

Abstract
Acquired renal scarring occurs in a subset of patients following febrile urinary tract infections and is associated with hypertension, proteinuria, and chronic kidney disease. Limited knowledge of histopathology, immune cell recruitment, and gene expression changes during pyelonephritis restricts the development of therapies to limit renal scarring. Here, we address this knowledge gap using immunocompetent mice with vesicoureteral reflux. Transurethral inoculation of uropathogenic Escherichia coli in C3H/HeOuJ mice leads to renal mucosal injury, tubulointerstitial nephritis, and cortical fibrosis. The extent of fibrosis correlates most significantly with inflammation at 7 and 28 days postinfection. The recruitment of neutrophils and inflammatory macrophages to infected kidneys is proportional to renal bacterial burden. Transcriptome analysis reveals molecular signatures associated with renal ischemia-reperfusion injury, immune cell chemotaxis, and leukocyte activation. This murine model recapitulates the cardinal histopathological features observed in humans with acquired renal scarring following pyelonephritis. The integration of histopathology, quantification of cellular immune influx, and unbiased transcriptional profiling begins to define potential mechanisms of tissue injury during pyelonephritis in the context of an intact immune response. The clear relationship between inflammatory cell recruitment and fibrosis supports the hypothesis that acquired renal scarring arises as a consequence of excessive host inflammation and suggests that immunomodulatory therapies should be investigated to reduce renal scarring in patients with pyelonephritis.
Keywords: urinary tract infection, pyelonephritis, inflammation, fibrosis, mucosal injury, vesicoureteral reflux
acute pyelonephritis (APN), defined by bacterial infection of the kidney, leads to 57,000 hospitalizations and $1.2 billion in annual hospital charges in the United States (20). Despite diagnosis of APN and institution of antibiotic therapy, a subset of patients develops chronic kidney disease due to renal scarring (2, 28). Strategies to predict which patients with APN are at risk for these long-term sequelae or to limit renal scarring remain to be determined. For instance, antibiotic prophylaxis reduced urinary tract infection (UTI) recurrence but not acquired renal scarring in a recent randomized, placebo-controlled clinical trial (21).
Risk factors for APN include urinary tract obstruction, bladder/bowel dysfunction, chronic cystitis, defects in innate immunity, and vesicoureteral reflux (VUR) (2, 29). Primary VUR is defined as retrograde urine flow toward the kidney, due to a congenital defect in the ureterovesical junction. In a large, modern cohort of US children with VUR and history of UTI, 10% had evidence of renal scars, of which near 79% were acquired over the 2-year study period (29). The incidence of recurrent UTI, APN, and acquired renal scarring in this subset of patients with VUR has prompted trials to prevent UTI through antibiotic prophylaxis and/or surgical repair of VUR (6, 10, 21). However, these interventions have not reduced acquired renal scarring (7, 21, 44).
The laboratory mouse is the best studied animal model for experimental UTI (24). Endotoxin-hyporesponsive C3H/HeJ mice are especially susceptible to cortical scars resembling those seen in children with acquired renal scarring after APN (5). Likewise, experimental UTI in mice deficient for the chemokine receptor Cxcr2, a key mediator of neutrophil recruitment, leads to increased rates of APN and acquisition of renal scars (5, 46). These landmark studies provide insight regarding potential roles for pattern recognition receptors and leukocyte recruitment during APN pathogenesis. However, since most published models of acquired cortical scarring rely on defects in host immunity, it is difficult to delineate the complete spectrum of immune responses, cell types, and inflammation that lead to renal fibrosis.
A greater understanding of the pathophysiology of APN and renal fibrosis in immunocompetent hosts will lead to strategies to prevent recurrent infection and parenchymal scarring. We hypothesized that C3H/HeOuJ mice are prone to renal scarring following APN, based on historical accounts of sustained renal bacterial burden and higher renal inflammatory scores in this strain, despite an intact response to bacterial endotoxin (22, 23). By developing a model of acquired cortical scarring in immunocompetent C3H/HeOuJ mice, we have begun to define the molecular processes that distinguish infections that lead to renal scars from those that eventually resolve through analysis of histopathology, immune cell influx, and transcriptional responses to infection.
METHODS
Experimental APN.
Experiments followed Institutional Animal Care and Use Committee (IACUC) regulations at Nationwide Children’s Hospital, Columbus, OH. Six-week-old female C3H/HeOuJ mice (Jackson Laboratories, Bay Harbor, ME) were acclimated in our facility for 1 wk and then inoculated transurethrally with 108 colony forming units (CFU) of uropathogenic Escherichia coli (UPEC) strain CFT073 in 50 µl phosphate-buffered saline (PBS) under isoflurane anesthesia (27). The CFT073 inoculate was isolated from the blood of a woman with APN (33). It is a type I fimbriated, hlyA+, cnf1− UPEC strain with serotype O6:H1 (31). Control animals underwent intravesical injection of PBS alone. Animals were observed daily for signs of morbidity but his did not occur in either infected or uninfected mice; in particular, there were no deaths following experimental APN. At necropsy at 7 or 28 dpi, organs were sterilely dissected under deep anesthesia, and animals were euthanized by cervical dislocation. Kidney and bladder homogenates were cultured and colonies enumerated as described (1).
Flow cytometry.
Single-cell kidney suspensions were stained with fluorophore-conjugated antibodies and analyzed on an LSRI II cytometer (BD Biosciences, San Jose, CA) (4). The following antibodies were used (BioLegend, San Diego, CA): anti-CD11b-Alexa Fluor 700, M1/70; anti-F4/80-APC-Cy7, BM8; anti-Ly6G-PerCP-Cy5.5, 1A8; anti-Ly6C−eFluor 450, HK1.4; and anti-CD45-PE, 30-F11. Viable cells were identified on the basis of exclusion of reactive dye (L23105; Invitrogen, Carlsbad, CA). Compensation and data analysis were conducted with FlowJo software (FlowJo, Ashland, OR).
Histopathology.
Kidneys were fixed in 4% paraformaldehyde, embedded in paraffin, sectioned serially at 4 µm, and stained with hematoxylin and eosin (H&E) or Masson’s trichrome. Inflammation and fibrosis grades were blindly evaluated by two veterinary pathologists (B. Becknell, R. Kohnken) using an established scale (5, 22, 26). Mucosal injury was scored using the scale in Table 1. Only sections with an intact renal pelvis were graded.
Table 1.
Grading system used to assess mucosal injury
| Score | Description |
|---|---|
| 0 | Within normal limits (stratified squamous epithelium, 1–3 cells thick) |
| 1 | One to several foci of hyperplasia (5+ cells in thickness) |
| 2 | Widespread hyperplasia with variable degree of superficial erosion |
| 3 | Extensive superficial to deep erosion with up to one small ulcer |
| 4 | Extensive deep erosion with multiple small ulcers |
Cystograms.
VUR status was evaluated by terminal cystogram as described previously in 6-wk-old female mice acquired from Jackson Laboratories (C57BL/6J and C3H/HeOuJ) or Taconic Biosciences (C3H/HeN; Albany, NY) (5, 15, 49).
Immunofluorescence microscopy.
Immunostaining was done on fixed, deparaffinized sections following citrate antigen retrieval (1, 3). The following primary stains were used: α-smooth muscle actin (α-SMA; Dako, UK); biotinylated Lotus tetragonolobus lectin (Vector Laboratories, Burlingame, CA); E. coli (US Biological, Salem, MA); Aqp2 (Santa Cruz Biotechnology, Dallas, TX); and Ly6G (BioLegend). Cy3 or Alexa Fluor 488-labeled secondary antibodies or streptavidin were used for detection (Jackson ImmunoResearch, West Grove, PA). Slides were mounted with VECTASHIELD containing DAPI for nuclear visualization (Vector Laboratories). Micrographs were captured using a BZ-II scope (Keyence, Itasca, IL). The α-SMA staining was quantified at ×40 using BZ-II software (Keyence) in bright extraction mode and reported as a percentage of the sample surface area.
Transcriptome analysis.
Tissues were disrupted with a TissueLyser II Instrument (Qiagen, Carlsbad, CA), and total RNA was isolated (mirVana Kit; Life Technologies). RNA was labeled and hybridized to the Aligent SurePrint G3 Mouse GE 8x60K Microarray, then scanned. Raw data were filtered for outliers and normalized to remove nonbiological variation. Genes with significant differential expression, defined by <10% false discovery rate (FDR) and P value < 0.05, were uploaded to Ingenuity Pathway Analysis (Qiagen). Microarray data are deposited at https://www.ncbi.nlm.nih.gov/geo/; accession # GSE76469.
qRT-PCR.
Quantitative (q) RT-PCR reactions were performed with SYBR Green using primers in Table 2 (45). PCR products were detected with the 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA). Expression changes were calculated by the 2−ΔΔCT method, normalizing to pooled, uninfected kidney cDNA (43).
Table 2.
qRT-PCR primers
| Gene | Forward | Reverse |
|---|---|---|
| Lcn2 | 5′-atatgcacaggtatcctcag-3′ | 5′-gaaacgttccttcagttcag-3′ |
| Gapdh | 5′-ctggagaaacctgccaagta-3′ | 5′-tgttgctgtagccgtattca-3′ |
| C1qb | 5′-agaacaccaggattccatac-3′ | 5′-catggagaaaacctagaagc-3′ |
| C1qc | 5′-aaacagtatgttcctgtctg-3′ | 5′-ccatcctgagggtgtaag-3′ |
| C3 | 5′-tagtgattgaggatggtgtg-3′ | 5′-ctaccatgtcactacctgag-3′ |
| Clu | 5′-cttaagagaaggtgaagatgac-3′ | 5′-caggattgttggttgaacag-3′ |
| Col3a1 | 5′-actcaagagtggagaatactg-3′ | 5′-aacatgtttcttctctgcac-3′ |
| Ctss | 5′-cattcctccttcttcttctac-3′ | 5′-ccttgatcaccaaagttaagg-3′ |
| Emr1 | 5′-tttcaaatggatccagaagg-3′ | 5′-cagaaggaagcataaccaag-3′ |
| Laptm5 | 5′-ctagacttctgtttgagtatcc-3′ | 5′-ggtaattcatgtggttcatgg-3′ |
| Ltbp2 | 5′-aaatcaaagtcgtcttcacc-3′ | 5′-gcagaaatagatacggaagc-3′ |
| Dpysl3 | 5′-ccaggaagaaaggaaatgtg-3′ | 5′-cttactccagtaatgagttcc-3′ |
| Lyz1 | 5′-caagatctaagaatgcctgtg-3′ | 5′-ttccgaatatactgggacag-3′ |
| Serpina3 g | 5′-tgctggtgaactacatctac-3′ | 5′-tttcctgtgtatttcagctc-3′ |
| Vcam1 | 5′-actgattatccaagtctctcc-3′ | 5′-ccatccacagactttaatacc-3′ |
Statistics.
qRT-PCR and flow cytometry data were compared by Mann-Whitney U-test (GraphPad, La Jolla, CA). Spearman’s coefficient measured the correlations between bacterial burden and histopathology scores. Gene expression network P values and Z-scores were generated by Ingenuity Pathway Analysis (Qiagen). P values of < 0.05 were considered significant.
RESULTS
C3H/HeOuJ mice develop sustained renal bacterial burden following transurethral inoculation with UPEC.
To establish experimental pyelonephritis, we transurethrally inoculated C3H/HeOuJ females with UPEC strain CFT073 (27). Animals were euthanized 1, 3, 7, 14, or 28 days postinoculation (dpi) for evaluation of bacterial burden, and compared with uninfected controls (n = 7–18/group/time point). We confirmed sterility of the urinary tract in the absence of experimental UTI, arguing against spontaneous UTI in C3H/HeOuJ. UPEC inoculation led to sustained renal and bladder bacterial burden (Fig. 1A), with ≥60% of these organs harboring UPEC 28 dpi (Fig. 1B).
Fig. 1.
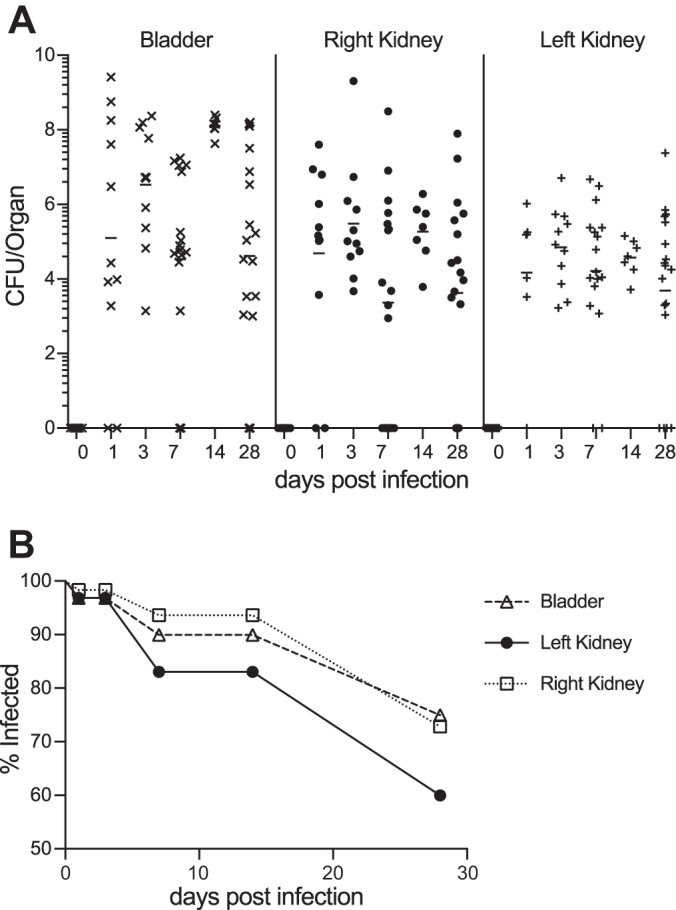
Persistent UPEC colonization of the C3H/HeOuJ mouse urinary tract. A: persistent urinary tract colonization in C3H/HeOuJ mice following transurethral UPEC inoculation. Bacterial recovery from each organ is shown for selected days postinfection (dpi). The horizontal bars indicate the geometric mean for each data set. The limit of detection was 100 CFU. B: the frequency (% infected) of urinary tract organs with UPEC recovery is depicted over time.
Close relatives of the C3H/HeOuJ strain exhibit 100% incidence of primary VUR, in part due to a shortened intravesical ureter (35). This observation led us to perform terminal cystograms in adult females (5, 17, 22). For comparison, we evaluated C57BL/6J and C3H/HeN mice, which have a reported VUR incidence of 0% and 100%, respectively (5, 35). We observed bilateral VUR in ~80% of C3H/HeOuJ and C3H/HeN adults, whereas VUR was absent in all C57BL/6J mice (P < 0.05, Fisher’s exact test; Table 3). Thus the susceptibility of C3H/HeOuJ mice to renal colonization may be due, in part, to the presence of VUR.
Table 3.
VUR Status (%) in mouse strains
Fisher exact test, P < 0.05 vs. C57BL/6J.
UPEC localize to medullary collecting ducts and cortical abscesses.
UPEC primarily occupied two distinct niches within the infected kidney at 7 dpi. In the medulla, we observed UPEC localized to the lumen of collecting tubules, either as individual bacteria (Fig. 2A) or embedded within cellular casts that filled the collecting tubule space (Fig. 2B). The majority of the host cells embedded within the casts were Ly6G+ (Fig. 2C), suggesting that casts were formed by degranulating neutrophils and luminal UPEC. In the renal cortex, UPEC colocalized with Ly6G+ neutrophils in interstitial abscesses (Fig. 2D).
Fig. 2.

UPEC localization within C3H/HeOuJ kidneys. A: UPEC (green) localize within collecting ducts indicated by aquaporin (red) staining. Cell nuclei are indicated in blue. B: UPEC aggregates accumulate in cellular casts (*) within medullary collecting ducts. Tubular epithelial cells are identified by E-cadherin (red) staining. C: UPEC are embedded in intraluminal casts (*) composed of cells expressing the neutrophil marker, Ly6G (red). D: UPEC (green) within a cortical abscess (outlined with white dashes) containing numerous neutrophils (Ly6G; red). u, Urinary space. All micrographs were taken using the ×60 objective. Scale bar indicates 60 µm.
Mucosal injury is a prominent feature in APN.
The detection of interstitial abscesses led us to hypothesize that APN leads to mucosal injury, compromising the urine permeability barrier and permitting UPEC to disseminate into the underlying interstitium of the renal cortex. We therefore investigated changes in renal mucosal integrity that develop during APN. The mucosal lining of the uninfected papilla and renal pelvis was cuboidal and 1–2 cell layers thick (Fig. 3A). In marked contrast, sustained infection led to submucosal nests of inflammatory cells and engorged peritubular capillaries 7 dpi (Fig. 3B), as well as mucosal hyperplasia and variable injury ranging from superficial erosion to ulceration (Fig. 3B). At 28 dpi, we identified widespread mucosal hyperplasia and infiltrating immune cells in the pelvic lining (Fig. 3C). Furthermore, the underlying interstitium contained an intense infiltration of mononuclear cells along with submucosal collagen deposition (Fig. 3C). Mucosal injury grades significantly increased in C3H/HeOuJ kidneys at both 7 and 28 dpi compared with uninfected kidneys (Mann-Whitney U-test, P < 0.05, Fig. 3D).
Fig. 3.

Mucosal injury and hyperplasia in UPEC infected C3H/HeOuJ kidneys. A: renal mucosa with 1–2 layers of cuboidal epithelium lining the pelvis (P) in the absence of UTI; the black dashed line demarcates the urothelial basement membrane. B: early APN (7 dpi), indicated by mucosal hyperplasia with superficial erosion, submucosal neutrophilic infiltrate, engorged peritubular capillaries (dotted white circle), and focus of mucosal ulceration (black arrow). C: chronic APN (28 dpi), demonstrated by increased mucosal hyperplasia, heavy submucosal mononuclear cell infiltrate, and collagen deposition. D: the extent of mucosal injury is significantly increased 7 and 28 dpi, compared with uninfected kidneys. Mucosal injury was graded using a scale (Table 5) as described in materials and methods. *P < 0.05, Mann-Whitney U-test. All micrographs were acquired with the ×40 objective. Scale bar indicates 100 µm.
Cortical tubulointerstitial nephritis assumes a wedge-shaped profile in mice, as observed in human APN.
UPEC infection caused significant renal inflammation at 7 and 28 dpi (Fig. 4). Twenty percent (5/25) of C3H/HeOuJ kidneys possessed cortical lesions with a triangular profile at these times, consistent with infectious spread to all nephrons exiting via a common collecting duct (Fig. 5A) (11, 12). In some kidneys, degenerating tubules in the papilla were surrounded by an eosinophilic rim of inflammatory cells consistent with papillary necrosis (Fig. 5B). Dilated collecting ducts were engorged with neutrophil-rich casts, with evidence of atrophy and occasional necrosis within the ductal epithelium (Fig. 5C). The cortex contained numerous collections of encapsulated neutrophils characteristic of an abscess (Fig. 5D). Intact tubules in necrotic zones contained granular hematoxylin-positive structures consistent with UPEC morphology (Fig. 5E), suggesting that UPEC forms biofilms within necrotic tubules. We observed congestion of peritubular capillaries and fibrillar eosinophilic plugs conforming to the shape of the venules, suggesting that thrombi can develop in vessels with sluggish blood flow near sites of APN (Fig. 5F).
Fig. 4.
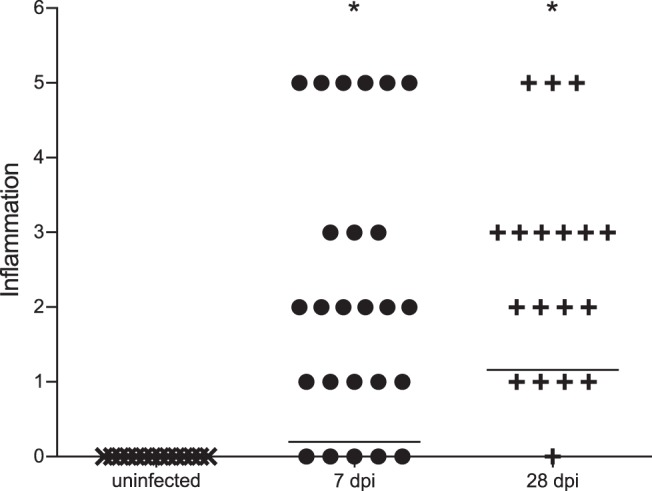
Increased inflammation score in kidneys of UPEC-infected C3H/HeOuJ mice 7 and 28 dpi, compared with uninfected control mice. Horizontal lines demonstrate the group mean values, indicating that the degree of inflammation increases over time. Inflammation was graded using an established scale as described in materials and methods. *P < 0.05, Mann-Whitney U-test.
Fig. 5.

UPEC infection leads to tubulointerstitial nephritis 7 dpi. A: H&E staining identifies a cortical lesion with triangular profile, outlined in white dashed lines, indicating ascending spread of infection to all nephrons that exit into a common collecting duct. Multiple cortical abscesses (x) are composed of intact and degenerating neutrophils (×4 objective). B: papillary necrosis (*) and tubular casts (arrows) are evident in the medulla (×4 objective). The necrotic core (pale pink) is surrounded by a dense rim of neutrophils (purple). C: neutrophil-rich casts (#) occupy dilated medullary collecting ducts (×20 objective). D: degenerating neutrophils cluster densely in a cortical abscess (×20 objective). E: cortical tubules are lined by necrotic (pink) epithelial cells (*) and surrounded by numerous neutrophils. Tubules in necrotic zones contain large UPEC colonies exhibiting a granular, pale blue appearance (arrowheads) (×20 objective). F: intravenous thrombus (T) formation in a venule within the markedly inflamed cortex (×4 objective). Scale bars: ×4, 300 µm; ×20, 100 µm.
Cellular immune responses to APN.
To elucidate cellular immune responses to sustained renal infection, we determined the type and magnitude of leukocyte recruitment in infected C3H/HeOuJ kidneys by flow cytometry. APN led to significant increases in total leukocytes and neutrophils at 7 and 28 dpi, compared with uninfected control kidneys (Fig. 6). Macrophage influx was observed at 7 dpi, with recruitment of both Ly6C+ and Ly6C− subsets, but these populations were not elevated at 28 dpi. We noted significant correlations between UPEC burden and absolute numbers of leukocytes, neutrophils (defined by Ly6G expression), and Ly6C+ macrophages at both 7 and 28 dpi (Table 4).
Fig. 6.

Leukocyte recruitment during APN. Flow cytometry demonstrates significant enrichment of neutrophils (PMN), as well as Ly6C+ and Ly6C− macrophage (Mac) subsets in kidneys during APN (n = 10), compared with control kidneys (n = 4). The y-axis depicts the absolute number of viable events/kidney. *P < 0.05, Mann-Whitney U-test vs. uninfected kidneys for each leukocyte population.
Table 4.
Bacterial burden correlates with renal leukocyte populations
| Comparison | 7 dpi |
28 dpi |
||
|---|---|---|---|---|
| R | P | R | P | |
| Log(renal CFU) vs. total leukocytes | 0.52001 | 0.00058 | 0.62334 | 0.00002 |
| Log(renal CFU) vs. neutrophils | 0.58233 | 0.00706 | 0.82047 | 0.00001 |
| Log(renal CFU) vs. total macrophages | 0.41843 | 0.06634 | 0.32266 | 0.16529 |
| Log(renal CFU) vs. Ly6C− macrophages | 0.23867 | 0.31087 | 0.08143 | 0.73288 |
| Log (renal CFU) vs. Ly6C+ macrophages | 0.4781 | 0.03299 | 0.74364 | 0.00017 |
R, Spearman’s Rho; P, 2-tailed P value from Spearman’s test; significant values are in bold; nonsignificant values are in italics.
Sustained renal infection leads to interstitial fibrosis.
Using Masson’s trichrome stain to identify collagen deposition, we observed a spectrum of fibrosis in APN, ranging from localized submucosal collagen deposits to large cortical scars (Fig. 7). The fibrosis grade was significantly higher in C3H/HeOuJ kidneys at 7 and 28 dpi, compared with uninfected kidneys (Mann-Whitney U-test, P < 0.05, Fig. 8A). Twenty percent (5/25) of C3H/HeOuJ kidneys contained cortical scars, which typically occurred along with increased periglomerular fibrosis. We next performed immunostaining for α-SMA, a marker of activated myofibroblasts, a cell type implicated in renal collagen production (47). Quantification of α-SMA reactivity demonstrated significant increases in infected C3H/HeOuJ kidneys at 7 and 28 dpi, compared with uninfected controls (Mann-Whitney U-test, P < 0.05, Fig. 8B). Cortical α-SMA expression was limited to vascular smooth muscle in uninfected C3H/HeOuJ kidneys (Fig. 8C), whereas severely affected C3H/HeOuJ kidneys exhibited expanded, confluent cortical α-SMA expression, accompanied by loss of the proximal tubule marker Lotus tetragonolobus lectin (Fig. 8D).
Fig. 7.

APN leads to renal fibrosis. A: trichrome staining of an uninfected kidney identifies rare areas of collagen (blue) associated with an artery (arrow) and adjacent to the renal pelvis (arrowhead; ×4 objective). B: submucosal collagen deposition (asterisks) 7 dpi first occurs in the interstitium immediately beneath the epithelial lining of the pelvis (×10 objective). C: the dotted black line delineates a wedge-shaped early renal scar extending through the cortex 14 dpi (×4 objective). D: higher magnification view (×10 objective) of the section in C, highlighting the spatial restriction of fibrosis within the inflamed cortical parenchyma during subacute APN.
Fig. 8.

Myofibroblast activation and cortical interstitial fibrosis in UPEC-infected kidneys. A: significant increase in renal fibrosis scores 7 and 28 dpi; *P < 0.05, Mann-Whitney U-test. B: significant increase in α-smooth muscle actin (α-SMA)-expressing myofibroblasts in kidneys 7 and 28 dpi; *P < 0.05, Mann-Whitney U-test. C: cortical α-SMA distribution (green) in uninfected kidneys reveals reactivity restricted to small arteries and rare interstitial myofibroblasts. LTL lectin (red) identifies proximal tubules. Cell nuclei are visualized with DAPI (blue). D: widespread increase in cortical α-SMA reactivity due to heavy proliferation of reactive myofibroblasts in a severely fibrotic kidney 28 dpi, with reduction in LTL+ proximal tubules. Both images were acquired using the ×20 objective. The scale bar represents 100 µm.
Inflammation positively correlates with fibrosis.
We evaluated the relationship between bacterial burden and histopathological grades of mucosal injury, inflammation, and fibrosis using Spearman’s correlation test. Significant positive correlations existed between each of these variables 7 dpi (Table 5). The strongest correlation at both 7 and 28 dpi was between renal inflammation and fibrosis, indicated by correlation coefficient (R) values of 0.81 and 0.90, respectively, at these two time points..
Table 5.
Bacterial burden correlates with mucosal injury, inflammation, and fibrosis
| Comparison | 7 dpi |
28 dpi |
||
|---|---|---|---|---|
| R | P | R | P | |
| Log(renal CFU) vs. fibrosis | 0.604 | 0.006 | 0.532 | 0.114 |
| Log(renal CFU) vs. inflammation | 0.539 | 0.017 | 0.599 | 0.067 |
| Inflammation vs. fibrosis | 0.817 | 0.00002 | 0.9 | 0.0004 |
| Inflammation vs. mucosal injury | 0.802 | 0.002 | 0.766 | 0.027 |
| Log(renal CFU) vs. mucosal injury | 0.697 | 0.012 | 0.467 | 0.244 |
R: Spearman’s Rho; P, 2-tailed P value from Spearman’s test; significant values are in bold; nonsignificant values are in italics.
Renal ischemia and reperfusion pathways are triggered by APN.
We performed renal transcriptome analysis to identify molecular pathways associated with APN. Total RNA from C3H/HeOuJ kidneys was analyzed from uninfected mice and from infected mice at 7 dpi by oligonucleotide microarray. We used Ingenuity Pathway Analysis software to identify gene expression networks regulated by APN. Four of the top 10 toxicity pathways most activated by APN contained genes associated with renal injury (Fig. 9A). The “persistent renal ischemia reperfusion injury pathway” was most significantly activated (P = 6.3 × 10−19, Fisher’s exact test). We confirmed increased expression of renal injury transcripts within this pathway at 7 and 28 dpi by qRT-PCR (Fig. 9B).
Fig. 9.

Pathway analysis identifies renal transcripts associated with APN. A: the top 10 most significant toxicity pathways in APN kidneys predicted by Ingenuity Pathway Analysis software. The height of each bar indicates the –log(P value) for each pathway (y-axis). B: qRT-PCR validates significant increases in renal expression of 13 transcripts belonging to the “persistent renal ischemia-reperfusion injury” toxicity pathway at 7 and 28 dpi, compared with uninfected controls (n = 4 kidneys/group; P < 0.05, Mann-Whitney U-test).
Innate immune response and leukocyte recruitment are the primary transcriptional signatures during APN.
Pathway analysis predicted activation of canonical pathways responsible for pathogen detection, leukocyte recruitment and activation, and communication between leukocytes during APN (Fig. 10A). The most activated canonical pathway was “Granulocyte adhesion and diapedesis” (P = 1.1 × 10−26, Fisher’s exact test). The highest-scoring transcriptional network was “Antimicrobial response, inflammatory response, and infectious diseases” (Table 6), consisting of 33 interacting gene products, all of which significantly increased during APN at 7 dpi (Fig. 10B). These findings led us to examine the expression profile of renal antimicrobial peptides (AMPs) during APN. We identified 11 renal AMP transcripts with ≥2-fold induction at 7 dpi (Table 7).
Fig. 10.
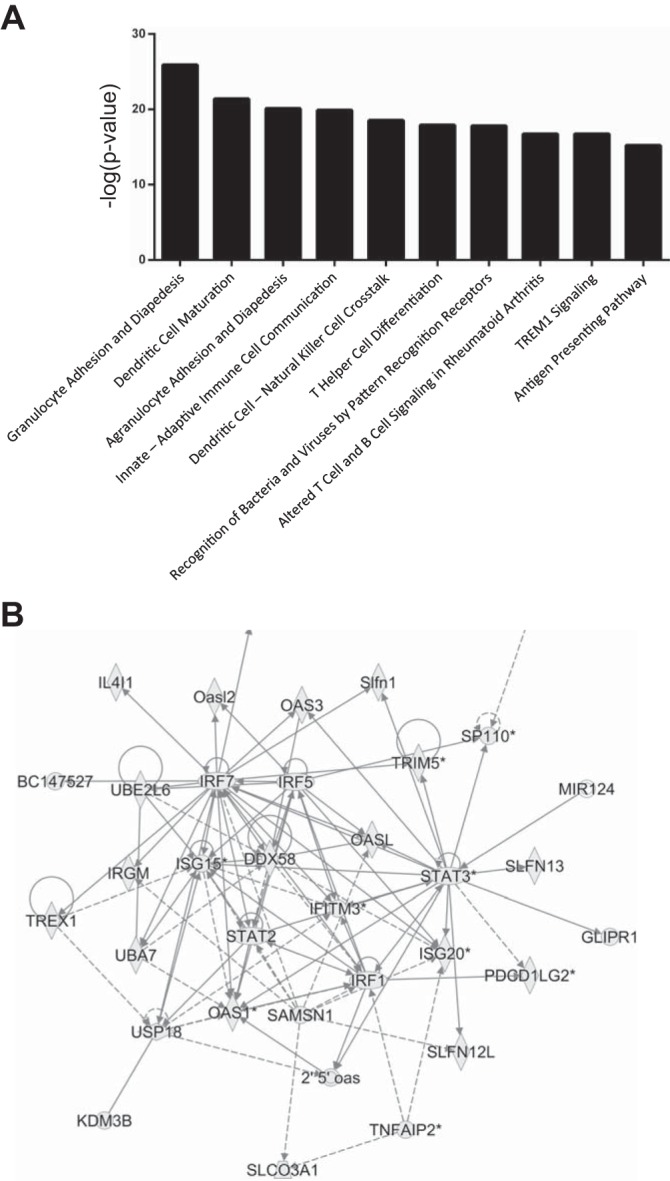
Pathway analysis predicts activation of biological processes and functional gene networks during APN. A: top 10 canonical pathways activated during APN according to Ingenuity Pathway Analysis software. B: “antimicrobial response, inflammatory response, infectious disease” is the top-ranked network during APN. Shapes indicate 33 transcripts with increased expression 7 dpi. Lines indicate regulatory relationships between proteins encoded by these transcripts.
Table 6.
The most activated transcriptional networks during APN
| Rank | Diseases and Functions | Score |
|---|---|---|
| 1 | Antimicrobial response, inflammatory response, infectious diseases | 41 |
| 2 | Cell death and survival, embryonic development, dermatological diseases and conditions | 39 |
| 3 | Developmental disorder, hematological disease, hereditary disorder | 34 |
| 4 | Cellular movement, hematological system development and function, immune cell Trafficking | 34 |
| 5 | Hematological Disease, Immunological Disease, Cardiovascular Disease | 34 |
Predictions based on Ingenuity Pathway Analysis of C3H/HeOuJ kidney transcriptomes 7 dpi vs. uninfected controls.
Table 7.
Kidney AMP mRNA Induction 7 dpi, Based on Microarray
| AMP | Fold | FDR | P value |
|---|---|---|---|
| S100a9 | 257.352 | 2.79 | 0.00001 |
| S100a8 | 134.098 | 3.65 | 0.00001 |
| Lcn2 | 125.625 | 2.82 | 0.00002 |
| Ltf | 65.522 | 1.77 | 0.00001 |
| Slpi | 41.647 | 5.23 | 0.00025 |
| Reg3g | 30.126 | 2.24 | 0.00002 |
| Lyz2 | 23.802 | 5.04 | 0.00001 |
| Lyz1 | 22.548 | 9.14 | 0.00191 |
| Ear2 | 7.876 | 3.2 | 0.00001 |
| Rnase6 | 3.166 | 2.41 | 0.00001 |
| Ear1 | 2.413 | 6.38 | 0.00046 |
Pathway analysis predicts activation of TFs during APN.
Pathway analysis also identified putative “upstream regulators” of transcription during APN on the basis of gene expression changes. Among the 10 transcription factors (TFs) with the highest predicted activation Z-scores during APN, six belonged to the Interferon regulatory factor (Irf) and Signal transducer and activator of transcription (Stat) families (Table 8). The highest activation Z-score was for Irf7, and Irf7 transcript levels were increased in infected C3H/HeOuJ kidneys at 7 dpi (Table 8). Pathway analysis predicted interactions between these TFs in a mechanistic network whereby Irf7 activation leads to type I interferon production and activation of multiple other Irf as well as Stat TFs (Fig. 11).
Table 8.
Predicted TF activation during APN
| TF | Fold Change | Activation Z-Score | P Value |
|---|---|---|---|
| Irf7 | 6.4 | 8.1 | 7.59E−43 |
| Stat1 | (n.s.) | 7.8 | 2.59E−58 |
| Stat3 | 3.7 | 6.3 | 4.43E−44 |
| Rela | (n.s.) | 6.2 | 7.96E−28 |
| Irf1 | 8 | 6 | 5.40E−35 |
| Irf3 | (n.s.) | 6 | 3.06E−35 |
| Spi1 | (n.s.) | 5.5 | 2.01E−27 |
| Stat4 | 2.4 | 5.3 | 9.36E−15 |
| Egr1 | (n.s.) | 4.9 | 2.27E−13 |
| Smarca4 | (n.s.) | 4.9 | 1.40E−20 |
Predictions based on Ingenuity Pathway Analysis of C3H/HeOuJ kidney transcriptomes 7 dpi vs. uninfected controls; n.s., not significant.
Fig. 11.
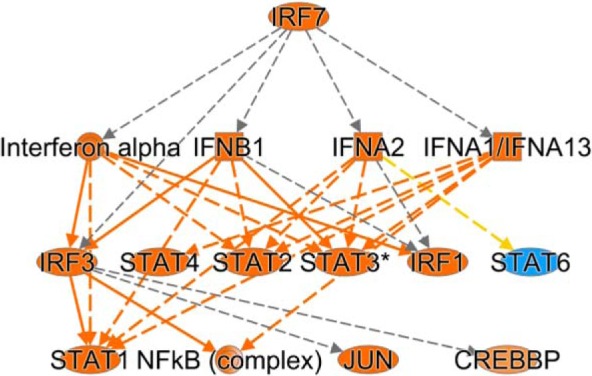
Irf7 mechanistic network. Transcriptome pathway analysis predicts a mechanistic network in which Irf7 activation and type I interferon production lead to activation of downstream Irf and Stat TFs. Orange predicts activation, while blue predicts inhibition. Yellow line, findings inconsistent with state of downstream molecule; gray line, effect not predicted.
DISCUSSION
Renal fibrosis is a known sequela of APN that is associated with hypertension, proteinuria, and progression of chronic kidney disease (28, 37, 39). Current clinical management of APN is not directed toward preventing these sequelae, and a greater understanding of APN pathogenesis is clearly required to achieve this goal. The ideal treatment of APN should eradicate the uropathogen responsible for infection, while preventing permanent renal injury. We conducted this study to provide mechanistic insight regarding renal inflammation and fibrosis during APN in an immunocompetent host.
Distinct origins of fibrosis during APN.
The ascending pattern of fibrosis associated with APN is distinct from that associated with renal injury paradigms such as unilateral ureteral obstruction and ischemia-reperfusion injury (9, 36). During APN, fibrosis initiates immediately deep to the renal urothelium. Expansive submucosal connective tissue deposition ensues. Making use of perivascular medullary rays, the collagenous deposition spreads through the interstitium to the outer medulla and cortex. In the setting of this unique pathology, C3H/HeOuJ mice with APN may be utilized to identify the cellular origin of the myofibroblasts responsible for renal fibrosis. In other paradigms of renal injury, renal tubular epithelial cells, pericytes, and circulating bone marrow-derived cells have been implicated in renal scarring (13, 30). Additionally, C3H/HeOuJ mice with APN may be used to evaluate which imaging studies are best suited for early detection of renal scars, as well as to test novel antifibrotic or anti-inflammatory agents. Thus experimental UTI in C3H/HeOuJ mice comprises a novel animal model of renal fibrosis that recapitulates the human condition of APN-associated scarring in an immunocompetent setting.
Renal localization of UPEC during APN.
We identified UPEC in cortical abscesses and medullary tubules. The most prominent UPEC localization was within the collecting duct lumen, where UPEC were observed individually or were embedded inside neutrophil casts. Our data suggest that UPEC forms a biofilm integrated within leukocyte casts; these biofilms serve as a potential niche to survive the otherwise hostile climate posed by the renal inner medulla. Biofilm formation may favor antibiotic resistance and serve as a UPEC reservoir for reinfection of the renal or bladder parenchyma. The association of UPEC within casts likely triggers neutrophil degranulation that contributes to tubular epithelial injury. Additional studies are required to determine how the interactions between UPEC and neutrophil casts impact UTI chronicity, recurrence, and renal scar formation.
APN associated fibrosis correlates with renal inflammation and mucosal injury.
We detected strong correlations between bacterial burden, inflammation, mucosal injury, and fibrosis in C3H/HeOuJ kidneys at 7 and 28 dpi. This observation supports the hypothesis that sustained renal colonization with UPEC provokes an exuberant inflammatory response, provoking epithelial damage and APN-associated renal scarring. Flow cytometry and gene expression studies provide further insight regarding inflammatory cell lineages and pathways governing their recruitment and activation.
Significance of myeloid cell populations during APN.
We observed recruitment of neutrophils, Ly6C+ macrophages, and Ly6C− macrophages during APN. Antibody-mediated depletion of neutrophils leads to impaired renal clearance of UPEC, attesting to their essential antimicrobial roles in the urinary tract (18, 19). However, neutrophils can also trigger tissue injury during APN. Genetic deletion of chemokines and chemokine receptors responsible for neutrophil recruitment to the urinary space leads to retention of neutrophils in the renal submucosa, chronic inflammation, and renal fibrosis (16, 46). Moreover, partial depletion of neutrophils reduces the incidence of chronic cystitis, and similar protective effects are conferred by systemic administration of nonselective anti-inflammatory agents (17, 18). Additional studies are required to determine whether neutrophil-directed therapeutic strategies are beneficial in preventing renal scarring as a result of APN.
Less is known regarding Ly6C+ and Ly6C− macrophage subsets during APN. Liposomal depletion of monocytes/macrophages reduces the incidence of chronic cystitis at 28 dpi (8, 18). During cystitis, recruitment of Ly6C+ inflammatory macrophages from the bloodstream triggers resident Ly6C− macrophages to promote transepithelial neutrophil migration into the urinary space (42). It will be insightful to determine whether similar cross talk between these cell populations occurs in the kidney during APN, and whether manipulation of macrophage subsets alters renal pathology in APN.
Pathway analysis predicts activation of biological networks during APN.
The persistent renal ischemia-perfusion injury network was the most activated toxicity pathway during APN, raising the hypothesis that APN leads to ischemia-reperfusion injury. This is an intriguing concept, given that Tlr4 signaling in intrinsic kidney cells is responsible, at least in part, for kidney damage during ischemia-reperfusion injury and APN (38, 48). Moreover, APN and renal infarction can be clinically and radiologically indistinct in human patients, suggesting shared pathophysiology (40). Indeed, certain histopathological features of the C3H/HeOuJ APN model overlap with ischemia-reperfusion injury as well, for example, the presence of interstitial inflammation, cellular casts in dilated tubules, and acute tubular necrosis (34). Moreover, direct microinjection of UPEC into the rat proximal tubule triggers increased epithelial oxygen consumption and clot formation within peritubular capillaries, causing local ischemia (32). This may serve a protective function, i.e., restricting dissemination of tubular bacteria to the bloodstream, since administration of enoxaparin to suppress clotting in these subjects led to fatal urosepsis (32). Additional studies of C3H/HeOuJ kidneys are required to determine the extent to which renal parenchymal damage during APN is due to ischemia-reperfusion injury.
Pathway analysis also predicted recruitment of leukocyte populations, findings supported by flow cytometry and histopathology, as well as prominent roles for Stat and Irf TFs as upstream activators of innate immune genes associated with “antimicrobial response, inflammatory response, and infectious disease.”
Emerging experimental evidence supports a central role for interferons and Irf TFs in APN. Mice lacking Ifn-β or Irf3 exhibit increased susceptibility to APN and renal abscess formation (14). A recent study identifies upregulation of Irf7 mRNA and protein in Irf3−/− mice and implicates the Irf7 TF as a driver of a hyperinflammatory phenotype in these animals (41). Importantly, Irf7−/− mice do not exhibit altered susceptibility to APN, and combined deficiency of Irf7 and Irf3 reverses APN susceptibility and renal injury in Irf3−/− mice (41). Finally, humans with APN exhibit promoter polymorphisms in IRF3 that confer reduced activity in reporter assays, whereas attenuating IRF7 promoter polymorphisms negatively correlate with recurrent APN (14, 41). Altogether, these studies argue that the balance of IRF7/IRF3 determines the extent of inflammatory injury during APN. Future studies in the C3H/HeOuJ mouse model of APN should test the hypothesis that Irf7 drives inflammatory renal injury, perhaps through the use of liposomal small interfering RNA directed against Irf7 as has been recently reported (41).
Proposed pathological sequence.
Based on our observations, we surmise that UPEC initially attach to the epithelial lining of renal collecting ducts, thereby triggering recruitment of circulating neutrophils. Degranulation of neutrophils within the lumen of collecting ducts drives epithelial injury. Infection of a medullary collecting duct ascends into all nephrons that drain into it, leading to a triangular spread of the APN lesion into the cortex. This histological finding in our mouse model parallels the wedge-shaped patterns of renal parenchymal injury observed in patients with APN (11, 12). Within the cortex, evolving defects in mucosal integrity result in UPEC translocation into the interstitium and eventual abscess formation. Widespread tubular injury and inflammatory cell recruitment prompt infiltration of myofibroblasts into the interstitium, followed by collagen deposition, and ultimately leading to interstitial fibrosis.
Conclusions.
This study provides a knowledge framework to investigate acquired renal scarring from APN in immunocompetent hosts. The histopathology observed in the kidneys of infected C3H/HeOuJ mice recapitulates the cardinal features of human APN, with progressive mucosal injury, tubulointerstitial inflammation, and interstitial fibrosis (11). The C3H/HeOuJ mouse model of APN will serve as a platform to dissect the molecular mechanisms promoting renal scar formation. Improved knowledge of APN pathogenesis will lead to innovative approaches to quell infection and curtail chronic tissue injury, thereby preserving renal function and substantially improving patient quality of life.
GRANTS
This work was supported by NIDDK 5R01DK085242-03 (K. M. McHugh); NIDDK R01-DK106286-01 (D. S. Hains and A. L. Schwaderer); NIDDK K08-DK094970 (J. D. Spencer); NIAID R01-AI092117 (S. Partida-Sanchez); and NIDDK K08-DK102594 (B. Becknell).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
B.L., B.H., H.C., and N.M. performed experiments; B.L., B.H., A.R.J., H.C., R.K., B. Bolon, K.M.M., A.L.S., C.B.C., and B. Becknell analyzed data; B.L., A.R.J., and B. Becknell prepared figures; H.C., C.B.C., S.S.J., S.P.-S., and B. Becknell interpreted results of experiments; R.K., B. Bolon, K.M.M., A.L.S., J.D.S., C.B.C., D.S.H., S.S.J., S.P.-S., and B. Becknell edited and revised manuscript; B. Becknell approved final version of manuscript.
REFERENCES
- 1.Becknell B, Eichler TE, Beceiro S, Li B, Easterling RS, Carpenter AR, James CL, McHugh KM, Hains DS, Partida-Sanchez S, Spencer JD. Ribonucleases 6 and 7 have antimicrobial function in the human and murine urinary tract. Kidney Int 87: 151–161, 2015. doi: 10.1038/ki.2014.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Becknell B, Schober M, Korbel L, Spencer JD. The diagnosis, evaluation and treatment of acute and recurrent pediatric urinary tract infections. Expert Rev Anti Infect Ther 13: 81–90, 2015. doi: 10.1586/14787210.2015.986097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becknell B, Spencer JD, Carpenter AR, Chen X, Singh A, Ploeger S, Kline J, Ellsworth P, Li B, Proksch E, Schwaderer AL, Hains DS, Justice SS, McHugh KM. Expression and antimicrobial function of beta-defensin 1 in the lower urinary tract. PLoS One 8: e77714, 2013. doi: 10.1371/journal.pone.0077714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolton M, Horvath DJ Jr, Li B, Cortado H, Newsom D, White P, Partida-Sanchez S, Justice SS. Intrauterine growth restriction is a direct consequence of localized maternal uropathogenic Escherichia coli cystitis. PLoS One 7: e33897, 2012. doi: 10.1371/journal.pone.0033897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowen SE, Watt CL, Murawski IJ, Gupta IR, Abraham SN. Interplay between vesicoureteric reflux and kidney infection in the development of reflux nephropathy in mice. Dis Model Mech 6: 934–941, 2013. doi: 10.1242/dmm.011650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brandström P, Jodal U, Sillén U, Hansson S. The Swedish reflux trial: review of a randomized, controlled trial in children with dilating vesicoureteral reflux. J Pediatr Urol 7: 594–600, 2011. doi: 10.1016/j.jpurol.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 7.Brandström P, Nevéus T, Sixt R, Stokland E, Jodal U, Hansson S. The Swedish reflux trial in children: IV. Renal damage. J Urol 184: 292–297, 2010. doi: 10.1016/j.juro.2010.01.060. [DOI] [PubMed] [Google Scholar]
- 8.Carey AJ, Sullivan MJ, Duell BL, Crossman DK, Chattopadhyay D, Brooks AJ, Tan CK, Crowley M, Sweet MJ, Schembri MA, Ulett GC. Uropathogenic Escherichia coli engages CD14-dependent signaling to enable bladder-macrophage-dependent control of acute urinary tract infection. J Infect Dis 213: 659–668, 2016. doi: 10.1093/infdis/jiv424. [DOI] [PubMed] [Google Scholar]
- 9.Chevalier RL, Forbes MS, Thornhill BA. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int 75: 1145–1152, 2009. doi: 10.1038/ki.2009.86. [DOI] [PubMed] [Google Scholar]
- 10.Craig JC, Simpson JM, Williams GJ, Lowe A, Reynolds GJ, McTaggart SJ, Hodson EM, Carapetis JR, Cranswick NE, Smith G, Irwig LM, Caldwell PH, Hamilton S, Roy LP; Prevention of Recurrent Urinary Tract Infection in Children with Vesicoureteric Reflux and Normal Renal Tracts (PRIVENT) Investigators . Antibiotic prophylaxis and recurrent urinary tract infection in children. N Engl J Med 361: 1748–1759, 2009. doi: 10.1056/NEJMoa0902295. [DOI] [PubMed] [Google Scholar]
- 11.Craig WD, Wagner BJ, Travis MD. Pyelonephritis: radiologic-pathologic review. Radiographics 28: 255–277, 2008. doi: 10.1148/rg.281075171. [DOI] [PubMed] [Google Scholar]
- 12.Demertzis J, Menias CO. State of the art: imaging of renal infections. Emerg Radiol 14: 13–22, 2007. doi: 10.1007/s10140-007-0591-3. [DOI] [PubMed] [Google Scholar]
- 13.Duffield JS. Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest 124: 2299–2306, 2014. doi: 10.1172/JCI72267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fischer H, Lutay N, Ragnarsdóttir B, Yadav M, Jönsson K, Urbano A, Al Hadad A, Rämisch S, Storm P, Dobrindt U, Salvador E, Karpman D, Jodal U, Svanborg C. Pathogen specific, IRF3-dependent signaling and innate resistance to human kidney infection. PLoS Pathog 6: e1001109, 2010. doi: 10.1371/journal.ppat.1001109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hains DS, Sims-Lucas S, Carpenter A, Saha M, Murawski I, Kish K, Gupta I, McHugh K, Bates CM. High incidence of vesicoureteral reflux in mice with Fgfr2 deletion in kidney mesenchyma. J Urol 183: 2077–2084, 2010. doi: 10.1016/j.juro.2009.12.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hang L, Frendéus B, Godaly G, Svanborg C. Interleukin-8 receptor knockout mice have subepithelial neutrophil entrapment and renal scarring following acute pyelonephritis. J Infect Dis 182: 1738–1748, 2000. doi: 10.1086/317599. [DOI] [PubMed] [Google Scholar]
- 17.Hannan TJ, Mysorekar IU, Hung CS, Isaacson-Schmid ML, Hultgren SJ. Early severe inflammatory responses to uropathogenic E. coli predispose to chronic and recurrent urinary tract infection. PLoS Pathog 6: e1001042, 2010. doi: 10.1371/journal.ppat.1001042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hannan TJ, Roberts PL, Riehl TE, van der Post S, Binkley JM, Schwartz DJ, Miyoshi H, Mack M, Schwendener RA, Hooton TM, Stappenbeck TS, Hansson GC, Stenson WF, Colonna M, Stapleton AE, Hultgren SJ. Inhibition of cyclooxygenase-2 prevents chronic and recurrent cystitis. EBioMedicine 1: 46–57, 2014. doi: 10.1016/j.ebiom.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haraoka M, Hang L, Frendéus B, Godaly G, Burdick M, Strieter R, Svanborg C. Neutrophil recruitment and resistance to urinary tract infection. J Infect Dis 180: 1220–1229, 1999. doi: 10.1086/315006. [DOI] [PubMed] [Google Scholar]
- 20.Healthcare Cost and Utilization Project. National Inpatient Sample. 2012. http://www.hcup-us.ahrq.gov/nisoverview.jsp.
- 21.RIVUR Trial Investigators; Hoberman A, Greenfield SP, Mattoo TK, Keren R, Mathews R, Pohl HG, Kropp BP, Skoog SJ, Nelson CP, Moxey-Mims M, Chesney RW, Carpenter MA. Antimicrobial prophylaxis for children with vesicoureteral reflux. N Engl J Med 370: 2367–2376, 2014. doi: 10.1056/NEJMoa1401811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hopkins W, Gendron-Fitzpatrick A, McCarthy DO, Haine JE, Uehling DT. Lipopolysaccharide-responder and nonresponder C3H mouse strains are equally susceptible to an induced Escherichia coli urinary tract infection. Infect Immun 64: 1369–1372, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hopkins WJ, Gendron-Fitzpatrick A, Balish E, Uehling DT. Time course and host responses to Escherichia coli urinary tract infection in genetically distinct mouse strains. Infect Immun 66: 2798–2802, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hung CS, Dodson KW, Hultgren SJ. A murine model of urinary tract infection. Nat Protoc 4: 1230–1243, 2009. doi: 10.1038/nprot.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaneto H, Morrissey J, McCracken R, Reyes A, Klahr S. Enalapril reduces collagen type IV synthesis and expansion of the interstitium in the obstructed rat kidney. Kidney Int 45: 1637–1647, 1994. doi: 10.1038/ki.1994.215. [DOI] [PubMed] [Google Scholar]
- 27.Kao JS, Stucker DM, Warren JW, Mobley HL. Pathogenicity island sequences of pyelonephritogenic Escherichia coli CFT073 are associated with virulent uropathogenic strains. Infect Immun 65: 2812–2820, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karlén J, Linné T, Wikstad I, Aperia A. Incidence of microalbuminuria in children with pyelonephritic scarring. Pediatr Nephrol 10: 705–708, 1996. doi: 10.1007/s004670050194. [DOI] [PubMed] [Google Scholar]
- 29.Keren R, Shaikh N, Pohl H, Gravens-Mueller L, Ivanova A, Zaoutis L, Patel M, deBerardinis R, Parker A, Bhatnagar S, Haralam MA, Pope M, Kearney D, Sprague B, Barrera R, Viteri B, Egigueron M, Shah N, Hoberman A. Risk factors for recurrent urinary tract infection and renal scarring. Pediatrics 136: e13–e21, 2015. doi: 10.1542/peds.2015-0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R. Origin and function of myofibroblasts in kidney fibrosis. Nat Med 19: 1047–1053, 2013. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lloyd AL, Rasko DA, Mobley HL. Defining genomic islands and uropathogen-specific genes in uropathogenic Escherichia coli. J Bacteriol 189: 3532–3546, 2007. doi: 10.1128/JB.01744-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melican K, Boekel J, Månsson LE, Sandoval RM, Tanner GA, Källskog O, Palm F, Molitoris BA, Richter-Dahlfors A. Bacterial infection-mediated mucosal signalling induces local renal ischaemia as a defence against sepsis. Cell Microbiol 10: 1987–1998, 2008. doi: 10.1111/j.1462-5822.2008.01182.x. [DOI] [PubMed] [Google Scholar]
- 33.Mobley HL, Green DM, Trifillis AL, Johnson DE, Chippendale GR, Lockatell CV, Jones BD, Warren JW. Pyelonephritogenic Escherichia coli and killing of cultured human renal proximal tubular epithelial cells: role of hemolysin in some strains. Infect Immun 58: 1281–1289, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Molitoris BA. Therapeutic translation in acute kidney injury: the epithelial/endothelial axis. J Clin Invest 124: 2355–2363, 2014. doi: 10.1172/JCI72269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murawski IJ, Maina RW, Malo D, Guay-Woodford LM, Gros P, Fujiwara M, Morgan K, Gupta IR. The C3H/HeJ inbred mouse is a model of vesico-ureteric reflux with a susceptibility locus on chromosome 12. Kidney Int 78: 269–278, 2010. doi: 10.1038/ki.2010.110. [DOI] [PubMed] [Google Scholar]
- 36.Oxburgh L, de Caestecker MP. Ischemia-reperfusion injury of the mouse kidney. Methods Mol Biol 886: 363–379, 2012. doi: 10.1007/978-1-61779-851-1_32. [DOI] [PubMed] [Google Scholar]
- 37.Park YS. Renal scar formation after urinary tract infection in children. Korean J Pediatr 55: 367–370, 2012. doi: 10.3345/kjp.2012.55.10.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patole PS, Schubert S, Hildinger K, Khandoga S, Khandoga A, Segerer S, Henger A, Kretzler M, Werner M, Krombach F, Schlöndorff D, Anders HJ. Toll-like receptor-4: renal cells and bone marrow cells signal for neutrophil recruitment during pyelonephritis. Kidney Int 68: 2582–2587, 2005. doi: 10.1111/j.1523-1755.2005.00729.x. [DOI] [PubMed] [Google Scholar]
- 39.Peters C, Rushton HG. Vesicoureteral reflux associated renal damage: congenital reflux nephropathy and acquired renal scarring. J Urol 184: 265–273, 2010. doi: 10.1016/j.juro.2010.03.076. [DOI] [PubMed] [Google Scholar]
- 40.Piccoli GB, Priola AM, Vigotti FN, Guzzo G, Veltri A. Renal infarction versus pyelonephritis in a woman presenting with fever and flank pain. Am J Kidney Dis 64: 311–314, 2014. doi: 10.1053/j.ajkd.2014.02.027. [DOI] [PubMed] [Google Scholar]
- 41.Puthia M, Ambite I, Cafaro C, Butler D, Huang Y, Lutay N, Rydström G, Gullstrand B, Swaminathan B, Nadeem A, Nilsson B, Svanborg C. IRF7 inhibition prevents destructive innate immunity-A target for nonantibiotic therapy of bacterial infections. Sci Transl Med 8: 336ra59, 2016. doi: 10.1126/scitranslmed.aaf1156. [DOI] [PubMed] [Google Scholar]
- 42.Schiwon M, Weisheit C, Franken L, Gutweiler S, Dixit A, Meyer-Schwesinger C, Pohl JM, Maurice NJ, Thiebes S, Lorenz K, Quast T, Fuhrmann M, Baumgarten G, Lohse MJ, Opdenakker G, Bernhagen J, Bucala R, Panzer U, Kolanus W, Gröne HJ, Garbi N, Kastenmüller W, Knolle PA, Kurts C, Engel DR. Crosstalk between sentinel and helper macrophages permits neutrophil migration into infected uroepithelium. Cell 156: 456–468, 2014. doi: 10.1016/j.cell.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108, 2008. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 44.Smellie JM, Barratt TM, Chantler C, Gordon I, Prescod NP, Ransley PG, Woolf AS. Medical versus surgical treatment in children with severe bilateral vesicoureteric reflux and bilateral nephropathy: a randomised trial. Lancet 357: 1329–1333, 2001. doi: 10.1016/S0140-6736(00)04520-7. [DOI] [PubMed] [Google Scholar]
- 45.Spencer JD, Jackson AR, Li B, Ching CB, Vonau M, Easterling RS, Schwaderer AL, McHugh KM, Becknell B. Expression and significance of the HIP/PAP and RegIIIγ antimicrobial peptides during mammalian urinary tract infection. PLoS One 10: e0144024, 2015. doi: 10.1371/journal.pone.0144024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Svensson M, Yadav M, Holmqvist B, Lutay N, Svanborg C, Godaly G. Acute pyelonephritis and renal scarring are caused by dysfunctional innate immunity in mCxcr2 heterozygous mice. Kidney Int 80: 1064–1072, 2011. doi: 10.1038/ki.2011.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsukada T, McNutt MA, Ross R, Gown AM. HHF35, a muscle actin-specific monoclonal antibody. II. Reactivity in normal, reactive, and neoplastic human tissues. Am J Pathol 127: 389–402, 1987. [PMC free article] [PubMed] [Google Scholar]
- 48.Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alexander SI, Sharland AF, Chadban SJ. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest 117: 2847–2859, 2007. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu OH, Murawski IJ, Myburgh DB, Gupta IR. Overexpression of RET leads to vesicoureteric reflux in mice. Am J Physiol Renal Physiol 287: F1123–F1130, 2004. doi: 10.1152/ajprenal.00444.2003. [DOI] [PubMed] [Google Scholar]