

In this manuscript, we show that VEGF levels associated with preeclampsia are a net negative contributor to potential vasodilator production in both a human ex vivo and in vitro endothelial cell model. Therefore, pharmacological targeting of VEGF-stimulated signaling pathways could be a novel treatment modality for preeclampsia-related hypertension.
Keywords: endothelium, pregnancy, nitric oxide, Ca2+, human
Abstract
The role increased vascular endothelial growth factor (VEGF) plays in vascular function during normal vs. preeclamptic pregnancy has been a source of some controversy of late. In this study, we seek to understand how VEGF165 influences vasodilator production via Ca2+ signaling mechanisms in human endothelial cells. We utilize human umbilical vein endothelial cells (HUVEC) as well as intact ex vivo human umbilical vein (HUV Endo) to address direct stimulation of Ca2+ and NO by VEGF165 alone, as well as the effect of VEGF165 on subsequent ATP-stimulated Ca2+ signaling and NO production. We show that VEGF165 stimulates Ca2+ responses in both HUVEC and HUV Endo, which results in a corresponding increase in NO production in HUV Endo. Longer-term VEGF165 pretreatment then inhibits sustained Ca2+ burst responses to ATP in HUVEC and HUV Endo. This is paralleled by a corresponding drop in ATP-stimulated NO production in HUV Endo, likely through inhibition of Cx43 gap-junction function. Thus, although VEGF165 makes a small initial positive impact on vasodilator production via direct stimulation of Ca2+ responses, this is outweighed by the greater subsequent negative impact on Ca2+ bursts and vasodilator production promoted by more potent agonists such as ATP. Overall, elevated levels of VEGF165 associated with preeclampsia could contribute to the endothelial dysfunction by preventing Ca2+ bursts to other agonists including but not limited to ATP.
NEW & NOTEWORTHY In this manuscript, we show that VEGF levels associated with preeclampsia are a net negative contributor to potential vasodilator production in both a human ex vivo and in vitro endothelial cell model. Therefore, pharmacological targeting of VEGF-stimulated signaling pathways could be a novel treatment modality for preeclampsia-related hypertension.
pregnancy is a time of dramatic changes in vascular physiology. Processes of angiogenesis and vasodilation both contribute to the decrease in vascular resistance and the redirection of blood to the uteroplacental unit. Vascular endothelial growth factor (VEGF) has been implicated to varying degrees in each of these processes. To further complicate matters, the exact role VEGF signaling plays in the vascular physiology of pregnancy is still poorly understood, not least because of the many isoforms of VEGF that may be present. Much of the confusion in the VEGF literature also has to do with imprecisely defined VEGF measurement criteria. Although “VEGF” is often used in shorthand for VEGFA, it should be noted that multiple variants of VEGFA occur naturally, including VEGF165 (the most abundant variant in humans), VEGF121, VEGF189, and VEGF206 (reviewed in Ref. 14). Often the literature on circulating VEGF in pregnancy and PE lack specifics on which forms of VEGF are detected and therefore reported.
A large proportion of more recent VEGF-related studies in the context of pregnancy adaptation and vascular disorders of pregnancy revolve around the soluble VEGFR1/FLT-1 receptor splice variant, known more commonly as sFLT-1, and its level relative to placental growth factor (PlGF), a member of the VEGF family of peptides. Although there is consensus that sFlt-1 is elevated, controversy still surrounds the specific role of sFLT-1 in preeclampsia (PE), a hypertensive disorder of pregnancy. High levels of circulating sFLT-1 have been correlated with PE (26, 44). Some believe that erroneous sFLT-1 production lowers circulating VEGF levels to an extent that inhibits the ability of VEGF to promote angiogenesis (33); others believe that sFLT-1 is a protective reaction to an overabundance of circulating VEGF in preeclampsia (13). There are also reports that VEGF production in preeclampsia outpaces sFLT-1 production (Ref. 27; reviewed in Ref. 18). Because of this, some have focused on “free VEGF” levels in PE, as opposed to total VEGF. Those studies, which have focused specifically on free VEGF, have shown that both free VEGF and PlGF levels are decreased in the circulation during PE pregnancies (24, 26, 31). Vascular pathologies related to decreased bioavailability of VEGF are currently being studied. However, there is evidence of greatly increased production and secretion of VEGF isoforms other than PlGF in certain tissues in PE, most notably in the decidua (13, 32, 43). Thus, although some tissues experience decreased VEGF bioavailability, others may simultaneously be experiencing local elevations of VEGF in the same patient. This raises the question of whether signaling mechanisms stimulated by excessive VEGF contributes to endothelial dysfunction of preeclampsia or whether local increases in VEGF are necessary to compensate for preexisting endothelial dysfunction caused by generally decreased circulating levels of free VEGF.
We have recently shown in an ovine model that uterine artery endothelial cells from pregnant ewes normally display a pregnancy adapted increase in Ca2+ signaling to heptahelical receptor agonists such as ATP that are linked to phospholipase C beta (39). Such pregnancy adaptation (reprogramming) enhances and sustains Ca2+ bursting and so nitric oxide (NO) output far above that in nonpregnancy, and this is dependent on increased connexin 43 (Cx43) gap-junction communication (40). An emerging role of VEGF165 signaling in preeclampsia is the role it plays in acutely phosphorylating Cx43 gap junctions at inhibitory residues, thus reducing cell-cell communication. We have recently shown that prior exposure to VEGF165 can reduce pregnancy-adapted function to levels equivalent to cells derived from non-pregnant ewes (6, 41), and this is apparently due to Cx43 inhibitory phosphorylation (6).
More recently, we have extended our studies to humans and shown enhanced Ca2+ bursting is also observable in human umbilical vein endothelium (HUV Endo) of cords from normal pregnancy. Of note, such bursting is blunted in cords from PE women, but we do not yet know whether VEGF165 exposure can reproduce this phenomenon or not. We also do not know to what extent VEGF165 may have a positive compensatory effect on these cells, since VEGF has been reported to be a vasodilator in its own right through activation of phospholipase C (PLC) gamma-mediated Ca2+ responses and NO production (1, 5, 17, 41).
In summary, to fully understand this dichotomy, we must be able to understand the relative contributions of VEGF to the inhibition of general vasodilator production capacity of the cell and VEGF-stimulated vasodilator production in human cells and tissues to be able to compare any inhibitory response to our prior data on PE subjects. Our translation of knowledge from former studies in animal models into human counterparts will also allow us to assess the possible impact of human clinical therapies based on VEGF signaling pathways in the future. We hypothesized that, in humans as in sheep, VEGF165 will stimulate Ca2+ responses in a subset of cells. We also hypothesized that VEGF165 will inhibit gap-junction function and therefore inhibit ATP-stimulated Ca2+ bursts responses far more, so the net effect is loss of function. If we are correct, then our study should support the notion that VEGF165 itself can be a damaging agent in certain circumstances and indirectly support the notion that sFLT-1 elevation in some PE subjects may be an attempt to deal with local VEGF excess but is insufficient to cope.
MATERIALS AND METHODS
Human Subjects Enrollment and Tissue Collection
The Institutional Review Boards of the University of Wisconsin Hospital and Clinics and Meriter Hospital (both located in Madison, WI) approved this study. All subjects signed a formal consent. Subjects used were the same control patients as in Krupp et al. (22). Data were obtained from intact HUV Endo of six subjects. Human umbilical vein endothelial cells (HUVEC) were pooled and cultured from five subjects for experimentation and data analyses.
Materials
Fura-2-AM, DAF2-DA, Minimum Essential Medium (MEM), Medium 199 (M199), fetal bovine serum (FBS), and all other general cell culture reagents were purchased from Life Technologies (Grand Island, NY) unless otherwise noted. The endothelial cell agonist ATP (disodium salt) and all other general laboratory chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless stated otherwise. Endothelial cell growth supplement was from Millipore (Billerica, MA), heparin (sodium salt, grade 1A from porcine intestinal mucosa, no. H3149) was from Sigma, and CaCl2 was from EMD Chemicals (San Diego, CA). Glass-bottom microwell dishes (35 mm) for [Ca2+]i imaging studies were from MatTek Corporation (Ashland, MA). Gap peptides GAP27(37,43) (sequence SRPTEKTIFII), GAP27 (37, 43) (sequence REKIITFIPT), GAP27(40) (sequence SRPTEKNVFIV), GAP26(43) (sequence VCYDKSFPISHVR), and GAP26(37,40) (sequence VCYDQAFPISHIR) were from Anaspec (San Jose, CA), purity of >95% by HPLC.
Methods
The protocols for isolating the intact human umbilical vein endothelium and for culturing the human umbilical vein endothelial cells were as in Krupp et al. (22). All standard culture media used for cell isolation or culture (MEM or M199) routinely contained 1% penicillin-streptomycin solution (Life Technologies) and 4 μg/ml gentamycin (Life Technologies). The methods of dual imaging of intracellular free Ca2+ concentration ([Ca2+]i) and simultaneous NO production utilized in intact UA Endo and the [Ca2+]i imaging alone were optimized in Krupp at el. (22) and repeated identically in this study.
Simultaneous imaging of [Ca2+]i and NO in intact HUV Endo.
Umbilical veins of ~6-mm length were loaded with both DAF2-DA (10 μM) and Fura-2-AM (10 μM) in standard Krebs buffer (in mM: 125 NaCl, 5 KCl, 1 MgSO4, 1 KH2PO4, 6 glucose, 2 CaCl2, 25 HEPES, pH 7.4) for 90 min. Dye hydrolysis was allowed to proceed for 30 min in Krebs buffer. The vessel placed on an inverted microscope (Diaphot 150; Nikon, Melville, NY) with the ×20 phase/fluor objective focused on the endothelium after mounting in imaging chamber. Individual endothelial cells (>30 cells/field) were visualized using dual excitation (switching at 340 and 380 nm) for Fura-2 and 485-nm excitation DAF-2 (high-speed Lambda 10-2; Sutter Instruments, Novato, CA). Emission was uniformly measured at 535 nm for all excitation wavelengths. Images were recorded in real time by a PixelFly camera (Cooke Corporation, Romulus, MI) using InCyt Im3 software (Intracellular Imaging, Cinncinati, OH). Relative fluorescence (F/F0) was then calculated as the relative measure of intracellular NO, whereas [Ca2+]i was calculated ratiometrically against a pre-recorded standard curve.
HUVEC cell isolation and culture.
HUVEC cells from normal (control) pregnancies were collected, cultured, and validated as in Krupp et al. (22) and Anaya et al. (2). Individual cell preps were isolated from freshly harvested umbilical cords and expanded to passage 2. Five individual preps were grown up and pooled to be frozen down for experimental usage at passage 3.
HUVEC imaging.
Cultured HUVEC frozen at passage 3 were plated to six 35-mm glass-bottom dishes in HEH media [MEM-based media containing 20% FBS, endothelial cell growth supplement (3.75 mg/100 ml), and porcine intestinal mucosa heparin (10 mg/100 ml)]. Cells were grown for 7 days and imaged at 95% confluence. HUVEC were incubated in 10 μM of Fura-2 AM with 0.05% Pluronic acid F127 (Life Technologies) dissolved in 1 ml HEH media for 1 h at 37°C. The cells were then incubated in Krebs buffer for 30 min at room temperature for ester hydrolysis. The dish was then placed in the field of view, and Fura-2 loading was verified by viewing at 380-nm UV excitation on a Nikon inverted microscope (above) and 70–90 cells were selected for imaging. An initial 5-min baseline recording was performed before subsequent addition of 100 μM ATP. The [Ca2+]i for each cell was calculated in real time against an established ratiometric standard curve using the InCyt Im2 software. ATP-stimulated Ca2+ bursts were counted before and after VEGF pretreatment.
Statistical analysis.
For vessels, data from six or more individual subject cell preparations were collected. For HUVEC, an average of ~80 cells/dish were analyzed for four to six dishes of pooled cells. For Ca2+ burst inhibition experiments, cells that initially responded with 3+ bursts were counted, since prior experiments have shown that complete conversion to capacitative entry has taken place by the third burst. Data were subsequently analyzed by Student’s t-test for single-stimulation experiments. For repeat ATP stimulation experiments, changes in Ca2+ burst numbers were analyzed by Rank sum test on differences against internal control. A value of P < 0.05 was considered statistically significant.
RESULTS
VEGF165-Stimulated Ca2+ Response in Primary HUVEC
Stimulation of primary HUVEC with 10 ng/ml VEGF165 revealed a slightly delayed onset but overall prolonged Ca2+ response. In Fig. 1, we show the mean Ca2+ response to VEGF165 stimulation. Consistent with what others have reported (3, 4, 12, 25), we see a moderately rapid rise in Ca2+ levels, followed by a sustained plateau phase, which continues for the duration of the 2,100-s experiment. This contrasts with the repetitive transient Ca2+ burst pattern resulting from a typical ATP stimulation (light gray dotted line). There was no significant difference in proportion of cells responding to VEGF165 (81.47 ± 5.94%) and ATP (90.56 ± 2.06%), P = 0.116. We also examined the role of gap junctions in VEGF165-stimulated Ca2+ responses using the classic pharmacological gap-junction inhibitor 12-O-tetradeconylphorbol 13-acetate (TPA). There was no change in proportion of cells responding to VEGF165 (86.46 ± 3.63%) and TPA+VEGF165 (90.64 ± 3.17%), P = 0.487; or area under the curve (AUC) of the VEGF165-stimulated Ca2+ response when pretreated with TPA (Fig. 1, inset).
Fig. 1.

Mean Ca2+ response to 30 min of VEGF165 treatment in human umbilical vein endothelial cells (HUVEC). Fura-2-loaded HUVEC were placed on an inverted fluorescent microscope. After a 5-min baseline intracellular free Ca2+ concentration ([Ca2+]i) recording, cells were exposed to 10 ng/ml vascular endothelial growth factor (VEGF)165 for 30 min. The mean response ± SE is depicted in black over the duration of the experiment. For comparison, a typical 100 μM ATP-stimulated HUVEC [Ca2+]i recording is shown in gray. For VEGF165 treatment, n = 6 dishes. Inset: mean area under the curve (AUC) of 10 ng/ml VEGF165-stimulated Ca2+ responses with and without 10 nM TPA pretreatment for 30 min, normalized to VEGF165 alone (VEGF165, n = 5 dishes; TPA + VEGF165, n = 5 dishes; P = 0.221).
VEGF165-Stimulated Ca2+ Response in HUV Endo
Intact HUV Endo from normal pregnancies respond to stimulation with 10 ng/ml VEGF165 with an initial transient Ca2+ peak followed by a relatively short (<10 min) sustained small elevation in [Ca2+]i (Fig. 2A). Figure 2A also shows that this is accompanied by modest NO production through the first 10–15 min of the 30-min recording, resulting in an increase amounting to 1.1-fold NO production compared with basal levels. Figure 2B shows there is no effect of TPA on VEGF165-stimulated Ca2+ or NO responses in HUV Endo, for mean peak Ca2+ (P = 0.640), mean Ca2+ at 900 s (P = 0.644), or total NO (P = 0.639).
Fig. 2.

Mean [Ca2+]i and DAF/nitric oxide (NO) recordings after VEGF165 stimulation in UV Endo. Dual imaging of [Ca2+]i and NO was performed in ex vivo intact UV Endo using simultaneous loading of Fura-2 and DAF-2 fluorescent dyes. A: 10 ng/ml VEGF165 was added at time 0 and recorded for 30 min. B: UV Endo are pretreated with 10 nM TPA for 30 min before 10 ng/ml VEGF165 addition at time 0. Values are means + SE (n = 6 vessels).
Role of Gap Junctions in Sustained Ca2+ Bursts in HUVEC
To test the role of gap junctions in regulating sustained ATP-stimulated Ca2+ bursts in primary HUVEC, we exposed cells to TPA. Figure 3, A and B, show representative single-cell Ca2+ tracings in response to 100 μM ATP treatment before and after exposure to TPA. Number of ATP-stimulated Ca2+ bursts are marked above each respective burst, showing inhibition of ATP-stimulated Ca2+ bursts. Figure 3C shows the quantification of the effect of 1 or 10 nM TPA on ATP-stimulated Ca2+ bursts. Pretreatment with TPA caused a significant reduction in Ca2+ bursts at 1 nM (58% of control) or 10 nM (32% of control). A repeat ATP stimulation control was carried out, and, in the absence of TPA pretreatment, no change in Ca2+ burst numbers were observed between first and second stimulation, so tachyphylaxis is not a concern.
Fig. 3.

TPA pretreatment inhibits sustained ATP-stimulated Ca2+ bursts in HUVEC. HUVEC were loaded with Fura-2, stimulated with 100 μM ATP for 30 min, and Ca2+ bursts were counted. Cells were then washed and pretreated with TPA for 30 min, with focus maintained on the cells initially recorded. After pretreatment, a 5-min baseline [Ca2+]i was recorded, and 100 μM ATP was added for an additional 30 min, with Ca2+ bursts counted a second time. A: a single-cell representative tracing shows the number of ATP-stimulated Ca2+ bursts before and after 1 nM TPA treatment in the same cell. B: the effect of 10 nM TPA is also shown for a single representative cell. C: the quantification of Ca2+ bursts for both TPA treatment types for all cells that gave three or more Ca2+ bursts upon initial ATP stimulation. For Control, n = 143 cells; for 1 nM TPA, n = 147 cells; for 10 nM TPA, n = 231 cells. Results are shown as means +SE, normalized to initial ATP stimulation. *Significant difference (P < 0.001).
To more specifically address which connexin proteins regulate ATP-stimulated Ca2+ bursts, we administered various short peptides designed to mimic extracellular regions of specific connexin proteins. Figure 4 shows the quantification of the effect of these treatments on ATP-stimulated Ca2+ bursts. Two peptides [Gap26(37,40) and Gap26(43)] significantly reduced Ca2+ bursts compared with control. One peptide [Gap27(40)] significantly improved Ca2+ burst responses.
Fig. 4.
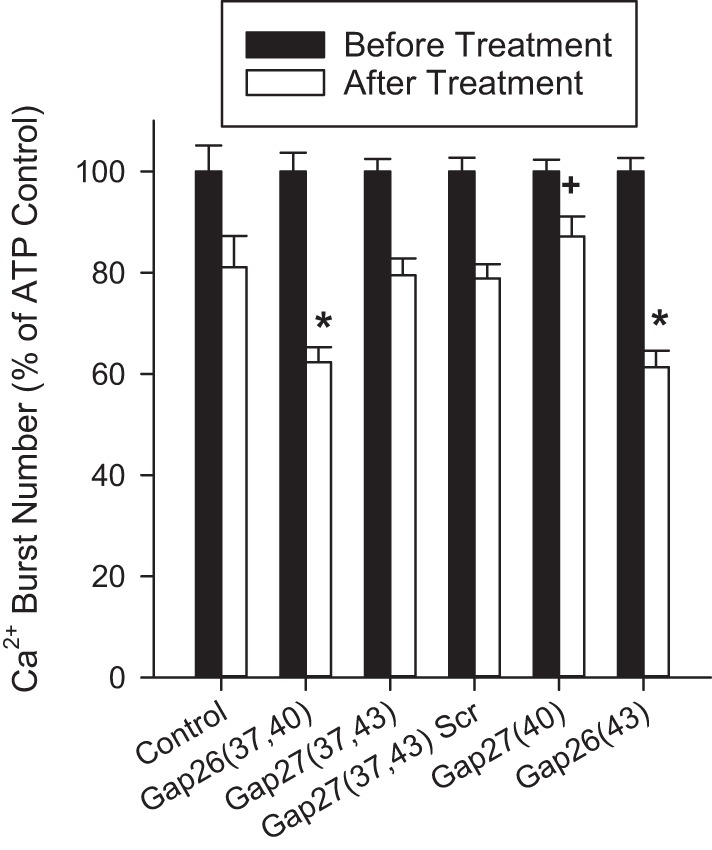
Connexin isoform specificity on ATP-stimulated Ca2+ burst inhibition using GAP peptides in HUVEC. Bar graph shows the effect of 2 h of pretreatment with 300 μM GAP peptide on subsequent ATP-stimulated Ca2+ bursts. Cells were treated initially with ATP for 30 min, and bursts were counted. Cells were then washed and incubated with GAP peptides before subsequent stimulation of the same cells for a second time with ATP. Ca2+ bursts were again counted. Initial ATP-stimulated Ca2+ burst numbers for cells giving 3+ bursts were taken as a mean and set to 100% in black bars. Any change in mean Ca2+ bursts after treatment was plotted in white bars. For Control, n = 66 cells; for Gap26(37,40), n = 149 cells; for Gap27(37,43), n = 119 cells; for Gap27(37,43) Scr, n = 147 cells; for Gap27(40), n = 101 cells; for Gap26(43), n = 116 cells. Data are means +SE. Statistics were performed on raw data. Significant differences with and without the corresponding Gap peptide are shown by *Inhibition vs. +Control stimulation (P < 0.05).
VEGF165 Inhibits Sustained Ca2+ Burst Responses in HUVEC
Pretreatment of HUVEC with 10 ng/ml VEGF165 for 30 min has an inhibitory effect on sustained ATP-stimulated Ca2+ bursts. Those cells, which produced three or more Ca2+ bursts upon initial ATP stimulation, were included in the analysis because the mechanistic switch between initial and sustained Ca2+ bursts has been fully realized by the third burst. Cells pretreated with VEGF165 produced only 55% (45% reduction in Ca2+ bursts) of initial control Ca2+ bursts upon subsequent ATP treatment (Fig. 5).
Fig. 5.
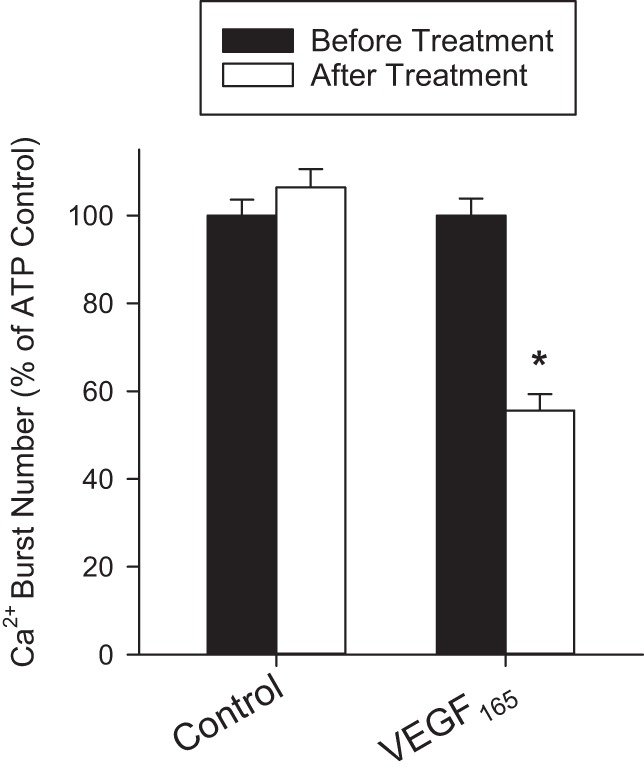
Effect of VEGF165 pretreatment on ATP-stimulated Ca2+ bursts in HUVEC. Cells were initially treated with ATP (100 μM) for 30 min, and Ca2+ bursts were counted. After being washed and pretreated with Krebs buffer (Control) or VEGF165 for 30 min, the same cells were again treated with ATP (100 μM), and Ca2+ bursts were counted. Initial Ca2+ burst numbers for those cells that gave three or more bursts was normalized to 100%. For Control, n = 143; for VEGF165, n = 112. Results are shown as means +SE. *Significant difference (P < 0.001).
VEGF165 Inhibits Sustained Ca2+ Burst Response in HUV Endo
When pretreated with 10 ng/ml VEGF165 for 30 min, 100 μM ATP-stimulated HUV Endo show fewer sustained Ca2+ bursts than those exposed to ATP alone. Mean tracings for ATP and VEGF165 + ATP are shown in Fig. 6, A and B, and quantification of the data is shown in Fig. 6C. VEGF165 pretreatment has no effect on initial ATP-stimulated Ca2+ peak height, but significant decreases of sustained Ca2+ and NO are apparent. At 900 s, ATP-stimulated Ca2+ responses remain ~40 nM above basal, whereas VEGF165 + ATP have returned to basal levels. DAF signal as a measure of NO production is at 0.25-fold above basal at 1,500 s in ATP-stimulated vessels, whereas VEGF165 + ATP-treated vessels fall short of 0.15-fold change above basal. Figure 6D shows a significant decrease in Ca2+ burst frequency by the second burst and is maintained through the third and fourth bursts.
Fig. 6.

Effect of VEGF165 pretreatment on ATP-stimulated Ca2+ and NO responses in HUV Endo. HUV Endo were loaded with Fura-2 and DAF-2 to simultaneously record changes in [Ca2+]i and NO. A: a 30-min recording is shown with mean levels of [Ca2+]i and NO for 100 μM ATP treatment. B: the vessels were pretreated with 10 ng/ml VEGF165 for 30 min before subsequent 100 μM ATP addition and recording for 30 min. C: the quantification of the data from A and B for peak and sustained (900 s) [Ca2+]i and total NO (at 1,500 s). Significant reductions after VEGF165 pretreatment occur at sustained [Ca2+]i and total NO. D: the proportion of cells yielding each successive Ca2+ burst (peaks) are plotted. For ATP, n = 6 vessels; for VEGF165 + ATP, n = 6 vessels. Results are shown as means +SE. *Significant difference (P < 0.05).
DISCUSSION
Regulation of vascular tone during pregnancy is critical for the health of both the mother and the developing fetus. A key regulator of vascular tone is vasodilator production in the vascular endothelium. Endothelial vasodilator production is controlled at many levels of cell signaling, but perhaps the most important and best understood is Ca2+ signaling driving NO production. Many circulating factors promote increases in [Ca2+]i, so examining the relative contributions of these factors is critical to understanding how they might support or impair pregnancy-related vasodilator production.
VEGF165 Stimulation of Ca2+ Responses
Although both ATP and VEGF165 promote increases in [Ca2+]i, they do so in different ways. Ultimately, this results in differing capacities for NO production. Both promote Ca2+ responses in a large number of cells exposed to each agonist, but the periodic and transient burst pattern typical of ATP-stimulated cells is less apparent in VEGF165-stimulated cells at the maximal dose of VEGF165 used. Instead, we observed a short initial Ca2+ peak followed by a sustained elevation of [Ca2+]i. No portion of the VEGF165-stimulated Ca2+ response appears to be gap junction dependent, since pretreatment with TPA had no effect on any portion of this response in either HUVEC or HUV Endo. In HUV Endo, there was also no effect of TPA on corresponding NO production. This is consistent with VEGF165 signaling coupling to PLCgamma and Ca2+ entry occurring via TRPC6 (5, 30). It should also be noted that, in our ovine model, PlGF was unable to reliably stimulate any Ca2+ response and therefore was not a focus of this study, although others have reported NO-dependent vasorelaxation in rat and human vessels when treated with PlGF (29). Further study into the role of PlGF in human endothelial cell Ca2+ responses may be warranted, but Cunningham et al. (12) failed to show any stimulation of Ca2+ responses in HUVEC at a very high dose of PlGF.
According to Tran et al. (35), the ability of endothelial NO synthase (eNOS) to produce NO is determined by a combination of eNOS phosphorylation state and [Ca2+]i elevation. Availability of substrate and cofactors such as l-arginine and tetrahydrobiopterin can also impact eNOS activity, but in our cell models and culture conditions, this has been shown not to be an issue (Refs. 9, 34 for UAEC; I. M. Bird, unpublished observations for HUVEC). Tran et al. showed that, in the case of eNOS failing to achieve optimal phosphorylation, a [Ca2+]i of 200 nM or higher is necessary for any significant degree of eNOS activation. In the event of optimal eNOS phosphorylation, only 100 nM [Ca2+]i is necessary to achieve 30% eNOS activation. We have previously shown in an ovine model that VEGF165 stimulates both s617 and s1179 phosphorylation on eNOS (16). Both VEGF165-stimulated phosphorylation events yielded only slight increases (even if statistically significant) over basal conditions, and therefore the majority of eNOS protein in any given cell would be unlikely to have both phosphorylation events occur simultaneously. This is in contrast to the effect of ATP stimulation of eNOS phosphorylation at s617 and s1179 in the same ovine model, where both residues are strongly phosphorylated after ATP treatment (34). Given that VEGF165 typically fails to stimulate [Ca2+]i above 200 nM in HUVEC, eNOS activation and therefore NO production will predictably be weak. This contrasts with ATP-stimulated Ca2+ signaling, where [Ca2+]i tends to remain elevated for only short periods of time as a series of bursts but easily crosses into the range of eNOS activation and does so when optimum phosphorylation is also achieved.
When the corresponding experiments are performed using intact HUV Endo such that NO production can be directly imaged with DAF-2, it is clear that this relationship holds true, and NO output is effected in parallel. As reported by Krupp et al. (22), HUV Endo Ca2+ signaling is optimized to sustain ATP-stimulated elevations in [Ca2+]i and maintain strong NO output, and this is lacking in PE subjects. Here, we show that, in comparison, VEGF165-stimulated Ca2+ signaling alone fails to sustain elevated [Ca2+]i in vessels when compared with the primary cells in culture at passage 4, and the resulting NO production is significantly less than for ATP-stimulated vessels. The period of time that corresponds to the greatest NO production in response to either agonist is when the mean ATP response is >150 nM [Ca2+]i, consistent with observations in our laboratory's ovine model (41, 42).
Role of Cx43 in Ca2+ Signaling
Apart from direct stimulation of Ca2+ signaling and NO production, VEGF165 also has been shown to activate signaling pathways (Src and MEK/ERK), which result in inhibitory phosphorylation of Cx43 (23, 38), and this has been confirmed in our ovine model (6). To that end, we first investigated the role of gap-junction subtypes in supporting ATP-stimulated Ca2+ bursting in our HUVEC model. Although confirmation that ATP-stimulated Ca2+ bursts in HUVEC are gap-junction-dependent is implied by TPA-mediated loss of Ca2+ bursts, it also appears to depend on a mix of gap-junction isoforms. By using GAP peptides designed to compete with the binding regions of the extracellular loop domains between adjacent cells, the effectiveness of multiple peptides gives us reason to believe Cx43 remains important, but Cx37 and Cx40 may also be involved in Ca2+ signaling in some capacity, perhaps through heteromeric or heterotypic gap junctions (10, 11, 21). Van Rijen et al. have previously shown that all three isoforms are expressed to some degree in cultured HUVEC (37), but electrical communication is predominantly mediated by Cx43. Certainly, the effectiveness of Gap26(43) to inhibit sustained ATP-stimulated Ca2+ bursts supports this conclusion. Surprisingly, Gap27(37,43) did not inhibit ATP-stimulated Ca2+ bursts, in contrast to the strong inhibition observed in the ovine model. Nonetheless, in the ovine model, Cx43 is the exclusive gap-junction peptide mediating sustained ATP-stimulated Ca2+ bursts, likely as a homodimer.
A key role for Cx43 in maintaining cell-cell communication would suggest VEGF165 will be able to inhibit Ca2+ bursting on subsequent ATP stimulation. VEGF165 treatment has been shown to phosphorylate Cx43 on multiple inhibitory residues by activation of MEK/ERK and Src kinase pathways in ovine uterine artery endothelial cells (6). Kinase regulation of the additional connexin isoforms present in HUVEC are still poorly understood compared with Cx43. It was thus unclear what effect VEGF165 may have on ATP-stimulated Ca2+ bursts on the mixed connexin isoform environment of HUVECs. Since VEGF165 pretreatment in HUVEC and HUV Endo did reduce ATP-stimulated Ca2+ bursts to a degree comparable to that observed in the ovine model (6, 41), our findings are consistent with Cx43 as the dominant isoform or, alternatively, imply kinase pathways stimulated by VEGF165 are similarly capable of inhibiting Cx37 and Cx40, despite our lack of understanding of these proteins. Given the report from van Rijen et al. (37), it would again appear that Cx43 is most likely playing a leading role in determining cell-cell communication, even if involved in heterotypic coupling with Cx37 and Cx40. Morchauser et al. (28) showed that, in ovine uterine arteries, like in the corresponding ovine primary UAEC (40), Cx43 is the mediator of sustained Ca2+ signaling and NO production.
VEGF165 Inhibition of ATP-Stimulated Ca2+ Responses
Yi et al. (41) observed that VEGF165 also inhibits ATP-stimulated Ca2+ bursts and NO production in the ovine uterine arteries. It therefore stands to reason that if VEGF165 acts through kinase signaling pathways to inhibit Cx43 function and Cx43 function is critical to ATP-stimulated Ca2+ bursts and NO production in HUV Endo, then VEGF165 will similarly block these Ca2+ and NO responses in HUV Endo. Our study now shows this is indeed the case. It is also important to consider that the degree to which VEGF165 inhibits ATP-stimulated Ca2+ and NO responses is greater than the degree to which VEGF165 stimulates Ca2+ and NO responses in its own right. The net result of VEGF165 presence locally in the vasculature at the doses tested would be a negative contribution toward vasodilation. These are, of course, prototypical agonists at prototypical doses used to tease out molecular mechanisms, but it should be noted that neither of these doses are beyond that which can be seen in a physiological or pathophysiological context (7, 8, 15, 19, 20, 36). Especially when the case of PE is considered, doses of 10 ng/ml total VEGF165 have been associated with the circulating range in the diseased state (although circulating sFLT-1 consistently reduces systemic bioavailability) and are likely to be easily achieved locally in tissues such as decidua. The data presented here also make the case that, since high circulating levels of VEGF165 can be deleterious to endothelial cell function (possibly even promoting endothelial dysfunction), elevated circulating sFLT-1 in PE may indeed be more of a response to high local VEGF concentrations rather than a cause of endothelial dysfunction through reduced systemic VEGF bioavailability but does not discount any further role reduced VEGF bioavailability may have on endothelial cell function. This is an important consideration, because even a local disruption in uterine perfusion, especially in the decidua, may instigate downstream hypoxia and upstream hypertension.
Ultimately, to translate these observations to therapy for hypertension associated with PE, we must either better understand how to adjust VEGF to levels more in line with normal pregnancies or better understand the signaling mechanisms downstream of VEGFR to allow us to target them pharmacologically. In the ovine cell culture model, we have shown that the specific isomer 10,12 CLA can target Src kinase signaling pathways to restore ATP-stimulated sustained Ca2+ signaling after VEGF165 pretreatment (6). However, we still lack sufficient knowledge in the human condition to know whether it will be effective in ameliorating hypertension associated with excessive VEGF in preeclampsia. Future studies will be needed to address that point. This study provides critical insight into subtle differences between ovine and human models, but, more importantly, it shows that the key regulators remain in place and behave in a similar fashion, such that therapies targeted to signaling pathways impacting on Cx43 function first elucidated in ovine will likely translate to human models and hopefully provide an option for clinical care for preeclampsia that target the endothelium directly.
GRANTS
This work was funded by an R&D Grant award from the Department of Obstetrics and Gynecology, University of Wisconsin Madison School of Medicine and Public Health and by National Institute of Child Health and Human Development Grants R21 HD-069181, P01 HD-38843, and R03 HD-079865.
DISCLOSURES
D.S.B. and I.M.B. have a patent pending for the therapeutic use of 10,12 CLA in preeclampsia.
AUTHOR CONTRIBUTIONS
D.S.B., J.K., F.-X.Y., and N.K. performed experiments; D.S.B., J.K., F.-X.Y., and N.K. analyzed data; D.S.B., J.K., F.-X.Y., D.M.S., and I.M.B. interpreted results of experiments; D.S.B. and F.-X.Y. prepared figures; D.S.B. drafted manuscript; D.S.B., J.K., D.M.S., and I.M.B. edited and revised manuscript; D.S.B., J.K., F.-X.Y., N.K., D.M.S., and I.M.B. approved final version of manuscript.
ACKNOWLEDGMENTS
This work was undertaken by J.K. and N.K. as part of their dissertations for an MS in the Endocrinology and Reproductive Physiology Graduate Training Program.
Current address for J.K.: Dean Health Clinic, Madison, WI.
REFERENCES
- 1.Abedi H, Zachary I. Vascular endothelial growth factor stimulates tyrosine phosphorylation and recruitment to new focal adhesions of focal adhesion kinase and paxillin in endothelial cells. J Biol Chem 272: 15442–15451, 1997. doi: 10.1074/jbc.272.24.15442. [DOI] [PubMed] [Google Scholar]
- 2.Anaya HA, Yi FX, Boeldt DS, Krupp J, Grummer MA, Shah DM, Bird IM. Changes in Ca2+ signaling and nitric oxide output by human umbilical vein endothelium in diabetic and gestational diabetic pregnancies. Biol Reprod 93: 60, 2015. doi: 10.1095/biolreprod.115.128645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrikopoulos P, Baba A, Matsuda T, Djamgoz MB, Yaqoob MM, Eccles SA. Ca2+ influx through reverse mode Na+/Ca2+ exchange is critical for vascular endothelial growth factor-mediated extracellular signal-regulated kinase (ERK) 1/2 activation and angiogenic functions of human endothelial cells. J Biol Chem 286: 37919–37931, 2011. doi: 10.1074/jbc.M111.251777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andrikopoulos P, Fraser SP, Patterson L, Ahmad Z, Burcu H, Ottaviani D, Diss JKJ, Box C, Eccles SA, Djamgoz MBA. Angiogenic functions of voltage-gated Na+ channels in human endothelial cells. J Biol Chem 286: 16846–16860, 2011. doi: 10.1074/jbc.M110.187559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boeldt DS, Grummer MA, Magness RR, Bird IM. Altered VEGF-stimulated Ca2+ signaling in part underlies pregnancy-adapted eNOS activity in UAEC. J Endocrinol 223: 1–11, 2014. doi: 10.1530/JOE-14-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boeldt DS, Grummer MA, Yi F, Magness RR, Bird IM. Phosphorylation of Ser-279/282 and Tyr-265 positions on Cx43 as possible mediators of VEGF-165 inhibition of pregnancy-adapted Ca2+ burst function in ovine uterine artery endothelial cells. Mol Cell Endocrinol 412: 73–84, 2015. doi: 10.1016/j.mce.2015.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bosio PM, Wheeler T, Anthony F, Conroy R, O’herlihy C, McKenna P. Maternal plasma vascular endothelial growth factor concentrations in normal and hypertensive pregnancies and their relationship to peripheral vascular resistance. Am J Obstet Gynecol 184: 146–152, 2001. doi: 10.1067/mob.2001.108342. [DOI] [PubMed] [Google Scholar]
- 8.Bussen S, Bussen D. Influence of the vascular endothelial growth factor on the development of severe pre-eclampsia or HELLP syndrome. Arch Gynecol Obstet 284: 551–557, 2011. doi: 10.1007/s00404-010-1704-x. [DOI] [PubMed] [Google Scholar]
- 9.Cale JM, Bird IM. Dissociation of endothelial nitric oxide synthase phosphorylation and activity in uterine artery endothelial cells. Am J Physiol Heart Circ Physiol 290: H1433–H1445, 2006. doi: 10.1152/ajpheart.00942.2005. [DOI] [PubMed] [Google Scholar]
- 10.Cottrell GT, Burt JM. Functional consequences of heterogeneous gap junction channel formation and its influence in health and disease. Biochim Biophys Acta 1711: 126–141, 2005. doi: 10.1016/j.bbamem.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 11.Cottrell GT, Burt JM. Heterotypic gap junction channel formation between heteromeric and homomeric Cx40 and Cx43 connexons. Am J Physiol Cell Physiol 281: C1559–C1567, 2001. [DOI] [PubMed] [Google Scholar]
- 12.Cunningham SA, Tran TM, Arrate MP, Bjercke R, Brock TA. KDR activation is crucial for VEGF165-mediated Ca2+ mobilization in human umbilical vein endothelial cells. Am J Physiol Cell Physiol 276: C176–C181, 1999. [DOI] [PubMed] [Google Scholar]
- 13.Fan X, Rai A, Kambham N, Sung JF, Singh N, Petitt M, Dhal S, Agrawal R, Sutton RE, Druzin ML, Gambhir SS, Ambati BK, Cross JC, Nayak NR. Endometrial VEGF induces placental sFLT1 and leads to pregnancy complications. J Clin Invest 124: 4941–4952, 2014. doi: 10.1172/JCI76864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 9: 669–676, 2003. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 15.Galazios G, Papazoglou D, Giagloglou K, Vassaras G, Koutlaki N, Maltezos E. Umbilical cord serum vascular endothelial growth factor (VEGF) levels in normal pregnancies and in pregnancies complicated by preterm delivery or pre-eclampsia. Int J Gynaecol Obstet 85: 6–11, 2004. doi: 10.1016/j.ijgo.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 16.Grummer MA, Sullivan JA, Magness RR, Bird IM. Vascular endothelial growth factor acts through novel, pregnancy-enhanced receptor signalling pathways to stimulate endothelial nitric oxide synthase activity in uterine artery endothelial cells. Biochem J 417: 501–511, 2009. doi: 10.1042/BJ20081013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He H, Venema VJ, Gu X, Venema RC, Marrero MB, Caldwell RB. Vascular endothelial growth factor signals endothelial cell production of nitric oxide and prostacyclin through flk-1/KDR activation of c-Src. J Biol Chem 274: 25130–25135, 1999. doi: 10.1074/jbc.274.35.25130. [DOI] [PubMed] [Google Scholar]
- 18.Hertig A, Liere P. New markers in preeclampsia. Clin Chim Acta 411: 1591–1595, 2010. doi: 10.1016/j.cca.2010.07.020. [DOI] [PubMed] [Google Scholar]
- 19.Holmsen H, Weiss HJ. Secretable storage pools in platelets. Annu Rev Med 30: 119–134, 1979. doi: 10.1146/annurev.me.30.020179.001003. [DOI] [PubMed] [Google Scholar]
- 20.Hunter A, Aitkenhead M, Caldwell C, McCracken G, Wilson D, McClure N. Serum levels of vascular endothelial growth factor in preeclamptic and normotensive pregnancy. Hypertension 36: 965–969, 2000. doi: 10.1161/01.HYP.36.6.965. [DOI] [PubMed] [Google Scholar]
- 21.Koval M, Molina SA, Burt JM. Mix and match: investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett 588: 1193–1204, 2014. doi: 10.1016/j.febslet.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krupp J, Boeldt DS, Yi FX, Grummer MA, Bankowski Anaya HA, Shah DM, Bird IM. The loss of sustained Ca(2+) signaling underlies suppressed endothelial nitric oxide production in preeclamptic pregnancies: implications for new therapy. Am J Physiol Heart Circ Physiol 305: H969–H979, 2013. doi: 10.1152/ajpheart.00250.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lampe PD, Lau AF. Regulation of gap junctions by phosphorylation of connexins. Arch Biochem Biophys 384: 205–215, 2000. doi: 10.1006/abbi.2000.2131. [DOI] [PubMed] [Google Scholar]
- 24.Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, Schisterman EF, Thadhani R, Sachs BP, Epstein FH, Sibai BM, Sukhatme VP, Karumanchi SA. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med 350: 672–683, 2004. doi: 10.1056/NEJMoa031884. [DOI] [PubMed] [Google Scholar]
- 25.Li J, Cubbon RM, Wilson LA, Amer MS, McKeown L, Hou B, Majeed Y, Tumova S, Seymour VA, Taylor H, Stacey M, O’Regan D, Foster R, Porter KE, Kearney MT, Beech DJ. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ Res 108: 1190–1198, 2011. doi: 10.1161/CIRCRESAHA.111.243352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 111: 649–658, 2003. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKeeman GC, Ardill JE, Caldwell CM, Hunter AJ, McClure N. Soluble vascular endothelial growth factor receptor-1 (sFlt-1) is increased throughout gestation in patients who have preeclampsia develop. Am J Obstet Gynecol 191: 1240–1246, 2004. doi: 10.1016/j.ajog.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 28.Morschauser TJ, Ramadoss J, Koch JM, Yi FX, Lopez GE, Bird IM, Magness RR. Local effects of pregnancy on connexin proteins that mediate Ca2+-associated uterine endothelial NO synthesis. Hypertension 63: 589–594, 2014. doi: 10.1161/HYPERTENSIONAHA.113.01171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Osol G, Celia G, Gokina N, Barron C, Chien E, Mandala M, Luksha L, Kublickiene K. Placental growth factor is a potent vasodilator of rat and human resistance arteries. Am J Physiol Heart Circ Physiol 294: H1381–H1387, 2007. doi: 10.1152/ajpheart.00922.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rameh LE, Rhee SG, Spokes K, Kazlauskas A, Cantley LC, Cantley LG. Phosphoinositide 3-kinase regulates phospholipase Cgamma-mediated calcium signaling. J Biol Chem 273: 23750–23757, 1998. doi: 10.1074/jbc.273.37.23750. [DOI] [PubMed] [Google Scholar]
- 31.Rios DR, Alpoim PN, Godoi LC, Perucci LO, de Sousa LP, Gomes KB, Dusse LM. Increased levels of sENG and sVCAM-1 and decreased levels of VEGF in severe preeclampsia. Am J Hypertens 29: 1307–1310. 2016. doi: 10.1093/ajh/hpv170. [DOI] [PubMed] [Google Scholar]
- 32.Sharma S, Godbole G, Modi D. Decidual control of trophoblast invasion. Am J Reprod Immunol 75: 341–350, 2016. doi: 10.1111/aji.12466. [DOI] [PubMed] [Google Scholar]
- 33.Sugimoto H, Hamano Y, Charytan D, Cosgrove D, Kieran M, Sudhakar A, Kalluri R. Neutralization of circulating vascular endothelial growth factor (VEGF) by anti-VEGF antibodies and soluble VEGF receptor 1 (sFlt-1) induces proteinuria. J Biol Chem 278: 12605–12608, 2003. doi: 10.1074/jbc.C300012200. [DOI] [PubMed] [Google Scholar]
- 34.Sullivan JA, Grummer MA, Yi FX, Bird IM. Pregnancy-enhanced endothelial nitric oxide synthase (eNOS) activation in uterine artery endothelial cells shows altered sensitivity to Ca2+, U0126, and wortmannin but not LY294002—evidence that pregnancy adaptation of eNOS activation occurs at multiple levels of cell signaling. Endocrinology 147: 2442–2457, 2006. doi: 10.1210/en.2005-0399. [DOI] [PubMed] [Google Scholar]
- 35.Tran QK, Leonard J, Black DJ, Nadeau OW, Boulatnikov IG, Persechini A. Effects of combined phosphorylation at Ser-617 and Ser-1179 in endothelial nitric-oxide synthase on EC50(Ca2+) values for calmodulin binding and enzyme activation. J Biol Chem 284: 11892–11899, 2009. doi: 10.1074/jbc.M806205200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem 140: 1–22, 1994. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 37.Van Rijen H, van Kempen MJ, Analbers LJ, Rook MB, van Ginneken AC, Gros D, Jongsma HJ. Gap junctions in human umbilical cord endothelial cells contain multiple connexins. Am J Physiol Cell Physiol 272: C117–C130, 1997. [DOI] [PubMed] [Google Scholar]
- 38.Warn-Cramer BJ, Lampe PD, Kurata WE, Kanemitsu MY, Loo LW, Eckhart W, Lau AF. Characterization of the mitogen-activated protein kinase phosphorylation sites on the connexin-43 gap junction protein. J Biol Chem 271: 3779–3786, 1996. doi: 10.1074/jbc.271.7.3779. [DOI] [PubMed] [Google Scholar]
- 39.Yi FX, Boeldt DS, Bird IM. Pregnancy induced reprogramming of endothelial function in response to ATP: evidence for post receptor Ca2+ signaling plasticity. In: Extracellular ATP and Adensosine as the Regulators of Endothelial Cell Function, edited by Gerasimovskaya E, Kaczmarek E. New York: Springer, 2010, p. 197. doi: 10.1007/978-90-481-3435-9_11. [DOI] [Google Scholar]
- 40.Yi FX, Boeldt DS, Gifford SM, Sullivan JA, Grummer MA, Magness RR, Bird IM. Pregnancy enhances sustained Ca2+ bursts and endothelial nitric oxide synthase activation in ovine uterine artery endothelial cells through increased connexin 43 function. Biol Reprod 82: 66–75, 2010. doi: 10.1095/biolreprod.109.078253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yi FX, Boeldt DS, Magness RR, Bird IM. [Ca2+]i signaling vs. eNOS expression as determinants of NO output in uterine artery endothelium: relative roles in pregnancy adaptation and reversal by VEGF165. Am J Physiol Heart Circ Physiol 300: H1182–H1193, 2011. doi: 10.1152/ajpheart.01108.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yi FX, Magness RR, Bird IM. Simultaneous imaging of [Ca2+]i and intracellular NO production in freshly isolated uterine artery endothelial cells: effects of ovarian cycle and pregnancy. Am J Physiol Regul Integr Comp Physiol 288: R140–R148, 2004. doi: 10.1152/ajpregu.00302.2004. [DOI] [PubMed] [Google Scholar]
- 43.Yong HE, Melton PE, Johnson MP, Freed KA, Kalionis B, Murthi P, Brennecke SP, Keogh RJ, Moses EK. Genome-wide transcriptome directed pathway analysis of maternal pre-eclampsia susceptibility genes. PLoS One 10: e0128230, 2015. doi: 10.1371/journal.pone.0128230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou Y, McMaster M, Woo K, Janatpour M, Perry J, Karpanen T, Alitalo K, Damsky C, Fisher SJ. Vascular endothelial growth factor ligands and receptors that regulate human cytotrophoblast survival are dysregulated in severe preeclampsia and hemolysis, elevated liver enzymes, and low platelets syndrome. Am J Pathol 160: 1405–1423, 2002. doi: 10.1016/S0002-9440(10)62567-9. [DOI] [PMC free article] [PubMed] [Google Scholar]