

Abstract
We examined renal Na and K transporters in mice with deletions in the gene encoding the aldosterone-induced protein SGK1. The knockout mice were hyperkalemic, and had altered expression of the subunits of the epithelial Na channel (ENaC). The kidneys showed decreased expression of the cleaved forms of the γENaC subunit, and the fully glycosylated form of the βENaC subunits when animals were fed a high-K diet. Knockout animals treated with exogenous aldosterone also had reduced subunit processing and diminished surface expression of βENaC and γENaC. Expression of the three upstream Na transporters NHE3, NKCC2, and NCC was reduced in both wild-type and knockout mice in response to K loading. The activity of ENaC measured as whole cell amiloride-sensitive current (INa) in principal cells of the cortical collecting duct (CCD) was minimal under control conditions but was increased by a high-K diet to a similar extent in knockout and wild-type animals. INa in the connecting tubule also increased similarly in the two genotypes in response to exogenous aldosterone administration. The activities of both ROMK channels in principal cells and BK channels in intercalated cells of the CCD were unaffected by the deletion of SGK1. Acute treatment of animals with amiloride produced similar increases in Na excretion and decreases in K excretion in the two genotypes. The absence of changes in ENaC activity suggests compensation for decreased surface expression. Altered K balance in animals lacking SGK1 may reflect defects in ENaC-independent K excretion.
Keywords: aldosterone-induced protein, amiloride-sensitive channels, surface expression, cortical collecting duct, connecting tubule
the serum and glucocorticoid-induced kinase SGK1 is encoded by a gene whose transcription is among those induced by the mineralocorticoid aldosterone in the mammalian kidney (4, 26, 27). Coexpression of SGK1 with the epithelial Na channel (ENaC) increased channel activity in exogenous expression systems (4, 27). Thus induction of the kinase could account at least in part for increased Na reabsorption from urine, a hallmark of aldosterone action. Furthermore stimulation of ENaC could promote K excretion as aldosterone regulates renal K+ secretion in part through depolarization of the apical membrane of principal cells, enhancing the driving force for ion movement from cell to urine (36).
Several mouse models lacking expression of SGK1 have been used to test the involvement of the kinase in Na and K homeostasis. Mice with a global knockout of SGK1 had increased plasma aldosterone levels and mild hyperkalemia (37). The mice showed transient Na wasting when fed with a low-Na diet, accompanied by a decreased amiloride-sensitive lumen-negative electrical potential in isolated cortical collecting ducts. Later studies with the same mouse strain documented K retention when animals were fed chronically with a diet rich in K (35) or administered an acute intravenous K load (21).
A second SGK1-deficient mouse model was created from animals homozygous for a floxed Sgk1 allele crossed with a strain having a global cre-deleter (9). Like the first strain, these mice exhibited increased aldosterone production judged by urinary excretion rates and increased urinary Na losses when fed a low-Na diet. However, amiloride-sensitive Na+ current (INa) measured under whole-cell clamp conditions in CCDs isolated from the animals was larger than those of wild-type mice after 2 days and no different from wild-type after 7 days on a low-Na diet. Despite the lack of demonstrable functional deficiencies, the γENaC subunits in the Sgk1−/− animals showed reduced levels of cleavage, suggesting impaired processing and perhaps trafficking of the channel protein.
The same floxed-Sgk1 mouse strain was subsequently used to produce a kidney-specific sgk1 knockout (8). These animals also exhibited Na wasting and hyperaldosteronism on a low-Na diet. A subsequent study using the same model documented hyperkalemia and defective K excretion in animals on a high-K diet (1). Thus the global and kidney-specific inducible knockouts share the basic phenotype involving alterations in electrolyte homeostasis.
The work reported here had two goals. The first was to examine the SGK1-dependence of the activities of ENaC as well as other channels and transporters involved in renal K secretion in mice on a diet rich in K, to see if defects in these activities could account for K retention under these conditions. The second aim was to test further the hypothesis that ENaC processing and trafficking is defective in animals lacking SGK1. Here we selected the original SGK1-deficient mouse strain for this work due to the extensive whole animal data available for these animals under conditions of K load (20, 35).
METHODS
Animals.
All procedures were approved by the Institutional Animal Care and Use Committee of Weill-Cornell Medical College. All studies used mice in which the Sgk1 gene was deleted as described previously (37) or their wild-type littermates. For K loading studies animals were fed a diet enriched in 5% K or a matched control diet containing 0.5% K (Harlan Teklad) for 7 days. In both diets K was added as 1/3 chloride, 1/3 carbonate, and 1/3 citrate salts; both diets contained 0.3% Na. In some cases mice were implanted with osmotic minipumps (Alzet model 2001) filled with aldosterone to infuse hormone at a rate of 12 μg/day for 7 days. Plasma aldosterone levels were not assessed, but similar doses in rats resulted in concentrations of ∼240 ng/dl (29). In one set of experiments animals were given amiloride (80 mg/l) in their drinking water for 5 days. In some experiments animals were injected ip with 0.05 μmol amiloride in 1 ml H2O or H2O alone. Mice were then kept in metabolic cages for 2.5 h. Blood was usually collected from the right ventricle to avoid hemolysis. In one group of experiments in which blood was obtained from the inferior vena cava, measured values of plasma K concentration were lower, possibly because the sample included a contribution from the renal veins which will have lower levels of the ion as a result of renal K+ secretion. Urine and plasma were analyzed for Na and K by flame photometry (Instrumentation Laboratory IL model 943).
Electrophysiology.
Kidneys were harvested and individual nephron segments dissected and assayed for activity of epithelial Na channels (ENaC), ROMK (Kir1.1) channels, or BK channels using electrophysiology described previously (15, 18, 30).
Western blots.
Kidneys were excised and cells were lysed using a Dounce homogenizer in 2 ml of lysis buffer (in mM): 250 sucrose, 10 triethanolamine HCl, 1.6 ethanolamine and 0.5 EDTA, and 15 μl protease inhibitors cocktail (Sigma-Aldrich) at pH 7.4. Samples of whole-kidney homogenates containing equal amounts of total protein were electrophoresed on 4–12% Bis-Tris gels (Invitrogen) and the proteins were transferred electrophoretically to PVDF membranes. After blocking, membranes were incubated overnight at 4°C with primary antibodies. Dilutions and sources for antibodies were αENaC [1:2,000; J Loffing, U. Zürich (31)] β and γENaC [1:500 (6)]; NCC [1:5,000, A.A. McDonough, U. Southern California (28)], NKCC2 (1:1,000; Chemicon), and NHE3 (1:1,000; Chemicon). Anti-rabbit IgG conjugated with alkaline phosphatase was used as a secondary antibody. Bound antibody was visualized on autoradiography film (HyBlot CL, Denville Scientific) using a chemiluminescence substrate (Western Breeze, Invitrogen). Films were scanned and semiquantitative densitometry of protein bands was carried out using a program designed to resolve closely spaced bands (25). Examples are shown in Figs. 1 and 4. In some cases the loading and transfer processes were checked by measuring overall staining of each lane with Ponceau Red. Data normalized to these values were not substantially different from those obtained assuming equal protein loading and transfer.
Fig. 1.

Expression of ENaC proteins in Sgk1+/+ and Sgk1−/− mice on control diet. A–C: Western blots of kidney homogenates probed with antibodies against αENaC, βENaC, and γENaC. Lanes were loaded with 80 μg (α and βENaC) or 45 μg (γENaC) of total protein. In C one lane is enlarged to illustrate the presence of two cleaved bands at 70 and 65 kDa, along with the densitometry trace used to quantitate their expression. D–F: quantitation of the blots in A–C. Data are represented as means ± SE for 4 animals in each group. **Significant difference compared with Sgk1+/+ with P < 0.01.
Fig. 4.

Expression of ENaC protein in Sgk1+/+ and Sgk1−/− mice fed a high-K diet. A–C: Western blots of kidney homogenates probed with antibodies against αENaC, βENaC, and γENaC. Lanes were loaded with 80 μg of total protein. In B, two lanes are enlarged to illustrate the presence of a diffuse, higher molecular mass material above the main band at 90 kDa, along with the densitometry traces used to quantitate their expression. D–F: quantitation of the blots in A–C. Data are represented as means ± SE for 4–5 animals in each group. *Significant difference compared with Sgk1+/+ with P < 0.05. **Significant difference compared with Sgk1+/+ with P < 0.01.
Biotinylation.
Biotinylation of mouse kidneys followed the procedures previously described for rats (6, 10, 16, 17) with some modifications. Briefly, mice were anesthetized with xylamine/ketamine, the abdominal cavity was opened, and the kidneys were superfused with ice-cold saline. A cannula was inserted into the aorta distal to the renal arteries. Perfusion was started and the left renal vein was opened to allow outflow. A clamp was placed occluding the thoracic aorta. No other vessels were tied. Urine continued to flow from the urethra until the end of the experiment. Kidneys were perfused for 15–20 min with 50 ml of PBS containing 50 mg sulfo-NHS-SS-biotin (Campbell Science, Rockford, IL). They were then perfused for 25–30 min with 120 ml of Tris-buffered saline to quench the biotinylation reaction. Both kidneys were homogenized in lysis buffer (see above) and centrifuged for 2 h at 100,000 g to obtain a membrane pellet. This was suspended in 2 ml of lysis buffer and stored frozen for later processing. The isolation of biotinylated proteins used Neutravidin Ultralink beads (Pierce) as described previously for rat kidneys (12). The biotinylation procedure was technically more difficult in the mouse, and more experiments had to be discarded because urine flow during the perfusion process slowed dramatically or even stopped entirely.
Statistical analysis.
Statistical significance between two groups was assessed by unpaired Student t-tests. P < 0.05 was considered significant.
RESULTS
SGK1 deficiency prevents normal γENaC protein processing in mice on a high-K diet.
Two of the hallmarks of the action of aldosterone on ENaC are increased amounts of αENaC protein, presumably secondary to induction of mRNA encoding the subunit (3, 7, 32), and the appearance of cleaved forms of both α and γENaC (6, 23). We therefore examined the effects of elimination of SGK1 on the expression of the various forms of the ENaC subunits.
WT and knockout animals had similar levels of full-length αENaC when animals ate a control diet; expression of the 30 kDa NH2-terminal cleaved form was low under these conditions and again there was no difference between the two groups (Fig. 1A). The overall content of βENaC was similar in WT and KO animals (Fig. 1B). The most obvious difference between the two genotypes was a diminished amount of full-length γENaC in the knockout animals (Fig. 1C). Unlike the rat, in the mouse the anti-γENaC antibody recognizes two cleaved forms of the subunit, with apparent molecular masses of about 65 and 70 kDa. Under control conditions most of the cleaved subunit was in the 70-kDa, presumably partially cleaved form. There were no differences in the expression of either of the cleaved forms in Sgk1−/− relative to Sgk1+/+ animals.
In the wild-type, the most dramatic effect of K-loading was on the γENaC subunit (Fig. 2). In animals on a high-K diet for 7 days, the amounts of both cleaved forms of the subunit increased, while that of the full-length form decreased slightly, similar to results reported previously in rats (6, 13). For βENaC, the density of the main band at ∼90 kDa decreased slightly but significantly with high K. In addition, there was a small shift in the apparent molecular mass of this band. We do not know the cause of this shift. In addition, high-molecular-mass material, presumably representing fully glycosylated subunit, appeared above the sharp band at 80 kDa in the samples from K-loaded animals; this diffuse band was difficult to discern under control conditions. For αENaC we observed little effect of K-loading on either the full-length or the cleaved form. This is consistent with one previous study in rats (6) but diverges from a second (13). The reasons for the variability are unclear.
Fig. 2.

Effect of a high-K diet on ENaC expression in Sgk1+/+ mice. A–C: Western blots of kidney homogenates probed with antibodies against αENaC, βENaC, and γENaC. Lanes were loaded with 30 μg (αENaC), 60 μg (βENaC), or 40 μg (γENaC) of total protein. D–F: quantitation of the blots in A–C. Data are represented as means ± SE for 4 animals in each group. *Significant difference compared with control diet with P < 0.05. **Significant difference compared with control diet with P < 0.01.
The Sgk1−/− mice responded quite differently to K loading. The absolute amounts of both the full-length and the 70-kDa cleaved forms of the γENaC subunit decreased (Fig. 3); the cleaved form diminished less, such that the ratio of cleaved/full-length forms was significantly higher. The 65-kDa form was weakly expressed and no difference was detected with K loading. The amount of cleaved αENaC in the KO mice also decreased in response to the high-K diet. The amount of the major βENaC species did not change dramatically. The higher molecular mass material was less prominent than in WT.
Fig. 3.

Effect of a high-K diet on ENaC expression in Sgk1−/− mice. A–C: Western blots of kidney homogenates probed with antibodies against αENaC, βENaC, and γENaC. Lanes were loaded with 40 μg (α and γENaC) or 60 (βENaC) of total protein. D–F: quantitation of the blots in A–C. Data are represented as means ± SE for 4 animals in each group. *Significant difference compared with control diet with P < 0.05. **Significant difference compared with control diet with P < 0.01.
Comparing ENaC expression in the two genotypes under high-K conditions (Fig. 4), the most obvious difference was the profoundly diminished amounts of both uncleaved and cleaved γENaC in the Sgk1−/− animals. The knockout mice also had increased expression of full-length αENaC but the amount of cleaved subunit was not significantly different. The high molecular mass βENaC component was more abundant in the wild-type compared with the knockout animals. Thus the expression of the fully processed forms of the β and γ, but not the α subunit were lower in the Sgk1−/− animals relative to Sgk1+/+ under conditions of high K intake.
A complication with these results is that both plasma K and aldosterone levels are higher in the knockout mice (21, 35, 37). We therefore also examined ENaC expression in mice given exogenous aldosterone via osmotic minipumps (Fig. 5). Under these conditions circulating levels of the hormone are expected to be maximal and comparable in the two groups. Levels of both full-length and cleaved αENaC were lower in the Sgk1−/− mice, although the decrease in the cleaved form was more pronounced. Expression of both the 90-kDa band of βENaC as well as that of the putative fully glycosylated species was diminished. Finally, the abundance of all forms of γENaC diminished, although again the largest effect was on the 65-kDa, fully cleaved subunit. Thus the amounts of completely processed forms of all the subunits were reduced in mutant mice relative to wild-type under conditions of exogenous mineralocorticoid administration.
Fig. 5.

Expression of ENaC protein in Sgk1+/+ and Sgk1−/− mice treated with aldosterone. A–C: Western blots of kidney homogenates probed with antibodies against αENaC, βENaC, and γENaC. Lanes were loaded with 60 μg (αENaC), 75 μg (βENaC), or 45 μg (γENaC)of total protein. D–F: quantitation of the blots in A–C. Data are represented as means ± SE for 4 animals in each group. *Significant difference compared with Sgk1+/+ with P < 0.05. **Significant difference compared with Sgk1+/+ with P < 0.01.
SGK1 deficiency reduces ENaC surface expression with aldosterone administration.
The cleaved form of γENaC correlates with ENaC surface expression under a variety of conditions (10, 12, 13, 16). We therefore adapted a biotinylation protocol used previously to assess surface expression of ENaC and other membrane proteins in rat kidney (10, 17) to the mouse. Controls for the procedure are shown in Fig. 6. γENaC was eluted from neutravidin beads in both full-length and cleaved form, but the cleaved form was relatively more abundant in the eluate than in the starting material. No γENaC could be detected in eluates when the kidneys were perfused without biotin (Fig. 6, top). Similar results were obtained another apical membrane protein, NCC (not shown). The ER-resident protein calnexin, which should not be biotinylated by the membrane-impermeant reagent, was observed in small amounts in the eluate (Fig. 6, bottom). Similar results were obtained with the endosomal membrane protein Rab11 (not shown). Figure 7 shows assays of biotinylated fractions from four Sgk1+/+ and four Sgk1−/− mice treated with aldosterone. Similar to findings in the rat, γENaC expressed at the surface was mostly in the cleaved form; only a single cleaved species, presumably corresponding to the fully cleaved subunit, could be resolved. The expression was robust in wild-type mice but strongly reduced in Sgk1−/− animals. βENaC at the surface was also lower in the KO mice, although to a lesser extent. We did not assess αENaC surface expression due to nonspecific binding to the neutravidin beads (17).
Fig. 6.
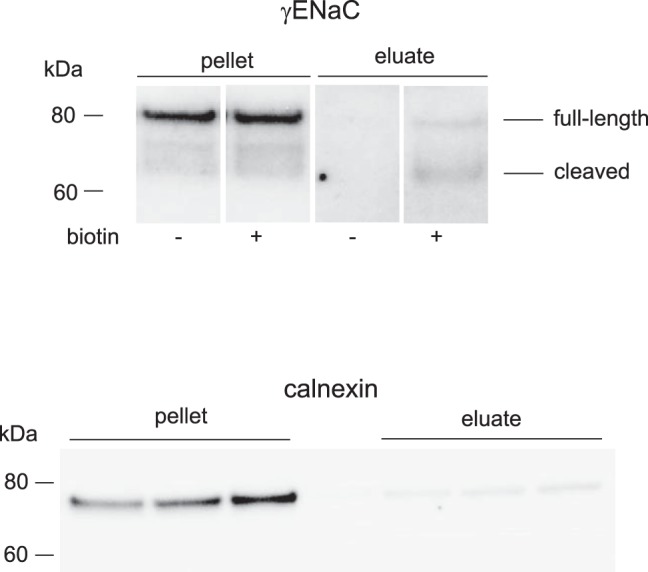
Biotinylation of mouse kidney surface proteins. Top: Western blot of kidney microsomes and neutravidin eluates from mouse kidneys perfused with or without biotin probed with antibodies against γENaC. Lanes were loaded with 50 μg of total microsomal protein, or eluates corresponding to 600 μg of total microsomal protein. Shown are 4 noncontiguous lanes of the same film of a single blot with uniform image processing. Intervening lanes are omitted for clarity. Bottom: Western blot of kidney microsomes and neutravidin eluates from mouse kidneys perfused with biotin probed with antibodies against calnexin. Lanes were loaded with 30 μg of total microsomal protein, or eluates corresponding to 600 μg of total microsomal protein.
Fig. 7.

Surface expression of ENaC protein in Sgk1+/+ and Sgk1−/− mice treated with aldosterone. A and B: Western blots of neutravidin eluates from 750 μg of total microsomal protein were probed with antibodies against βENaC and γENaC. C and D: quantitation of the blots in A and B. Data are represented as means ± SE for 4 animals in each group. **Significant difference compared with Sgk1+/+ with P < 0.01.
Upstream Na transporters are downregulated by high K intake in the presence or absence of SGK1.
We also assessed the expression of other apical Na transporters in the kidneys of Sgk1+/+ and Sgk1−/− mice under control and K-loaded conditions. These transporters can influence renal K secretion by determining the rate of Na+ delivery to K+-secreting segments. Under basal conditions the amounts of NHE3 and NCC were similar in the two genotypes, while the expression of the TALH triple-cotransporter NKCC2 was slightly (30%) but significantly higher in the knockout animals. The abundance of all three of these transporters decreased in response to the high-K diet in both genotypes. The reductions in NHE3 (Fig. 8) and NKCC2 (Fig. 9) were similar for the two groups. In the case of NCC (Fig. 10), the fall was more profound in the KO animals, in agreement with previous results (35). Thus the downregulation of upstream transporters in response to high K intake remains intact, or is even magnified, in the Sgk1−/− mice.
Fig. 8.
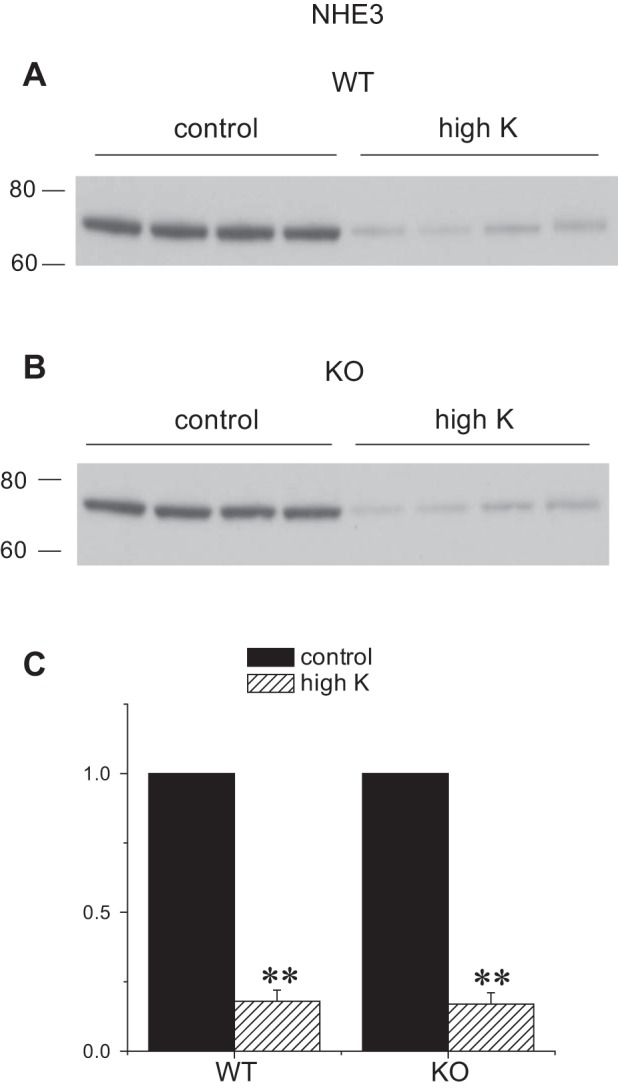
Expression of NHE3 protein in Sgk1+/+ and Sgk1−/− mice under control conditions and fed a high-K diet. A and B: Western blots of kidney homogenates probed with antibodies against NHE3. Lanes were loaded with 60 μg of total protein. C: Quantitation of the blots in A and B. Data are represented as means ± SE for 4 animals in each group. **Significant difference compared with control diet with P < 0.01.
Fig. 9.
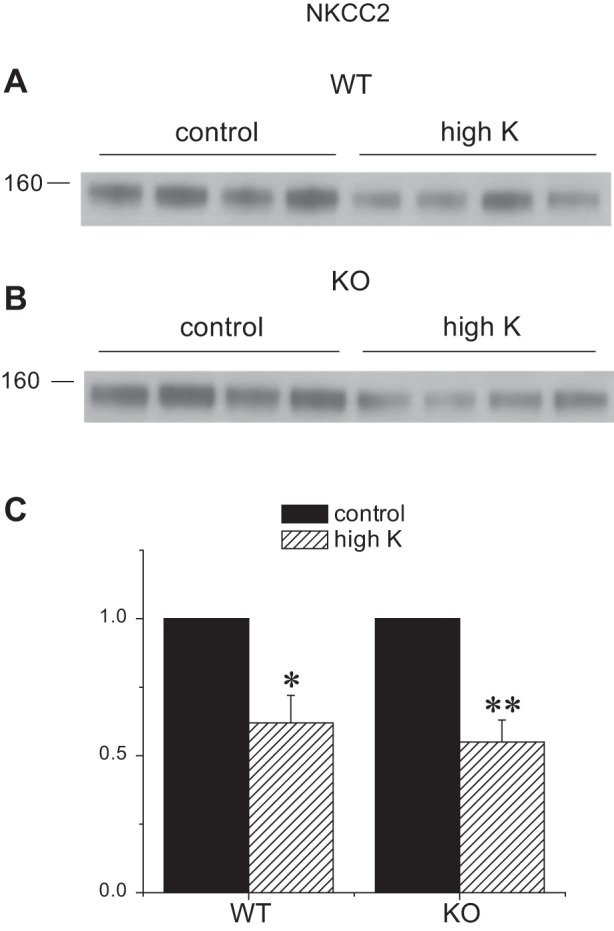
Expression of NKCC2 protein in Sgk1+/+ and Sgk1−/− mice under control conditions and fed a high-K diet. A and B: Western blots of kidney homogenates probed with antibodies against NKCC2. Lanes were loaded with 35 μg of total protein. C: quantitation of the blots in A and B. Data are represented as means ± SE for 4 animals in each group. *Significant difference compared with control diet with P < 0.05. **Significant difference compared with control diet with P < 0.01.
Fig. 10.
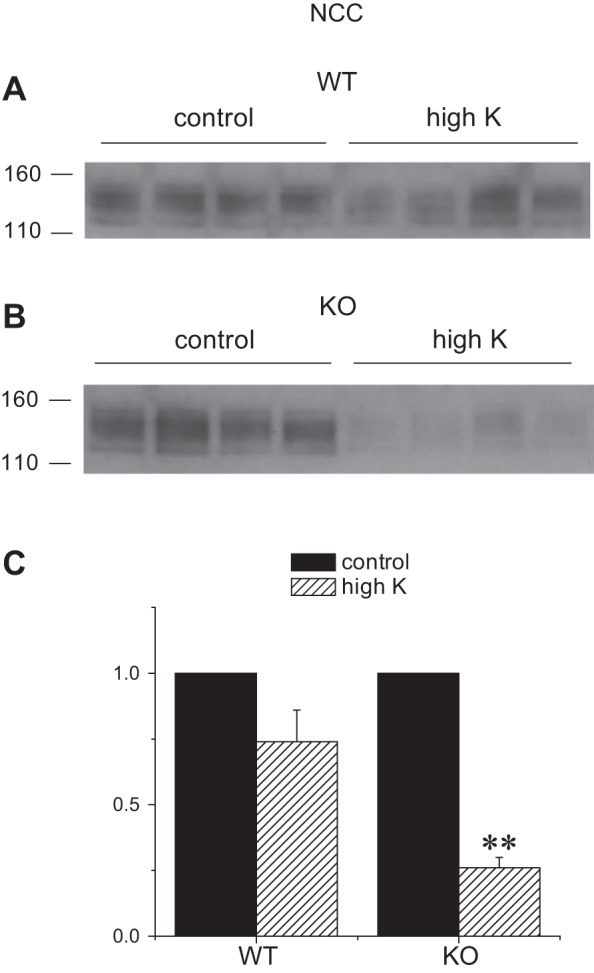
Expression of NCC protein in Sgk1+/+ and Sgk1−/− mice under control conditions and fed a high-K diet. A and B: Western blots of kidney homogenates probed with antibodies against NCC. Lanes were loaded with 40 μg of total protein. C: quantitation of the blots in A and B. Data are represented as means ± SE for 4 animals in each group. **Significant difference compared with control diet with P < 0.01.
ENaC activity is normal in the absence of SGK1.
We also assessed the activity of Na+ channels using whole-cell clamp techniques to measure amiloride-sensitive currents (INa) in principal cells of the mouse CCD. In mice under control conditions these currents were small and in most cells undetectable. In contrast when animals were fed a high-K diet INa was robust in both Sgk1+/+ and Sgk1−/− animals (Fig. 11, A–D). Although under these K loaded conditions there was considerable variation in the magnitude of the currents from animal to animal and from cell to cell within the same animal, there was no significant difference among these two groups when a number of observations from several different mice were compared (Fig. 11I).
Fig. 11.

Amiloride-sensitive currents in Sgk1+/+ and Sgk1−/− mice. A: whole cell currents ± amiloride (10 μM) from a principal cell of a CCD in an Sgk1+/+ mouse fed a high-K diet. B: current-voltage (I-V) relationships for the cell in A. INa represents amiloride-sensitive currents. C: whole cell currents ± amiloride (10 μM) from a principal cell of a CCD in an Sgk1−/− mouse fed a high-K diet. D: I-V relationships for the cell in C. E: whole cell currents ± amiloride (10 μM) from a principal cell of a CNT from an Sgk1+/+ mouse infused with aldosterone (12 μg/day). F: I-V relationships for the cell in E. G: whole cell currents ± amiloride (10 μM) from a principal cell of a CNT from an Sgk1−/− infused with aldosterone (12 μg/day). H: I-V relationships for the cell in G. I: quantitation of amiloride-sensitive currents measured at a −100 mV. Data are represented as means ± SE for 7–24 cells.
As discussed above, a caveat to the interpretation of these results is that both plasma K and plasma aldosterone levels are higher in the knockout mice. Thus although ENaC activity in these animals was similar to that measured in wild-types, it was associated with stronger signals to upregulate the channels. We therefore compared INa in the two genotypes after infusion of animals on a normal diet with aldosterone to equalize the stimuli in the two groups. In this set of experiments we measured currents in principal cells of the connecting tubule, since this is likely to be the most important site of aldosterone-dependent Na reabsorption (15, 24) and we had not previously examined this segment in the SGK1-deficient mice. As shown in Fig. 11, E–H, again there was no significant difference in INa from wild-type and knockout animals.
K channel activity is normal in the absence of SGK1.
K secretion could also be reduced in the Sgk1−/− animals if apical K+-channel conductance is inadequate. To test the activity of Kir1.1 (ROMK) channels we measured currents (ISK) sensitive to TPNQ, a specific blocker of these channels (18). As shown in Fig. 12, principal cells of the CCD of both genotypes had increased ISK when animals were fed a high-K diet. There was no significant difference in the value of the currents between the wild-type and knockout animals under either condition.
Fig. 12.

TPNQ-sensitive currents in Sgk1+/+ and Sgk1−/− mice. Whole cell currents were recorded at a membrane potential of 0 mV from principal cell of CCDs. TPNQ was added at the arrows. A: Sgk1+/+ mouse fed a control diet. B: Sgk1−/− mouse fed a control diet. C: Sgk1+/+ mouse fed a high-K diet D: Sgk1−/− mouse fed a high-K diet. E: quantitation of TPNQ-sensitive currents (ISK). Data are represented as means ± SE for 16–23 cells.
We also assessed the activity of Ca2+-activated BK channels in intercalated cells of these mice. This was estimated as described previously (30) as the whole-cell current activated by depolarization and inhibited by TEA (5 mM). Again there was no measurable difference in the magnitude of the currents in Sgk1+/+ and Sgk1−/− mice fed a high-K diet (Fig. 13).
Fig. 13.

TEA-sensitive whole cell currents in Sgk1+/+ and Sgk1−/− mice. A, top: currents with and without 5 mM TEA from an intercalated cell of a CCD in an Sgk1+/+ mouse fed a high-K diet. A, bottom: I-V relationships. Solid symbols represent TEA-sensitive currents. B, top: currents with and without 5 mM TEA from an intercalated cell of a CCD in an Sgk1+/+ mouse fed a high-K diet. B, bottom: I-V relationships. IBK represents TEA-sensitive currents. C: quantitation of TEA-sensitive currents, measured at +60 mV. Data are represented as means ± SE for 8–10 cells.
Amiloride-sensitive Na excretion is normal in the absence of SGK1.
The electrophysiological assays indicated that apical Na+ and K+ channels in the Sgk1−/− mice have normal activity measured in vitro. To test for changes in channel activity in vivo, we used acute treatment of mice with amiloride to assess the effects of blocking ENaC on Na and K excretion.
As shown in Fig. 14, a single administration of amiloride increased Na excretion and decreased K excretion in both genotypes, although in the WT mice the latter was not statistically significant. In mice fed a high-K diet, the natriuretic effects were larger and in this case were accompanied by marked anti-kaliuresis. The response to the diuretic was not diminished in the knockout animals, consistent with the notion that ENaC activity is close to normal in the Sgk1−/− mice in vivo as well as in vitro.
Fig. 14.

Amiloride-sensitive urinary Na and K excretion in Sgk1+/+ and Sgk1−/− mice under control conditions or fed a high-K diet. Excretion rates were measured with and without amiloride administration 0.05 μmol/mouse together with 1 ml saline in animals fed control (A and B) or high-K (C and D) diets. E and F: changes in excretion rates with amiloride. *Significant difference compared with control with P < 0.05. **Significant difference compared with control with P < 0.01.
Mice lacking SGK1 are hyperkalemic.
Previous studies on Sgk1−/− mice found varying degrees of hyperkalemia (21, 35, 37). We confirmed this basic result (Table 1); plasma K was significantly higher in knockout animals under both control and high-K fed conditions. This pattern differs somewhat from previous findings of a more profound hyperkalemia in K-loaded animals (35). The reasons for this difference are unclear.
Table 1.
Plasma sodium and potassium levels in wild-type and Sgk1−/− mice under different treatment conditions
| Treatment | Genotype | Plasma Na+, meq/l | Plasma K+, meq/l | Animals |
|---|---|---|---|---|
| Control | WT | 156 ± 2 | 3.66 ± 0.28 | 4 |
| KO | 157 ± 1 | 5.96 ± 0.34† | 4 | |
| High-K diet | WT | 156 ± 3 | 3.80 ± 0.29 | 10 |
| KO | 149 ± 2* | 5.46 ± 0.36† | 7 | |
| Aldosterone | WT | 142 ± 3 | 4.01 ± 0.32 | 4 |
| KO | 146 ± 3 | 5.06 ± 0.83 | 4 | |
| Control‡ | WT | 166 ± 1 | 2.80 ± 0.18 | 4 |
| KO | 165 ± 1 | 3.69 ± 0.28* | 4 | |
| Amiloride‡ | WT | 148 ± 3 | 4.26 ± 0.38 | 3 |
| KO | 151 ± 2 | 5.84 ± 0.11* | 3 |
Blood collected from the vena cava.
WT, wild type; KO, Sgk1 knockout. Statistical significance vs. WT:
P < 0.05,
P < 0.01.
To test the idea that hyperkalemia results from defective ENaC activity in vivo we treated animals chronically with amiloride (5). We reasoned that if the hyperkalemia of the knockout animals resulted from defective ENaC activity in vivo, blocking the channels in both genotypes should equalize K+ secretion and plasma K concentration. As expected, plasma K levels were higher in the chronically treated animals (Table 1), but the difference between the genotypes was maintained. A caveat to this experiment is that the knockout animals lost considerable weight under these conditions, 6.6 ± 0.7 g, compared with 0.8 ± 0.5 g for wild-type animals. The animals became sluggish and probably reduced food intake, which would if anything diminish the observed hyperkalemia. We do not have a full explanation for this increased sensitivity, although a similar finding with triamterene was reported previously (2). It could entail impairment of other Na-retaining mechanisms such as the NCC (8, 35) in the knockout animals.
DISCUSSION
ENaC processing and trafficking are altered by SGK1 deletion.
The reduction in the cleaved form of γENaC in Sgk1−/− mice fed a K-rich diet or treated with aldosterone observed here is consistent with a previous study using a different Sgk1-deficient mouse line that reported decreased ratios of cleaved to full-length forms of the subunit in animals undergoing dietary Na depletion (9). We also observed diminished expression of the processed forms of αENaC (assessed by subunit cleavage) and of βENaC (assessed by glycosylation) in knockout compared with wild-type mice.
In studies with rats, the abundance of the cleaved form of γENaC correlates strongly with both surface expression and activity of subunits under a variety of experimental conditions including dietary Na restriction, aldosterone administration, acute Na loading, and increased dietary K intake (10, 12, 13, 16). A likely reason behind this correlation is that the subunits are cleaved in the Golgi apparatus, and this cleavage reflects the rate of trafficking through the Golgi to the apical membrane (11, 22). Direct measurements of surface proteins using biotinylation confirmed that lack of SGK1 diminished γENaC and βENaC surface expression in aldosterone-treated animals (Fig. 7). Previous studies showed similar increases in apical staining of αENaC in wild-type and SGK1-deficient mice on a high-K diet (21). We could not assess αENaC surface expression by biotinylation for technical reasons (10). The mechanisms underlying the reduced processing and surface expression appear to be more complex than simple reversal of aldosterone-dependent processes; in mutant animals the amount of full-length γENaC subunit is decreased relative to wild-type, whereas inhibiting the effects of aldosterone should lead to an increase in this species. The decrease in overall γENaC protein could involve diminished gene transcription, although this point was not tested.
ENaC activity is normal in SGK1-knockout mice.
In these mice normal ENaC activity measured in vitro did not correlate with the impaired processing and surface expression of the channel subunits. We measured normal activity in the CCD of K-loaded mice or the CNT of aldosterone-treated animals. In a previous study using a different knockout mouse line we reported normal activity in CCDs of animals that were chronically Na depleted or treated with aldosterone (9). Thus we found no evidence for decreased ENaC function in two different nephron segments with 3 different experimental treatments using two independent mouse lines. This contrasts with measurements of amiloride-sensitive transepithelial voltage differences which were reduced in the knockout animals compared with controls fed a high-K diet (21).
The dissociation of activity from surface expression implies compensation in the knockout animals by increased transport per channel at the surface. We do not know at this point if this compensation entails an increase in the open probability of active channels or a higher percentage of channels at the surface recruited into the active state. The pathways leading to the compensation also remain to be clarified.
Mice lacking SGK1 are hyperkalemic.
Both previous studies (21, 35, 37) and the work reported here revealed hyperkalemia in the Sgk1−/− mice, although the extent of the changes and the conditions under which they were observed were somewhat variable. Increased plasma K may account, at least in part, for the elevated levels of plasma aldosterone also found in these mice (9, 21, 35, 37). Since SGK1 was ablated globally in these animals, it is possible that hyperkalemia could reflect events outside the kidney. This possibility seems unlikely since a kidney-specific Sgk1 conditional knockout line also exhibits high plasma K (1). Thus it seems that the altered K balance reflects a defect in the renal K+ secretory processes.
Although decreased ENaC activity could produce defects in K+ secretion and increased plasma K, we were unable to document difference in the activity of the channels in vivo or in vitro. Neither were there deficiencies in the apical K+ channels ROMK and BK thought to mediate K secretion in the distal nephron. A previous study found that ROMK protein expression was actually higher in knockout animals compared with wild-type under K-loaded conditions (21). In both cases plasma K+, a signal that likely regulates the apical K+ channels, was higher in the Sgk1−/− mice. Thus it is possible that the hyperkalemia is the result of a reduced sensitivity of ENaC and ROMK to stimuli (plasma K+ and aldosterone) that mediate increased K+ secretion. This idea, however, predicts a decreased response of the channels to a fixed stimulus. At least in the case of aldosterone administration (Fig. 11) the activation of ENaC was similar in the two genotypes.
Reduced K+ secretion can also result from inadequate delivery of Na+ to the K+-secreting parts of the nephron. For example, hyperactivity of the NaCl cotransporter NCC due to mutations in WNK pathways is thought to underlie the hyperkalemia of pseudo-hypoaldosteronism type II (33). Conversely, hypokalemia can result from block of NCC by thiazide diuretics. However, the lack of SGK1 did not impair the downregulation of Na transporters upstream of the K+-secreting segments, at least at the protein level. Expression of both NHE3 and NKCC2 decreased to similar extents in the wild-type and knockout animals on a high-K diet. NCC expression actually decreased much more in the Sgk1−/− mice than in wild-type in response to K loading, confirming previous results (35).
We were therefore unable to identify defects in a transport process that could account for elevated plasma K through classical mechanisms of secretion. However, it is likely that other noncanonical pathways contribute to K homeostasis. In rats, a component of K+ excretion is insensitive to amiloride, which should abolish the driving force for K+ secretion in the aldosterone-sensitive distal nephron, and this component varies with physiological conditions (14). A large fraction of K excretion is amiloride-resistant in the mouse (Fig. 14), and at least under control conditions this fraction tended to be smaller in the Sgk1−/− animals. It is possible that the mechanisms underlying this process, which have not been identified, may be compromised by the absence of SGK1.
Conclusions.
SGK1 is a crucial component in the response of the aldosterone-sensitive distal nephron to process ENaC and to insert the channels into the apical membrane under conditions of extracellular volume depletion, K loading or hyperaldosteronism. However, SGK1 is not required for a robust response of these cells to the mineralocorticoid. This conclusion is consistent with previous findings showing similar increases in blood pressure in response to DOCA + high-salt treatment in wild-type and knockout animals (34), although the gene was required for hypertension associated with insulin-dependent Na retention (19). This indicates that other aldosterone-induced proteins are involved in the process of ENaC activation. These likely work in concert with SGK1 to effect mineralocorticoid-dependent regulation of renal electrolyte excretion.
GRANTS
This work was supported by National Institutes of Health Grants R01-DK-27847 and R01-DK-099284 (to L. G. Palmer) and R01-DK-106102 and P30-DK-079337 (to V. Vallon) and by the Department of Veterans Affairs (V. Vallon).
DISCLOSURES
V. Vallon has received within the past 12 mo research grant support for basic science studies from Boehringer Ingelheim Pharma, AstraZeneca, Fresenius, Bayer Pharma, and Janssen Pharmaceutical. V. Vallon has served as a consultant and received honoraria from Boehringer Ingelheim, Janssen Pharmaceutical, Lilly, and Merck.
AUTHOR CONTRIBUTIONS
L.Y., G.F., F.L., D.K., V.V., and L.G.P. conception and design of research; L.Y., G.F., and L.G.P. performed experiments; L.Y., G.F., and L.G.P. analyzed data; L.Y., G.F., V.V., and L.G.P. interpreted results of experiments; L.Y. prepared figures; L.Y., G.F., F.L., V.V., and L.G.P. edited and revised manuscript; L.Y., G.F., F.L., D.K., V.V., and L.G.P. approved final version of manuscript; L.G.P. drafted manuscript.
REFERENCES
- 1.Al-Qusairi L, Basquin D, Roy A, Stifanelli M, Rajaran RD, Debonneville A, Nita I, Maillard MP, Loffing J, Subramanya AR, Staub O. Renal-tubular SGK1 deficiency causes impaired K+ excretion via the loss of regulation of NEDD4-2/WNK1 and ENaC. Am J Physiol Renal Physiol 311: F330–F342, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Artunc F, Ebrahim A, Siraskar B, Nasir O, Rexhepaj R, Amann K, Friedrich B, Risler T, Lang F. Responses to diuretic treatment in gene-targeted mice lacking serum- and glucocorticoid-inducible kinase 1. Kidney Blood Press Res 32: 119–127, 2009. [DOI] [PubMed] [Google Scholar]
- 3.Asher C, Wald H, Rossier BC, Garty H. Aldosterone-induced increase in the abundance of Na+ channel subunits. Am J Physiol Cell Physiol 271: C605–C611, 1996. [DOI] [PubMed] [Google Scholar]
- 4.Chen SY, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, Firestone GL, Verrey F, Pearce D. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci USA 96: 2514–2519, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cremades A, Vicente-Ortega V, Penafiel R. Influence of murine renal sexual dimorphism on amiloride-induced hyperkalemia. Nephron Physiol 95: p57–p66, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Ergonul Z, Frindt G, Palmer LG. Regulation of maturation and processing of ENaC subunits in the rat kidney. Am J Physiol Renal Physiol 291: F683–F693, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Escoubet B, Coureau C, Bonvalet JP, Farman N. Noncoordinate regulation of epithelial Na channel and Na pump subunit mRNAs in kidney and colon by aldosterone. Am J Physiol Cell Ph ysiol 272: C1482–C1491, 1997. [DOI] [PubMed] [Google Scholar]
- 8.Faresse N, Lagnaz D, Debonneville A, Ismailji A, Maillard M, Fejes-Toth G, Naray-Fejes-Toth A, Staub O. Inducible kidney-specific Sgk1 knockout mice show a salt-losing phenotype. Am J Physiol Renal Physiol 302: F977–F985, 2012. [DOI] [PubMed] [Google Scholar]
- 9.Fejes-Tóth G, Frindt G, Náray-Fejes-Tóth A, Palmer LG. Epithelial Na+ channel activation and processing in mice lacking SGK1. Am J Physiol Renal Physiol 294: F1298–F1305, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Frindt G, Ergonul Z, Palmer LG. Surface expression of epithelial Na channel protein in rat kidney. J Gen Physiol 131: 617–627, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frindt G, Gravotta D, Palmer LG. Regulation of ENaC trafficking in rat kidney. J Gen Physiol 147: 217–227, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frindt G, Palmer LG. Acute effects of aldosterone on the epithelial Na channel in rat kidney. Am J Physiol Renal Physiol 308: F572–F578, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frindt G, Palmer LG. Effects of dietary K on cell-surface expression of renal ion channels and transporters. Am J Physiol Renal Physiol 299: F890–F897, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frindt G, Palmer LG. K+ secretion in the rat kidney: Na+ channel-dependent and -independent mechanisms. Am J Physiol Renal Physiol 297: F389–F396, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frindt G, Palmer LG. Na channels in the rat connecting tubule. Am J Physiol Renal Physiol 286: F669–F674, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Frindt G, Palmer LG. Regulation of epithelial Na+ channels by adrenal steroids: Mineralocorticoid and glucocorticoid effects. Am J Physiol Renal Physiol 302: F20–F26, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frindt G, Palmer LG. Surface expression of sodium channels and transporters in rat kidney: effects of dietary sodium. Am J Physiol Renal Physiol 297: F1249–F1255, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frindt G, Shah A, Edvinsson JM, Palmer LG. Dietary K regulates ROMK channels in connecting tubule and cortical collecting duct of rat kidney. Am J Physiol Renal Physiol 296: F347–F354, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang DY, Boini KM, Friedrich B, Metzger M, Just L, Osswald H, Wulff P, Kuhl D, Vallon V, Lang F. Blunted hypertensive effect of combined fructose and high-salt diet in gene-targeted mice lacking functional serum- and glucocorticoid-inducible kinase SGK1. Am J Physiol Regul Integr Comp Physiol 290: R935–R944, 2006. [DOI] [PubMed] [Google Scholar]
- 20.Huang DY, Boini KM, Osswald H, Friedrich B, Artunc F, Ullrich S, Rajamanickam J, Palmada M, Wulff P, Kuhl D, Vallon V, Lang F. Resistance of mice lacking the serum- and glucocorticoid-inducible kinase SGK1 against salt-sensitive hypertension induced by a high-fat diet. Am J Physiol Renal Physiol 291: F1264–F1273, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Huang DY, Wulff P, Volkl H, Loffing J, Richter K, Kuhl D, Lang F, Vallon V. Impaired regulation of renal K+ elimination in the sgk1-knockout mouse. J Am Soc Nephrol 15: 885–891, 2004. [DOI] [PubMed] [Google Scholar]
- 22.Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD, Johnson JP, Stockand JD, Kleyman TR. Epithelial sodium channels are activated by furin-dependent proteolysis. J Biol Chem 279: 18111–18114, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest 104: R19–R23, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meneton P, Loffing J, Warnock DG. Sodium and potassium handling by the aldosterone-sensitive distal nephron: the pivotal role of the distal and connecting tubule. Am J Physiol Renal Physiol 287: F593–F601, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Mitov MI, Greaser ML, Campbell KS. GelBandFitter—a computer program for analysis of closely spaced electrophoretic and immunoblotted bands. Electrophoresis 30: 848–851, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller OG, Parnova RG, Centeno G, Rossier BC, Firsov D, Horisberger JD. Mineralocorticoid effects in the kidney: correlation between alphaENaC, GILZ, and Sgk-1 mRNA expression and urinary excretion of Na+ and K+. J Am Soc Nephrol 14: 1107–1115, 2003. [DOI] [PubMed] [Google Scholar]
- 27.Náray-Fejes-Tóth A, Canessa C, Cleaveland G, Aldrich G, Fejes-Tóth G. sgk is an aldosterone-induced kinase in the renal collecting duct Effects on epithelial Na+ channels. J Biol Chem 274: 16973–16978, 1999. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen MT, Lee DH, Delpire E, McDonough AA. Differential regulation of Na+ transporters along nephron during ANG II-dependent hypertension: distal stimulation counteracted by proximal inhibition. Am J Physiol Renal Physiol 305: F510–F519, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmer LG, Antonian L, Frindt G. Regulation of apical K and Na channels and Na/K pumps in rat cortical collecting tubule by dietary K. J Gen Physiol 104: 693–710, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmer LG, Frindt G. High-conductance K channels in intercalated cells of the rat distal nephron. Am J Physiol Renal Physiol 292: F966–F973, 2007. [DOI] [PubMed] [Google Scholar]
- 31.Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int 83: 811–824, 2013. [DOI] [PubMed] [Google Scholar]
- 32.Stokes JB, Sigmund RD. Regulation of rENaC mRNA by dietary NaCl and steroids: organ, tissue, and steroid heterogeneity. Am J Physiol Cell Physiol 274: C1699–C1707, 1998. [DOI] [PubMed] [Google Scholar]
- 33.Subramanya AR, Yang CL, McCormick JA, Ellison DH. WNK kinases regulate sodium chloride and potassium transport by the aldosterone-sensitive distal nephron. Kidney Int 70: 630–634, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Vallon V, Huang DY, Grahammer F, Wyatt AW, Osswald H, Wulff P, Kuhl D, Lang F. SGK1 as a determinant of kidney function and salt intake in response to mineralocorticoid excess. Am J Physiol Regul Integr Comp Physiol 289: R395–R401, 2005. [DOI] [PubMed] [Google Scholar]
- 35.Vallon V, Schroth J, Lang F, Kuhl D, Uchida S. Expression and phosphorylation of the Na+-Cl− cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol 297: F704–F712, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weinstein AM. Potassium excretion during antinatriuresis: perspective from a distal nephron model. Am J Physiol Renal Physiol 302: F658–F673, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wulff P, Vallon V, Huang DY, Volkl H, Yu F, Richter K, Jansen M, Schlunz M, Klingel K, Loffing J, Kauselmann G, Bosl MR, Lang F, Kuhl D. Impaired renal Na+ retention in the sgk1-knockout mouse. J Clin Invest 110: 1263–1268, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]