

Abstract
Mutations in the renal sodium-dependent phosphate cotransporters NPT2a and NPT2c have been reported in patients with renal stone disease and nephrocalcinosis. Oral phosphate supplementation is currently thought to reduce risk by reversing the hypercalciuria, but the exact mechanism remains unclear and the relative contribution of modifiers of mineralization such as osteopontin (Opn) to the formation of renal mineral deposits in renal phosphate wasting disorders has not been studied. We observed a marked decrease of renal gene expression and urinary excretion of Opn in Npt2a−/− mice, a mouse model of these disorders, at baseline. Following supplementation with phosphate Opn gene expression was restored to wild-type levels in Npt2a−/− mice; however, urine excretion of the protein remained low. To further investigate the role of Opn, we used a double-knockout strategy, which provides evidence that loss of Opn worsens the nephrocalcinosis and nephrolithiasis observed in these mice on a high-phosphate diet. These studies suggest that impaired Opn gene expression and urinary excretion in Npt2a−/− mice may be an additional risk factor for nephrolithiasis, and normalizing urine Opn levels may improve the therapy of phosphaturic disorders.
Keywords: hypophosphatemia, nephrocalcinosis, NPT2a, osteopontin, rickets
mutations in the sodium phosphate cotransporters NPT2a (18, 21, 23) and NPT2c (6, 17) are associated with intraluminal stones (nephrolithiasis) and mineral deposits in the renal parenchyma (nephrocalcinosis) in patients with familial forms of hypophosphatemia. In genome-wide association studies, NPT2a has also been associated with nephrolithiasis (3) and altered renal function (7, 20). With both mutations, affected individuals show renal phosphate wasting, high circulating levels of 1,25(OH)2D, and absorptive hypercalciuria as a result of increased intestinal uptake of calcium (6, 17, 18, 21, 23, 25, 26). Oral phosphate supplements are currently thought to reduce the risk for renal mineralization in carriers of NPT2a and NPT2c mutations by lowering circulating levels of 1,25(OH)2D and absorptive hypercalciuria. However, since phosphate supplementation, despite reducing urine calcium excretion, may contribute to the formation of renal mineralization under certain conditions, it is unclear whether other factors play a role in preventing renal mineralization.
Renal calcium phosphate deposits, for example, are observed in X-linked hypophosphatemia treated with oral phosphate supplements given multiple times throughout the day (2, 9) and in otherwise healthy individuals following treatment with phosphate enema (13), despite the absence of hypercalciuria. Furthermore, we recently observed in a survey of 27 kindreds with hereditary hypophosphatemic rickets with hypercalciuria that a 10% decrease in TRP predicts a twofold increase in renal mineralization, independent of NPT2c mutation carrier status (11).
Intraluminal and interstitial renal mineralization can be modulated by osteopontin (Opn) (8). Opn is a phosphorylated protein that has high affinity to calcium and thereby inhibits intraluminal crystal growth. Via integrin-binding RGD motifs, it also mediates attachment of crystals to the renal tubular epithelium resulting in transport of crystals to the interstitium. Opn is produced by cells of the thick ascending limbs of the loop of Henle and by osteoblasts in the skeleton, and its expression is upregulated by phosphate (28) and pyrophosphate (15). It was also recently shown to be upregulated by fibroblast growth factor 23 (FGF23) via FGFR3 in a klotho-independent fashion (19). Opn appears to be a substrate for Phex, and accumulation of Opn fragments in Hyp bone may be responsible for the mineralization deficit surrounding osteocyte lacunae in these mice (4). ASARM peptides derived from Opn and other sibling proteins appear in the circulation of Hyp mice and may contribute to the Phex-dependent hypophosphatemia in Hyp.
Hypophosphatemia and reduced cellular uptake of phosphorus in the absence of Npt2a may reduce Opn levels and its protective effect in the kidneys. To test this hypothesis, we used Npt2a−/− (10) mice, which like human patients with homozygous or compound heterozygous NPT2a mutations, develop renal mineral ion deposits at intraluminal and interstitial sites which contain calcium, phosphate, and osteopontin, and resemble the composition of Randall’s plaques (10, 16). Our results suggest that Opn gene expression and urinary excretion are reduced at baseline in Npt2a−/− mice. Furthermore, ablation of Opn increases mineralization size in Opn−/− and Npt2a−/− double-knockout mice when compared with Npt2a−/− mice. These data suggest that Opn may protect wild-type (WT) mice from nephrolithiasis and nephrocalcinosis, and its absence in the urine of Npt2a−/− mice may be a thus far unrecognized risk factor. On the basis of these findings, normalizing urine Opn levels could be considered as a therapeutic target in phosphaturic disorders.
MATERIALS AND METHODS
Animals.
Male and female C57BL/6 mice were obtained from the Charles River Laboratory. Male and female Npt2a−/− mice (B6.129S2-Slc34a1tm1Hten/J, Stock No: 004802). Opn−/− mice were kindly provided by Dr. Caren Gundberg, Dept. Orthopedics and Rehabilitation, Yale School of Medicine, who received them originally from Dr. Susan Rittling, Dept. of Cell Biology and Neuroscience, Rutgers University, Piscataway, NJ (22). Mice were genotyped by polymerase chain reaction (PCR) amplification of genomic DNA extracted from tail clippings and amplified by PCR, as described previously (1, 5, 22, 24). Mice were weaned at 3 wk of age and allowed free access to water and normal [1.0% calcium, 0.7% phosphate (nonphytate bioavailable phosphorus 4%), and TD.2018S] or special diets. Specialized mouse diets were purchased from Harlan Teklad (Madison, WI) and used egg whites as protein source. High-phosphate (HP) diet (TD.85349) contained 0.6% Ca and 1.2% Pi, and control diet (CO) diet (TD.09803) contained 0.6% Ca and 0.3% Pi. In all diets caloric content was 3.7 kcal/g, vitamin D content was 2 IU/g, and the magnesium content was 0.2%. The background of all mouse lines is C57Bl6; use of littermates for controls further reduced bias based on genetic background. Mice were placed on special diets immediately after weaning and killed after 10 wk on these diets.
All animals were maintained in facilities operated by the Yale Center for Comparative Research and all experimental procedures were approved by the institution's Subcommittee on Research Animal Care.
Blood and urine parameters.
Biochemical analysis was done on blood samples collected by cardiac puncture or orbital exsanguination following an overnight fast. Concentrations of serum and urinary total Ca, Pi, and blood urea nitrogen were determined by Stanbio Laboratories (0155, 0830, and 0580, respectively; Boerne, TX). The concentration of urinary creatinine (Cr), urine Opn, and plasma 1,25-dihydroxyvitamin D (1,25(OH)2D) was determined by quantitative sandwich enzyme immunoassays from R&D Systems (KGE005, MOST00, and AC-62F1, respectively; Minneapolis, MN). Concentrations of plasma intact parathyroid hormone and COOH-terminal FGF23 were determined by quantitative sandwich enzyme immunoassays from Immutopics (60-2305 and 60-6300, respectively; San Clemente, CA).
Kidney histology.
Left kidneys were fixed in 4% formalin/PBS at 4°C for 12 h and then dehydrated with increasing concentration of ethanol and xylene, followed by paraffin embedding. Mineral deposits were determined on 10-μm von Kossa–stained sections counterstained with 1% methyl green. Hematoxyline and eosin was used as counterstain for morphological evaluation. Histomorphometric evaluation was performed with an Osteomeasure System (Osteometrics, Atlanta, GA). Percent of calcified area was determined by the formula 5 calc. area = 100 × calcified area/total area, and mineralization size was determined by the formula calc. size = calcified area/number of mineralizations.
Renal gene expression analysis.
Right kidneys were used for preparation of total RNA using Trizol (Thermo Fisher Scientific, Waltham, MA). Quantitative reverse transcription PCR (Omniscript, QuantiTect, Qiagen, Valencia, CA) was performed with the mouse β-actin forward primer GGCTGTATTCCCCTCCATCG and reverse primer CCAGTTGGTAACAATGCCATGT and the mouse osteopontin forward primer AGCAAGAAACTCTTCCAAGCAA and reverse primer GTGAGATTCGTCAGATTCATCCG using the ABI-Step One Plus Cycler (Fisher, Life Technologies, Waltham, MA).
Statistical analysis.
Data are expressed as means ± SE and were analyzed in Graphpad Prism 7.0. Differences between groups were considered significant if P values, by using the univariate linear regression analysis, one-way or two-way ANOVA, and Tukey’s test for multiple comparisons, were less than 0.05 unless indicated otherwise.
RESULTS
Humans with loss-of-function NPT2a (18, 21, 23) and NPT2c (6, 17) develop renal mineralization either during early childhood before specific therapy, while inappropriately receiving active vitamin D analogs, or throughout life (25). To model these kidney abnormalities, following weaning Npt2a−/− and WT mice were placed for 10 wk on diets containing differing amounts of phosphate [HP diet (1.20% Pi) or CO diet (0.3 % Pi)] and equal nutritional, vitamin D, calcium, and magnesium contents.
Consistent with previous reports (5, 10), when compared with WT mice plasma PTH and FGF23 were decreased in Npt2a−/− mice on the CO diet, while urine phosphorus excretion was increased (Table 1). Also, serum 1,25(OH)2D and urine calcium excretion were increased, albeit not significantly. Phosphaturia markedly increased in Npt2a−/− mice on the HP diet, while the hypercalciuria resolved along with a reduction in serum 1,25(OH)2D when compared with CO diet. Blood urea nitrogen levels of all the groups were in the normal range, indicating that the kidney function was normal. Furthermore, size and body weight of mice in each diet group were indistinguishable, and the animals appeared to be thriving well.
Table 1.
Biochemical parameters
| Diet | N | Ca, mg/dl | P, mg/dl | U-Opn/U-Crea, ng/mg | U-Ca/U-Crea, mg/mg | U-P/U-Crea, mg/mg | iPTH, pg/ml | FGF23, pg/ml | 1,25(OH)2-D, pmol/l |
|---|---|---|---|---|---|---|---|---|---|
| Npt2a−/− mice | |||||||||
| HP | 23 | 12.3 ± 0.7 | 8.5 ± 0.9 | 412.14 ± 56.95d | 0.29 ± 0.05 | 12.55 ± 1.0c,d | 93.6 ± 10.8c,d | 225 ± 22c | 291 ± 58c |
| CO | 21 | 9.8 ± 0.4 | 8.3 ± 0.6 | 498.38 ± 44.43d | 0.44 ± 0.07 | 5.69 ± 0.6c,d | 42.7 ± 4c,d | 42 ± 6c | 317 ± 57 |
| Wild-type mice | |||||||||
| HP | 7 | 8.4 ± 0.6 | 14.3 ± 0.2 | 549.50 ± 19.85d | 0.10 ± 0.01 | 10.32 ± 1.01d | 618 ± 134d | 464 ± 32 | 449 ± 5d |
| CO | 10 | 10.0 ± 0.2 | 12.5 ± 0.4 | 755.45 ± 77.76 | 0.22 ± 0.03 | 9.13 ± 1.47 | 498 ± 90 | 278 ± 27 | 275 ± 43 |
Data represent the means ± SE; comparisons were performed using a two-way ANOVA and Tukey’s test for multiple comparisons. P, plasma phosphorus; Ca, serum calcium; 1,25(OH)2-D, serum 1,25(OH)2-vitamin D; iPTH, plasma intact PTH; FGF23, plasma c-terminal FGF23; U-Opn, urine osteopontin; U-Crea, creatinine; U-P, phosphorus; U-Ca, calcium. All diets are egg-white based: HP, high-phosphate diet, 0.6% calcium, 1.20% phosphate; CO, control diet; 0.6% calcium, 0.3% phosphate.
P < 0.05 vs. Npt2a−/− HP;
P < 0.05 vs. Npt2a−/− CO;
P < 0.05 vs. WT HP;
P < 0.05 vs. WT CO.
Npt2a-null mice form renal mineralizations on HP diet.
Following 10 wk on the respective diets the animals were killed, left kidneys were paraffin-embedded, and sections evaluated for phosphate deposits using von Kossa staining and histomorphometry to determine calcified area (Fig. 1A) and mineralization size (Fig. 1B). Npt2a−/− mice fed CO diet (n = 21) showed 0.27 ± 0.18% calcified area (vs. 0.05 ± 0.11% WT). Mineralized area was similar in Npt2a−/− mice fed a HP diet (0.23 ± 0.08% calcified area, n = 23), despite improvement of hypercalciuria. No mineralization was observed in WT mice on HP diet, but mineralization was seen in 2 of 10 WT mice on CO diet, albeit less than in Npt2a−/− mice on the same diet.
Fig. 1.
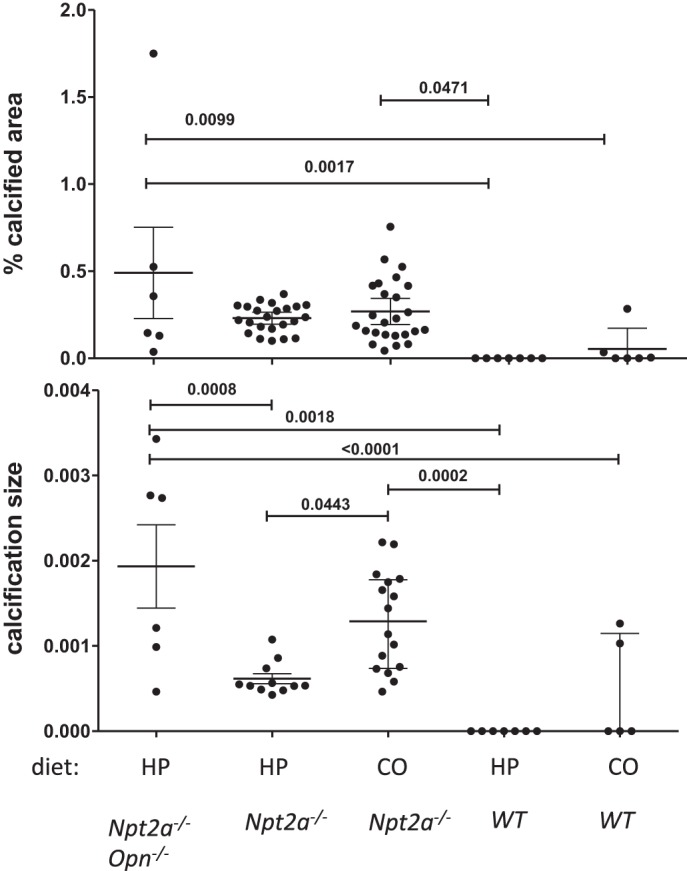
Ablation of Opn worsens renal mineralization size seen in Npt2a−/− mice on HP diet. Histomorphometric analysis of renal mineralization [% calcified area = 100 × mineralization area/tissue area (A) and mineralization size = mineralization area/number of mineralizations, μm2 (B)] in 10 μm sections of kidneys from mice feed different diets for 10 wk (see legend of Table 1 for composition of diets). The data represent individual animals (●) with the means ± SE. P values shown above the lines of comparisons were calculated by one-way ANOVA and Tukey’s test for multiple comparisons.
Renal gene expression and urinary excretion of Opn is decreased in Npt2a−/− mice.
Opn is detected in the calcium phosphorus deposits of Npt2a−/− mice (16). To test whether Opn contributes to the formation of mineral deposits in Npt2a−/− mice, we next measured Opn gene expression by quantitative reverse transcription PCR in total RNA prepared from the right kidneys of our experimental animals and found it to be ~100-fold reduced in Npt2a−/− mice (0.00078 ± 0.00014, n = 9 when compared with 0.108 ± 0.024 in WT mice, n = 10, P = 0.0159) (Fig. 2A). Opn gene expression was induced by HP diet in Npt2a−/− mice (Fig. 2A); however, urinary excretion of Opn protein corrected for urine creatinine (U-Opn/U-Crea) remained reduced to 498 ± 44 pg/mg when compared with 755 ± 78 in WT mice (P < 0.0001, Fig. 2B and Table 1), and this reduction was independent of dietary phosphate. Furthermore, calcified area inversely correlated with urine Opn excretion in a pooled analysis of all experimental WT and Npt2a−/− mice from Table 1 (n = 61, P = 0.017, R2 = 0.1365) (Fig. 3). Taken together, these observations suggest that urine Opn may protect kidneys from forming renal mineral deposits in the setting of renal phosphate wasting.
Fig. 2.
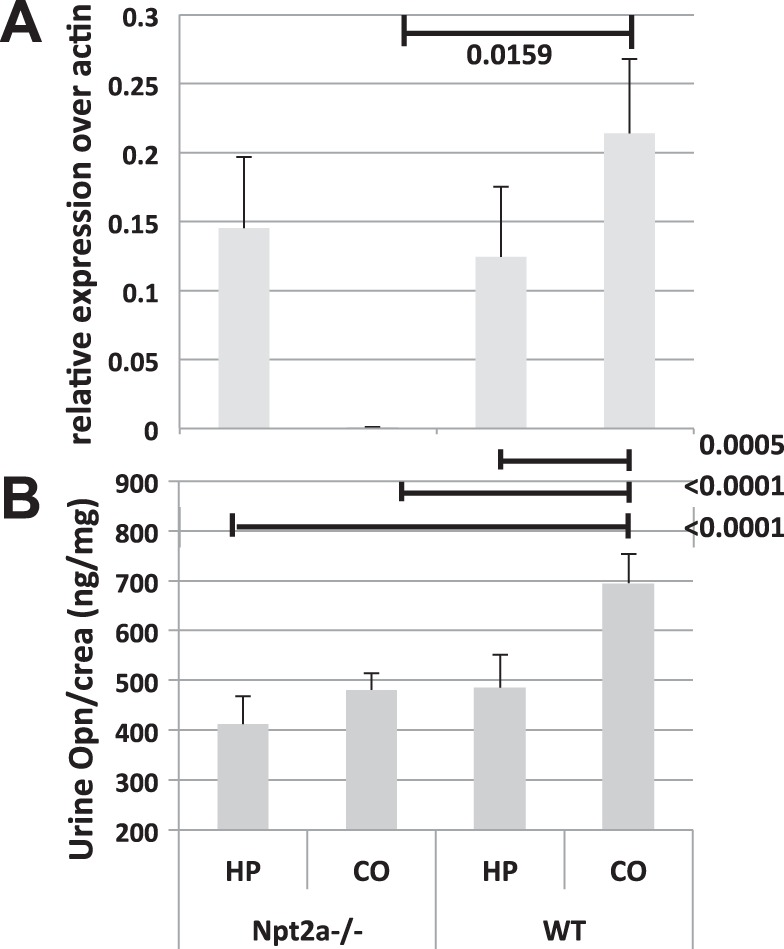
Renal osteopontin mRNA expression and urinary excretion are decreased in Npt2a−/− mice. A: Opn gene expression measured by quantitative reverse transcription PCR corrected over β-actin in total kidney RNA. B: urinary excretion of Opn protein (U-Opn) corrected for urine creatinine (U-Crea) from mice feed different diets for 10 wk (see legend of Table 1 for composition of diets). The data represent means ± SE. P values shown above the lines of comparisons were calculated by one-way ANOVA and Tukey’s test for multiple comparisons.
Fig. 3.
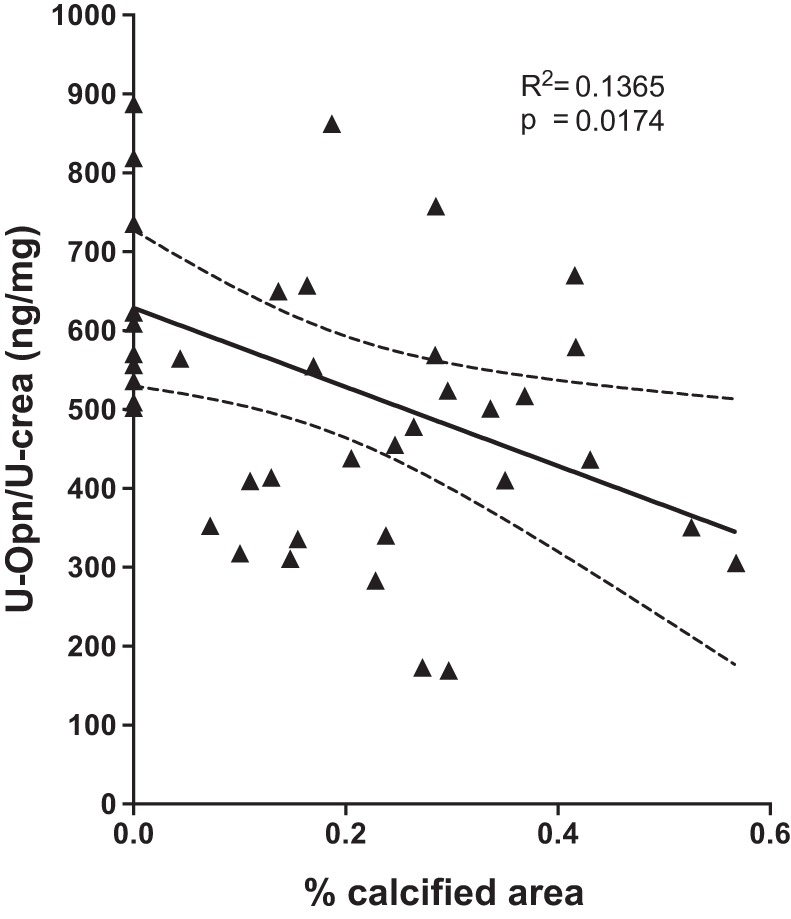
Urinary Opn excretion is significantly associated with renal mineralization in a combined bivariate linear regression analysis of WT and Npt2a−/− mice fed different diets. WT and Npt2a−/− mice from Table 1 (n = 42) were evaluated by linear regression analysis to determine the association of renal mineralization with the ratio of urine osteopontin/urine creatinine (U-Opn/U-crea). Data points represent values of individual animals. Results of the linear regression analysis are shown as solid line with 95% confidence interval (stippled lines), R2, and P values.
Ablation of Opn worsens renal mineralization in Npt2a−/− mice.
If urine Opn prevents the formation of nephrocalcinosis in the setting of renal phosphate wasting, then ablation of Opn in Opn−/−/Npt2a−/− mice would be expected to worsen renal mineral deposits. No significant change in renal mineralization was observed in Opn−/−/Npt2a−/− on CO diet when compared with Opn−/− or Npt2a−/− single-knockout mice on CO diet, presumably because Opn excretion was already reduced (data not shown). However, on HP diet percent of calcified area was increased (albeit nonsignificantly) (Fig. 1A), and the size of renal mineralization tripled from 0.00062 ± 0.00006 μm2 in Npt2a−/− to 0.0019 ± 0.0005 μm2 in Opn−/−/Npt2a−/− mice (P = 0.0008) (Fig. 1B), consistent with an inhibitory role of Opn in crystal growth in Npt2a−/− mice.
Histological evaluation furthermore showed small intraluminal mineral deposits in cortical and medullary tubular segments of the kidneys of Npt2a−/− and double-knockout mice (Fig. 4A). In addition, we observed large interstitial mineral deposits that displaced the surrounding renal tubules (Fig. 4B). No mineralization was observed in renal vasculature or in the renal pelvis. No difference in location of mineralization was appreciated between Npt2a−/− mice on different diets and between genotypes. Ultrastructural analysis using transmission electron microscopy revealed thin submicrometer, plate-like crystals starting at center and organizing into microspheres in double-knockout mice similar to what has been described in Npt2a−/− mice (16) (Fig. 5).
Fig. 4.

Cortical and medullary renal mineralization. Light micrographs of 10-μm renal sections, prepared from paraffin-embedded kidneys: von Kossa, methylene green staining, 4X (A); von Kossa, hematoxylin and eosin staining, 40X (B). See legend of Table 1 for composition of CO and HP diets.
Fig. 5.

Transmission electron microscopy of mouse kidneys reveals thin submicrometer, plate-like crystals starting at center and organizing into microspheres. Cross section shows concentrically arranged microspherules microspherules that appear to be growing by addition of crystal layers at the periphery. White box indicates approximate location of the image with next higher magnification. Letters indicate the following: a–c Npt2a−/− CO diet; d–f Npt2a−/− and Opn−/− HP diet.
DISCUSSION
Oral phosphate supplements are currently thought to reduce risk for renal mineralization in human carriers of NPT2a and NPT2c mutations. However, as mentioned in the introduction, there is concern that this therapy might contribute to the formation of renal mineralization despite reduced 1,25(OH)2D levels and thus reduced urinary calcium excretion under certain conditions. When evaluating Npt2a−/− (10) mice, we similarly found that mineralization persists and/or reappears in mice supplemented with phosphorus despite resolution of hypercalciuria.
Opn is detected in the calcium phosphorus deposits of Npt2a−/− mice (16) and is thought to affect intraluminal and interstitial renal mineralization in two ways (8). As phosphorylated protein, Opn has high affinity to calcium and thereby inhibits intraluminal crystal growth. Via integrin-binding RGD motifs, it also mediates attachment of crystals to the renal tubular epithelium resulting in transport of crystals to the interstitium.
On the basis of these studies we hypothesized that hypophosphatemia and reduced cellular uptake of phosphorus in the absence of Npt2a may reduce urinary Opn levels, which may (as shown in Fig. 2A) be due to reduced renal Opn gene expression. We furthermore found that urine Opn levels inversely correlate with percent of calcified area in kidney sections obtained from our mice (Fig. 2). Opn ablation increased mineralization size in double-knockout mice when compared with Npt2a−/− mice (Fig. 4), consistent with the known role of Opn in calcium phosphate crystal growth. However, similar ultrastructural composition in single and double Npt2a and Opn knockout mice (Fig. 5) is consistent with direct effects of Opn in the development of renal calcification in Npt2a−/− mice.
Taken together, these findings may suggest that absence of Npt2a may reduce phosphate “sensing,” resulting in downregulation of Opn. Reduced Opn levels may permit luminal crystal growth in the setting of phosphaturia and hypercalciuria and, despite the low interstitial Pi in Npt2a−/− mice, be sufficient to mediate transepithelial transport and interstitial crystal growth.
Opn is produced by cells of the thick ascending limbs of the loop of Henle and by osteoblasts in the skeleton, and its expression is positively regulated by phosphate (28) and pyrophosphate (15). It was also recently shown to be regulated by FGF23 via FGFR3 in a klotho-independent fashion (19). Finally, Opn appears to be a substrate for Phex, and accumulation of Opn fragments in Hyp bone may be responsible for the mineralization deficit surrounding osteocyte lacunae in these mice (4). Interestingly, oral phosphate supplementation of Npt2a−/− mice with HP diet is able to restore Opn gene expression, while urine excretion of the protein remained below the levels seen in WT mice, which may indicate that Opn is regulated differentially on the levels of gene expression and secretion, both requiring the presence of Npt2a.
A limitation of this study is that Npt2a−/− mice exhibit a milder biochemical phenotype than that seen in humans with sodium phosphate cotransporter mutations. Renal mineralization resolves after weaning (10, 16) in this mouse model, and composition may differ between mice and humans who carry NPT2a mutations.
Future studies are required to determine whether bone-origin in addition to renal-origin Opn contributes to urine Opn protein levels. Studies using mice that transgenically express mutant Opn and/or use of antibodies to inhibit Opn may be able to further evaluate the causal relationship of Opn deficiency to nephrolithiasis and nephrocalcinosis in Npt2a−/− mice (14). Other dietary components such as calcium, magnesium, oxalate and vitamin D, urinary pH and osmolarity, and urinary levels of citrate, oxalate, and magnesium may contribute to the development of renal calcification in Npt2a−/− mice. It may also be of interest to study the role and regulation of uromodulin (Tamm-Horsfall protein, THP), like Opn a potent mineralization inhibitor that is abundantly present in renal mineralizations (8), and to determine the time course and spatial distribution of renal stone formation in Npt2a−/− mice and compare its natural history to other models of nephrolithiasis, i.e., NHERF-1 and THP-null (12) mice. In contrast to Npt2a−/− mice that form intratubular deposits that translocate to the interstitium (16, 27), NHERF-1 and THP-null (12) mice produce interstitial apatite plaques in the renal papillae similar to human plaques.
In summary, we have shown for the first time that Npt2a−/− mice have impaired Opn gene expression and urinary excretion of the protein. The loss of Opn expression and excretion may be risk factors for nephrolithiasis. Normalizing urine Opn levels could be explored as a therapeutic target in phosphaturic disorders.
GRANTS
Support for this study was provided by National Institute of Diabetes and Digestive and Kidney Diseases Grant 5K08-DK-078361 (to C. Bergwitz), Young Investigator Awards by the National Kidney Foundation, and the American Association for Clinical Investigation (to C. Bergwitz), National Natural Science Foundation of China Grant 81271713 (to C. Zhu), and Scientific research fund of Anhui Medical University 2011xkj074 to Yuwen Li.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
D.C., Y.L., J.P., C.Z., and C.B. performed experiments; D.C., Y.L., J.P., C.Z., and C.B. analyzed data; D.C., Y.L., J.P., C.Z., and C.B. approved final version of manuscript; Y.L. and C.B. interpreted results of experiments; Y.L. and C.B. drafted manuscript; Y.L. and C.B. edited and revised manuscript; C.B. conceived and designed research; C.B. prepared figures.
REFERENCES
- 1.Albright RA, Stabach P, Cao W, Kavanagh D, Mullen I, Braddock AA, Covo MS, Tehan M, Yang G, Cheng Z, Bouchard K, Yu ZX, Thorn S, Wang X, Folta-Stogniew EJ, Negrete A, Sinusas AJ, Shiloach J, Zubal G, Madri JA, De La Cruz EM, Braddock DT. ENPP1-Fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy. Nat Commun 6: 10006, 2015. doi: 10.1038/ncomms10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alon U, Donaldson DL, Hellerstein S, Warady BA, Harris DJ. Metabolic and histologic investigation of the nature of nephrocalcinosis in children with hypophosphatemic rickets and in the Hyp mouse. J Pediatr 120: 899–905, 1992. doi: 10.1016/S0022-3476(05)81957-2. [DOI] [PubMed] [Google Scholar]
- 3.Arcidiacono T, Mingione A, Macrina L, Pivari F, Soldati L, Vezzoli G. Idiopathic calcium nephrolithiasis: a review of pathogenic mechanisms in the light of genetic studies. Am J Nephrol 40: 499–506, 2014. doi: 10.1159/000369833. [DOI] [PubMed] [Google Scholar]
- 4.Barros NM, Hoac B, Neves RL, Addison WN, Assis DM, Murshed M, Carmona AK, McKee MD. Proteolytic processing of osteopontin by PHEX and accumulation of osteopontin fragments in Hyp mouse bone, the murine model of X-linked hypophosphatemia. J Bone Miner Res 28: 688–699, 2013. doi: 10.1002/jbmr.1766. [DOI] [PubMed] [Google Scholar]
- 5.Beck L, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, Tenenhouse HS. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc Natl Acad Sci USA 95: 5372–5377, 1998. doi: 10.1073/pnas.95.9.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, Frappier D, Burkett K, Carpenter TO, Anderson D, Garabedian M, Sermet I, Fujiwara TM, Morgan K, Tenenhouse HS, Jüppner H. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet 78: 179–192, 2006. doi: 10.1086/499409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Böger CA, Gorski M, Li M, Hoffmann MM, Huang C, Yang Q, Teumer A, Krane V, O’Seaghdha CM, Kutalik Z, Wichmann HE, Haak T, Boes E, Coassin S, Coresh J, Kollerits B, Haun M, Paulweber B, Köttgen A, Li G, Shlipak MG, Powe N, Hwang SJ, Dehghan A, Rivadeneira F, Uitterlinden A, Hofman A, Beckmann JS, Krämer BK, Witteman J, Bochud M, Siscovick D, Rettig R, Kronenberg F, Wanner C, Thadhani RI, Heid IM, Fox CS, Kao WH, CKDGen Consortium . Association of eGFR-related loci identified by GWAS with incident CKD and ESRD. PLoS Genet 7: e1002292, 2011. doi: 10.1371/journal.pgen.1002292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Canales BK, Anderson L, Higgins L, Ensrud-Bowlin K, Roberts KP, Wu B, Kim IW, Monga M. Proteome of human calcium kidney stones. Urology 76: 1017.e13–1017.e20, 2010. doi: 10.1016/j.urology.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 9.Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. A clinician’s guide to X-linked hypophosphatemia. J Bone Miner Res 26: 1381–1388, 2011. doi: 10.1002/jbmr.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chau H, El-Maadawy S, McKee MD, Tenenhouse HS. Renal calcification in mice homozygous for the disrupted type IIa Na/Pi cotransporter gene Npt2. J Bone Miner Res 18: 644–657, 2003. doi: 10.1359/jbmr.2003.18.4.644. [DOI] [PubMed] [Google Scholar]
- 11.Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ, Sharma A, Schlingmann KP, Janner M, Biggin A, Lazier J, Gessner M, Chrysis D, Tuchman S, Baluarte HJ, Levine MA, Tiosano D, Insogna K, Hanley DA, Carpenter TO, Ichikawa S, Hoppe B, Konrad M, Sävendahl L, Munns CF, Lee H, Jüppner H, Bergwitz C. Mutations in SLC34A3/NPT2c are associated with kidney stones and nephrocalcinosis. J Am Soc Nephrol 25: 2366–2375, 2014. doi: 10.1681/ASN.2013101085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evan AP, Weinman EJ, Wu XR, Lingeman JE, Worcester EM, Coe FL. Comparison of the pathology of interstitial plaque in human ICSF stone patients to NHERF-1 and THP-null mice. Urol Res 38: 439–452, 2010. doi: 10.1007/s00240-010-0330-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonlusen G, Akgun H, Ertan A, Olivero J, Truong LD. Renal failure and nephrocalcinosis associated with oral sodium phosphate bowel cleansing: clinical patterns and renal biopsy findings. Arch Pathol Lab Med 130: 101–106, 2006. doi: 10.1043/1543-2165(2006)130[101:RFANAW]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 14.Hamamoto S, Yasui T, Okada A, Hirose M, Matsui Y, Kon S, Sakai F, Kojima Y, Hayashi Y, Tozawa K, Uede T, Kohri K. Crucial role of the cryptic epitope SLAYGLR within osteopontin in renal crystal formation of mice. J Bone Miner Res 26: 2967–2977, 2011. doi: 10.1002/jbmr.495. [DOI] [PubMed] [Google Scholar]
- 15.Harmey D, Hessle L, Narisawa S, Johnson KA, Terkeltaub R, Millán JL. Concerted regulation of inorganic pyrophosphate and osteopontin by Ak p2, Enpp1, and Ank: an integrated model of the pathogenesis of mineralization disorders. Am J Pathol 164: 1199–1209, 2004. doi: 10.1016/S0002-9440(10)63208-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khan SR, Canales BK. Ultrastructural investigation of crystal deposits in Npt2a knockout mice: are they similar to human Randall’s plaques? J Urol 186: 1107–1113, 2011. doi: 10.1016/j.juro.2011.04.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, Gershoni-Baruch R, Albers N, Lichtner P, Schnabel D, Hochberg Z, Strom TM. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet 78: 193–201, 2006. doi: 10.1086/499410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Magen D, Berger L, Coady MJ, Ilivitzki A, Militianu D, Tieder M, Selig S, Lapointe JY, Zelikovic I, Skorecki K. A loss-of-function mutation in NaPi-IIa and renal Fanconi’s syndrome. N Engl J Med 362: 1102–1109, 2010. doi: 10.1056/NEJMoa0905647. [DOI] [PubMed] [Google Scholar]
- 19.Murali SK, Roschger P, Zeitz U, Klaushofer K, Andrukhova O, Erben RG. FGF23 regulates bone mineralization in a 1,25(OH)2 D3 and Klotho-independent manner. J Bone Miner Res 31: 129–142, 2016. doi: 10.1002/jbmr.2606. [DOI] [PubMed] [Google Scholar]
- 20.Pattaro C, Teumer A, Gorski M, Chu AY, Li M, Mijatovic V, Garnaas M, Tin A, Sorice R, Li Y, Taliun D, Olden M, Foster M, Yang Q, Chen MH, Pers TH, Johnson AD, Ko YA, Fuchsberger C, Tayo B, Nalls M, Feitosa MF, Isaacs A, Dehghan A, d’Adamo P, Adeyemo A, Dieffenbach AK, Zonderman AB, Nolte IM, van der Most PJ, Wright AF, Shuldiner AR, Morrison AC, Hofman A, Smith AV, Dreisbach AW, Franke A, Uitterlinden AG, Metspalu A, Tonjes A, Lupo A, Robino A, Johansson Å, Demirkan A, Kollerits B, Freedman BI, Ponte B, Oostra BA, Paulweber B, Krämer BK, Mitchell BD, Buckley BM, Peralta CA, Hayward C, Helmer C, Rotimi CN, Shaffer CM, Müller C, Sala C, van Duijn CM, Saint-Pierre A, Ackermann D, Shriner D, Ruggiero D, Toniolo D, Lu Y, Cusi D, Czamara D, Ellinghaus D, Siscovick DS, Ruderfer D, Gieger C, Grallert H, Rochtchina E, Atkinson EJ, Holliday EG, Boerwinkle E, Salvi E, Bottinger EP, Murgia F, Rivadeneira F, Ernst F, Kronenberg F, Hu FB, Navis GJ, Curhan GC, Ehret GB, Homuth G, Coassin S, Thun GA, Pistis G, Gambaro G, Malerba G, Montgomery GW, Eiriksdottir G, Jacobs G, Li G, Wichmann HE, Campbell H, Schmidt H, Wallaschofski H, Völzke H, Brenner H, Kroemer HK, Kramer H, Lin H, Leach IM, Ford I, Guessous I, Rudan I, Prokopenko I, Borecki I, Heid IM, Kolcic I, Persico I, Jukema JW, Wilson JF, Felix JF, Divers J, Lambert JC, Stafford JM, Gaspoz JM, Smith JA, Faul JD, Wang JJ, Ding J, Hirschhorn JN, Attia J, Whitfield JB, Chalmers J, Viikari J, Coresh J, Denny JC, Karjalainen J, Fernandes JK, Endlich K, Butterbach K, Keene KL, Lohman K, Portas L, Launer LJ, Lyytikäinen LP, Yengo L, Franke L, Ferrucci L, Rose LM, Kedenko L, Rao M, Struchalin M, Kleber ME, Cavalieri M, Haun M, Cornelis MC, Ciullo M, Pirastu M, de Andrade M, McEvoy MA, Woodward M, Adam M, Cocca M, Nauck M, Imboden M, Waldenberger M, Pruijm M, Metzger M, Stumvoll M, Evans MK, Sale MM, Kähönen M, Boban M, Bochud M, Rheinberger M, Verweij N, Bouatia-Naji N, Martin NG, Hastie N, Probst-Hensch N, Soranzo N, Devuyst O, Raitakari O, Gottesman O, Franco OH, Polasek O, Gasparini P, Munroe PB, Ridker PM, Mitchell P, Muntner P, Meisinger C, Smit JH, Kovacs P, Wild PS, Froguel P, Rettig R, Mägi R, Biffar R, Schmidt R, Middelberg RP, Carroll RJ, Penninx BW, Scott RJ, Katz R, Sedaghat S, Wild SH, Kardia SL, Ulivi S, Hwang SJ, Enroth S, Kloiber S, Trompet S, Stengel B, Hancock SJ, Turner ST, Rosas SE, Stracke S, Harris TB, Zeller T, Zemunik T, Lehtimäki T, Illig T, Aspelund T, Nikopensius T, Esko T, Tanaka T, Gyllensten U, Völker U, Emilsson V, Vitart V, Aalto V, Gudnason V, Chouraki V, Chen WM, Igl W, März W, Koenig W, Lieb W, Loos RJ, Liu Y, Snieder H, Pramstaller PP, Parsa A, O’Connell JR, Susztak K, Hamet P, Tremblay J, de Boer IH, Böger CA, Goessling W, Chasman DI, Köttgen A, Kao WH, Fox CS, ICBP Consortium, AGEN Consortium, CARDIOGRAM, CHARGe-Heart Failure Group, ECHOGen Consortium . Genetic associations at 53 loci highlight cell types and biological pathways relevant for kidney function. Nat Commun 7: 10023, 2016. doi: 10.1038/ncomms10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prié D, Huart V, Bakouh N, Planelles G, Dellis O, Gérard B, Hulin P, Benqué-Blanchet F, Silve C, Grandchamp B, Friedlander G. Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. N Engl J Med 347: 983–991, 2002. doi: 10.1056/NEJMoa020028. [DOI] [PubMed] [Google Scholar]
- 22.Rittling SR, Matsumoto HN, McKee MD, Nanci A, An XR, Novick KE, Kowalski AJ, Noda M, Denhardt DT. Mice lacking osteopontin show normal development and bone structure but display altered osteoclast formation in vitro. J Bone Miner Res 13: 1101–1111, 1998. doi: 10.1359/jbmr.1998.13.7.1101. [DOI] [PubMed] [Google Scholar]
- 23.Schlingmann KP, Ruminska J, Kaufmann M, Dursun I, Patti M, Kranz B, Pronicka E, Ciara E, Akcay T, Bulus D, Cornelissen EA, Gawlik A, Sikora P, Patzer L, Galiano M, Boyadzhiev V, Dumic M, Vivante A, Kleta R, Dekel B, Levtchenko E, Bindels RJ, Rust S, Forster IC, Hernando N, Jones G, Wagner CA, Konrad M. Autosomal-recessive mutations in SLC34A1 encoding sodium-phosphate cotransporter 2A cause idiopathic infantile hypercalcemia. J Am Soc Nephrol 27: 604–614, 2015. doi: 10.1681/ASN.2014101025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segawa H, Onitsuka A, Furutani J, Kaneko I, Aranami F, Matsumoto N, Tomoe Y, Kuwahata M, Ito M, Matsumoto M, Li M, Amizuka N, Miyamoto K. Npt2a and Npt2c in mice play distinct and synergistic roles in inorganic phosphate metabolism and skeletal development. Am J Physiol Renal Physiol 297: F671–F678, 2009. doi: 10.1152/ajprenal.00156.2009. [DOI] [PubMed] [Google Scholar]
- 25.Tieder M, Arie R, Bab I, Maor J, Liberman UA. A new kindred with hereditary hypophosphatemic rickets with hypercalciuria: implications for correct diagnosis and treatment. Nephron 62: 176–181, 1992. doi: 10.1159/000187029. [DOI] [PubMed] [Google Scholar]
- 26.Tieder M, Modai D, Samuel R, Arie R, Halabe A, Bab I, Gabizon D, Liberman UA. Hereditary hypophosphatemic rickets with hypercalciuria. N Engl J Med 312: 611–617, 1985. doi: 10.1056/NEJM198503073121003. [DOI] [PubMed] [Google Scholar]
- 27.Wu XR. Interstitial calcinosis in renal papillae of genetically engineered mouse models: relation to Randall’s plaques. Urolithiasis 43, Suppl 1: 65–76, 2015. doi: 10.1007/s00240-014-0699-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie Y, Sakatsume M, Nishi S, Narita I, Arakawa M, Gejyo F. Expression, roles, receptors, and regulation of osteopontin in the kidney. Kidney Int 60: 1645–1657, 2001. doi: 10.1046/j.1523-1755.2001.00032.x. [DOI] [PubMed] [Google Scholar]