized volume of skeletal muscle when systemic levels of have not been elevated upstream (i.e., drug infusion or oral administration). Future investigations into various delivery methods of NaNO2 (i.e., injectable pellets or cutaneous patches) may be warranted considering that the administration of augments Po2mv at rest and during the rest-exercise transition.
Muscle contractile function was not directly measured in the current investigation yet previous studies have utilized the same contraction protocol to elicit a moderate metabolic stress of approximately three- and sevenfold elevation in blood flow and V̇o2, respectively (4, 5). At this level of exercise contractile performance and V̇o2 (or V̇o2 kinetics) are not O2 delivery limited. Accordingly, contractile performance and V̇o2 would not be expected to increase with administration but may be manifested with higher metabolic rates or under conditions of pathologically-reduced O2 delivery as seen in heart failure patients during high-intensity exercise, for example (61). A higher rate of stimulation could have been used to create higher metabolic stress; however, any evoked increases in MAP would have complicated the interpretation of the effect of on cardiovascular control. Thus the current contraction protocol and level of metabolic stress was selected to demonstrate proof-of-principle that the local NaNO2 application to the spinotrapezius muscle would elevate Po2mv significantly in the absence of altered MAP. Additionally, while was investigated herein due to the selective reduction to NO in low Po2 and pH environments found in ischemic muscle and elsewhere, other endogenous and exogenous vasodilators (i.e., ADP, cAMP, lidocaine, cyclooxygenase, or indomethacin) may potentially exert an elevation of Po2mv without impacting central hemodynamic regulation (MAP or HR). Future investigations into these vasodilators may also serve to benefit patient populations.
A potential limitation to the Po2mv measurement herein is assuring that the entirety of phosphorescent signal emanates from capillaries and associated microvessels. Micrograph images demonstrate that greater than 90% of the vascular volume in skeletal muscle [rat soleus and plantaris (49, 50), diaphragm (48), and mouse tibialis anterior (47)] is comprised of capillaries. Additionally, the light guide is positioned away from large vessels to minimize the potential for macrovascular influence.
Conclusion
NaNO2 serves therapeutically as a hypoxic vasodilator with efficacy for improving exercise performance in patients with cardiovascular disease (1, 7, 38, 43, 44). The present investigation demonstrates the ability of NaNO2 to locally elevate skeletal muscle Po2mv at rest and following the onset of contractions in the healthy rat without altering MAP. Enhancing the muscle vascular O2 driving pressure via NaNO2 would provide a fast-acting modality to potentially improve metabolic control and thus delay fatigue and/or hypoxic damage. Improving Po2mv dynamics alongside what remains of the endogenous NOS system under these conditions, NaNO2 may ameliorate perturbations in the Q̇o2-to-V̇o2 ratio commonly found in disease states such as CHF and PAD. Fast-acting improvements in both resting and exercise Po2mv dynamics may add to the current standard of care in populations with limited exercise tolerance along with individuals that may be suffering from focal tissue hypoxia and/or ischemia (i.e., frostbite, PAD, stroke).
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant HL-108328 (to D. C. Poole) and American Heart Association (AHA) Grant 10GRNT4350011 (to D. C. Poole).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
T.D.C., S.K.F., C.T.H., J.C.C., T.I.M., and D.C.P. performed experiments; T.D.C., S.K.F., C.T.H., J.C.C., T.I.M., and D.C.P. analyzed data; T.D.C., S.K.F., C.T.H., J.C.C., T.I.M., and D.C.P. interpreted results of experiments; T.D.C., S.K.F., C.T.H., J.C.C., T.I.M., and D.C.P. prepared figures; T.D.C., C.T.H., J.C.C., T.I.M., and D.C.P. drafted manuscript; T.D.C., C.T.H., J.C.C., T.I.M., and D.C.P. edited and revised manuscript; T.D.C., S.K.F., C.T.H., J.C.C., T.I.M., and D.C.P. approved final version of manuscript.
REFERENCES
- 1.Bailey JC, Feelisch M, Horowitz JD, Frenneaux MP, Madhani M. Pharmacology and therapeutic role of inorganic nitrite and nitrate in vasodilatation. Pharmacol Ther 144: 303–320, 2014. doi: 10.1016/j.pharmthera.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 2.Bailey JK, Kindig CA, Behnke BJ, Musch TI, Schmid-Schoenbein GW, Poole DC. Spinotrapezius muscle microcirculatory function: effects of surgical exteriorization. Am J Physiol Heart Circ Physiol 279: H3131–H3137, 2000. [DOI] [PubMed] [Google Scholar]
- 3.Basireddy M, Isbell TS, Teng X, Patel RP, Agarwal A. Effects of sodium nitrite on ischemia-reperfusion injury in the rat kidney. Am J Physiol Renal Physiol 290: F779–F786, 2006. doi: 10.1152/ajprenal.00334.2005. [DOI] [PubMed] [Google Scholar]
- 4.Behnke BJ, Barstow TJ, Kindig CA, McDonough P, Musch TI, Poole DC. Dynamics of oxygen uptake following exercise onset in rat skeletal muscle. Respir Physiol Neurobiol 133: 229–239, 2002. doi: 10.1016/S1569-9048(02)00183-0. [DOI] [PubMed] [Google Scholar]
- 5.Behnke BJ, Kindig CA, Musch TI, Koga S, Poole DC. Dynamics of microvascular oxygen pressure across the rest-exercise transition in rat skeletal muscle. Respir Physiol 126: 53–63, 2001. doi: 10.1016/S0034-5687(01)00195-5. [DOI] [PubMed] [Google Scholar]
- 6.Behnke BJ, McDonough P, Padilla DJ, Musch TI, Poole DC. Oxygen exchange profile in rat muscles of contrasting fibre types. J Physiol 549: 597–605, 2003. doi: 10.1113/jphysiol.2002.035915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borlaug BA, Koepp KE, Melenovsky V. Sodium nitrite improves exercise hemodynamics and ventricular performance in heart failure with preserved ejection fraction. J Am Coll Cardiol 66: 1672–1682, 2015. doi: 10.1016/j.jacc.2015.07.067. [DOI] [PubMed] [Google Scholar]
- 8.Brown GC. Nitric oxide as a competitive inhibitor of oxygen consumption in the mitochondrial respiratory chain. Acta Physiol Scand 168: 667–674, 2000. doi: 10.1046/j.1365-201x.2000.00718.x. [DOI] [PubMed] [Google Scholar]
- 9.Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci USA 95: 7631–7636, 1998. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Copp SW, Hirai DM, Ferguson SK, Holdsworth CT, Musch TI, Poole DC. Effects of chronic heart failure on neuronal nitric oxide synthase-mediated control of microvascular O2 pressure in contracting rat skeletal muscle. J Physiol 590: 3585–3596, 2012. doi: 10.1113/jphysiol.2012.235929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Copp SW, Hirai DM, Schwagerl PJ, Musch TI, Poole DC. Effects of neuronal nitric oxide synthase inhibition on resting and exercising hindlimb muscle blood flow in the rat. J Physiol 588: 1321–1331, 2010. doi: 10.1113/jphysiol.2009.183723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Copp SW, Hirai DM, Sims GE, Fels RJ, Musch TI, Poole DC, Kenney MJ. Neuronal nitric oxide synthase inhibition and regional sympathetic nerve discharge: implications for peripheral vascular control. Respir Physiol Neurobiol 186: 285–289, 2013. doi: 10.1016/j.resp.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO III, Gladwin MT. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med 9: 1498–1505, 2003. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 14.Dejam A, Hunter CJ, Tremonti C, Pluta RM, Hon YY, Grimes G, Partovi K, Pelletier MM, Oldfield EH, Cannon RO III, Schechter AN, Gladwin MT. Nitrite infusion in humans and nonhuman primates: endocrine effects, pharmacokinetics, and tolerance formation. Circulation 116: 1821–1831, 2007. doi: 10.1161/CIRCULATIONAHA.107.712133. [DOI] [PubMed] [Google Scholar]
- 15.Delp MD, Duan C. Composition and size of type I, IIA, IID/X, and IIB fibers and citrate synthase activity of rat muscle. J Appl Physiol (1985) 80: 261–270, 1996. [DOI] [PubMed] [Google Scholar]
- 16.DeVan AE, Johnson LC, Brooks FA, Evans TD, Justice JN, Cruickshank-Quinn C, Reisdorph N, Bryan NS, McQueen MB, Santos-Parker JR, Chonchol MB, Bassett CJ, Sindler AL, Giordano T, Seals DR. Effects of sodium nitrite supplementation on vascular function and related small metabolite signatures in middle-aged and older adults. J Appl Physiol (1985) 120: 416–425, 2015. doi: 10.1152/japplphysiol.00879.2015. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=26607249&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diederich ER, Behnke BJ, McDonough P, Kindig CA, Barstow TJ, Poole DC, Musch TI. Dynamics of microvascular oxygen partial pressure in contracting skeletal muscle of rats with chronic heart failure. Cardiovasc Res 56: 479–486, 2002. doi: 10.1016/S0008-6363(02)00545-X. [DOI] [PubMed] [Google Scholar]
- 18.Dunphy I, Vinogradov SA, Wilson DF. Oxyphor R2 and G2: phosphors for measuring oxygen by oxygen-dependent quenching of phosphorescence. Anal Biochem 310: 191–198, 2002. doi: 10.1016/S0003-2697(02)00384-6. [DOI] [PubMed] [Google Scholar]
- 19.Duranski MR, Greer JJM, Dejam A, Jaganmohan S, Hogg N, Langston W, Patel RP, Yet S-F, Wang X, Kevil CG, Gladwin MT, Lefer DJ. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest 115: 1232–1240, 2005. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferguson SK, Glean AA, Holdsworth CT, Wright JL, Fees AJ, Colburn TD, Stabler T, Allen JD, Jones AM, Musch TI, Poole DC. Skeletal muscle vascular control during exercise: impact of nitrite infusion during nitric oxide synthase inhibition in healthy rats. J Cardiovasc Pharmacol Ther 21: 201–208, 2016. doi: 10.1177/1074248415599061. [DOI] [PubMed] [Google Scholar]
- 21.Ferguson SK, Hirai DM, Copp SW, Holdsworth CT, Allen JD, Jones AM, Musch TI, Poole DC. Impact of dietary nitrate supplementation via beetroot juice on exercising muscle vascular control in rats. J Physiol 591: 547–557, 2013. doi: 10.1113/jphysiol.2012.243121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferguson SK, Hirai DM, Copp SW, Holdsworth CT, Allen JD, Jones AM, Musch TI, Poole DC. Effects of nitrate supplementation via beetroot juice on contracting rat skeletal muscle microvascular oxygen pressure dynamics. Respir Physiol Neurobiol 187: 250–255, 2013. doi: 10.1016/j.resp.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferreira LF, Hageman KS, Hahn SA, Williams J, Padilla DJ, Poole DC, Musch TI. Muscle microvascular oxygenation in chronic heart failure: role of nitric oxide availability. Acta Physiol (Oxf) 188: 3–13, 2006. doi: 10.1111/j.1748-1716.2006.01598.x. [DOI] [PubMed] [Google Scholar]
- 24.Ferreira LF, Padilla DJ, Williams J, Hageman KS, Musch TI, Poole DC. Effects of altered nitric oxide availability on rat muscle microvascular oxygenation during contractions. Acta Physiol (Oxf) 186: 223–232, 2006. doi: 10.1111/j.1748-1716.2006.01523.x. [DOI] [PubMed] [Google Scholar]
- 25.Glean AA, Ferguson SK, Holdsworth CT, Colburn TD, Wright JL, Fees AJ, Hageman KS, Poole DC, Musch TI. Effects of nitrite infusion on skeletal muscle vascular control during exercise in rats with chronic heart failure. Am J Physiol Heart Circ Physiol 309: H1354–H1360, 2015. doi: 10.1152/ajpheart.00421.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greenway FL, Predmore BL, Flanagan DR, Giordano T, Qiu Y, Brandon A, Lefer DJ, Patel RP, Kevil CG. Single-dose pharmacokinetics of different oral sodium nitrite formulations in diabetes patients. Diabetes Technol Ther 14: 552–560, 2012. doi: 10.1089/dia.2011.0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirai DM, Copp SW, Ferguson SK, Holdsworth CT, Musch TI, Poole DC. The NO donor sodium nitroprusside: evaluation of skeletal muscle vascular and metabolic dysfunction. Microvasc Res 85: 104–111, 2013. doi: 10.1016/j.mvr.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirai DM, Musch TI, Poole DC. Exercise training in chronic heart failure: improving skeletal muscle O2 transport and utilization. Am J Physiol Heart Circ Physiol 309: H1419–H1439, 2015. doi: 10.1152/ajpheart.00469.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirai T, Musch TI, Morgan DA, Kregel KC, Claassen DE, Pickar JG, Lewis SJ, Kenney MJ. Differential sympathetic nerve responses to nitric oxide synthase inhibition in anesthetized rats. Am J Physiol Regul Integr Comp Physiol 269: R807–R813, 1995. [DOI] [PubMed] [Google Scholar]
- 30.Hirai T, Visneski MD, Kearns KJ, Zelis R, Musch TI. Effects of NO synthase inhibition on the muscular blood flow response to treadmill exercise in rats. J Appl Physiol (1985) 77: 1288–1293, 1994. [DOI] [PubMed] [Google Scholar]
- 31.Hogan MC, Arthur PG, Bebout DE, Hochachka PW, Wagner PD. Role of O2 in regulating tissue respiration in dog muscle working in situ. J Appl Physiol (1985) 73: 728–736, 1992. [DOI] [PubMed] [Google Scholar]
- 32.Jung KH, Chu K, Ko SY, Lee ST, Sinn DI, Park DK, Kim JM, Song EC, Kim M, Roh JK. Early intravenous infusion of sodium nitrite protects brain against in vivo ischemia-reperfusion injury. Stroke 37: 2744–2750, 2006. doi: 10.1161/01.STR.0000245116.40163.1c. [DOI] [PubMed] [Google Scholar]
- 33.Kenjale AA, Ham KL, Stabler T, Robbins JL, Johnson JL, Vanbruggen M, Privette G, Yim E, Kraus WE, Allen JD. Dietary nitrate supplementation enhances exercise performance in peripheral arterial disease. J Appl Physiol (1985) 110: 1582–1591, 2011. doi: 10.1152/japplphysiol.00071.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larsen FJ, Schiffer TA, Borniquel S, Sahlin K, Ekblom B, Lundberg JO, Weitzberg E. Dietary inorganic nitrate improves mitochondrial efficiency in humans. Cell Metab 13: 149–159, 2011. doi: 10.1016/j.cmet.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 35.Larsen FJ, Weitzberg E, Lundberg JO, Ekblom B. Effects of dietary nitrate on oxygen cost during exercise. Acta Physiol (Oxf) 191: 59–66, 2007. doi: 10.1111/j.1748-1716.2007.01713.x. [DOI] [PubMed] [Google Scholar]
- 36.Lauer T, Preik M, Rassaf T, Strauer BE, Deussen A, Feelisch M, Kelm M. Plasma nitrite rather than nitrate reflects regional endothelial nitric oxide synthase activity but lacks intrinsic vasodilator action. Proc Natl Acad Sci USA 98: 12814–12819, 2001. doi: 10.1073/pnas.221381098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leek BT, Mudaliar SRD, Henry R, Mathieu-Costello O, Richardson RS. Effect of acute exercise on citrate synthase activity in untrained and trained human skeletal muscle. Am J Physiol Regul Integr Comp Physiol 280: R441–R447, 2001. [DOI] [PubMed] [Google Scholar]
- 38.Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov 7: 156–167, 2008. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- 39.Maher AR, Arif S, Madhani M, Abozguia K, Ahmed I, Fernandez BO, Feelisch M, O’Sullivan AG, Christopoulos A, Sverdlov AL, Ngo D, Dautov R, James PE, Horowitz JD, Frenneaux MP. Impact of chronic congestive heart failure on pharmacokinetics and vasomotor effects of infused nitrite. Br J Pharmacol 169: 659–670, 2013. doi: 10.1111/bph.12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maher AR, Milsom AB, Gunaruwan P, Abozguia K, Ahmed I, Weaver RA, Thomas P, Ashrafian H, Born GV, James PE, Frenneaux MP. Hypoxic modulation of exogenous nitrite-induced vasodilation in humans. Circulation 117: 670–677, 2008. doi: 10.1161/CIRCULATIONAHA.107.719591. [DOI] [PubMed] [Google Scholar]
- 41.McDonagh STJ, Wylie LJ, Winyard PG, Vanhatalo A, Jones AM. The effects of chronic nitrate supplementation and the use of strong and weak antibacterial agents on plasma nitrite concentration and exercise blood pressure. Int J Sports Med 36: 1177–1185, 2015. doi: 10.1055/s-0035-1554700. [DOI] [PubMed] [Google Scholar]
- 42.McDonough P, Behnke BJ, Padilla DJ, Musch TI, Poole DC. Control of microvascular oxygen pressures in rat muscles comprised of different fibre types. J Physiol 563: 903–913, 2005. doi: 10.1113/jphysiol.2004.079533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Omar SA, Webb AJ. Nitrite reduction and cardiovascular protection. J Mol Cell Cardiol 73: 57–69, 2014. doi: 10.1016/j.yjmcc.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 44.Ormerod JO, Arif S, Mukadam M, Evans JD, Beadle R, Fernandez BO, Bonser RS, Feelisch M, Madhani M, Frenneaux MP. Short-term intravenous sodium nitrite infusion improves cardiac and pulmonary hemodynamics in heart failure patients. Circ Heart Fail 8: 565–571, 2015. doi: 10.1161/CIRCHEARTFAILURE.114.001716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poole DC, Behnke BJ, McDonough P, McAllister RM, Wilson DF. Measurement of muscle microvascular oxygen pressures: compartmentalization of phosphorescent probe. Microcirculation 11: 317–326, 2004. doi: 10.1080/10739680490437487. [DOI] [PubMed] [Google Scholar]
- 46.Poole DC, Hirai DM, Copp SW, Musch TI. Muscle oxygen transport and utilization in heart failure: implications for exercise (in)tolerance. Am J Physiol Heart Circ Physiol 302: H1050–H1063, 2012. doi: 10.1152/ajpheart.00943.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poole DC, Mathieu-Costello O. Effects of hypoxia on capillary orientation in anterior tibialis muscle of highly active mice. Respir Physiol 82: 1–10, 1990. doi: 10.1016/0034-5687(90)90019-U. [DOI] [PubMed] [Google Scholar]
- 48.Poole DC, Mathieu-Costello O. Capillary and fiber geometry in rat diaphragm perfusion fixed in situ at different sarcomere lengths. J Appl Physiol (1985) 73: 151–159, 1992. [DOI] [PubMed] [Google Scholar]
- 49.Poole DC, Mathieu-Costello O. Relationship between fiber capillarization and mitochondrial volume density in control and trained rat soleus and plantaris muscles. Microcirculation 3: 175–186, 1996. doi: 10.3109/10739689609148286. [DOI] [PubMed] [Google Scholar]
- 50.Poole DC, Mathieu-Costello O, West JB. Capillary tortuosity in rat soleus muscle is not affected by endurance training. Am J Physiol Heart Circ Physiol 256: H1110–H1116, 1989. [DOI] [PubMed] [Google Scholar]
- 51.Rumsey WL, Vanderkooi JM, Wilson DF. Imaging of phosphorescence: a novel method for measuring oxygen distribution in perfused tissue. Science 241: 1649–1651, 1988. doi: 10.1126/science.3420417. [DOI] [PubMed] [Google Scholar]
- 52.Schrage WG, Joyner MJ, Dinenno FA. Local inhibition of nitric oxide and prostaglandins independently reduces forearm exercise hyperaemia in humans. J Physiol 557: 599–611, 2004. doi: 10.1113/jphysiol.2004.061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shiotani I, Sato H, Sato H, Yokoyama H, Ohnishi Y, Hishida E, Kinjo K, Nakatani D, Kuzuya T, Hori M. Muscle pump-dependent self-perfusion mechanism in legs in normal subjects and patients with heart failure. J Appl Physiol (1985) 92: 1647–1654, 2002. doi: 10.1152/japplphysiol.01096.2000. [DOI] [PubMed] [Google Scholar]
- 54.Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L, Wang X, MacArthur PH, Shoja A, Raghavachari N, Calvert JW, Brookes PS, Lefer DJ, Gladwin MT. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med 204: 2089–2102, 2007. doi: 10.1084/jem.20070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tripatara P, Patel NS, Webb A, Rathod K, Lecomte FM, Mazzon E, Cuzzocrea S, Yaqoob MM, Ahluwalia A, Thiemermann C. Nitrite-derived nitric oxide protects the rat kidney against ischemia/reperfusion injury in vivo: role for xanthine oxidoreductase. J Am Soc Nephrol 18: 570–580, 2007. doi: 10.1681/ASN.2006050450. [DOI] [PubMed] [Google Scholar]
- 56.Webb A, Bond R, McLean P, Uppal R, Benjamin N, Ahluwalia A. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia-reperfusion damage. Proc Natl Acad Sci USA 101: 13683–13688, 2004. doi: 10.1073/pnas.0402927101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilson DF, Erecińska M, Drown C, Silver IA. Effect of oxygen tension on cellular energetics. Am J Physiol Cell Physiol 233: C135–C140, 1977. [DOI] [PubMed] [Google Scholar]
- 58.Wilson JR, Hoyt RW, Ferraro N, Janicki JS, Weber KT. Effect of hydralazine on nutritive flow to working canine gracilis skeletal muscle. J Am Coll Cardiol 4: 529–534, 1984. doi: 10.1016/S0735-1097(84)80097-2. [DOI] [PubMed] [Google Scholar]
- 59.Wilson JR, Martin JL, Ferraro N, Weber KT. Effect of hydralazine on perfusion and metabolism in the leg during upright bicycle exercise in patients with heart failure. Circulation 68: 425–432, 1983. doi: 10.1161/01.CIR.68.2.425. [DOI] [PubMed] [Google Scholar]
- 60.Wylie LJ, Kelly J, Bailey SJ, Blackwell JR, Skiba PF, Winyard PG, Jeukendrup AE, Vanhatalo A, Jones AM. Beetroot juice and exercise: pharmacodynamic and dose-response relationships. J Appl Physiol (1985) 115: 325–336, 2013. doi: 10.1152/japplphysiol.00372.2013. [DOI] [PubMed] [Google Scholar]
- 61.Zamani P, Rawat D, Shiva-Kumar P, Geraci S, Bhuva R, Konda P, Doulias P-T, Ischiropoulos H, Townsend RR, Margulies KB, Cappola TP, Poole DC, Chirinos JA. Effect of inorganic nitrate on exercise capacity in heart failure with preserved ejection fraction. Circulation 131: 371–380, 2015. doi: 10.1161/CIRCULATIONAHA.114.012957. [DOI] [PMC free article] [PubMed] [Google Scholar]