

We assessed the estrogen receptor (ER) dependence of female-specific cardioprotection using a rat model of chronic volume-overload stress. ER antagonism worsened ventricular wall stress, ventricular dilation, and cardiac dysfunction induced by volume overload. Further, blocking ERs resulted in cardiac remodeling and functional changes similar to that previously found in ovariectomized rats.
Keywords: collagen, estrogen, extracellular matrix, heart, receptor
Abstract
We have previously demonstrated the cardioprotective effects of ovarian hormones against adverse ventricular remodeling imposed by chronic volume overload. Here, we assess the estrogen receptor dependence of this cardioprotection. Four groups of female rats were studied: sham-operated (Sham), volume overloaded [aortocaval fistula (ACF)], Sham treated with estrogen receptor antagonist ICI 182,780 (Sham + ICI), and ACF treated with ICI. Cardiac function was assessed temporally using echocardiogram, and tissue samples were collected at 5 days and 6 wk postsurgery. All rats with volume overload had significantly increased cardiac output (96 ± 32 ml/min for ACF and 108 ± 11 ml/min for ACF + ICI vs. 31 ± 2 for Sham, P < 0.05). At 6 wk, volume overload induced significant left ventricular (LV) hypertrophy in both untreated and treated ACF groups. Both ACF groups developed significantly increased LV end-diastolic diameter (LVEDD), indicating LV dilatation, with the ACF + ICI group having the greatest increase (340%, relative to Sham). Ejection fraction was significantly reduced in the ACF + ICI group (23% reduction) at 6 wk postsurgery compared with untreated ACF (P < 0.05). Interstitial collagen staining was significantly reduced by volume overload, with estrogen receptor antagonism causing greater collagen loss at both 5 days and 6 wk postsurgery. Furthermore, volume overload induced a significant increase in LV wall stress only in rats treated with estrogen antagonist. These data indicate that estrogen receptor signaling is essential for sex hormone-dependent cardioprotection against adverse remodeling. The maintenance of myocardial extracellular matrix collagen appears to play a key role in this cardioprotection.
NEW & NOTEWORTHY We assessed the estrogen receptor (ER) dependence of female-specific cardioprotection using a rat model of chronic volume-overload stress. ER antagonism worsened ventricular wall stress, ventricular dilation, and cardiac dysfunction induced by volume overload. Further, blocking ERs resulted in cardiac remodeling and functional changes similar to that previously found in ovariectomized rats.
the cardioprotective role of ovarian-derived estrogen in heart disease remains unclear. We have previously reported sex differences in the progression of heart failure using the aortocaval fistula rat model of chronic volume overload (VO) (16). VO cardiac stress in humans is found in cases of aortic and mitral valve regurgitation, as well as ventricular septal defects, chronic heart failure, and following myocardial infarction (19). We have shown that male rats subjected to 8 wk of VO have significantly higher mortality rates than intact females (24.5% vs. 2.5%, respectively) (16). Further, male rats with VO develop greater ventricular dilatation and a more rapid decline in cardiac function and progression to heart failure than females (17). Our laboratory also found that female rats are able to more successfully adapt to VO-induced myocardial stress by developing compensatory concentric hypertrophy (17). These marked sex differences are ovarian hormone dependent, as ovariectomy abrogates this apparent sex-specific cardioprotection (17). Additionally, we found that estrogen replacement partially restores cardioprotection in ovariectomized rats (17). Our current study sought to determine the estrogen receptor (ER) dependence of the sex-specific cardioprotection.
ICI 182,780 was developed as a pure ER antagonist and has been show to block both endogenous and exogenous estrogen signaling (5, 50). This potent anti-estrogen binds ERs at a high affinity and downregulates ER levels by reducing intracellular half-life (10, 50). We therefore hypothesized that ICI 182,780 would attenuate female cardioprotection against adverse ventricular remodeling and dysfunction produced by chronic VO.
METHODS
Studies were performed using 9- to 10-wk-old female Sprague-Dawley (Harlan Hsd:SD) rats weighing ~190–200 g at surgery. Rats were housed under standard environmental conditions and maintained on commercial rat chow (Harlan Teklad 2018) and tap water ad libitum. All studies conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by our Institution’s Animal Care and Use Committee.
Rat model of chronic VO stress.
Anesthesia for surgical procedures was induced with isoflurane (4% induction with 3% maintenance, balance oxygen). Infrarenal aortocaval fistula (ACF) was surgically created to induce chronic VO, as previously described by our laboratory (14, 48). Briefly, a ventral laparotomy was performed to expose the abdominal aorta and vena cava. A short-bevel 18-gauge needle was inserted into the abdominal aorta and advanced through the medial wall of the vena cava, creating a shunt below the renal arteries. The needle was withdrawn and the aortic puncture sealed with cyanoacrylate. Shunting of aortic blood into the vena cava was visually evident and indicated successful creation of VO. Postoperative analgesia was provided by buprenorphine hydrochloride.
Experimental timeline.
The effects of ER antagonism on cardiac remodeling and dysfunction induced by VO were studied in four age-matched groups: sham-operated controls (Sham) and fistula-induced volume overload (ACF), both with and without ER antagonism by ICI 182,780. To ensure complete ER blockade and prevent any transient effects of the drug, treatment with ICI was initiated 1 wk before surgery and was delivered by intraperitoneal injection in the acute 5-day study, or by osmotic minipump in the chronic 6-wk study (Alzet models 2004 and 2002), at a dose of 0.1 mg·kg−1·day−1. These time points were chosen to represent the early and compensated phases of ventricular remodeling (21). At the experimental end points, each rat was weighed and anesthetized. The heart, lungs, and uterus were removed and weighed. The left ventricle (LV), including septum, and right ventricle (RV) were separated and weighed. A section of the mid-LV region was fixed with 4% paraformaldehyde, and the remainder snap-frozen in liquid nitrogen and stored at −80°C for further analysis.
Temporal progression of adverse ventricular remodeling and dysfunction assessed by echocardiogram.
In vivo LV chamber and wall dimensions and function were assessed in sedated (isoflurane 1.0%) rats by echocardiography (VEVO 770; Visualsonics, Toronto, Ontario, Canada) before surgery and every 2 wk thereafter. B-mode images were used to obtain LV short-axis view at the midventricular level, and then M-mode recordings were collected using a two-dimensional reference sector. The leading edge method was used for processing the M-mode images (28). LV end-diastolic diameter (LVEDD), end-systolic diameter (LVESD), and posterior wall thickness (LVPW) at diastole (D) and systole (S) were measured. All measurements were performed on a minimum of three cardiac cycles, and group averages were calculated for each time point.
End point cardiac LV pressure-volume catheterization.
At the chronic experimental end point of 6 wk, rats were weighed and anesthetized with isoflurane (3%). Rats were then intubated and attached to a ventilator. The left jugular vein was cannulated. A skin incision was made along the throat, and the right carotid artery was isolated via blunt dissection. Following the method described by Pacher et al. (36), a Millar pressure-volume catheter (model SPR-838; Millar Instruments, Houston, TX) was introduced into the right carotid artery, and advanced into the aorta and LV (36). After 5 min of stabilization, LV pressure and volume data were collected. Blood-filled cuvettes of known volume were used for calibration. Ventricular volumes were adjusted using the saline-bolus, parallel-conductance method. All measures of cardiac function were evaluated from a minimum of 10 consecutive pressure-volume loops. After collection of functional data, the chest cavity was opened to expose the heart, and cardiac output was measured using a Doppler flow probe (model 3SB; Transonic Systems, Ithaca, NY) placed on the ascending aorta. The cardiac output provided by the Doppler flow probe was used to confirm or correct the catheter volume estimates.
Calculation of LV wall stress.
Two different methods were utilized to estimate LV wall stress. The first method is a calculation of the ratio between LV end-diastolic volume (EDV) and LV mass. This ratio is a useful indicator of the balance between ventricular hypertrophy and dilatation. If LV chamber volume increases without a relative increase in LV mass, more specifically wall thickness, then LV stress is elevated. The second method estimates LV wall stress using the following equation:
| (1) |
where P is LV pressure (mmHg), V is LV volume (ml), and M is LV mass (g). LV wall stress was calculated at both systole and diastole (19).
Analysis of the collagen matrix.
Interstitial collagen volume fraction (CVF) was measured in mid-LV sections at the experimental end points of 5 days and 6 wk. Left ventricular sections were fixed in 4% paraformaldehyde overnight and embedded in paraffin. Five-micron sections were cut, attached to slides, and stained with collagen-specific Picrosirius Red (PSR), as previously described (4, 48). The CVF for each LV section was determined by analyzing a minimum of 15 interstitial regions from two sections of each heart. Perivascular collagen was excluded from these measurements. Images were captured (20×) and processed using a Nikon Eclipse model TE2000-U fluorescence microscope with NIS Elements Software. The CVF was expressed as a percentage of the total area for each LV sections and then group averaged.
Data and statistical analysis.
Statistical analyses were performed using Graphpad software (Prism 5.0, San Diego, CA). Grouped data comparisons were made by one-way analysis of variance. When a significant F ratio (P < 0.05) was obtained, intergroup comparisons were made using a Dunnett’s posttest. Two-way analysis of variance was used to assess interaction. Statistical significance was taken to be P < 0.05.
RESULTS
VO-induced ventricular hypertrophy.
At the 6-wk end point, both ACF groups had significantly increased cardiac output (CO), confirming the VO condition (109 ± 5 vs. 30 ± 1 ml/min, average of combined ACF and Sham groups, respectively). ICI treatment did not alter CO in either the Sham or ACF groups (Table 1). Confirming our previous studies, 6 wk of VO caused significant LV and RV hypertrophy in untreated female rats (71% increase and 101% increase vs. Sham, respectively; P < 0.05; Table 1). ICI treatment did not significantly alter the VO-induced LV or RV hypertrophy (62% increase and 105% increase vs. Sham, respectively; Table 1). Both the ACF and ACF + ICI groups developed increased lung-to-body weight ratio (16.6% increase and 23.5% increase vs. Sham, respectively; Table 1); the increase was greatest in the ACF + ICI group (P < 0.05 vs. ACF). There were no significant differences in body or uterine weights in the volume-overloaded groups compared with Sham rats. No differences were detected in body weight, LV, RV, or lung-to-body weight ratios in the ICI treated Shams with respect to their untreated Sham counterparts.
Table 1.
Morphological parameters and cardiac output
| Sham | Sham + ICI | ACF | ACF + ICI | |
|---|---|---|---|---|
| 5 days postsurgery | n = 6 | n = 5 | n = 7 | n = 5 |
| BW, g | 217 ± 4 | 227 ± 2 | 202 ± 8 | 199 ± 3 |
| LV/BW, mg/g | 2.43 ± 0.11 | 2.54 ± 0.04 | 2.72 ± 0.15 | 2.90 ± 0.13* |
| RV/BW, mg/g | 0.62 ± 0.02 | 0.60 ± 0.03 | 0.83 ± 0.08* | 0.81 ± 0.05* |
| Lung/BW, mg/g | 5.67 ± 0.09 | 5.74 ± 0.03 | 6.12 ± 0.51 | 6.87 ± 0.30* |
| 6 wk postsurgery | n = 8 | n = 6 | n = 8 | n = 7 |
| CO, ml/min | 31.1 ± 2.4 | 28.5 ± 2.7 | 110 ± 12* | 108 ± 11* |
| BW, g | 242 ± 21 | 228 ± 3 | 242 ± 4 | 245 ± 16 |
| LV/BW, mg/g | 2.40 ± 0.11 | 2.29 ± 0.06 | 4.11 ± 0.07* | 4.06 ± 0.09* |
| RV/BW, mg/g | 0.59 ± 0.02 | 0.60 ± 0.02 | 1.19 ± 0.07* | 1.26 ± 0.07* |
| Lung/BW, mg/g | 5.72 ± 0.20 | 5.51 ± 0.20 | 6.67 ± 0.18* | 7.40 ± 0.18*† |
| Uterus/BW, mg/g | 2.54 ± 0.40 | 2.04 ± 0.15 | 2.25 ± 0.24 | 1.85 ± 0.18 |
Values are means ± SD. BW, body weight; Sham, sham-operated control; ICI, estrogen receptor antagonist ICI-182,780; ACF, aortocaval fistula volume overloaded; LV, left ventricle; RV, right ventricle; CO, cardiac output.
P < 0.05 vs. Sham;
P < 0.05 vs. ACF.
ER antagonism exacerbated VO-induced ventricular dilatation and cardiac dysfunction.
Confirming our previous studies, VO caused significant LV dilatation in both ACF groups, as indicated by an increase in LVEDD (Fig. 1A) (48). ICI did not alter LVEDD in Sham animals; however, ICI did worsen ventricular dilatation in the ACF + ICI group relative to untreated ACF at the 6-wk end point (340% vs. 218% increase in LVEDD relative to Sham, respectively). This exacerbation of LV dilation was reflected by reduced ejection fraction (EF%) in the ACF + ICI group (Fig. 1B) by as early as 4 wk postsurgery. ER antagonism had no significant effect on EF% in Sham animals. Further evidence of the negative effects of ER antagonism was provided by LV pressure-volume conductance catheterization (Table 2; Fig. 2). After 6 wk, VO caused a significant increase in both end-systolic and end-diastolic volume, resulting in an increased stroke volume (Table 2). These changes are evident in the representative LV pressure-volume loops depicted in Fig. 2 for the ACF and ACF + ICI groups. Stroke work was increased by volume overload, and arterial elastance was decreased. Consistent with our echo data, ICI treatment alone did not alter cardiac functional parameters assessed by catheterization in Sham animals. However, ICI treatment did produce greater LV dilatation in the treated ACF group, which developed significantly increased end-diastolic and -systolic LV volumes relative to untreated ACF (Fig. 2; Table 2; P < 0.05). ICI treatment did not alter stroke volume, stroke work, or arterial elastance in either the ACF or Sham groups.
Fig. 1.
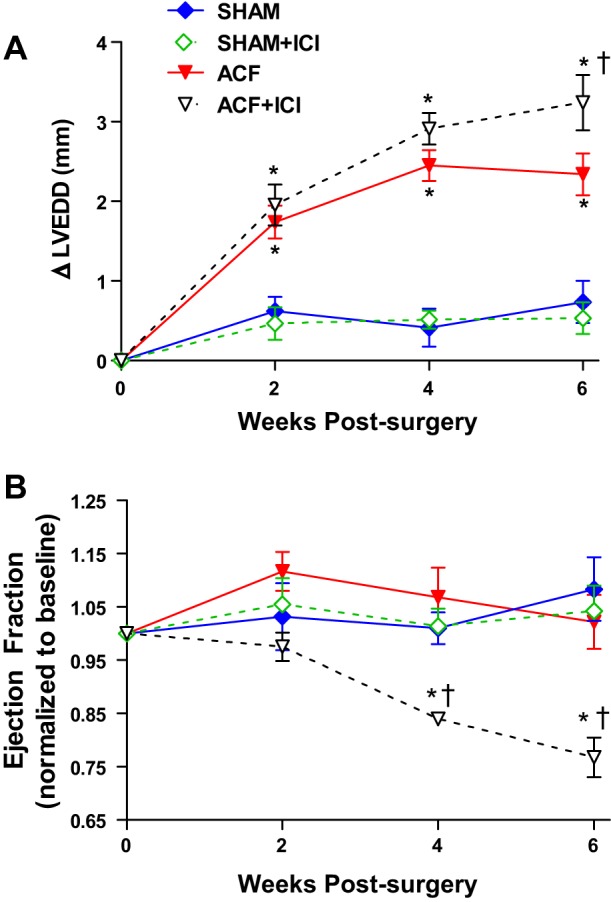
Left ventricular structural and functional changes are worsened by estrogen receptor (ER) antagonism. Temporal echocardiogram data demonstrating that ER antagonism with ICI-182,780 (ICI) exacerbated volume overload (VO)-induced remodeling. A: ICI treatment led to increased left ventricular (LV) end-diastolic diameter (LVEDD), indicating dilatation. Data are presented as increase above presurgery baseline measures for each animal. Baseline LVEDD for all groups combined was 6.18 ± 0.41 mm with no significant difference between groups. B: this increase in chamber size was associated with a significant decrease in ejection fraction (EF%) in the ICI-treated aortocaval fistula (ACF) group. Baseline EF% for all groups combined was 68 ± 2% with no significant difference between groups (n = 6–8 per group; *P < 0.05 vs. Sham; †vs. ACF).
Table 2.
Cardiac functional parameters at 6 wk postsurgery, as assessed by pressure-volume conductance catheterization
| Sham | Sham + ICI | ACF | ACF + ICI | |
|---|---|---|---|---|
| HR, beats/min | 350 ± 11 | 333 ± 18 | 351 ± 9 | 335 ± 61 |
| EDV, µl | 180 ± 33 | 185 ± 39 | 487 ± 80* | 694 ± 119*† |
| ESV, µl | 91 ± 42 | 92 ± 37 | 271 ± 118* | 353 ± 37*† |
| EDP, mmHg | 7 ± 1 | 6 ± 1 | 6 ± 4 | 15 ± 2 |
| ESP, mmHg | 123 ± 18 | 110 ± 5 | 117 ± 23 | 130 ± 28 |
| SV, µl | 90 ± 9 | 93 ± 5 | 276 ± 96* | 335 ± 89* |
| SW, mmHg × ml | 9 ± 2 | 8 ± 1 | 28 ± 12* | 36 ± 13* |
| Ea, mmHg/µl | 1.50 ± 0.10 | 1.38 ± 0.11 | 0.47 ± 0.17* | 0.43 ± 0.19* |
Values are means ± SE. HR, heart rate; EDV, end-diastolic volume; ESV, end-systolic volume; EDP, end-diastolic pressure; ESP, end-systolic pressure; SV, stroke volume; SW, stroke work; Ea, arterial elastance.
P < 0.05 vs. Sham;
P < 0.05 vs. ACF.
Fig. 2.

Representative LV pressure-volume (PV) loops at 6 wk postsurgery. Both ACF groups exhibited LV dilatation, as indicated by the rightward shift of the PV loops. ER antagonism (ICI) had no significant effect on the PV loop of sham-operated animals, but caused a greater LV dilation in animals with volume overload (i.e., ACF + ICI).
ER antagonism exacerbated VO-induced LV wall stress.
ER antagonism with ACF led to a greater increase in volume-to-mass ratio compared with untreated Sham and ACF animals (40% increase relative to Sham; 67% increase relative to ACF alone; P < 0.05) (Fig. 3A). LV wall stress was calculated for both systole and diastole (Fig. 3B). At 6 wk, ACF did not induce a significant increase in LV wall stress compared with Sham. However, ACF + ICI had a pronounced increase in both systolic and diastolic wall stress (404% and 438%, respectively, compared with Sham; P < 0.05). Although there was a trend for increasing wall stress, no significant differences were found between Sham and Sham + ICI groups.
Fig. 3.

ER antagonism led to inappropriate LV hypertrophy and elevated LV wall stress. A: LV end-diastolic volume (EDV)-to-mass (V/M) ratio for each group. Volume overload (VO) significantly increased LV V/M. ICI treatment led to further increase of LV V/M in the rats with VO, suggesting greater wall stress and less successful compensatory remodeling. B: LV wall stress at diastole and systole was significantly increased in rats with VO treated with ER antagonist (i.e., ACF + ICI). (*P < 0.05 vs. Sham; †vs. ACF; n = 4–5 per group).
ER antagonism worsened VO-induced extracellular matrix remodeling.
Interstitial collagen volume fraction (CVF) was calculated from PSR-stained midventricular sections at 5 days and 6 wk postsurgery (Fig. 4). Six weeks of chronic VO caused the disruption and redistribution of fibrillar collagens in both ACF groups (Fig. 4A). Interstitial CVF was significantly reduced in the untreated ACF group (58% decrease vs. Sham), with a greater loss of collagen found in the ICI-treated ACF group (73% decrease vs. Sham; P < 0.05; Fig. 4B). Histologically, the hearts of the ICI-treated ACF group exhibited less collagen staining and greater disorganization (i.e., less uniform distribution), relative to untreated ACF. The initial stage of extracellular matrix remodeling in response to VO stress is associated with a dramatic loss of interstitial collagen due largely to increased MMP activity (21). Using a male rat model of VO, we have previously demonstrated that estrogen can reduce this early collagen remodeling (18); therefore, we assessed CVF following 5 days of VO in our treatment groups. Five days of VO caused a significant reduction in collagen staining in the untreated ACF group (30% decrease vs. Sham; Fig. 4), with an even greater loss of collagen found in the ICI-treated ACF group (57% decrease and 40% decrease vs. Sham and ACF, respectively; P < 0.05; Fig. 4B).
Fig. 4.

LV collagen loss due to volume overload was increased by ER antagonism. A: representative collagen staining of LV sections by Picrosirius Red (PSR) 5 days and 6 wk after surgery. All volume-overloaded groups exhibited a loss of interstitial collagen. ER antagonism with ICI exacerbated the loss of interstitial collagen seen in the volume-overloaded groups. Image brightness was increased for presentation (scale bar = 300 μm). B: collagen volume fraction (CVF) calculated from PSR-stained ventricular sections. Interstitial CVF was significantly reduced at 5 days and 6 wk postsurgery in both volume-overloaded groups (ACF and ACF + ICI) relative to Sham. Treatment with an ER antagonist (ICI) led to further loss of collagen (n = 4–5 in all groups; *P < 0.05 vs. Sham; †vs. ACF).
DISCUSSION
Congestive heart failure is a pervasive clinical syndrome with an increasing incidence in the U.S. population (National Heart, Lung, and Blood Institute, National Institutes of Health Report). The protective effects of estrogen on the cardiovascular system are thought to be mediated through ER-dependent mechanisms. Estrogen-mediated gene transcription occurs through the binding of estrogen to ERs. ERs can directly alter gene transcription in the nucleus or activate kinase signaling in the cytosol. The downstream effects of ER signaling include modulation of vascular function, inflammation, metabolism, insulin sensitivity, cardiac myocyte survival, and cardiac hypertrophy (12). We have shown that estrogen supplementation reduces VO-induced structural myocardial remodeling, indicating that this hormone may have anti-fibrotic effects (18, 48). Further, our previous findings suggest that young, cycling female rats adapt more favorably to chronic VO than male rats (16). We hypothesize that a possible mechanism of this apparent female and ovarian-dependent cardioprotection is the influence of estrogen working through estrogen receptors. Accordingly, the purpose of the present study was to test the hypothesis that the cardioprotective effects of female sex hormones are mediated through an ER-dependent mechanism.
The rat ACF model of chronic VO mimics myocardial remodeling seen in human heart failure. Sustained, chronic VO causes progressive ventricular dilatation and hypertrophy with extracellular matrix (ECM) alterations and subsequent systolic and diastolic dysfunction (19). We have previously demonstrated the cardioprotective effects of estrogen treatment against chronic VO-induced ventricular remodeling in both male and ovariectomized females rats (18, 48). In those studies, estrogen treatment attenuated LV hypertrophy and dilatation caused by chronic VO, which resulted in improved diastolic and systolic function. These studies also established the rat ACF model of heart failure as a suitable model for the evaluation of hormonal contributions to sex-specific differences in ventricular remodeling. In agreement with our studies, several investigators have shown that estrogen limits cardiac hypertrophy in multiple models of cardiac stress (2, 13, 47). More recently, Li et al. found that estrogen replacement in ovariectomized rats prevents adverse cardiac remodeling and hypertrophy caused by pressure overload, potentially through reduction of mast cell-derived chymase, thereby limiting the activation of TGF-β and angiotensin (20, 30). In the present study, we used an estrogen receptor antagonist, ICI 182,780 (i.e., Fulvestrant; Faslodex), to assess cardiac remodeling and functional compensatory adaptations to chronic VO in intact females without ER signaling. We found that ICI-treated animals developed cardiac dysfunction by 4 wk of VO, and that this dysfunction was associated with significant alterations in LV interstitial collagen, first apparent by 5 days postsurgery. In this study, ICI treatment exacerbated VO-induced ventricular remodeling, causing greater LV chamber dilatation and reduced ejection fraction. These findings suggest that estrogen attenuates LV remodeling in chronic VO via ER-dependent signaling. Acutely, the early response to VO has been shown to be mediated by inflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-1β (9, 22, 29). Coinciding with this altered inflammatory state, genes promoting cardiac oxidative stress are upregulated in the absence of estrogen in the volume-overloaded heart (34) and may contribute to the adverse remodeling found in our study.
In chronic heart dysfunction, myocardial stress and injury induce structural remodeling to compensate for elevated wall stress and to maintain cardiac function and mechanical support (15). Female VO rats with intact ER signaling did not exhibit significant increases in LV wall stress. This apparent normalization of wall stress was abated by ER antagonism, as evidenced by significant increases in LV wall stress in the ICI-treated group. Wall stress was estimated using two methods. First, LV volume-to-mass ratio was calculated. A greater increase in volume-to-mass indicates greater wall stress, because wall thickness has not increased proportionally to compensate for an increase in chamber volume. Second, wall stress was calculated using an equation that takes into account ventricular pressure (Eq. 1). Both methods yielded similar results, indicating increased wall stress in the ACF + ICI group vs. both Sham and ACF. The LV volume-to-mass method is much simpler and can be estimated using noninvasive echocardiography data. Our finding of relatively modest changes in wall stress for the untreated ACF group is consistent with our previous study which showed that cycling females, unlike their male counterparts, are able to achieve a stable compensated state of cardiac function in response to VO (16). However, ACF rats treated with ER antagonist developed significant increases in wall stress, indicating a less successful remodeling and failure to normalize the wall stress imposed by chronic VO. The unabated increase in wall stress with ER blockade overpowers the intrinsic compensatory remodeling that occurs in the heart, thus accelerating the progression of adverse remodeling and dysfunction, as indicated by the significant decrease in ejection fraction in the ACF + ICI group. The increased wall stress and deterioration of systolic function can be attributed to maladaptive remodeling exacerbated by a lack of estrogen signaling. These data underscore the vital role of estrogen signaling in modulating cardiac remodeling and normalizing chronic wall stress.
Accumulation and degradation of fibrillar collagens play a pivotal role in the remodeling process. Collagen is a vital component of the cardiac ECM, providing a structural framework for cellular attachment and alignment, as well as diastolic stiffness. During sustained increases in preload or afterload, the cardiac ECM undergoes remodeling to compensate for the additional stress on the heart. A loss of extracellular collagen leads to weakening and dilation of the ventricular wall (45). In accordance with previous studies, VO caused progressive ventricular remodeling including significant LV and RV hypertrophy (48). Our data indicate that rats with VO exhibit an acute loss of interstitial collagen. These data are consistent with previous studies demonstrating that ACF-induced VO causes acute inflammation, resulting in the activation of matrix metalloproteinases (MMP) and subsequent interstitial collagen degradation (21, 35, 44). Alterations in collagen distribution in disease states are brought about by changes in the balance of ventricular MMPs and their tissue inhibitors (TIMPs) (11, 23, 26, 32, 42, 44). Others have reported that the increase of MMPs and subsequent collagen loss occurs within 12 h of creating the aortocaval fistula, characterized by a marked decrease in cardiac collagen by 3 days postsurgery. In male rats, we have shown that estrogen treatment prevents MMP-2 and MMP-9 activation and subsequent breakdown of interstitial collagen during the acute phase of VO (18). Concordantly, here we show that a loss of estrogen signaling, due to ER antagonism, exacerbated acute loss of LV interstitial collagen in response to VO compared with intact females. These adverse effects of ER antagonism on the cardiac ECM were also evident in chronic VO. Our data implicate the essential role of estrogen and ERs in maintaining cardiac interstitial collagen levels and the integrity of the ECM in both the acute and chronic phases of VO.
Several studies, including our own, have utilized ovariectomized rodents to study the effect of estrogen on TIMP/MMP expression and thus collagen distribution (7, 8, 27, 31, 37, 48, 51). Our previous studies have shown that a decrease in interstitial myocardial collagen in ovariectomized rats corresponds to increased protein expression of MMP-2 and MMP-9 (15, 48). However, estrogen replacement to ovariectomized rats leads to a decrease in MMP-9 expression and an increase in TIMP-1 expression (15, 48). These studies demonstrate that the ratio between TIMP-1 and MMP-9 increased in the estrogen-treated rats, which caused a subsequent increase in collagen expression. Furthermore, studies have demonstrated that treatment of dermal fibroblasts with estrogen stimulated increases in collagen type I protein expression and simultaneously caused a significant decrease in MMP-1 expression (38). Other studies support this by demonstrating the positive effect of an ER agonist in upregulating ER expression and inducing collagen biosynthesis, while simultaneously inhibiting the expression of MMP-9 (46). Taken together, these data elaborate the importance of estrogen and ERs in the regulation of MMP/TIMP activity.
Our data indicate that ER antagonism exacerbated VO-induced cardiac ECM remodeling, leading to further ventricular dilatation and accelerated progression to heart failure, marked by a rapid decline in cardiac function (Fig. 5). These findings demonstrate that estrogen and ER signaling play a pivotal role in maintaining the myocardial collagenous ECM and preserving systolic function in VO-stressed hearts of females. As such, these results implicate estrogen as the primary ovarian hormone responsible for the sex-specific cardioprotection observed in the ACF model of VO-induced heart failure.
Fig. 5.

ER antagonism blocks appropriate compensatory remodeling mechanisms in the heart by potentiating cardiac stress and collagen degradation. Volume overload (VO) increases ventricular wall stress, activating compensatory mechanisms that include collagen degradation and cardiomyocyte hypertrophy. Eventually, these compensatory mechanisms become maladaptive leading to adverse cardiac remodeling and dysfunction. Our studies and others have shown that males and ovariectomized females are more susceptible to this adverse remodeling than cycling, intact females. This apparent cardioprotection is likely due to estrogen receptor (ER)-dependent signaling, as ER antagonism led to increased ventricular wall stress and greater collagen degradation, which accelerated the development of cardiac dysfunction.
One in four female deaths were caused by heart disease in 2009 (25), making it the leading cause of death in women in the United States. Sex differences play a significant role in the incidence of cardiovascular diseases, with premenopausal women exhibiting better outcomes in response to aortic stenosis, hypertrophic cardiomyopathy, chronic hypertension, and acute myocardial ischemia compared with their male counterparts (33, 39). These disparities indicate an apparent cardioprotective mechanism in premenopausal females. However, this favorable cardiac profile gradually disappears after menopause, with postmenopausal women exhibiting a higher cardiovascular risk than their male counterparts (1, 24). The paradoxical sex-dependent shift in cardiovascular outcomes can be attributed to ovarian hormones, with reduced levels significantly increasing the incidence of coronary artery disease (3, 40, 41, 49) and left ventricular hypertrophy (2, 43, 47). Further understanding of the role of estrogen in prevention of pathological cardiac remodeling is needed to help elucidate downstream mediators, which could be targeted therapeutically, while avoiding the side effects of systemic estrogen delivery. Development of such therapies would have important clinical implications in treating aging or postmenopausal women without the adverse side effects of hormone replacement therapy.
GRANTS
This study was supported by National Institute on Alcohol Abuse and Alcoholism (NIAAA) Grant 1R21-AA-022690–01A1 (J. D. Gardner) and NIAAA Grant 5-T32-AA007577–14 (P. E. Molina).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
M.C.E.H., V.K.N., E.C.E.H., J.M.B., and J.D.G. performed experiments; M.C.E.H., V.K.N., E.C.E.H., J.M.B., and J.D.G. analyzed data; M.C.E.H., E.C.E.H., and J.D.G. interpreted results of experiments; M.C.E.H., E.C.E.H., V.K.N., and J.D.G. prepared figures; M.C.E.H. drafted manuscript; M.C.E.H., E.C.E.H., V.K.N., and J.D.G. edited and revised manuscript; M.C.E.H., V.K.N., E.C.E.H., J.M.B., and J.D.G. approved final version of manuscript; J.D.G. conception and design of research.
ACKNOWLEDGMENTS
We thank J. Clary and C. Corll for technical assistance.
REFERENCES
- 1.Anderson RD, Pepine CJ. Gender differences in the treatment for acute myocardial infarction: bias or biology? Circulation 115: 823–826, 2007. doi: 10.1161/CIRCULATIONAHA.106.685859. [DOI] [PubMed] [Google Scholar]
- 2.Babiker FA, Lips D, Meyer R, Delvaux E, Zandberg P, Janssen B, van Eys G, Grohé C, Doevendans PA. Estrogen receptor beta protects the murine heart against left ventricular hypertrophy. Arterioscler Thromb Vasc Biol 26: 1524–1530, 2006. doi: 10.1161/01.ATV.0000223344.11128.23. [DOI] [PubMed] [Google Scholar]
- 3.Bittner V. Menopause, age, and cardiovascular risk: a complex relationship. J Am Coll Cardiol 54: 2374–2375, 2009. doi: 10.1016/j.jacc.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 4.Bradley JM, Nguyen JB, Fournett AC, Gardner JD. Cigarette smoke exacerbates ventricular remodeling and dysfunction in the volume overloaded heart. Microsc Microanal 18: 91–98, 2012. doi: 10.1017/S1431927611012207. [DOI] [PubMed] [Google Scholar]
- 5.Branham WS, Fishman R, Streck RD, Medlock KL, De George JJ, Sheehan DM. ICI 182,780 inhibits endogenous estrogen-dependent rat uterine growth and tamoxifen-induced developmental toxicity. Biol Reprod 54: 160–167, 1996. doi: 10.1095/biolreprod54.1.160. [DOI] [PubMed] [Google Scholar]
- 6.Brower GL, Chancey AL, Thanigaraj S, Matsubara BB, Janicki JS. Cause and effect relationship between myocardial mast cell number and matrix metalloproteinase activity. Am J Physiol Heart Circ Physiol 283: H518–H525, 2002. doi: 10.1152/ajpheart.00218.2000. [DOI] [PubMed] [Google Scholar]
- 7.Cao J, Zhu T, Lu L, Geng L, Wang L, Zhang Q, Yang K, Wang H, Shen W. Estrogen induces cardioprotection in male C57BL/6J mice after acute myocardial infarction via decreased activity of matrix metalloproteinase-9 and increased Akt-Bcl-2 anti-apoptotic signaling. Int J Mol Med 28: 231–237, 2011. doi: 10.3892/ijmm.2011.681. [DOI] [PubMed] [Google Scholar]
- 8.Dai Q, Lin J, Craig T, Chou YM, Hinojosa-Laborde C, Lindsey ML. Estrogen effects on MMP-13 and MMP-14 regulation of left ventricular mass in Dahl salt-induced hypertension. Gend Med 5: 74–85, 2008. doi: 10.1016/S1550-8579(08)80010-1. [DOI] [PubMed] [Google Scholar]
- 9.Dai RP, Dheen ST, He BP, Tay SS. Differential expression of cytokines in the rat heart in response to sustained volume overload. Eur J Heart Fail 6: 693–703, 2004. doi: 10.1016/j.ejheart.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 10.Dauvois S, White R, Parker MG. The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling. J Cell Sci 106: 1377–1388, 1993. [DOI] [PubMed] [Google Scholar]
- 11.Deleon-Pennell KY, Altara R, Yabluchanskiy A, Modesti A, Lindsey ML. The circular relationship between matrix metalloproteinase-9 and inflammation following myocardial infarction. IUBMB Life 67: 611–618, 2015. doi: 10.1002/iub.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest 116: 561–570, 2006. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donaldson C, Eder S, Baker C, Aronovitz MJ, Weiss AD, Hall-Porter M, Wang F, Ackerman A, Karas RH, Molkentin JD, Patten RD. Estrogen attenuates left ventricular and cardiomyocyte hypertrophy by an estrogen receptor-dependent pathway that increases calcineurin degradation. Circ Res 104: 265–275, 2009. doi: 10.1161/CIRCRESAHA.108.190397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El Hajj EC, El Hajj MC, Ninh VK, Gardner JD. Cardioprotective effects of lysyl oxidase inhibition against volume overload-induced extracellular matrix remodeling. Exp Biol Med (Maywood) 241: 539–549, 2016. doi: 10.1177/1535370215616511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gardner JD, Brower GL, Janicki JS. Effects of dietary phytoestrogens on cardiac remodeling secondary to chronic volume overload in female rats. Journal of Applied Physiology 99: 1378–1383, 2005. doi: 10.1152/japplphysiol.01141.2004. [DOI] [PubMed] [Google Scholar]
- 16.Gardner JD, Brower GL, Janicki JS. Gender differences in cardiac remodeling secondary to chronic volume overload. J Card Fail 8: 101–107, 2002. doi: 10.1054/jcaf.2002.32195. [DOI] [PubMed] [Google Scholar]
- 17.Gardner JD, Brower GL, Voloshenyuk TG, Janicki JS. Cardioprotection in female rats subjected to chronic volume overload: synergistic interaction of estrogen and phytoestrogens. Am J Physiol Heart Circ Physiol 294: H198–H204, 2008. doi: 10.1152/ajpheart.00281.2007. [DOI] [PubMed] [Google Scholar]
- 18.Gardner JD, Murray DB, Voloshenyuk TG, Brower GL, Bradley JM, Janicki JS. Estrogen attenuates chronic volume overload induced structural and functional remodeling in male rat hearts. Am J Physiol Heart Circ Physiol 298: H497–H504, 2010. doi: 10.1152/ajpheart.00336.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hutchinson KR, Stewart JA Jr, Lucchesi PA. Extracellular matrix remodeling during the progression of volume overload-induced heart failure. J Mol Cell Cardiol 48: 564–569, 2010. doi: 10.1016/j.yjmcc.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jalil JE, Ocaranza MP. Estrogens and myocardial chymase: new insights into pathological hypertrophy and remodeling. Hypertension 65: 271–272, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04375. [DOI] [PubMed] [Google Scholar]
- 21.Janicki JS, Brower GL, Gardner JD, Forman MF, Stewart JA Jr, Murray DB, Chancey AL. Cardiac mast cell regulation of matrix metalloproteinase-related ventricular remodeling in chronic pressure or volume overload. Cardiovasc Res 69: 657–665, 2006. doi: 10.1016/j.cardiores.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 22.Jobe LJ, Meléndez GC, Levick SP, Du Y, Brower GL, Janicki JS. TNF-alpha inhibition attenuates adverse myocardial remodeling in a rat model of volume overload. Am J Physiol Heart Circ Physiol 297: H1462–H1468, 2009. doi: 10.1152/ajpheart.00442.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jourdan-Lesaux C, Zhang J, Lindsey ML. Extracellular matrix roles during cardiac repair. Life Sci 87: 391–400, 2010. doi: 10.1016/j.lfs.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim ES, Menon V. Status of women in cardiovascular clinical trials. Arterioscler Thromb Vasc Biol 29: 279–283, 2009. doi: 10.1161/ATVBAHA.108.179796. [DOI] [PubMed] [Google Scholar]
- 25.Kochanek KD, Xu J, Murphy SL, Miniño AM, Kung HC. Deaths: final data for 2009. Natl Vital Stat Rep 60: 1–116, 2011. [PubMed] [Google Scholar]
- 26.Kwon JS, Kim YS, Cho AS, Cho HH, Kim JS, Hong MH, Jeong HY, Kang WS, Hwang KK, Bae JW, Jeong MH, Cho MC, Ahn Y. Regulation of MMP/TIMP by HUVEC transplantation attenuates ventricular remodeling in response to myocardial infarction. Life Sci 101: 15–26, 2014. doi: 10.1016/j.lfs.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 27.Lam KK, Cheng PY, Hsiao G, Chen SY, Shen HH, Yen MH, Lee YM. Estrogen deficiency-induced alterations of vascular MMP-2, MT1-MMP, and TIMP-2 in ovariectomized rats. Am J Hypertens 22: 27–34, 2009. doi: 10.1038/ajh.2008.306. [DOI] [PubMed] [Google Scholar]
- 28.Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ; Chamber Quantification Writing Group; American Society of Echocardiography’s Guidelines and Standards Committee; European Association of Echocardiography . Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 18: 1440–1463, 2005. doi: 10.1016/j.echo.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 29.Levick SP, Gardner JD, Holland M, Hauer-Jensen M, Janicki JS, Brower GL. Protection from adverse myocardial remodeling secondary to chronic volume overload in mast cell deficient rats. J Mol Cell Cardiol 45: 56–61, 2008. doi: 10.1016/j.yjmcc.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J, Jubair S, Janicki JS. Estrogen inhibits mast cell chymase release to prevent pressure overload-induced adverse cardiac remodeling. Hypertension 65: 328–334, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Liao EY, Dai RC, Wei QY, Luo XH. Effects of 17 beta-estradiol on the expression of interstitial collagenases-8 and −13 (MMP-8 and MMP-13) and tissue inhibitor of metalloproteinase-1 (TIMP-1) in ovariectomized rat osteoblastic cells. J Mol Histol 35: 723–731, 2004. doi: 10.1007/s10735-004-6206-3. [DOI] [PubMed] [Google Scholar]
- 32.Lindsey ML, Yabluchanskiy A, Ma Y. Tissue Inhibitor of Metalloproteinase-1: Actions beyond Matrix Metalloproteinase Inhibition. Cardiology 132: 147–150, 2015. doi: 10.1159/000433419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luczak ED, Leinwand LA. Sex-based cardiac physiology. Annu Rev Physiol 71: 1–18, 2009. doi: 10.1146/annurev.physiol.010908.163156. [DOI] [PubMed] [Google Scholar]
- 34.McLarty JL, Meléndez GC, Levick SP, Bennett S, Sabo-Attwood T, Brower GL, Janicki JS. Estrogenic modulation of inflammation-related genes in male rats following volume overload. Physiol Genomics 44: 362–373, 2012. doi: 10.1152/physiolgenomics.00146.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murray DB, Gardner JD, Brower GL, Janicki JS. Endothelin-1 mediates cardiac mast cell degranulation, matrix metalloproteinase activation, and myocardial remodeling in rats. Am J Physiol Heart Circ Physiol 287: H2295–H2299, 2004. doi: 10.1152/ajpheart.00048.2004. [DOI] [PubMed] [Google Scholar]
- 36.Pacher P, Nagayama T, Mukhopadhyay P, Bátkai S, Kass DA. Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat Protoc 3: 1422–1434, 2008. doi: 10.1038/nprot.2008.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pechenino AS, Lin L, Mbai FN, Lee AR, He XM, Stallone JN, Knowlton AA. Impact of aging vs. estrogen loss on cardiac gene expression: estrogen replacement and inflammation. Physiol Genomics 43: 1065–1073, 2011. doi: 10.1152/physiolgenomics.00228.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Philips N, Devaney J. Beneficial regulation of type I collagen and matrixmetalloproteinase-1 expression by estrogen, progesterone, and its combination in skin fibroblasts. J Am Aging Assoc 26: 59–62, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piro M, Della Bona R, Abbate A, Biasucci LM, Crea F. Sex-related differences in myocardial remodeling. J Am Coll Cardiol 55: 1057–1065, 2010. doi: 10.1016/j.jacc.2009.09.065. [DOI] [PubMed] [Google Scholar]
- 40.Reckelhoff JF, Maric C. Sex and gender differences in cardiovascular-renal physiology and pathophysiology. Steroids 75: 745–746, 2010. doi: 10.1016/j.steroids.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 41.Rosano GM, Vitale C, Marazzi G, Volterrani M. Menopause and cardiovascular disease: the evidence. Climacteric 10, Suppl 1: 19–24, 2007. doi: 10.1080/13697130601114917. [DOI] [PubMed] [Google Scholar]
- 42.Roten L, Nemoto S, Simsic J, Coker ML, Rao V, Baicu S, Defreyte G, Soloway PJ, Zile MR, Spinale FG. Effects of gene deletion of the tissue inhibitor of the matrix metalloproteinase-type 1 (TIMP-1) on left ventricular geometry and function in mice. J Mol Cell Cardiol 32: 109–120, 2000. doi: 10.1006/jmcc.1999.1052. [DOI] [PubMed] [Google Scholar]
- 43.Skavdahl M, Steenbergen C, Clark J, Myers P, Demianenko T, Mao L, Rockman HA, Korach KS, Murphy E. Estrogen receptor-beta mediates male-female differences in the development of pressure overload hypertrophy. Am J Physiol Heart Circ Physiol 288: H469–H476, 2005. doi: 10.1152/ajpheart.00723.2004. [DOI] [PubMed] [Google Scholar]
- 44.Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev 87: 1285–1342, 2007. doi: 10.1152/physrev.00012.2007. [DOI] [PubMed] [Google Scholar]
- 45.Spinale FG, Coker ML, Krombach SR, Mukherjee R, Hallak H, Houck WV, Clair MJ, Kribbs SB, Johnson LL, Peterson JT, Zile MR. Matrix metalloproteinase inhibition during the development of congestive heart failure: effects on left ventricular dimensions and function. Circ Res 85: 364–376, 1999. doi: 10.1161/01.RES.85.4.364. [DOI] [PubMed] [Google Scholar]
- 46.Surazynski A, Jarzabek K, Haczynski J, Laudanski P, Palka J, Wolczynski S. Differential effects of estradiol and raloxifene on collagen biosynthesis in cultured human skin fibroblasts. Int J Mol Med 12: 803–809, 2003. [PubMed] [Google Scholar]
- 47.van Eickels M, Grohé C, Cleutjens JP, Janssen BJ, Wellens HJ, Doevendans PA. 17beta-estradiol attenuates the development of pressure-overload hypertrophy. Circulation 104: 1419–1423, 2001. doi: 10.1161/hc3601.095577. [DOI] [PubMed] [Google Scholar]
- 48.Voloshenyuk TG, Gardner JD. Estrogen improves TIMP-MMP balance and collagen distribution in volume-overloaded hearts of ovariectomized females. Am J Physiol Regul Integr Comp Physiol 299: R683–R693, 2010. doi: 10.1152/ajpregu.00162.2010. [DOI] [PubMed] [Google Scholar]
- 49.Wake R, Yoshiyama M. Gender differences in ischemic heart disease. Recent Pat Cardiovasc Drug Discov 4: 234–240, 2009. doi: 10.2174/157489009789152249. [DOI] [PubMed] [Google Scholar]
- 50.Wakeling AE, Bowler J. ICI 182,780, a new antioestrogen with clinical potential. J Steroid Biochem Mol Biol 43: 173–177, 1992. doi: 10.1016/0960-0760(92)90204-V. [DOI] [PubMed] [Google Scholar]
- 51.Xu Y, Arenas IA, Armstrong SJ, Davidge ST. Estrogen modulation of left ventricular remodeling in the aged heart. Cardiovasc Res 57: 388–394, 2003. doi: 10.1016/S0008-6363(02)00705-8. [DOI] [PubMed] [Google Scholar]