

Abstract
Previously, we documented that activation of protein kinase C-ε (PKC-ε) mediates mitochondrial dysfunction in cultured renal proximal tubule cells (RPTC). This study tested whether deletion of PKC-ε decreases dysfunction of renal cortical mitochondria and improves kidney function after renal ischemia. PKC-ε levels in mitochondria of ischemic kidneys increased 24 h after ischemia. Complex I- and complex II-coupled state 3 respirations were reduced 44 and 27%, respectively, in wild-type (WT) but unchanged and increased in PKC-ε-deficient (KO) mice after ischemia. Respiratory control ratio coupled to glutamate/malate oxidation decreased 50% in WT but not in KO mice. Activities of complexes I, III, and IV were decreased 59, 89, and 61%, respectively, in WT but not in KO ischemic kidneys. Proteomics revealed increases in levels of ATP synthase (α-subunit), complexes I and III, cytochrome oxidase, α-ketoglutarate dehydrogenase, and thioredoxin-dependent peroxide reductase after ischemia in KO but not in WT animals. PKC-ε deletion prevented ischemia-induced increases in oxidant production. Plasma creatinine levels increased 12-fold in WT and 3-fold in KO ischemic mice. PKC-ε deletion reduced tubular necrosis, brush border loss, and distal segment damage in ischemic kidneys. PKC-ε activation in hypoxic RPTC in primary culture exacerbated, whereas PKC-ε inhibition reduced, decreases in: 1) complex I- and complex II-coupled state 3 respirations and 2) activities of complexes I, III, and IV. We conclude that PKC-ε activation mediates 1) dysfunction of complexes I and III of the respiratory chain, 2) oxidant production, 3) morphological damage to the kidney, and 4) decreases in renal functions after ischemia.
Keywords: acute kidney injury, ischemia and reperfusion, hypoxia, proteomics, renal proximal tubular cells, mitochondria, electron transport chain
protein kinase C-ε (PKC-ε) is a novel PKC isozyme that is present at low levels in renal proximal tubules of the kidney but is induced by ischemic injury to this organ (29, 42). PKC-ε plays an important role in supporting cell survival and suppressing apoptosis in several cell types, including cardiac myocytes and neuronal and cancer cells (7, 8, 11, 18, 24, 25). PKC-ε activation has been implicated in protection of the heart and brain against ischemic injury and in reduction of myocardial infarct (1, 2, 6, 10, 15–17, 22, 33). Furthermore, it has been shown that PKC-ε activation is a pivotal signaling event in the cardioprotective mechanisms of ischemic preconditioning (2, 6, 16, 16, 17, 27, 32, 32, 32, 32, 33, 43, 44, 44–46). Ischemic preconditioning induces translocation of PKC-ε to the mitochondria, and it is thought that the protection it offers is mediated through mitochondrial and transport mechanisms (6, 15, 55). Inhibition of PKC-ε activation eliminates protection offered by ischemic preconditioning in the heart (16).
Very little is known about involvement of PKC-ε activation in the physiological and pathological processes of the kidney. DeCoy and colleagues reported that PKC-ε negatively regulates vasopressin-stimulated Na+ reabsorption in the cortical collecting duct (9). LaPorta and Comolli (29) and Padanilam (42) reported activation of PKC-ε in the kidney cortex during and after renal ischemia. It has been shown that PKC-dependent signaling is involved in protection against ischemic injury in the kidney, but the PKC isoform responsible for this protection has not been identified (12). Renal proximal tubular cells (RPTC) are a major target of ischemia-reperfusion injury within the kidney because of their active oxidative metabolism and high demands for ATP. RPTC dysfunction is a major factor responsible for the pathophysiological and clinical presentations of the acute renal failure (5, 31). We have shown that hypoxia and oxidant exposure activate PKC-ε in RPTC and that PKC-ε activation mediates mitochondrial dysfunction, including decreases in complex I-coupled respiration and reductions in activity of complex I (36, 40). Furthermore, sustained activation of PKC-ε in noninjured RPTC produces mitochondrial dysfunction, hyperpolarization of the mitochondrial membrane, and mitochondrial fission as well as oxidant generation and cell death (40). Inhibition of PKC-ε activation offers protection against mitochondrial dysfunction and decreases in active Na+ transport in oxidant-injured RPTC in primary culture (36). Thus, we have shown that, in contrast to the protective effects of PKC-ε activation in cardiomyocytes, sustained activation of PKC-ε in RPTC is detrimental to mitochondrial function and cell viability (40).
It has been recently shown that deletion of the PKC-ε gene attenuates ischemic injury in kidney allografts through inhibition of the production of reactive oxygen species (ROS), inflammatory response, and apoptosis (49). However, the role of PKC-ε in upstream events leading to ischemia-induced decreases in renal bioenergetics, such as mitochondrial dysfunction, and the mitochondrial targets of PKC-ε in vivo are not known. Therefore, the aim of this study was to determine whether PKC-ε mediates decreases in respiratory function of mitochondria in the ischemic kidney. The second goal of this study was to identify mitochondrial proteins that are involved in the effects of PKC-ε on mitochondrial function during ischemic kidney injury.
MATERIALS AND METHODS
Animals.
All animal procedures (including breeding) were carried out in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of Arkansas for Medical Sciences. Heterozygous C57BL/6J mice carrying a deletion of the PKC-ε gene (Prkcetm1Msg/Prkcetm1Msg) have been purchased from The Jackson Laboratory (Bar Harbor, ME). PKC-ε deletion was obtained by using a recombination vector containing neomycin resistance and herpes simplex virus thymidine kinase genes. The recombination vector was used to disrupt a 1.2-kb region of the PKC-ε gene, and the construct was electroporated into embryonic stem cells to generate PKC-ε mutant mice by homologous recombination (26). The heterozygous animals were bred to obtain homozygous PKC-ε-deficient mice (PKC-ε KO) and wild-type homozygous littermates (WT). Tail snips obtained from 3-wk-old offspring were lysed using 0.2 ml DirectPCR Lysis Reagent (Viagen Biotech, Los Angeles, CA) containing 12 μg proteinase K at 55°C overnight. PKC-ε-deficient mice were selected from the offspring of heterozygous matings based on results of polymerase chain reaction of the tail DNA. Wild-type littermates were used as controls. Female New Zealand White rabbits (2.5–3.0 kg) were purchased from Harlan Laboratories (Oxford, MI), and their kidneys were used to isolate renal proximal tubules used for primary culture of RPTC.
Ischemia-reperfusion injury.
Male mice 12–16 wk of age were used for the study. Mice were anesthetized using pentobarbital sodium (50 mg/kg body wt), and kidneys were exposed under sterile conditions through a midline incision of the abdomen as described previously (34). The kidneys were decapsulated, both renal hila were clamped for 50 min using small vascular clamps, and mice were transferred to a heating pad that maintained a temperature of 37°C. Mice received ∼0.5 ml sterile saline intraperitoneally during operation. After the clamps were removed, reperfusion of both kidneys was confirmed visually, and the abdomen was closed with silk suture. The animals were returned to their cages and allowed free access to food and water. Sham operations were also performed during which kidneys were manipulated as described, but ischemia was not induced. At 24 h of reperfusion, blood and kidneys were harvested to assess renal functions and tissue morphology and to isolate mitochondria from renal cortex.
Morphological assessment.
Harvested kidneys were immersed and fixed in 4% neutral-buffered formaldehyde. The tissues were embedded in paraffin, and thin (4–8 μm) sections were cut, processed for light microscopy, and stained with hematoxylin-eosin and periodic acid-Schiff. The degree of morphological damage to the kidney after ischemia was evaluated by light microscopy using the following histological criteria to assess morphological damage to the kidney: tubular necrosis, brush border loss, red blood cell extravasation, tubular dilatation, distal nephron damage, tubular cast formation, and inflammation. These parameters were evaluated on a scale of 0 to 4: not present (0), mild (1), moderate (2), severe (3), and very severe (4) on three different animals. The images were captured using a Nikon Eclipse E800 microscope and the objectives Nikon Plan Apo ×4 and Nikon Plan Apo ×20. The power of the phototube lens of the camera was ×12.2.
Creatinine and blood urea nitrogen levels.
Creatinine and blood urea nitrogen were determined spectrophotometrically using the Creatinine Reagent Set and Blood Urea Nitrogen kit from Biotron Diagnostics (Hemet, CA) following the manufacturer's protocol.
Isolation of RPTC mitochondria.
At 24 h after ischemia, mice were killed, kidneys were removed, and renal cortexes were dissected and homogenized in the ice-cold isolation buffer [10 mM N-2-hydroethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 225 mM mannitol, 75 mM sucrose, 2 mM EGTA, and 0.1% BSA, fatty acid free, pH 7.4]. The homogenate was spun down at 1,000 g for 5 min at 4°C, and the resulting supernatant was centrifuged at 15,000 g for 15 min at 4°C. The mitochondrial pellet was washed two times using the isolation buffer and centrifuged at 15,000 g for 10 min at 4°C. The final mitochondrial pellet was resuspended in the isolation buffer and used to measure state 3 respiration or in the assay buffer (10 mM KH2PO4, 5 mM MgCl2, pH 7.2) used to determine the activity of respiratory complexes.
Isolation and culture of renal proximal tubular cells.
Renal proximal tubules were isolated from rabbit kidneys by the iron-oxide perfusion method and cultured in 35-mm culture dishes in improved conditions as previously described (38, 39). The culture medium was a 50:50 mixture of DMEM and Ham's F-12 nutrient mix without phenol red, pyruvate, and glucose, supplemented with 15 mM NaHCO3, 15 mM HEPES, and 6 mM lactate (pH 7.4, 295 mosmol/kg).
Adenoviral constructs and amplification.
Adenoviral vector encoding dominant-negative (inactive) PKC-ε (dnPKC-ε) was constructed as previously described, and an aliquot of the adenovirus kindly provided by Dr. Peipei Ping, University of California at Los Angeles (Los Angeles, CA) (44). PKC-ε was made constitutively active by deletion of residues 154–163 of its inhibitory pseudosubstrate domain as described previously (56). Adenoviral vector encoding the constitutively active mutant of PKC-ε (caPKC-ε) was constructed as described previously (54) and an aliquot of the adenovirus kindly provided by Dr. Allen M. Samarel, Loyola University Medical Center (Maywood, IL). Adenoviruses were amplified and purified as we described previously (51).
PKC-ε activation and inhibition in primary culture.
All transfections were carried out using an adenoviral vector delivery technique in confluent quiescent cultures of RPTC as described earlier (40). PKC-ε was selectively activated by expressing caPKC-ε mutant [multiplicity of infection (MOI) = 50]. Selective inhibition of PKC-ε was obtained by expressing dnPKC-ε (MOI = 100). Infection with adenoviral particles encoding an empty pShuttle vector (MOI = 100) was used as a control. Culture media were changed 24 h after infections. Exposure to hypoxia and measurement of mitochondrial functions were carried out 48 h after infection.
Hypoxia exposure.
Hypoxia was induced in RPTC by aspirating culture media, overlaying cells with fresh and warm (37°C) media pregassed with 95% N2-5% CO2, and placing the dishes in an air-tight hypoxia chamber (equipped with the Proox oxygen controller; BioSpherix, Lacona, NY) that was gassed with 95% N2-5% CO2 to maintain the atmosphere of 94% N2-5% CO2-1% O2 and housed in a regular cell culture incubator at 37°C. Hypoxia was terminated by returning cultures to air-5% CO2 atmosphere (reoxygenation).
State 3 respiration.
State 3 respiration was determined by measuring oxygen consumption in isolated mitochondria in a buffer containing 20 mM HEPES (pH 7.4), 137 mM KCl, 2 mM KH2PO4, 0.5 mM EGTA, 5 mM MgCl2, and the respiratory substrates 5 mM glutamate and 5 mM malate as donors of electrons to complex I or 10 mM succinate plus 0.1 μM rotenone as a donor of electrons to complex II of the electron transport chain. State 3 respiration in cultured RPTC was measured as described earlier (37). State 3 respiration was initiated by adding ADP (0.4 mM final concentration). State 4 respiration was measured in the presence of oligomycin (5 μg/ml).
Activity of complexes of the electron chain.
Activities of complexes I and II of the electron transport chain were measured in isolated mitochondria as described previously (50). Complex I (NADH-ubiquinone oxidoreductase) activity was assayed spectrophotometrically by following the oxidation of NADH (0.25 mM) at 340 nm (30°C) in the assay buffer (10 mM KH2PO4, 5 mM MgCl2, pH 7.2) containing 62.5 μM ubiquinone, 0.25% BSA, 2 μg/ml antimycin A, and mitochondria. The decrease in absorbance (NADH oxidation) was recorded for 3 min, 10 μg/ml rotenone was added, and the absorbance was recorded for another 2 min. Complex I activity was calculated as the rotenone-sensitive NADH-ubiquinone oxidoreductase activity. Activity of complex II (succinate-ubiquinone oxidoreductase) was measured by following the reduction of dichlorophenolindophenol (DCCP, 0.25 mM) at 590 nm (30°C) in the presence of 20 mM succinate, 2 μg/ml antimycin A, 10 μg/ml rotenone, 0.25% BSA, and 62.5 μM ubiquinone. Complex III (ubiquinol-cytochrome c oxidoreductase) activity was assessed by following the reduction of cytochrome c (60 μM) at 550 nm (30°C) in the assay buffer containing 10 μg/ml rotenone, 10% BSA, 50 μM decyl ubiquinol, and 0.24 mM KCN. The increase in absorbance (reduction of cytochrome c) was recorded in the absence and presence of 2 μg/ml antimycin A. Complex III activity was calculated as the antimycin A-sensitive ubiquinol-cytochrome c oxidoreductase activity. Complex IV (cytochrome oxidase) activity was assessed by following the oxidation of reduced 90 μM cytochrome c at 550 nm (30°C) in the assay buffer containing 10% BSA and 2 μg/ml antimycin A in the presence and absence of 0.24 mM KCN. Complex IV activity was calculated as KCN-sensitive cytochrome oxidase activity.
Oxidant generation.
The carboxy derivative of fluorescein, 5-(and-6)-carboxy-2′,7′-dichlorodihydro-fluorescein (carboxy-H2DCFDA), was used to assess oxidant generation in renal cortical slices. The dye is taken up by the cells and is nonfluorescent until the acetate groups are removed by intracellular esterases and oxidation occurs in the cell. Renal cortexes were cut into thin slices and incubated at 37°C in 48-well plates containing the respiration buffer with either 5 mM glutamate and 5 mM malate or 10 mM succinate plus 0.1 μM rotenone. Tissues were loaded with 5 μM carboxy-H2DCFDA and incubated at 37°C for 30 min with gentle swirling. Following incubation, fluorescence in individual wells of the plate was analyzed using excitation at 485 nm and emission at 535 nm (Tecan SpectraFluor Plus microplate reader, Research Triangle Park, NC). Initial experiments performed using 1, 2, 5, and 10 μM carboxy-H2DCFDA determined that 5 μM carboxy-H2DCFDA was the optimal dye concentration in these conditions. Unstained renal cortical slices suspended in the respiration buffer containing substrates were assessed for autofluorescence and were included in each experiment. Following measurements of fluorescence, the tissue slices were blotted on the blotting paper and weighed. All results were corrected for autofluorescence and expressed as arbitrary fluorescence units per milligram of wet tissue.
Proteomic analysis.
To assess the effect of ischemia and reperfusion on the levels of mitochondrial proteins in wild-type and PKC-ε-deficient mice, mitochondria were isolated from renal cortexes at 24 h of reperfusion and subjected to proteomic analysis. Mitochondrial proteins were solubilized using a buffer containing 2 M thiourea, 7 M urea, 4% CHAPS, and 30 mM Tris·HCl, pH 8.8. The eluates were analyzed by two-dimensional differential in-gel electrophoresis performed at Applied Biomics (Hayward, CA) as described previously (35). In brief, the samples were covalently linked to green or red cyanine dye fluors and separated in the horizontal direction by isoelectric focusing (isoelectric point 3–10) followed by SDS-PAGE in the vertical direction (150-10 kDa). Image acquisition and in-gel analysis of protein fold changes were performed using DeCyder software (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK). The gel was washed multiple times, and protein spots of interest were trypsin digested in gels at 37°C. Digested peptide fragments were identified by mass spectrometry (MS) analysis using matrix-assisted laser desorption ionization/time of flight. Protein identification was based on peptide fingerprint mass mapping (using MS data) and peptide fragmentation mapping (using MS/MS data). Ten to 20 most abundant peptides were subjected to fragmentation (MS/MS), and both MS and MS/MS spectra were subjected to database search using GPS explorer equipped with the MASCOT search engine using the National Center for Biotechnology Information protein database.
Immunoblotting.
Phosphorylation and levels of proteins of interest in RPTC homogenates and mitochondria and renal cortex were assessed by immunoblot analysis as described previously (36).
Statistical analysis.
Differences between mouse genotypes were assessed by the two-sided Student's t-test for independent samples or by ANOVA. Multiple means were compared using Fisher's protected least-significance difference test. The level of significance was set at P < 0.05.
RESULTS
Ischemia induces PKC-ε translocation to mitochondria.
The increase in PKC-ε levels in renal cortical homogenates of WT mice at 24 h after ischemia was accompanied by increased levels of PKC-ε protein in renal cortical mitochondria (Fig. 1, A and B). These data show that reperfusion after ischemia is associated with translocation of PKC-ε to mitochondria. No PKC-ε was present in homogenates or mitochondria of PKC-ε KO mice (Fig. 1A).
Fig. 1.
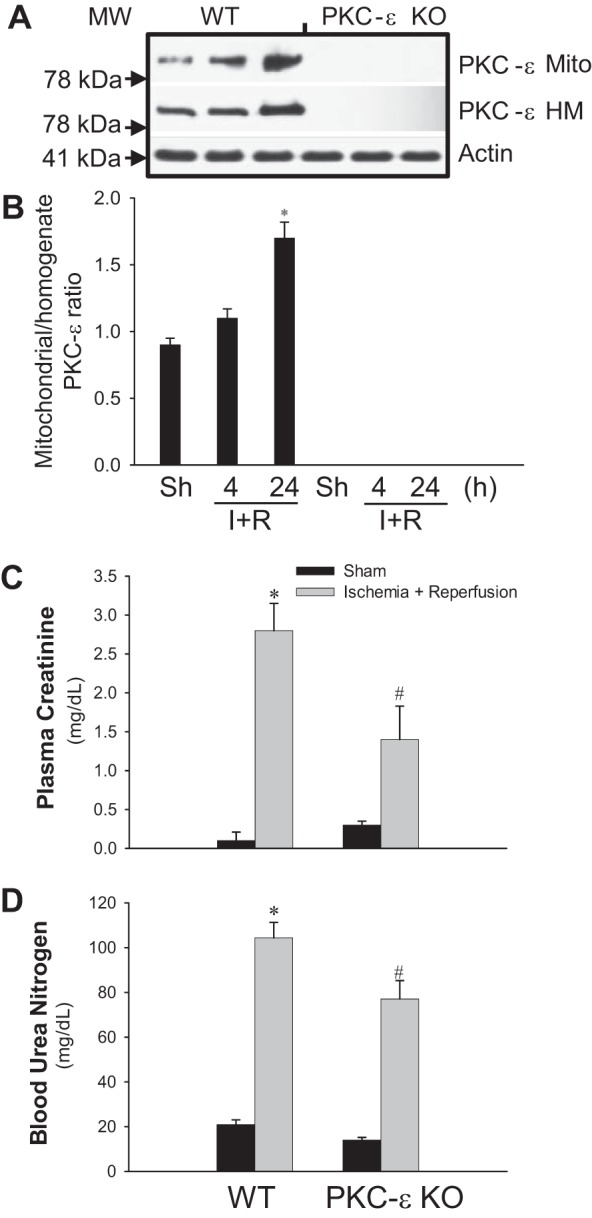
Deletion of protein kinase C-ε (PKC-ε) reduces decreases in renal functions after ischemia. A: PKC-ε levels in renal cortical mitochondria (Mito) and homogenates (HM) at 4 and 24 h after renal ischemia in wild-type (WT) and PKC-ε-deficient (PKC-ε KO) male mice. B: ratio of mitochondrial/total (homogenate) PKC-ε levels in renal cortexes at 4 and 24 h after renal ischemia in WT and PKC-ε KO male mice. Quantification of the PKC-ε levels was carried out by densitometric analysis of immunoblots. Renal function was evaluated using creatinine (C) and urea nitrogen (D) levels in mouse plasma collected at 24 h of reperfusion after renal ischemia. Results are averages ± SE of data obtained from 7–11 animals. *, #Values significantly different (P < 0.05) from respective controls. The immunoblot results are representative of 3 different experiments.
Deletion of PKC-ε improves renal function after ischemia.
Serum creatinine levels in WT mice increased 28-fold at 24 h of reperfusion after ischemia (Fig. 1C). Deletion of PKC-ε reduced ischemia-induced increases in creatinine levels by 50% (Fig. 1C). Serum blood urea nitrogen levels increased fivefold at 24 h of reperfusion in WT mice, and this increase was reduced in PKC-ε KO mice (Fig. 1D).
Deletion of PKC-ε improves renal morphology after ischemia.
Figure 2 shows that ischemia produced changes in renal morphology at the cortical-medullary junction, including brush border loss in the proximal and distal tubules, severe confluent necrosis in the proximal tubules, and tubular cast formation (Fig. 2, C and D). Assessment of seven criteria of kidney morphological damage revealed two- to fourfold increases in the degree of injury in ischemic kidneys from WT animals (Fig. 2I). Tubular necrosis and damage of the distal part of the nephron were accompanied by loss of the brush border, cast formation, and an increase in the number of inflammatory cells in WT mice (Fig. 2, C, D, and I). PKC-ε deletion reduced ischemia-induced morphological damage to proximal and distal tubules. The loss of brush border and proximal tubular necrosis were reduced, whereas damage to the distal portions of the nephron was blocked in PKC-ε KO mice (Fig. 2, G, H, and I). Confluent necrosis, cast formation, and the number of inflammatory cells were also reduced at 24 h after ischemia in the PKC-ε KO animals (Fig. 2, G, H, and I). Thus, these data demonstrate that PKC-ε is involved in the mechanisms of kidney injury and decreases in renal functions due to ischemia and that blocking PKC-ε reduces ischemia-induced kidney injury.
Fig. 2.

Deletion of PKC-ε improves renal morphology during reperfusion after ischemia. Renal histology of the cortical-medullary junction assessed in periodic acid-Schiff (PAS)-stained kidney sections from WT (A–D) and KO (E–H) mice. Sections represent kidneys from sham-operated mice (A, B, E, and F) and mice killed at 24 h of reperfusion after 50 min of renal ischemia (C, D, G, and H). Magnification: ×48.8 (A, C, E, and G) and ×244 (B, D, F, and H). The sections are representative of WT animals (n = 5) and KO animals (n = 4). I: quantitative evaluation of morphological kidney injury at 24 h of reperfusion after 50 min of renal ischemia expressed relatively according to materials and methods. The images were captured using a Nikon Eclipse E800 microscope using objectives Nikon Plan Apo ×4 (A, C, E, and G) and Nikon Plan Apo ×20 (B, D, F, and H). Morphology was scored according to proximal tubule necrosis, brush border loss, cast formation within tubules, inflammation (infiltration of the inflammatory cells), tubule dilatation, distal nephron damage, and red blood cell extravasation. Yellow arrows show severe confluent necrosis in the proximal tubules, and green stars indicate tubular cast formation. Results are averages ± SE of data obtained from kidney sections from WT (n = 5) and KO (n = 4) mice. Values with dissimilar superscripts (a, b, c) within a group are significantly different (P < 0.05) from each other.
PKC-ε mediates ischemia-induced changes in mitochondrial function.
State 3, state 4, and uncoupled respirations were used to assess mitochondrial function associated with oxidative capacity and phosphorylation (Fig. 3). State 3 and uncoupled respirations in mitochondria energized by substrates oxidized via complex I of the electron transfer chain (glutamate and malate) were reduced by 44 and 42%, respectively, at 24 h after ischemia in WT mice (Fig. 3, A and C). Deletion of PKC-ε increased complex I-coupled state 3 respiration in sham kidneys and attenuated the decreases in complex I-coupled state 3 and uncoupled respirations in mitochondria of ischemic kidneys (Fig. 3, A and C). State 4 respiration in mitochondria energized by glutamate and malate was not altered by ischemia and/or deletion of PKC-ε (Fig. 3B).
Fig. 3.
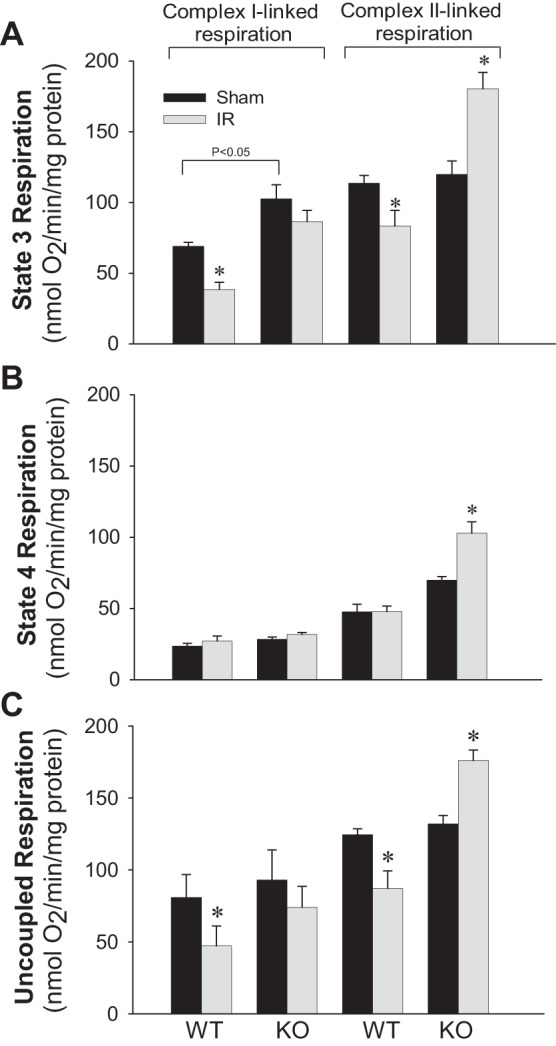
Deletion of PKC-ε restores mitochondrial respiration in renal cortical tissue injured by ischemia. State 3 (A), state 4 (B), and uncoupled (C) respirations coupled to the oxidation of electron donors to complex I (5 mM glutamate + 5 mM malate) and complex II (10 mM succinate + 0.1 μM rotenone) in renal cortical mitochondria isolated at 24 h of reperfusion after 50 min of renal ischemia in WT and KO mice. Results are averages ± SE of data obtained from 4–8 animals. *Values significantly different (P < 0.05) from respective controls.
State 3 and uncoupled respirations in mitochondria energized by succinate (the donor of electrons to complex II) decreased by 27 and 30%, respectively, at 24 h after ischemia in WT animals (Fig. 3, A and C). Deletion of PKC-ε had no effect on succinate-energized state 3 and uncoupled respirations but increased state 4 respiration by 46% in renal mitochondria of sham animals (Fig. 3, A–C). Ischemia increased succinate-energized respirations by 51% (state 3), 48% (state 4), and 33% (uncoupled) in kidneys of PKC-ε KO mice (Fig. 3, A–C). Respiratory control ratio (RCR) coupled to complex I decreased from 3.7 ± 0.4 in controls to 1.7 ± 0.1 in mitochondria of ischemic kidneys from WT animals but not in PKC-ε KO mice (3.7 ± 0.3 in sham-operated vs. 3.1 ± 0.4 in ischemic mice). Complex II-coupled RCR in sham WT kidneys (2.5 ± 0.1) was decreased compared with complex I-coupled RCR. Ischemia further decreased complex II-coupled RCR to 1.7 ± 0.1. Deletion of PKC-ε had no effect on complex II-coupled RCR in sham kidneys (2.7 ± 0.3), but it prevented decreases in RCR in ischemic kidneys (2.4 ± 0.2). These results show that PKC-ε activation is involved in the ischemia-induced decreases in the oxidative capacity of the respiratory chain and that deletion of PKC-ε ameliorates these decreases.
PKC-ε mediates ischemia-induced decreases in the activities of complexes of the respiratory chain.
Activities of respiratory complexes I, III, and IV in mitochondria isolated from ischemic kidneys of WT mice decreased 59, 89, and 77%, respectively, whereas the activity of complex II was not changed (Fig. 4). Deletion of PKC-ε reduced activities of complex I and complex III (33 and 38%, respectively) but not activities of complex II and complex IV in sham-operated kidney cortexes (Fig. 4). Deletion of PKC-ε prevented the decreases in activities of complexes I and III and reduced the decrease in activity of complex IV at 24 h after ischemia (Fig. 4, A, C, and D). These data demonstrate that deletion of PKC-ε attenuates ischemia-induced decreases in activities of complexes I, III, and IV of the mitochondrial electron transport chain.
Fig. 4.

Deletion of PKC-ε restores activities of complexes of the electron transport chain in mitochondria of renal cortical tissue injured by ischemia. Activities of NADH-ubiquinone oxidoreductase (complex I) (A), succinate-ubiquinone oxidoreductase (complex II) (B), ubiquinol-cytochrome c oxidoreductase (C), and cytochrome oxidase (complex IV) (D) in mitochondria isolated from renal cortexes of WT and KO mice at 24 h of reperfusion after 50 min renal ischemia. Results are averages ± SE of data obtained from 4–8 animals. Values with dissimilar superscripts (a, b, c) are significantly different (P < 0.05) from each other.
Deletion of PKC-ε alters the response of mitochondrial proteome to ischemia.
To test the effects of PKC-ε deletion on the response of mitochondrial proteins involved in oxidative phosphorylation, we analyzed mitochondrial proteome of renal cortexes of WT and PKC-ε KO mice. Proteomics revealed lower levels of several crucial mitochondrial proteins of fuel metabolism and oxidative phosphorylation in PKC-ε KO sham kidneys, including 2-oxoglutarate (α-ketoglutarate) dehydrogenase (spot 2), acetyltransferase component of pyruvate dehydrogenase complex, the α-subunit of ATP synthase (spot 3), subunits 1 and 2 of the cytochrome b-c1 complex (complex III) (spot 4), subunit 5B of cytochrome c oxidase (spot 6), iron-sulfur protein of NADH dehydrogenase (complex I) (spot 8), and branched-chain α-keto acid dehydrogenase complex (spot 10) (Fig. 5, A, D, G, J, and P). The decreases in subunits 1 and 2 of the cytochrome b-c1 complex and iron-sulfur protein of NADH dehydrogenase levels were consistent with lower activities of complex I and complex III in mitochondria from sham kidneys of PKC-ε KO mice than those in WT mice (Figs. 5, D and J, and 4, A and C). Ischemia had a profoundly different effect on the levels of these proteins in WT and PKC-ε KO mice. At 4 h of reperfusion, the levels of α-subunit of ATP synthase and subunits 1 and 2 of complex III were decreased in WT but not in PKC-ε KO kidneys, whereas the levels of β-subunit of ATP synthase were not different in WT and PKC-ε KO kidneys (Fig. 5P). At 24 h of reperfusion in WT mice, the levels of these proteins in ischemic kidneys were not different from sham kidneys (Fig. 5, B, E, H, K, and N). In contrast, renal cortical mitochondria from ischemic PKC-ε KO mice had severalfold higher levels of α-ketoglutarate dehydrogenase (3.8-fold), the α-subunit of ATP synthase (6.7-fold), subunit 2 of complex III (13.7-fold), subunit 5B of cytochrome c oxidase (2.6-fold), iron-sulfur protein of NADH dehydrogenase (2-fold), branched-chain α-keto acid dehydrogenase complex (2-fold), and acetyltransferase component of pyruvate dehydrogenase complex (4-fold) than mitochondria from sham kidneys (Fig. 5, C, F, I, and L, and Table 1). In addition, deletion of PKC-ε increased protein levels of thioredoxin-dependent peroxide reductase fivefold (Fig. 5L). These results: 1) show that ischemia in PKC-ε KO kidneys does not cause damage and degradation of mitochondrial proteins of energy metabolism that are observed during reoxygenation in WT kidneys and 2) suggest that deletion of PKC-ε maintains the capacity of renal cortical mitochondria to synthesize ATP.
Fig. 5.

Deletion of PKC-ε upregulates the levels of crucial proteins associated with oxidative phosphorylation in mitochondria of renal cortical tissue injured by ischemia and reperfusion. Mitochondria were isolated from sham-operated and ischemic WT and KO mice at 24 h of reperfusion after 50 min renal ischemia as described in materials and methods. Proteins present in mitochondria were labeled using Cy3 and Cy5 dyes, resolved using 2-dimensional differential in-gel electrophoresis (2D-DIGE: horizontally, by isoelectric focusing point from pH 3 to 10; vertically, by molecular weight from 150 to 10 kDa), excised and eluted from the gel, and identified by mass spectrometry (MS)/MS analysis. Top (WT vs. KO, sham), proteins from WT mice were labeled green (Cy5), whereas proteins from KO mice were labeled red (Cy3). Middle [WT, sham vs. ischemia-reperfusion (IR)], proteins from sham-operated mice were labeled green (Cy5), whereas proteins from ischemic mice were labeled red (Cy3). Bottom (KO, sham vs. IR), proteins from sham-operated mice were labeled green (Cy5), whereas proteins from ischemic mice were labeled red (Cy3). A–C: mitochondrial pyruvate carboxylase (spot 1) and 2-oxoglutarate dehydrogenase (spot 2). D–F: α-subunit of ATP synthase (spot 3), subunit 2 of cytochrome b-c1 complex (complex III, spot 4), and mitochondrial medium-chain specific acyl-CoA dehydrogenase (spot 5). G–I: subunit 5B of cytochrome oxidase (spot 6). J–L: thioredoxin-dependent peroxide reductase (spot 7) and NADH dehydrogenase (ubiquinone) iron-sulfur protein 3 (complex I, spot 8). M–O: lipoamide acyltransferase component of branched-chain α-keto acid dehydrogenase (spot 9) and succinate dehydrogenase (ubiquinone) iron-sulfur subunit (complex II, spot 10). P: left, protein levels of subunits (core proteins) 1 and 2 of cytochrome b-c1 complex and subunits-α and -β of ATP synthase (FoF1-ATPase) in renal cortical tissues of sham-operated and ischemic WT and PKC-ε KO mice at 4 and 24 h of reperfusion after 50 min renal ischemia. Immunoblot results are representative of 3 different experiments involving 3 WT mice and 3 KO mice each. Right, densitometric analysis of immunoblot bands shown on left. *Values significantly different (P < 0.05) from respective sham controls.
Table 1.
Identification of proteins of energy metabolism that are changed by the deletion of PKC-ε and ischemia in mouse renal cortical mitochondria
| Spot No. | Top-Ranked Protein Name | Accession No. | Protein Mol Wt | Protein PI | No. of Unique Peptides | Protein Score | Fold Change after Ischemia |
|---|---|---|---|---|---|---|---|
| 1 | Pyruvate carboxylase, mitochondrial | PYC_Mouse | 129,603 | 6.25 | 41 | 986 | +1.9 |
| 2 | 2-Oxoglutarate dehydrogenase, mitochondrial | ODO1_Mouse | 116,375 | 6.36 | 37 | 698 | +3.8* |
| 3 | ATP synthase, subunit-α, mitochondrial; ATP5a1 | ATPA_Mouse | 59,717 | 9.22 | 26 | 1140 | +6.7* |
| 4 | Cytochrome b-c1 complex subunit 2, mitochondrial | QCR2_Mouse | 48,205 | 9.26 | 14 | 405 | +13.7* |
| 5 | Medium-chain specific acyl-CoA dehydrogenase, mitochondrial | ACADM_Mouse | 46,452 | 8.60 | 25 | 780 | −1.6 |
| 6 | Cytochrome c oxidase subunit 5B, mitochondrial | COX5B_Mouse | 13,804 | 8.69 | 6 | 243 | +2.6* |
| 7 | Thioredoxin-dependent peroxide reductase, mitochondrial | PRDX3_Mouse | 28,109 | 7.15 | 10 | 628 | +4.9* |
| 8 | NADH dehydrogenase [ubiquinone] iron-sulfur protein | NDUS3_Mouse | 30,131 | 6.67 | 9 | 118 | +2.0* |
| 9 | Succinate dehydrogenase [ubiquinone] iron-sulfur subunit | DHSB_Mouse | 31,793 | 8.96 | 17 | 307 | −1.5 |
| 10 | Lipoamide acyltransferase component of branched-chain α-keto acid dehydrogenase complex, mitochondrial | ODB2_Mouse | 53,213 | 8.78 | 11 | 108 | +2.0* |
| 11 | Dihydrolipoyllysine-residue acetyltransferase component of pyruvate dehydrogenase complex, mitochondrial | ODP2_MOUSE | 67,898 | 8.81 | 13 | 438 | +4.0* |
Mitochondria from renal cortex were isolated from sham and ischemic mice at 24 h of reperfusion after bilateral ischemia. Proteins were identified using two-dimensional differential in-gel electrophoresis followed by mass spectrometry analysis using matrix-assisted laser desorption ionization/time of flight. Proteins were identified by GPS explorer equipped with MASCOT search engine using SwissProt database. Image acquisition and in-gel analysis of protein fold changes were performed using DeCyder software as described in materials and methods. PKC-ε, protein kinase C-ε.
Values significantly different (P < 0.05) from those in wild-type mice.
PKC-ε mediates renal oxidant production after ischemia.
Oxidant generation in renal cortex of WT animals was increased at 24 h of reperfusion after ischemia. This increase occurred in the presence of the substrates oxidized through complex I (glutamate + malate) and complex II (succinate + rotenone) of the respiratory chain (Fig. 6). In contrast, oxidant generation in kidneys of PKC-ε-deficient mice was not altered by ischemia (Fig. 6).
Fig. 6.
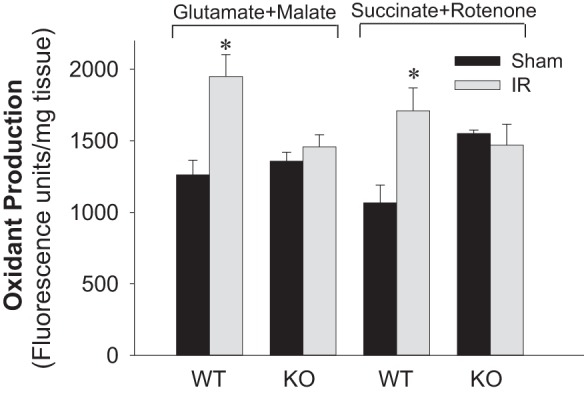
Deletion of PKC-ε decreases oxidant production in renal tissue after ischemia. Oxidant production was assessed using 5 μM 5-(and-6)-carboxy-2′,7′-dichlorodihydro-fluorescein (carboxy-H2DCFDA) in renal tissue slices energized with electron donors to the respiratory complex I (5 mM glutamate + 5 mM malate) and complex II (10 mM succinate + 0.1 μM rotenone) in WT and KO mice at 24 h of reperfusion after 50 min renal ischemia. Data are expressed as %sham controls. Results are averages ± SE of data obtained from 4–12 animals. *Values significantly different (P < 0.05) from respective sham controls.
PKC-ε deletion improves survival after renal ischemia.
Eighty six percent of WT mice survived 24 h of reperfusion following renal ischemia. In contrast, 100% of PKC-ε-deficient mice subjected to renal ischemia were alive at 24 h of reperfusion. No mortality was observed in sham-operated mice.
PKC-ε mediates hypoxia-induced decreases in mitochondrial function.
Renal cortex is composed of several different cell types, with the RPTC making up ∼80% of the renal cortex mass. To test whether 1) RPTC are the target of PKC-ε in the kidney cortex and 2) activation of PKC-ε changes respiratory functions of mitochondria in injured RPTC, we used primary cultures of RPTC grown in improved culture conditions (38, 39) and exposed them to hypoxia. Hypoxia induced PKC-ε translocation to RPTC mitochondria (Fig. 7, A and B). State 3 respiration in hypoxic RPTC was decreased 67 and 54% in mitochondria energized by glutamate and malate, and succinate, respectively (Fig. 7, C-and D). Transfection of RPTC with caPKC-ε increased the levels of phosphorylated PKC-ε in RPTC mitochondria (data not shown) and decreased state 3 respiration during normoxia to the levels observed during hypoxia (Fig. 7, C and D). In contrast, overexpressing the inactive mutant of PKC-ε (dnPKC-ε) had no effect on state 3 respiration during normoxia and improved state 3 respiration (36% increase in complex I- and 34% increase in complex II-coupled respirations) after hypoxia in RPTC (Fig. 7, C and D).
Fig. 7.
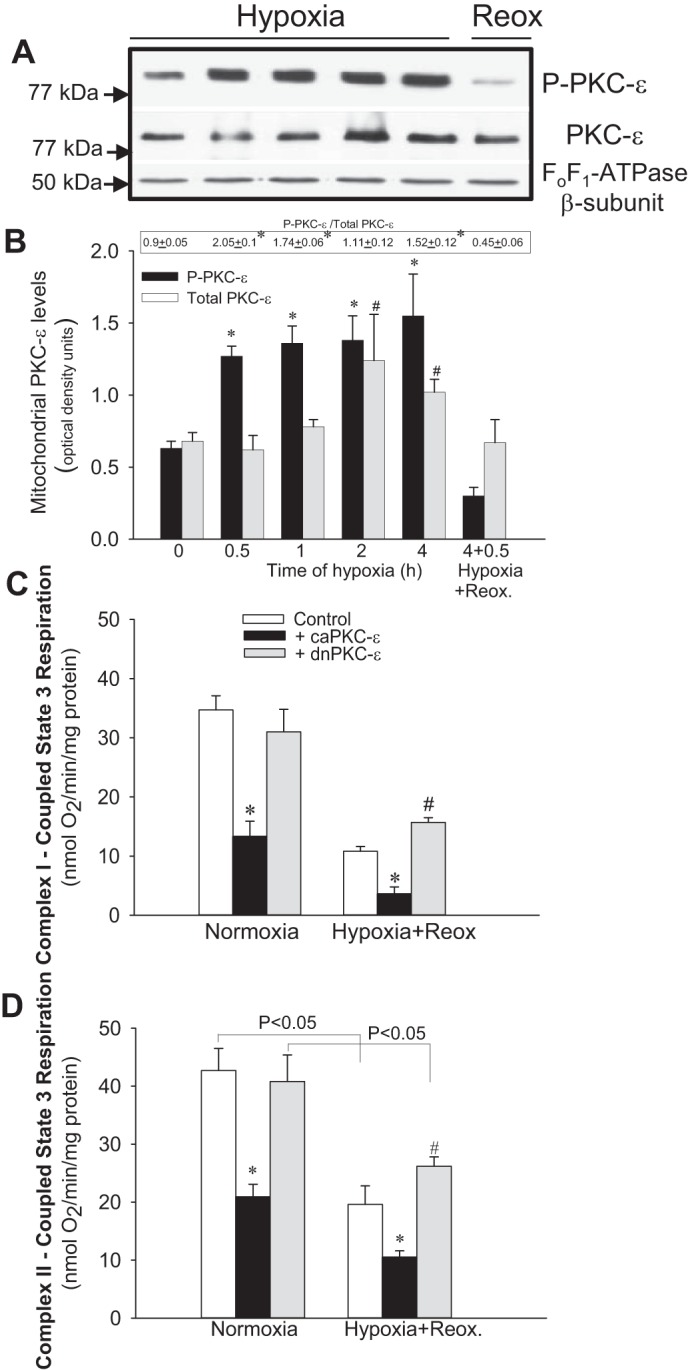
Activation of PKC-ε exacerbates hypoxia-induced decreases in mitochondrial respiration in renal proximal tubule cells (RPTC). A: protein levels of phosphorylated PKC-ε (P-PKC-ε) and total PKC-ε (PKC-ε) in RPTC mitochondria at different time points of hypoxia and reoxygenation. Levels of the β-subunit of FoF1-ATPase were used as loading controls. B: densitometric analysis of the phosphorylated and total PKC-ε levels at different time points of hypoxia (0.5–4 h) and at 0.5 h of reoxygenation after 4 h of hypoxia in mitochondria isolated from renal proximal tubular cells in primary culture. Quantification of the PKC-ε levels was carried out by densitometric analysis of immunoblots. The ratio of phosphorylated and total PKC-ε levels is shown above the bar graph in B. C and D: state 3 respiration coupled to the oxidation of electron donors to complex I (5 mM glutamate + 5 mM malate) and complex II (10 mM succinate + 0.1 μM rotenone) in cultured RPTC overexpressing the constitutively active (caPKC-ε) and inactive [dominant-negative (dn) PKC-ε] mutants of PKC-ε. RPTC were subjected to hypoxia for 4 h, and state 3 respiration was assessed at 0.5 h of reoxygenation after hypoxia. PKC-ε was activated in RPTC by expressing caPKC-ε mutant [multiplicity of infection (MOI) = 50] or inhibited by expressing dnPKC-ε (MOI = 100). Results are averages ± SE of data obtained from 5–7 independent experiments (RPTC isolations). *, #Values significantly different (P < 0.05) from respective controls.
PKC-ε mediates hypoxia-induced decreases in activities of complexes of the respiratory chain.
Hypoxia decreased activities of complexes I, III, and IV (Fig. 8) but not the activity of complex II (35.3 ± 5.4 vs. 32.5 ± 5.7 nmol DCCP·min−1·mg protein−1 in hypoxic vs. normoxic RPTC, respectively). Overexpression of caPKC-ε reduced activities of complexes I and III in normoxic RPTC, exacerbated the decrease in complex I activity, and induced a 24% decrease in complex II activity in hypoxic RPTC (Fig. 8, A and B). In contrast, overexpression of dnPKC-ε restored activities of complexes I, III, and IV in hypoxic RPTC (Fig. 8). These data show that PKC-ε is involved in hypoxia-induced decreases in mitochondrial respiration and activities of complexes I, III, and IV of the respiratory chain in RPTC.
Fig. 8.
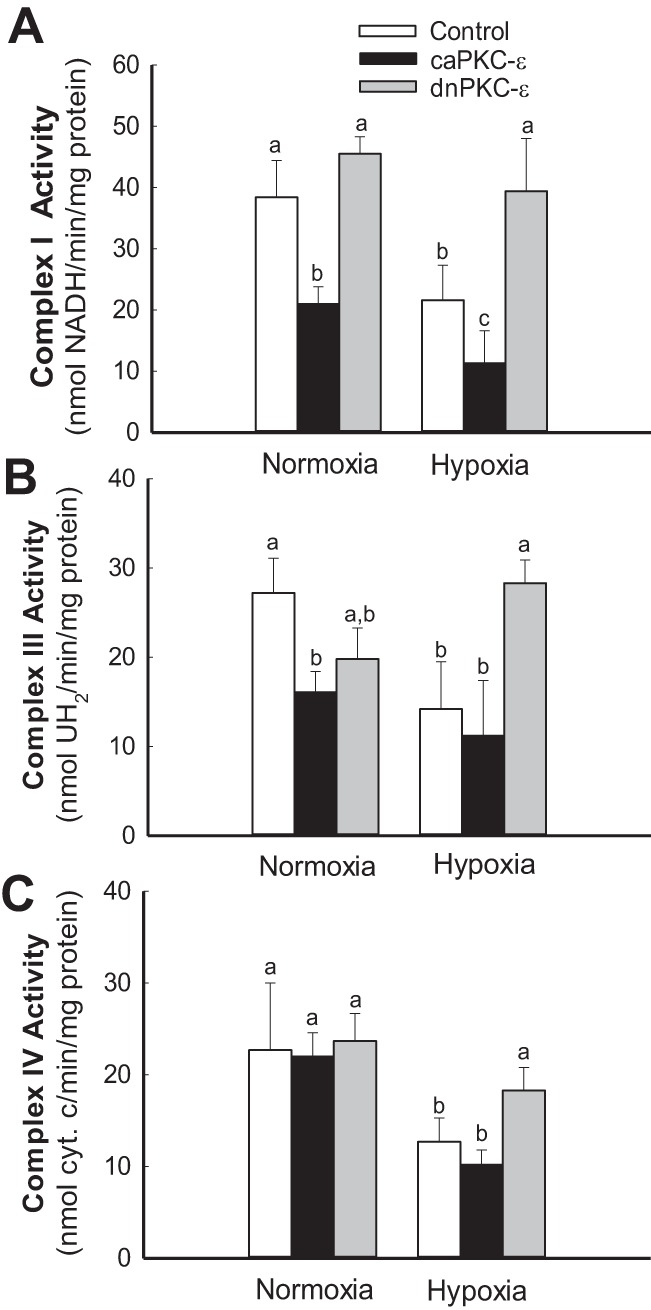
Activation of PKC-ε exacerbates hypoxia-induced decreases in activities of complexes of the electron transport chain in RPTC mitochondria. Activities of NADH-ubiquinone oxidoreductase (complex I) (A), ubiquinol-cytochrome c oxidoreductase (B), and cytochrome oxidase (complex IV) (C) in mitochondria isolated from cultured RPTC overexpressing caPKC-ε and dnPKC-ε mutants of PKC-ε. PKC-ε was activated in RPTC by expressing caPKC-ε mutant (MOI = 50) or inhibited by expressing dnPKC-ε mutant (MOI = 100) at 48 h before the exposure to hypoxia. RPTC were subjected to hypoxia for 4 h, and mitochondria were isolated at 0.5 h of reoxygenation. Results are averages ± SE of data obtained from 4–8 independent experiments (RPTC isolations). Values with dissimilar superscripts (a, b, c) are significantly different (P < 0.05) from each other.
DISCUSSION
Kinase prediction analysis shows an important role for PKA and PKC in mitochondria of major tissues (3). PKC-ε is present at low levels in healthy renal cortex but increases during reperfusion after ischemia, and returns to low levels during kidney recovery (29, 42). We have shown that PKC-ε is also activated by hypoxia and oxidant-induced injury in primary cultures of RPTC and that the inhibition of PKC-ε activation improves RPTC survival after injury (36). Injury to RPTC activates PKC-ε in mitochondria, induces mitochondrial dysfunction and energy deficits, and leads to mitochondrial fission and RPTC death (36, 40). In addition, active PKC-ε increases oxidant generation in cultured RPTC (40). Our data showed that blocking PKC-ε activation protects against energy deficits, oxidative stress, and cell injury in cultured RPTC and suggested that inhibition of PKC-ε may be renoprotective against ischemic injury in vivo. This study tested this hypothesis in an in vivo model of renal ischemia using mice deficient in PKC-ε gene.
Renal ischemia in vivo decreased integrity of the respiratory chain and targeted complexes I and III in renal cortical mitochondria. There was a marked decrease in protein levels of subunits 1 and 2 of complex III at 4 h after ischemia. Deletion of PKC-ε decreased protein levels of subunits of complex I (Fig. 5J) and complex III (Fig. 5, D and P) and reduced activity of these complexes in sham kidneys without compromising mitochondrial respiration or kidney functions (Figs. 3 and 4, A and C). This suggests that the levels of complexes I and III in sham kidneys were not limiting factors in maintaining higher complex I-coupled respiration in PKC-ε KO mice. Furthermore, PKC-ε deletion blocked ischemia-induced decreases in protein levels of critical subunits of complexes I and III (Fig. 5, F, L, and P) and improved activity of these complexes compared with activities in WT mice (Fig. 4, A and C). These results suggest that active PKC-ε negatively regulates integrity of the electron transport chain in renal mitochondria after ischemia and that respiratory complexes of the renal cortex are major targets of PKC-ε in the injured kidney.
Taken together, our data suggest that PKC-ε activation in the ischemic kidney stimulates degradation of the core proteins 1 and 2 of complex III and α-subunit of the ATP synthase during early reperfusion (4 h) and that the deletion of PKC-ε prevents the loss of these peptides in ischemic kidney (Fig. 5P). We have found no evidence of PKC-ε-mediated phosphorylation of complexes I and III or subunits of ATP synthase in renal cortical mitochondria, but many phosphorylations are unstable and lost during isolation of mitochondria and/or their processing for proteomic analysis. Phosphorylations of several subunits of complexes I and III (in a cAMP-dependent manner) have been documented previously, including the studies by He and Lemasters, who showed that Rieske iron-sulfur protein of complex III is regulated by phosphorylation (20). However, the kinase that mediates this modification has not been identified. PKC-ε-mediated phosphorylation of complexes I and III has not been reported. Thus, the mechanism responsible for maintaining activity of these two complexes in PKC-ε-deficient animals appears to be through preservation of mitochondrial integrity and the key proteins of oxidative phosphorylation.
Other factors, including increased import of substrates into mitochondria and/or enhanced substrate oxidation to yield more NADH and FADH2, could contribute to increased respiration in mitochondria of PKC-ε-deficient mice. Respiration coupled to the oxidation of succinate was higher in ischemic kidneys of PKC-ε KO mice than in respective sham controls, which suggests that 1) integrity of complexes III and IV was not compromised and 2) substrate import and/or oxidation in mitochondria was increased in these kidneys. Active PKC-ε directly interacts with and phosphorylates the voltage-dependent anion channel (VDAC-1) in cardiac mitochondria, which prevents the onset of mitochondrial permeability transition, mitochondrial injury, and apoptosis (1). This mechanism is thought to contribute to protective function of active PKC-ε in cardiac tissue (6, 10, 16, 17). VDAC has multiple phosphorylation sites in different tissues, including those within the consensus site for PKC-ε. We have shown that PKC-ε activation in RPTC significantly increases mitochondrial membrane potential, decreases substrate oxidation through the electron transport chain, and induces mitochondrial dysfunction (40). This suggests that PKC-ε is involved in VDAC closure in renal tubular mitochondria, which limits the access of solutes, substrates, and ADP to mitochondria and decreases respiration and oxidative phosphorylation. We hypothesize that the deletion of PKC-ε prevents VDAC phosphorylation and closure during ischemic injury and improves import of respiratory substrates and their oxidation in renal tubular mitochondria.
Phosphorylations of cardiac cytochrome oxidase (complex IV) regulate its sensitivity to allosteric regulation by ATP and the affinity for its substrate, cytochrome c (4, 19, 21, 30, 41, 52). Phosphorylation of cytochrome oxidase by PKC-ε in cardiac tissue promotes complex IV activity (13, 19, 21, 41). Specifically, phosphorylation of the subunit 5B by PKC-ε increases affinity of the cardiac cytochrome oxidase for cytochrome c, improves activity of this complex, and is involved in cardioprotection against ischemia (19, 30, 41). Recent data show that mitochondria from different cell types significantly differ in the pattern of phosphorylation of their proteins. For example, mitochondria from the skeletal and the heart muscles share only 39% of the protein phosphorylation sites (3). The difference in protein phosphorylation sites is even larger between mitochondria from tissues with distinct metabolism and functions. For example, liver mitochondria only share 19% of phosphorylation sites with the skeletal muscle and 25% of phosphorylation sites with heart muscle (3). Therefore, mitochondrial substrates of PKC-ε in renal and cardiac mitochondria may be different or the same substrates may be phosphorylated on different serine or threonine residues and produce different outcomes for the stability and/or activity of these proteins. Although we have not identified any PKC-ε-dependent phosphorylations of cytochrome oxidase in ischemic kidneys or hypoxic RPTC, our data show that the levels of subunit 5B and the activity of cytochrome oxidase are improved in ischemic kidneys from PKC-ε KO mice. Thus, the activation of PKC-ε that is protective in cardiac mitochondria is detrimental to the function and integrity of renal mitochondria.
Ischemia increases ROS production in renal cortical tissue. Dysfunctional electron transport chain and generation of superoxide are major mechanisms that contribute to oxidative stress, lipid peroxidation, and oxidative damage in renal tissue during reperfusion. Our results show that PKC-ε activation contributes to oxidative stress in injured kidney in vivo, since the deletion of PKC-ε blocks increases in oxidant generation in renal cortex regardless of the substrate used to energize mitochondria. These data are consistent with maintained activity of complex I and complex III (two major sites of electron escape within the electron transport chain) during reperfusion after ischemia in PKC-ε KO mice. In addition, our data show increased levels of thioredoxin-dependent peroxide reductase in ischemic kidneys from PKC-ε-deficient mice, which suggests improved antioxidant capacity of mitochondria in PKC-ε KO kidneys. Previously, we documented that sustained activation of PKC-ε leads to persistent increases in ROS production in noninjured RPTC (40). Collectively, our data are consistent with the report of Rong and colleagues (49) who demonstrated decreased generation of ROS during reperfusion in ischemic kidneys of PKC-ε-deficient mice. Because increased ROS production and elevated oxidative stress are major mechanisms leading to inflammation, the deletion of PKC-ε reduced infiltration of inflammatory cells in ischemic kidneys (Fig. 2I), consistent with a previous report demonstrating decreased infiltration of monocytes and macrophages in the renal tissue after ischemia (49). Activation of PKC-ε also contributes to the chronic immune response in cardiac allografts, and inhibition of PKC-ε suppresses chronic inflammation and prolongs cardiac graft survival (28).
Our data also show that renal tissue morphology after ischemia is greatly improved in PKC-ε-deficient mice. Blocking PKC-ε signaling reduces energy deficits and cell death, loss of the brush border, confluent tubular necrosis, and cast formation and blocks the damage to the distal nephron. The improvements in renal morphology are associated with increased renal functions and greater survival after ischemia in PKC-ε KO mice. Similar outcomes of PKC-ε deletion on renal morphology, functions, and survival during reperfusion after ischemia were reported by Rong and colleagues (49). Furthermore, Fuller and colleagues demonstrated that inhibition of PKC by sotrastaurin ameliorates renal damage and promotes cell survival in a model of renal cold preservation and transplantation (14). Thus, blocking PKC-ε-dependent signaling attenuates mitochondrial dysfunction and tissue damage and improves renal functions.
Our observations on the detrimental role of PKC-ε activation in the ischemic injury to the kidney are in stark contrast to numerous reports documenting a protective role of PKC-ε activation against ischemic injury in cardiac tissue and vascular endothelial and neuronal cells and in improving heart recovery from ischemia (23, 25, 48, 53, 55). Likewise, mitochondria have been proposed as a target organelle of the active PKC-ε and the mediator of protective actions of PKC-ε in these cell types (6, 15). Consequently, PKC-ε-selective activators have been proposed as a therapy against ischemia-reperfusion injury in the heart (6). Interestingly, the protective effects of PKC-ε activation may not extend to ischemia associated with cardiac transplantation. It has been reported that pharmacological inhibition of PKC-ε suppresses inflammatory reaction associated with tissue injury during heart transplantation and sepsis due to altered macrophage signaling (28). Thus, the effects of PKC-ε activation appear to be tissue/cell type dependent.
In conclusion, we present evidence that the deletion of PKC-ε gene improves mitochondrial function and decreases ROS production in the kidney during reperfusion after renal ischemia in vivo. These results suggest that PKC-ε activation mediates mitochondrial dysfunction and ROS generation after ischemia. The electron transport chain, specifically complexes I, III, and IV of the chain, are major mitochondrial targets of active PKC-ε, which decreases activity of these complexes, possibly through increased protein degradation. Deletion of PKC-ε increases protein levels of critical subunits of complexes I, III, and IV and restores their activity and mitochondrial respiration after ischemia. Blocking PKC-ε activation also prevents increases in ROS levels and decreases infiltration of inflammatory cells in the injured renal tissues. These changes result in reduced tissue damage and improved overall kidney functions after ischemia. Our data suggest that therapies that block or decrease PKC-ε activation may prove beneficial in reducing acute kidney injury.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-059558 and American Heart Association Grant No. 0151416Z to G. Nowak.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.N. conception and design of research; G.N., D.T.B., and J.M. performed experiments; G.N. and J.M. analyzed data; G.N. and J.M. interpreted results of experiments; G.N. prepared figures; G.N. drafted manuscript; G.N., D.T.B., and J.M. edited and revised manuscript; G.N., D.T.B., and J.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Allen M. Samarel (Loyola University Medical Center, Maywood, IL) for providing an aliquot of adenoviral vector coding the constitutively active PKC-ε and Dr. Peipei Ping (University of California at Los Angeles, Los Angeles, CA) for an aliquot of adenovirus coding the dominant-negative (inactive) mutant of PKC-ε.
REFERENCES
- 1.Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, Guo Y, Bolli R, Cardwell EM, Ping P. Protein kinase Cε interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ Res 92: 873–880, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baines CP, Zhang J, Wang GW, Zheng YT, Xiu JX, Cardwell EM, Bolli R, Ping P. Mitochondrial PKCε and MAPK form signaling modules in the murine heart: enhanced mitochondrial PKCε-MAPK interactions and differential MAPK activation in PKCε-induced cardioprotection. Circ Res 90: 390–397, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Bak S, León I, Jensen O, Højlund K. Tissue specific phosphorylation of mitochondrial proteins isolated from rat liver, heart muscle, and skeletal muscle. J Proteome Res 12: 4327–4339, 2013. [DOI] [PubMed] [Google Scholar]
- 4.Bender E, Kadenbach B. The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. FEBS Lett 466: 130–134, 2000. [DOI] [PubMed] [Google Scholar]
- 5.Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol 14: 2199–2210, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Budas GR, Mochly-Rosen D. Mitochondrial protein kinase Cε (PKCε): emerging role in cardiac protection from ischaemic damage. Biochem Soc Trans 35: 1052–1054, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Cacace AM, Guadagno SN, Krauss RS, Fabbro D, Weinstein IB. The epsilon isoform of protein kinase C is an oncogene when overexpressed in rat fibroblasts. Oncogene 8: 2095–2104, 1993. [PubMed] [Google Scholar]
- 8.Castrillo A, Pennington DJ, Otto F, Parker PJ, Owen MJ, Bosca L. Protein kinase Cε is required for macrophage activation and defense against bacterial infection. J Exp Med 194: 1231–1242, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeCoy DL, Snapper JR, Breyer MD. Anti sense DNA down-regulates proteins kinase C-ε and enhances vasopressin-stimulated Na+ absorption in rabbit cortical collecting duct. J Clin Invest 95: 2749–2756, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di-Capua N, Sperling O, Zoref-Shani E. Protein kinase C-ε is involved in the adenosine-activated signal transduction pathway conferring protection against ischemia-reperfusion injury in primary rat neuronal cultures. J Neurochem 84: 409–412, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Ding L, Wang H, Lang W, Xiao L. Protein kinase C-ε promotes survival of lung cancer cells by suppressing apoptosis through dysregulation of the mitochondrial caspase pathway. J Biol Chem 277: 35305–35313, 2002. [DOI] [PubMed] [Google Scholar]
- 12.Eldaif SM, Deneve JA, Wang N, Jiang R, Mosunjac M, Mutrie CJ, Guyton RA, Zhao Z, Vinten-Johansen J. Attenuation of renal ischemia/reperfusion injury by postconditioning involves adenosine receptor and protein kinase C activation. Transplant Int 23: 217–226, 2010. [DOI] [PubMed] [Google Scholar]
- 13.Fang J, Prabu SK, Sepuri NB, Raza H, Anandatheerthavarada HK, Galati D, Spear J, Avadhani NG. Site specific phosphorylation of cytochrome c oxidase subunits I, IVi1 and Vb in rabbit hearts subjected to ischemia/reperfusion. FEBS Lett 581: 1302–1310, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuller TF, Kusch A, Chaykovska L, Catar R, Pützer J, Haller M, Troppmair J, Hoff U, Dragun D. Protein kinase C inhibition ameliorates posttransplantation preservation injury in rat renal transplants. Transplantation 94: 679–686, 2012. [DOI] [PubMed] [Google Scholar]
- 15.Garlid KD, Costa ADT, Quinlan CL, Pierre SV, Dos Santos P. Cardioprotective signaling to mitochondria. J Mol Cell Cardiol 46: 858–866, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gray MO, Karliner JS, Mochly-Rosen D. A selective epsilon-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. J Biol Chem 272: 30945–30951, 1997. [DOI] [PubMed] [Google Scholar]
- 17.Gray MO, Zhou HZ, Schafhalter-Zoppoth I, Zhu P, Mochly-Rosen D, Messing RO. Preservation of base-line hemodynamic function and loss of inducible cardioprotection in adult mice lacking protein kinase C ε. J Biol Chem 279: 3596–3604, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer 7: 281–294, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Guo D, Nguyen T, Ogbi M, Tawfik H, Ma G, Yu Q, Caldwell RW, Johnson JA. Protein kinase C-ε coimmunoprecipitates with cytochrome oxidase subunit IV and is associated with improved cytochrome-c oxidase activity and cardioprotection. Am J Physiol Heart Circ Physiol 293: H2219–H2230, 2007. [DOI] [PubMed] [Google Scholar]
- 20.He L, Lemasters JJ. Dephosphorylation of the Rieske iron-sulfur protein after induction of the mitochondrial permeability transition. Biochem Biophys Res Commun 334: 829–837, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Helling S, Vogt S, Rhiel A, Ramzan R, Wen L, Marcus K, Kadenbach B. Phosphorylation and kinetics of mammalian cytochrome c oxidase. Mol Cell Proteomics 7: 1714–1724, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu K, Mochly-Rosen D, Boutjdir M. Evidence for functional role of εPKC isozyme in the regulation of cardiac Ca(2+) channels. Am J Physiol Heart Circ Physiol 279: H2658–H2664, 2000. [DOI] [PubMed] [Google Scholar]
- 23.Inagaki K, Hahn HS, Dorn GW, Mochly-Rosen D. Additive protection of the ischemic heart ex vivo by combined treatment with delta-protein kinase C inhibitor and ε-protein kinase C activator. Circulation 108: 869–875, 2003. [DOI] [PubMed] [Google Scholar]
- 24.Ivaska J, Whelan RD, Watson R, Parker PJ. PKCε controls the traffic of β1 integrins in motile cells. EMBO J 21: 3608–3619, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang J. Protein kinase C (PKC) isozymes and cancer. New J Sci 2014: 2014. [Google Scholar]
- 26.Khasar SG, Lin Y, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A Novel nociceptor signaling pathway revealed in protein kinase C ε mutant mice. Neuron 24: 253–260, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korzick DH, Kostyak JC, Hunter JC, Saupe KW. Local delivery of PKCε-activating peptide mimics ischemic preconditioning in aged hearts through GSK-3β but not F1-ATPase inactivation. Am J Physiol Heart Circ Physiol 293: H2056–H2063, 2007. [DOI] [PubMed] [Google Scholar]
- 28.Koyanagi T, Noguchi K, Ootani A, Inagaki K, Robbins RC, Mochly-Rosen D. Pharmacological inhibition of εPKC suppresses chronic inflammation in murine cardiac transplantation model. J Mol Cell Cardiol 43: 517–522, 2007. [DOI] [PubMed] [Google Scholar]
- 29.Laporta CAM, Comolli R. Biochemical and immunological characterization of calcium-dependent and -independent PKC isoenzymes in renal ischemia. Biochem Biophys Res Commun 191: 1124–1130, 1993. [DOI] [PubMed] [Google Scholar]
- 30.Lee I, Salomon AR, Ficarro S, Mathes I, Lottspeich F, Grossman LI, Hüttemann M. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J Biol Chem 280: 6094–6100, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Lieberthal W, Nigam SK. Acute renal failure. I. Relative importance of proximal vs distal tubular injury. Am J Physiol Renal Physiol 275: F623–F632, 1998. [DOI] [PubMed] [Google Scholar]
- 32.Liu H, McPherson BC, Yao Z. Preconditioning attenuates apoptosis and necrosis: role of protein kinase C ε and -δ isoforms. Am J Physiol Heart Circ Physiol 281: H404–H410, 2001. [DOI] [PubMed] [Google Scholar]
- 33.McCarthy J, McLeod CJ, Minners J, Essop MF, Ping P, Sack MN. PKCε activation augments cardiac mitochondrial respiratory post-anoxic reserve - a putative mechanism in PKCε cardioprotection. J Mol Cell Cardiol 38: 697–700, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Megyesi J, Andrade L, Vieira JMJ, Safirstein RL, Price PM. Positive effect of the induction of p21WAF1/CIP1 on the course of ischemic acute renal failure. Kidney Int 60: 2164–2172, 2001. [DOI] [PubMed] [Google Scholar]
- 35.Nowak G, Soundararajan S, Mestril R. Protein kinase C-α interaction with iHSP70 in mitochondria promotes recovery of mitochondrial function after injury in renal proximal tubular cells. Am J Physiol Renal Physiol 305: F764–F776, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nowak G, Bakajsova D, Clifton GL. Protein kinase C-ε modulates mitochondrial function and active Na+ transport after oxidant injury in renal cells. Am J Physiol Renal Physiol 286: F307–F316, 2004. [DOI] [PubMed] [Google Scholar]
- 37.Nowak G, Clifton GL, Godwin ML, Bakajsova D. Activation of ERK1/2 pathway mediates oxidant-induced decreases in mitochondrial function in renal cells. Am J Physiol Renal Physiol 291: F840–F855, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nowak G, Schnellmann RG. l-Ascorbic acid regulates growth and metabolism of renal cells: improvements in cell culture. Am J Physiol Cell Physiol 271: C2072–C2080, 1996. [DOI] [PubMed] [Google Scholar]
- 39.Nowak G, Schnellmann RG. Improved culture conditions stimulate gluconeogenesis in primary cultures of renal proximal tubule cells. Am J Physiol Cell Physiol 268: C1053–C1061, 1995. [DOI] [PubMed] [Google Scholar]
- 40.Nowak G, Bakajsova D, Samarel AM. Protein kinase C-ε activation induces mitochondrial dysfunction and fragmentation in renal proximal tubules. Am J Physiol Renal Physiol 301: F197–F208, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ogbi M, Johnson JA. Protein kinase Cε interacts with cytochrome c oxidase subunit IV and enhances cytochrome c oxidase activity in neonatal cardiac myocyte preconditioning. Biochem J 393: 191–199, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Padanilam BJ. Induction and subcellular localization of protein kinase C isozymes following renal ischemia. Kidney Int 59: 1789–1797, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Ping P, Song C, Zhang J, Guo Y, Cao X, Li RC, Wu W, Vondriska TM, Pass JM, Tang XL, Pierce WM, Bolli R. Formation of protein kinase Cε-Lck signaling modules confers cardioprotection. J Clin Invest 109: 499–507, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ping P, Takano H, Zhang J, Tang XL, Qiu Y, Li RC, Banerjee S, Dawn B, Balafonova Z, Bolli R. Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rabbits: a signaling mechanism for both nitric oxide-induced and ischemia-induced preconditioning. Circ Res 84: 587–604, 1999. [DOI] [PubMed] [Google Scholar]
- 45.Ping P, Zhang J, Huang S, Cao X, Tang XL, Li RC, Zheng YT, Qiu Y, Clerk A, Sugden P, Han J, Bolli R. PKC-dependent activation of p46/p54 JNKs during ischemic preconditioning in conscious rabbits. Am J Physiol Heart Circ Physiol 277: H1771–H1785, 1999. [DOI] [PubMed] [Google Scholar]
- 46.Ping P, Zhang J, Pierce WM Jr, Bolli R. Functional proteomic analysis of protein kinase C ε signaling complexes in the normal heart and during cardioprotection. Circ Res 88: 59–62, 2001. [DOI] [PubMed] [Google Scholar]
- 47.Prabu SK, Anandatheerthavarada HK, Raza H, Srinivasan S, Spear JF, Avadhani NG. Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J Biol Chem 281: 2061–2070, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Restivo M, Kozhevnikov DO, Qu YS, Yue Y, Rosen D, El-Sherif N, Boutjdir M. Activation of εPKC reduces reperfusion arrhythmias and improves recovery from ischemia: Optical mapping of activation patterns in the isolated guinea-pig heart. Biochem Biophys Res Commun 426: 237–241, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rong S, Hueper K, Kirsch T, Greite R, Klemann C, Mengel M, Meier M, Menne J, Leitges M, Susnik N, Meier M, Haller H, Shushakova N, Gueler F. Renal PKC-ε deficiency attenuates acute kidney injury and ischemic allograft injury via TNF-α-dependent inhibition of apoptosis and inflammation. Am J Physiol Renal Physiol 307: F718–F726, 2014. [DOI] [PubMed] [Google Scholar]
- 50.Shaik ZP, Fifer EK, Nowak G. Akt activation improves oxidative phosphorylation in renal proximal tubular cells following nephrotoxicant injury. Am J Physiol Renal Physiol 294: F423–F432, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shaik ZP, Fifer EK, Nowak G. Protein kinase B/Akt modulates nephrotoxicant-induced necrosis in renal cells. Am J Physiol Renal Physiol 292: F292–F303, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steenaart NAE, Shore GC. Mitochondrial cytochrome c oxidase subunit IV is phosphorylated by an endogenous kinase. FEBS Lett 415: 294–298, 1997. [DOI] [PubMed] [Google Scholar]
- 53.Steinberg R, Harari OA, Lidington EA, Boyle JJ, Nohadani M, Samarel AM, Ohba M, Haskard DO, Mason JC. A Protein kinase Cε-anti-apoptotic kinase signaling complex protects human vascular endothelial cells against apoptosis through Induction of Bcl-2. J Biol Chem 282: 32288–32297, 2007. [DOI] [PubMed] [Google Scholar]
- 54.Strait JB, Samarel AM. Isoenzyme-specific protein kinase C and c-Jun N-terminal kinase activation by electrically stimulated contraction of neonatal rat ventricular myocytes. J Mol Cell Cardiol 32: 1553–1566, 2000. [DOI] [PubMed] [Google Scholar]
- 55.Sun X, Budas GR, Xu L, Barreto GE, Mochly-Rosen D, Giffard RG. Selective activation of protein kinase Cε in mitochondria is neuroprotective in vitro and reduces focal ischemic brain injury in mice. J Neurosci Res 91: 799–807, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wotton D, Ways DK, Parker PJ, Owen MJ. Activity of both Raf and Ras is necessary for activation of transcription of the human T cell receptor beta gene by protein kinase C, Ras plays multiple roles. J Biol Chem 268: 17975–17982, 1993. [PubMed] [Google Scholar]