

Abstract
Elevated levels of reactive oxygen species and intracellular Ca2+ play a key role in endothelial barrier dysfunction in acute lung injury. We previously showed that H2O2-induced increases in intracellular calcium concentrations ([Ca2+]i) in lung microvascular endothelial cells (LMVECs) involve the membrane Ca2+ channel, transient receptor potential vanilloid-4 (TRPV4) and that inhibiting this channel attenuated H2O2-induced barrier disruption in vitro. We also showed that phosphorylation of TRPV4 by the Src family kinase, Fyn, contributes to H2O2-induced Ca2+ influx in LMVEC. In endothelial cells, Fyn is tethered to the cell membrane by CD36, a fatty acid transporter. In this study, we assessed the effect of genetic loss or pharmacological inhibition of CD36 on Ca2+ responses to H2O2. H2O2-induced Ca2+ influx was attenuated in LMVEC isolated from mice lacking CD36 (CD36−/−). TRPV4 expression and function was unchanged in LMVEC isolated from wild-type (WT) and CD36−/− mice, as well as mice with deficiency for Fyn (Fyn−/−). TRPV4 immunoprecipitated with Fyn, but this interaction was decreased in CD36−/− LMVEC. The amount of phosphorylated TRPV4 was decreased in LMVEC from CD36−/− mice compared with WT controls. Loss of CD36 altered subcellular localization of Fyn, while inhibition of CD36 fatty acid transport with succinimidyl oleate did not attenuate H2O2-induced Ca2+ influx. Lastly, we found that CD36−/− mice were protected from ischemia-reperfusion injury in vivo. In conclusion, our data suggest that CD36 plays an important role in H2O2-mediated lung injury and that the mechanism may involve CD36-dependent scaffolding of Fyn to the cell membrane to facilitate TRPV4 phosphorylation.
Keywords: acute lung injury, calcium, ROS
acute lung injury (ALI), a life-threatening disease with high morbidity and no effective pharmacological therapies, is characterized by increased lung microvascular endothelial permeability, leading to alveolar flooding and hypoxemia. Multiple signaling pathways in lung microvascular endothelial cells (LMVECs) have been implicated as contributors to barrier permeability in ALI. In particular, increases in reactive oxygen species (ROS) and intracellular Ca2+ concentration ([Ca2+]i) are key signaling events in the pathogenesis of ALI (7, 16, 37). For example, in ischemia-reperfusion (IR) models of experimental lung injury, generation of endothelial ROS and elevations in [Ca2+]i through Ca2+ influx from plasma membrane channels have been shown to promote endothelial permeability (39, 41). Moreover, [Ca2+]i is critical for initiating endothelial dysfunction and promoting formation of paracellular gaps in LMVEC (11, 12, 29). Specifically, members of the transient receptor potential (TRP) family of Ca2+ channels, such as TRPC6, have been implicated in the pathogenesis of lung IR injury (41). The primary source of injury in IR models is production of ROS, and ROS have been shown to increase [Ca2+]i in various vascular beds (5, 6, 25, 28, 42). We recently showed that H2O2-induced Ca2+ influx in LMVEC involves the vanilloid-4 TRP channel (TRPV4) (36). TRPV4 is a multifunctional Ca2+ channel responsive to various mechanical stimuli, such as heat and shear stress. In fact, TRPV4 has been shown to be critical for mediating lung injury in heart failure, stretch-induced ventilator injury, and more recently, acid-induced models of lung injury (3, 17, 38). We recently showed that inhibition of TRPV4 attenuated H2O2-induced LMVEC barrier disruption in vitro and that H2O2-induced Ca2+ influx in LMVEC may be regulated by phosphorylation of TRPV4 by Fyn, a member of the Src family of kinases (SFKs). SFKs are a multifunctional set of enzymes with several family members including Fyn, Lyn, Yes, and c-Src. Changes in [Ca2+]i following H2O2 challenge were absent in LMVEC isolated from Fyn-deficient (Fyn−/−) mice or in human LMVEC (HLMVEC) following genetic knockdown of Fyn. Furthermore, our data suggested that phosphorylation of TRPV4 was decreased when Fyn was pharmacologically inhibited (36).
The kinase activity of SFKs is posttranslationally regulated by several mechanisms (1, 31). Importantly, association with membrane anchor proteins retains Fyn at the cell membrane, allowing Fyn to exert its kinase activity on nearby membrane proteins (8). In the endothelium, Fyn associates with CD36, a fatty acid transporter (18, 32, 33). The role of CD36 in lung disease is under active investigation, with evidence showing that endothelial CD36 contributes to LPS-induced barrier dysfunction (4). Though principally involved in fatty acid transport across the cell membrane, CD36 has been recently implicated in many other cellular processes including angiogenesis and apoptosis (32). While the mechanism by which CD36 mediated these effects is still under investigation, there is growing evidence to suggest that CD36 participates in cell signaling events independent of fatty acid transport. As mentioned above, CD36 can act as a membrane anchor for the Src kinase Fyn. In addition, following activation by ligands such as thrombospondin, CD36 has been shown to serve as a nidus for recruitment and activation of various intracellular kinases (10, 23, 35). Moreover, the presence of CD36 on the membrane has been shown to be important for basal Ca2+ dynamics, with loss of CD36 attenuating Ca2+ influx in CHO cells following thapsigargin-induced store depletion (20). Collectively, these data suggest a critical role for CD36 in Ca2+ signaling and kinase activation, although the specifics of these two functions in LMVECs are not known.
CD36 has also been implicated in IR pathobiology. Loss of CD36 attenuates IR injury in various vascular beds including the brain and the heart (9, 26) and, in the lung, we recently showed that loss of CD36 is protective against H2O2-induced barrier dysfunction in LMVEC in vitro and malaria-induced ARDS in vivo (2). However, to our knowledge, a role for CD36 in lung IR injury has not been previously described in vivo. Given the involvement of CD36 in IR injury in other tissues, our data showing that endothelial dysfunction due to increased H2O2 is dependent on elevations in intracellular Ca2+ and prior data linking CD36 to intracellular Ca2+ flux, we hypothesized that CD36 participates in IR injury by modulating H2O2-induced Ca2+ influx. Specifically, we hypothesized that CD36-mediated tethering of Fyn to the cell membrane facilitates phosphorylation of TRPV4, a step we previously showed was necessary for subsequent activation of TRPV4 by H2O2 (36). Thus in this study we determined whether loss of CD36 alters H2O2-induced Ca2+ influx in vitro and whether loss of CD36 is protective against IR injury in vivo.
METHODS
All procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Animal Care and Use Committee of The Johns Hopkins University School of Medicine.
Isolation and culture of mouse lung microvascular endothelial cells.
Adult (8–10 wk), wild-type (WT; C57/B6) and Fyn−/− (B6.129S7-Fyntm1Sor/J) mice were purchased from Jackson Laboratories. CD36-deficient C57/B6 (CD36−/−) male mice were generously provided by Dr. Alan Scott (Johns Hopkins University). All mice were subjected to genotyping before use. Mice were euthanized by cervical dislocation, and the lungs were quickly removed by dissection and immersed in DMEM (GIBCO). As described by us previously (36), peripheral mouse tissue was obtained by dissection, minced, and digested in complete medium using collagenase (Type 1A; 1 mg/ml). Following centrifugation and resuspension of the cell pellet, the cell suspension was incubated with CD31-conjugated beads (Invitrogen) for 30 min at room temperature. The beads and cells were then cultured. Following growth of bead-selected cells, cells were trypsinized and incubated with beads conjugated with Griffonia simplicifolia lectin (GS-II; EY Laboratories). The cells that survived dual bead selection were expanded and repeat staining for microvascular endothelial markers CD31 (anti-CD31; Abcam) and GS-II was performed at passage three to ensure that only LMVECs were selected. Staining for smooth muscle-specific α-actin (SMA; Sigma) was also performed to rule out contamination from smooth muscle cells or fibroblasts. Only cells that were negative for SMA and positive for CD31 and GS-II lectin (>95%) were used in studies.
Intracellular Ca2+ measurements.
Cells were plated at 50–60% confluence on glass coverslips and loaded with 5 μM Fura 2-AM (Life Biotechnology) for 1 h at 37°C before being placed in a temperature-controlled (37°C) laminar flow chamber in a live cell Ca2+ imaging system as described previously (36). Cells loaded in the flow chamber were perfused with warmed modified Krebs solution containing (in mM) 118 NaCl, 4.7 KCl, 0.57 MgSO4, 1.18 KH2PO4, 25 NaHCO3, 2.5 CaCl2, and 10 glucose bubbled with 16% O2 gas at 37°C. The flow chamber and perfusates were maintained at 37°C. Reservoirs containing Krebs solution with and without drugs were connected via a manifold to the inlet of the laminar flow chamber, allowing for rapid switching between solutions with and without inhibitors. At the beginning of each experiment, cells were perfused for 15 min to allow for establishment of stable baseline. Hydrogen peroxide (Sigma) was freshly prepared in warmed Krebs before each experiment. [Ca2+]i was estimated from F340/F380 measured in calibration solutions with Ca2+ concentrations of 0–1,350 nM (Molecular Probes, Eugene, OR).
Immunofluorescence.
For acetylated LDL (AcLDL) uptake experiments, cells grown on glass coverslips were incubated with 2.5 µl AcLDL (Alexa Fluor; Life Technologies) with or without succinimidyl oleate (SSO; 100 µM; Cayman Biochemicals) for 1 h, after which cells were fixed with formalin (4%) and permeabilized with Triton X (0.5%). Cells were blocked in 2% BSA for 1 h, washed, and then incubated for 5–10 min with DAPI nuclear stain (1:1,000 in PBS; Thermo). After three washes with TBS-T (Tris-buffered saline containing Tween 20), coverslips were mounted with Prolong Anti-fade solution (Invitrogen). Images were obtained and analyzed using an Olympus IX51 inverted fluorescent microscope (×60 oil objective) and a Cooke digital camera connected to a computer running Micro-Manager. Total fluorescence was determined using ImageJ software. In addition, the number of cells staining positive for AcLDL were counted by an investigator blinded to condition and genotype.
Image analysis.
Analysis of immunofluorescence images for Fyn localization was performed using ImageJ and Fiji. Fluorescence values below a set threshold were excluded to decrease background. A Voronoi algorithm was used to automatically determine cell borders, and the distance between a pixel with fluorescence value above the set threshold and the center of the nucleus was calculated. The distances were then plotted as a histogram and the mean distance to the nucleus was calculated using GraphPad Prism software. To determine cell size, phase-contrast bright-field photomicrographs were taken of WT and CD36−/− mouse lung microvascular endothelial cells (MLMVEC). After scaling, cell edges were manually outlined and cell area was calculated using ImageJ. For radial profile measurements, cells from different coverslips were chosen at random. The nuclear shape was first outlined in the DAPI channel and transferred over the channel containing Fyn fluorescence, and integrated density was measured for this area. The ROI comprising the nucleus was automatically expanded radially by (change in radius = 30 pixels) and a second integrated density measurement was obtained. The entire cell was then selected and a total density measurement was taken. The perinuclear fraction was defined as the difference between the expanded ROI and the nuclear ROI divided by total cell integrated density.
Western blotting.
Cell lysates were prepared and protein concentrations determined as reported previously (36). Equal amounts of protein (40 µg) were loaded into each well of an 8% gel and subjected to electrophoresis. Following separation, proteins were transferred to nitrocellulose membranes, which were then blocked with blocking buffer (5% BSA in TBS-T), incubated with primary antibodies [anti-phosphotyrosine 1:3,000; Millipore; anti-TRPV4 1:500; Alomone (total cell lysates) or Millipore [immunoprecipitation (IP) lysates]; anti-Fyn 1:2,000; Santa Cruz] at room temperature for 1 h (phosphotyrosine, Fyn) or at 4°C overnight (TRPV4). Membranes were then washed and incubated with secondary antibodies (goat anti-mouse 1:10,000; Bio-Rad or goat anti-rabbit 1:10,000; Bio-Rad) for 1 h. Bands were visualized by enhanced chemiluminescence. Membranes were then stripped and reprobed with anti-GAPDH horseradish peroxidase-conjugated antibody (1:25,000; Bio-Rad). Densitometry was performed to quantify the amount of protein of interest and this value was normalized to the housekeeping protein using ImageJ software.
Immunoprecipitation.
Cell lysates were prepared using solubilization buffer containing 20 mM Tris, 200 mM NaCl, 1% Triton, 1% deoxycholate, 0.1% SDS, 5 mM EDTA, 5 mM EGTA, phosphatase inhibitors, and protease inhibitor cocktail. Protein concentrations were determined as reported previously (36). Samples were precleared by adding 1 µl IgG (rabbit IgG for TRPV4 IP, mouse IgG for p-Tyr IP; Bio-Rad) plus 100 µl A/G or A+ beads (Thermo Scientific). This solution was rocked for 30 min at 4°C. After incubation, the lysates were centrifuged (3,000 g; 4°C) and supernatants collected. Primary antibodies (anti-Fyn 5 µl; Santa Cruz; anti-phosphotyrosine 5 µl; Millipore) were added and the solutions incubated overnight. Next, 100 µl of A/G (phosphotyrosine IP) or A+ (TRPV4 IP) beads were added and incubation continued for another 4 h at 4°C. The beads were pelleted by centrifugation (4°C, 2,000 rpm, 2 min), washed three times with solubilization buffer (with rocking for 5 min at room temperature and centrifugation with each wash). A final wash was performed with salt-free wash buffer (20 mM Tris, 1% Triton, 5 mM EDTA, 5 mM EGTA). Lastly, proteins were eluted by incubation for 30 min at 37°C with elution buffer (10 mM Tris, 3% SDS, 10% glycerol, 0.01% bromophenol blue, and 0.5 M DTT). The supernatant containing eluted proteins as well as input lysates (1:10 dilution) were boiled and Western blotted as described above.
ECIS.
Cells were seeded on gold electrodes in 0.5-ml electrode wells (Applied BioPhysics) and grown to confluence. Baseline transmembrane electrical resistance (TER) was measured 24–48 h after initial seeding of cells. The medium was changed to serum-free basal medium (GIBCO). Following equilibration in serum-free medium, GSK1016790A (GSK) (1.5 µM) or diluent was added to each well. Data analysis was performed using the electrical cell impedance sensing (ECIS) ZTheta system (Applied BioPhysics).
IR injury.
Six- to 8-wk-old male C57/B6 mice were anesthetized, intubated, and mechanically ventilated (Harvard mouse ventilator). Adequate anesthesia was ensured by paw pinch. As previously described (13), a left thoracotomy was performed and the pulmonary artery was visualized. Using suture, the left pulmonary artery was occluded with a slipknot. One end of the slipknot was exteriorized for later release. The thoracotomy was then closed after verification of lung reexpansion. The mouse was extubated, and the pulmonary artery tie was released after 30 min of vessel occlusion. The mice were euthanized at the end of the reperfusion time (180 min). Evans blue dye (EBD; 30 mg/kg; Sigma) was administered intravenously during the period of left pulmonary artery ischemia. After reperfusion, mice were reanesthetized, intubated and ventilated. Intravascular EBD was removed with saline flush, and the left upper lobe and the right lungs were weighed and incubated in formamide (1 ml formamide/100 mg lung) for 24 h. EBD content was evaluated by calorimetric evaluation with a spectrophotometer (620 nm). The ratio of EBD measured in the left (injured) lung to EBD measured in the right (uninjured) lung allowed for assessment of lung permeability produced as a result of IR injury to the left lung. For sham surgical controls, the surgery was initiated with anesthesia and a left thoracotomy, but no pulmonary artery occlusion, was performed.
Data analysis/statistics.
All values are expressed as means ± SE. For [Ca2+]i measurements, data were collected from up to 30 cells and the values were averaged to obtain a single value for each experiment. Change in [Ca2+]i was computed by subtracting the average basal [Ca2+]i, determined from 1 min of data collected immediately before challenge with agonists, from the peak [Ca2+]i measured within the first 5 min after beginning agonist challenge. For ECIS data, resistance measurements (R) from each ECIS well were normalized to the resistance value at the beginning of the experiment (R0). Experiments were performed using cells isolated from at least three different mice. To quantify differences in amount of barrier dysfunction, area under the curve (AUC) was calculated for individual ECIS traces. Data were compared by unpaired Student’s t-test or by one-way ANOVA with a Holm-Sidak post hoc test to determine differences between groups. A P value < 0.05 was accepted as statistically significant.
RESULTS
Effect of CD36 deletion on H2O2-induced Ca2+ influx.
We have previously shown that 1 mM H2O2 produces significant barrier dysfunction in vitro, as measured by ECIS (2). To determine whether loss of CD36 interrupts H2O2-induced Ca2+ influx, we exposed WT and CD36−/− MLMVEC to 1 mM H2O2. Baseline [Ca2+]i was not significantly different between WT and CD36−/− MLMVEC (Fig. 1A). Application of H2O2 increased [Ca2+]i in WT MLMVEC as expected, but this response was significantly attenuated in CD36−/− MLMVEC (Fig. 1, B–D).
Fig. 1.

Effect of CD36 on H2O2-induced increase in intracellular calcium concentration ([Ca2+]i) in mouse lung microvascular endothelial cells (MLMVEC). A: scatterplot and mean ± SE of baseline intracellular calcium ([Ca2+]i) in wild-type (WT) and CD36-deficient (CD36−/−) MLMVEC. Representative traces from WT (B) and CD36−/− (C) MLMVEC show change in [Ca2+]i following exposure to 1 mM H2O2. D: scatterplot and mean ± SE values for peak change in [Ca2+]i from baseline in MLMVEC exposed to 1 mM H2O2. *Significant difference from WT; n = 5–13 experiments/group.
Expression and function of TRPV4 in CD36−/− MLMVEC.
We hypothesized that CD36 mediates H2O2-induced Ca2+ influx by scaffolding Fyn to the cell membrane, thereby facilitating phosphorylation of TRPV4 by Fyn, which is necessary for H2O2-induced Ca2+ influx, but not Ca2+ influx in response to chemical agonists of TRPV4. We first determined whether loss of CD36 or Fyn would alter TRPV4 levels or function. No significant difference was observed in total TRPV4 protein levels in lysates of LMVEC from WT, CD36−/−, and Fyn−/− mice (Fig. 2, A–B). To assess TRPV4 function, we exposed WT, CD36−/−, and Fyn−/− MLMVEC to GSK (1.5 µM), a specific TRPV4 agonist (38). The peak GSK-induced Ca2+ influx was not significantly different between WT, CD36−/−, and Fyn−/− MLMVEC (Fig. 2, C–F). The slope of [Ca2+]i rise from baseline to peak was calculated to determine the rate constant of calcium increase, and this was also not significantly different between WT, CD36−/−, and Fyn−/− MLMVEC (Fig. 2G).
Fig. 2.

Transient receptor potential-vanilloid 4 (TRPV4) calcium channel expression and function in WT, CD36−/−, and Fyn-deficient (Fyn−/−) MLMVEC. A: immunoblot images showing expression of TRPV4 in WT, CD36−/−, and Fyn−/− MLMVEC. B: bar graph showing mean ± SE fold change in TRPV4 expression in WT, CD36−/−, and Fyn−/− MLMVEC lysates. Representative traces show change in [Ca2+]i in WT (C), CD36−/− (D), and Fyn−/− (E) MLMVEC following exposure to the TRPV4 agonist GSK 1016790A (GSK; 1.5 µM). F: scatterplot and mean ± SE change in peak [Ca2+]i from in WT, CD36−/−, and Fyn−/− MLMVEC following exposure to GSK. *Significant difference from WT; n = 6–8 experiments/group.
We have previously shown that GSK-induced TRPV4 activation leads to loss of barrier function, as measured by decreased TER (36). To determine whether loss of CD36 or Fyn impaired barrier dysfunction mediated by TRPV4, TER was assessed in WT, CD36−/−, and Fyn−/− MLMVEC following exposure to GSK (Fig. 3). Baseline TER values were similar across all three groups (WT: 1,439 ± 103 Ω; CD36−/−: 1,167 ± 21 Ω; Fyn−/−: 1,267 ± 180 Ω). The maximal drop in TER (ΔTERmax) in response to GSK was also similar in WT, Fyn−/− and CD36−/− MLMVEC (Fig. 3B). However, the overall TER profile in CD36−/− and Fyn−/− MLMVEC appeared to reflect a more prolonged recovery than was observed in WT cells. To quantify potential changes in GSK-induced barrier dysfunction between WT, CD36−/−, and Fyn−/− MLMVEC, we assessed the AUC for individual ECIS traces; the AUC for CD36−/− and Fyn−/− MLMVEC exposed to GSK was significantly higher than WT controls (Fig. 3C), confirming an increase in total amount of barrier permeability due to slower recovery.
Fig. 3.
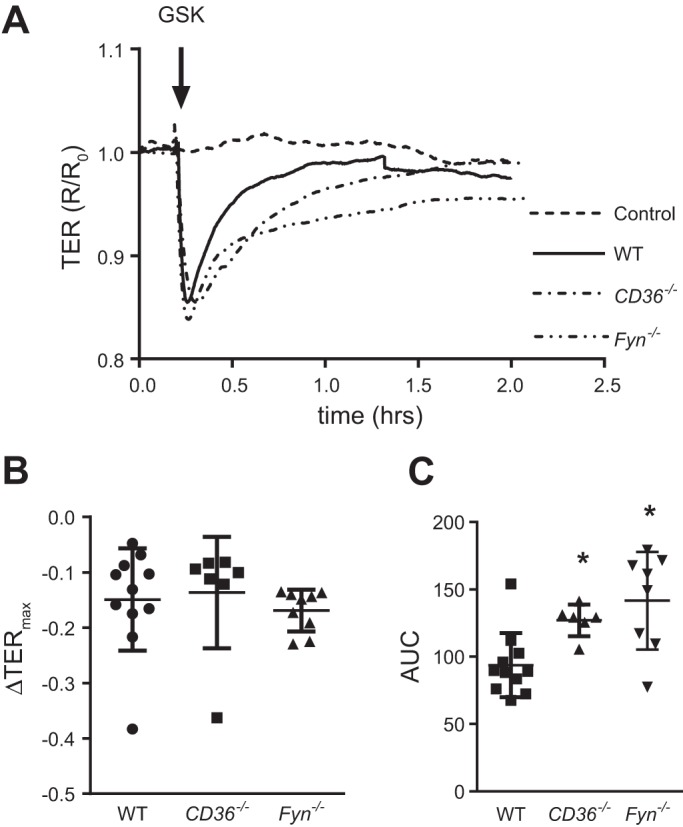
TRPV4 mediated barrier dysfunction in WT, CD36−/−, and Fyn−/− MLMVEC. A: representative traces of transmembrane electrical resistance (TER) measurements normalized to baseline in WT, CD36−/−, and Fyn−/− MLMVEC following exposure to GSK1016790 (GSK; 1.5 µM). Scatterplots showing maximum decrease in TER (B) and area under the curve (C) for WT, CD36−/−, and Fyn−/− MLMVEC. *Significant difference from WT; n = 7–12 experiments/group.
Subcellular localization of Fyn in CD36−/− MLMVEC.
We performed immunofluorescence studies to visualize Fyn cellular localization in WT and CD36−/− MLMVEC. Fyn localization in WT MLMVEC (Fig. 4A) appeared diffuse, whereas in CD36−/− MLMVEC Fyn expression was perinuclear (Fig. 4B). No significant difference in cell size was measured between WT and CD36−/− MLMVEC (WT: 178 ± 21.71 µm2; CD36−/−: 200 ± 24.13 µm2). To determine whether inhibition of CD36 altered Fyn localization, we also assessed Fyn localization in WT MLMVEC treated with SSO (100 µM; 1 h). Fyn localization appeared similar in WT MLMVEC with and without SSO treatment (Fig. 4C). To quantify changes in Fyn localization, we determined the distance between pixels with fluorescence values above a set threshold (i.e., positive fluorescence) and the cell center. As shown in Fig. 4, D–E, Fyn fluorescence in CD36−/− MLMVEC localized close to the nucleus while in WT and SSO-treated MLMVEC, the mean distance from the nucleus was greater, reflecting more Fyn present near the membrane. To validate our image analysis findings and to account for any bleed through from the nuclear channel (DAPI) and differences in cell size and shape, we quantified changes in Fyn localization by calculating a perinuclear fluorescence percentage. These measurements were made using a modified radial profile approach as described previously (22, 30). These measurements exclude any residual nuclear fluorescence and account for cell size/shape by measuring total cell fluorescence. As shown in Fig. 4F, loss of CD36 but not treatment with SSO increased perinuclear fluorescence percentage and the mean fluorescence in the radial area just outside the nucleus.
Fig. 4.

Subcellular localization of Fyn in CD36−/− MLMVEC. Representative immunofluorescence images of WT (A), CD36−/− (B), and SSO-treated WT (C) MLMVEC stained with anti-Fyn antibody (green) and nuclear counterstain (DAPI; blue). D: representative histograms showing distance of fluorescent pixels from the center of the cell nucleus in WT CD36−/− and SSO-treated MLMVEC. E: scatterplot showing mean ± SE distance from nucleus in WT, SSO-treated, and CD36−/− MLMVEC for the histogram shown in D. F: scatterplot showing mean ± SE perinuclear fraction in WT, SSO-treated, and CD36−/− MLMVEC. *Significant difference from WT; n = 3 independent experiments, consisting of 7–12 images for each condition.
Phosphorylation of TRPV4 in CD36−/− MLMVEC.
Given our hypothesis that CD36 serves as a scaffold for Fyn, and our prior work showing a critical role for Fyn-mediated TRPV4 phosphorylation in H2O2-induced Ca2+ influx, we reasoned that reduced H2O2-induced Ca2+ influx in CD36−/− MLMVEC could be due to movement of Fyn away from the cell membrane when CD36 was absent, leading to loss of TRPV4 phosphorylation/sensitivity to H2O2. To determine the role of CD36 in TRPV4 phosphorylation, we assessed the amount of TRPV4 bound to Fyn and amount of phosphorylated TRPV4 (p-TRPV4) in WT and CD36−/− MLMVEC. We first confirmed that our antibody reacted with the correct protein; the band of interest (∼75 kDa) was absent in TRPV4−/− MLMVEC (Fig. 5A). As shown in Fig. 5B, TRPV4 immunoprecipitated with Fyn in WT MLMVEC lysates. The amount of TRPV4 that immunoprecipitated with Fyn was significantly decreased in CD36−/− MLMVEC, suggesting that loss of CD36 decreased the interaction between Fyn and TRPV4. Immunoprecipitation with phosphotyrosine antibody revealed decreased TRPV4 in CD36−/− lysates, suggesting loss of CD36 reduced phosphorylation of TRPV4 (Fig. 5, C and D)
Fig. 5.

Interactions between CD36, Fyn, and TRPV4. A: representative immunoblot (IB) images showing TRPV4 and housekeeping control (GAPDH) expression in WT and TRPV4−/− MLMVEC lysates. B: immunoblot images showing TRPV4 and Fyn expression in WT and CD36−/− MLMVEC lysates before (input) and after immunoprecipitation with either anti-Fyn (IP: Fyn) or IgG control (IP: IgG) antibody. C: immunoblot images showing TRPV4 expression in WT and CD36−/− MLMVEC lysates before (input) and after immunoprecipitation with either anti-phosphotyrosine (IP: P-Tyr) or IgG control (IP: IgG) antibody. D: densitometry scatterplot showing fold change in TRPV4 in Fyn (IP: Fyn) and phosphotyrosine (IP: pTyr) immunoprecipitate lysates. *Significant difference from WT; n = 3 experiments/group.
Role of direct activation of CD36 by fatty acid binding.
CD36 is known to bind AcLDL and transport it across the cell membrane (43). To further delineate the mechanism by which CD36 attenuated H2O2-induced Ca2+ influx, we tested the hypothesis that direct binding of CD36 by lipid peroxide byproducts was not necessary for H2O2-induced Ca2+ influx using SSO, an antagonist that selectively prevents CD36 fatty acid transport (21). First, to confirm adequate inhibition of CD36, control and SSO (100 µM; 1 h)-treated MLMVEC were incubated with fluorescent AcLDL. As shown in Fig. 6A, there was robust uptake of AcLDL in untreated MLMVEC. In contrast, AcLDL uptake was nearly absent in SSO-treated MLMVEC (Fig. 6B). Similarly, very little uptake of AcLDL was seen in CD36−/− MLMVEC (Fig. 6C). However, as shown in Fig. 6C, AcLDL uptake was unchanged in Fyn−/− MLMVEC. Next, we measured the change in [Ca2+]i following challenge with H2O2 in untreated and SSO-treated MLMVEC. As shown in Fig. 7, no difference in H2O2-induced Ca2+ influx was seen in MLMVEC with and without SSO treatment.
Fig. 6.
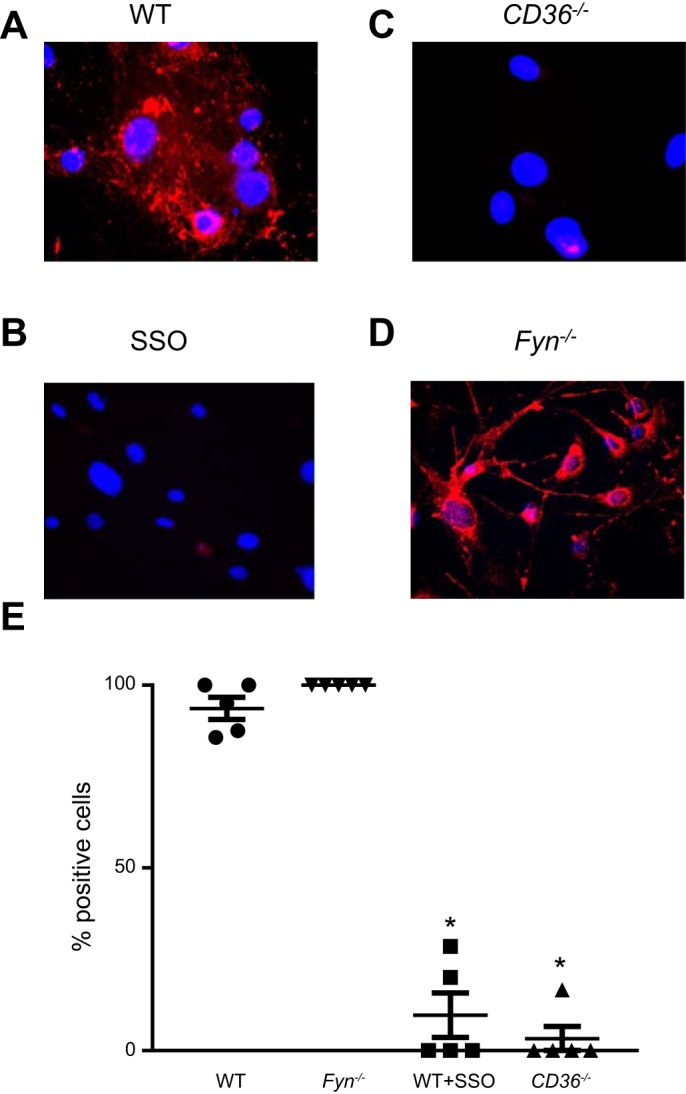
Effect of SSO on AcLDL uptake in MLMVEC. Representative immunofluorescence images of untreated (A) and SSO-treated (100 µM, 1 h; B) WT MLMVEC or CD36−/− MLMVEC. All cells were stained for AcLDL uptake (Alexa Fluor 598; red) and nuclear counterstain (DAPI; blue). D: scatterplot showing mean ± SE values for number of cells staining positive for AcLDL in WT, SSO-treated, and CD36−/− MLMVEC. *Significant difference from control; n = 5 images/group from at least 3 independent experiments.
Fig. 7.
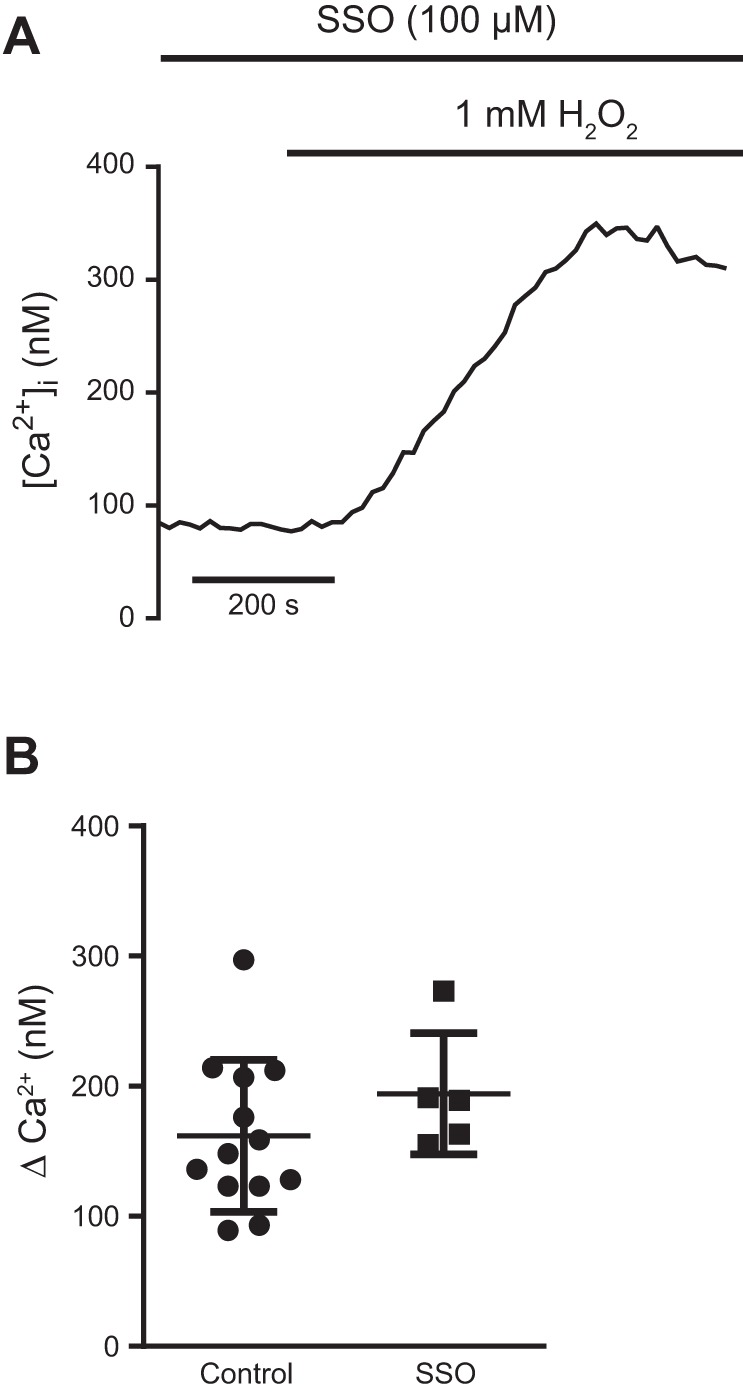
Effect of SSO on H2O2-induced Ca2+ influx in MLMVEC. A: representative trace showing change in peak [Ca2+]i from baseline following exposure to 1 mM H2O2 in MLMVEC treated with SSO. B: scatterplot and mean ± SE change in peak [Ca2+]i from baseline in control and SSO-treated MLMVEC; n = 5–13 experiments/group.
Effect of CD36 deletion on lung IR injury.
The results of the in vitro MLMVECs experiments predicted that CD36 deletion would result in protection from IR-induced lung injury in vivo. We employed a mouse model of IR lung injury with exclusion of EBD as a measure of change in barrier integrity. As shown in Fig. 8, performing sham surgery did not result in an influx of dye into the lung parenchyma, an indicator of vascular injury and permeability, in WT or CD36−/− mice. As expected, EBD ratio was increased in WT mice undergoing IR injury; however, EBD ratio was significantly attenuated in CD36−/− mice.
Fig. 8.
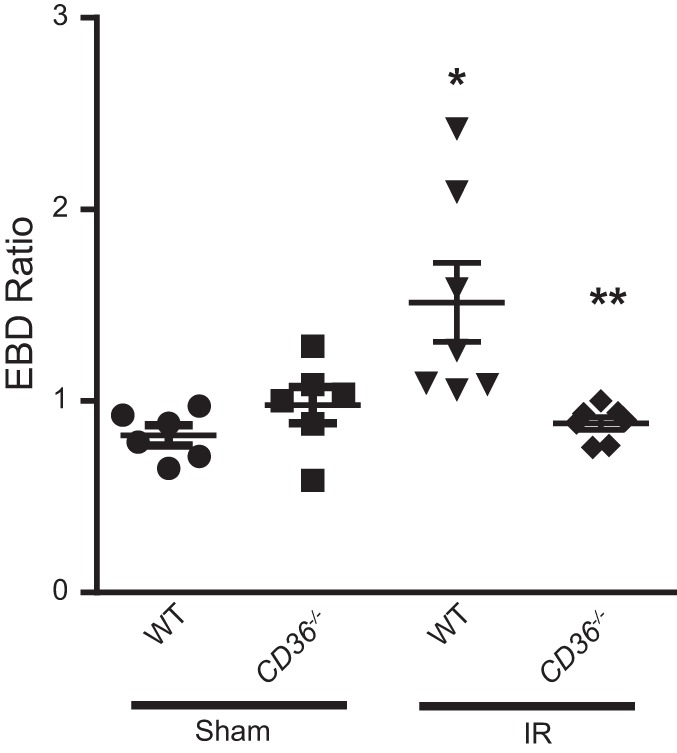
Lung ischemia-reperfusion (IR) injury is attenuated in CD36−/− mice. Left lung/right lung Evans blue dye (EBD) ratio in WT and CD36−/− mice following sham or IR surgery; n = 4–6 mice per group, *Significant difference from WT Sham; **Significant difference from WT IR.
DISCUSSION
In this study, we show that the fatty acid transporter CD36 plays an important role in ROS and Ca2+ signaling in the lung microvasculature. With the loss of CD36, there was a significant attenuation of H2O2-induced Ca2+ influx and endothelial permeability in vitro and protection from IR-induced lung injury in vivo. Our present findings also suggest a novel role for CD36 in modulating H2O2-induced endothelial dysfunction by acting as a membrane-associated docking molecule for Fyn, thus enabling efficient phosphorylation of TRPV4.
In MLMVEC from mice deficient for CD36, we observed significant attenuation of H2O2-induced Ca2+ influx, which we previously showed was mediated by TRPV4 (36). To determine the link between CD36 and TRPV4, we examined the effect of loss of CD36 on TRPV4 expression and function. Our data suggest that the attenuation of H2O2-induced Ca2+ influx in CD36−/− MLMVEC does not involve changes in overall TRPV4 expression, as total protein expression of TRPV4 was unaltered in CD36−/− MLMVEC. Moreover, no significant difference in peak change in [Ca2+]i was noted in CD36−/− MLMVEC when TRPV4 was activated by the chemical agonist GSK, indicating that attenuation of TRPV4-mediated H2O2-induced Ca2+ influx in CD36−/− was not due to TRPV4 inactivation and would seem to suggest that loss of CD36 does not prevent TRPV4 membrane localization; however, further experiments would be needed to verify this point. Though the representative trace showing GSK-induced Ca2+ influx in WT MLMVEC appears different in profile compared with CD36−/− and Fyn−/− MLMVEC, when the rate constant of the rise in [Ca2+]i was analyzed, there was no significant difference between WT, CD36−/−, and Fyn−/−. Interestingly, in WT MLMVEC, GSK-induced Ca2+ influx appears to be lower than H2O2-induced Ca2+ influx. One possibility for this difference is that H2O2-induced Ca2+ influx via TRPV4 may lead to sequential activation of other membrane and store-operated channels, contributing to elevations in [Ca2]i that are higher than with TRPV4 agonism alone. Lastly, although our data show that interruption of Fyn-mediated TRPV4 phosphorylation does not affect GSK-induced TRPV4 activation, TRPV4 can be activated by a myriad of other stimuli. The exact role of Fyn and CD36 in modulating TRPV4 responses to many of the other known stimuli (such as heat, tonicity, and stretch) still requires investigation.
Although CD36 is most widely known as a lipid scavenger, our data using SSO show that CD36 does not mediate H2O2-induced activation of TRPV4 via lipid peroxide binding of CD36. That we achieved effective inhibition of fatty acid uptake by CD36 in LMVECs following treatment with SSO is consistent with prior reports showing similar inhibition of CD36 by SSO in other cell types (21).
We have previously observed attenuation of H2O2-induced Ca2+ influx in 1) Fyn−/− MLMVEC; 2) HLMVEC treated the Src kinase inhibitor SU-6656; and 3) HLMVEC transfected with Fyn-targeted siRNA (36). Since the effect of loss of CD36 on H2O2-induced Ca2+ influx did not appear to involve changes in TRPV4 expression or CD36 lipid transport, we investigated the role of CD36 as a membrane-associated tether for Fyn. Under normal conditions, Fyn is widely distributed across cells, with primary localization at the cell membrane (2, 8, 27). This distribution was consistent with our observations, as was our observation of altered subcellular localization of Fyn in the absence of CD36 (2, 21). To quantitatively analyze changes in Fyn fluorescence between WT and CD36−/− MLMVEC, we analyzed the number of fluorescent pixels as a function of distance from the center of the nucleus and showed that Fyn staining in CD36−/− MLMVEC was largely concentrated in the perinuclear region. To ensure that our distance measurements were not affected by bleedthrough of nuclear dye or differences in cell size, we measured change in Fyn fluorescence localization using an alternate method of measuring perinuclear fluorescence, with similar results. It should be noted that the changes in Fyn fluorescence between WT and CD36−/− MLMVEC are unlikely to be due to decreased overall Fyn expression given that our laboratory previously demonstrated, using the same cells and antibodies as our present experiments, that total Fyn expression was unchanged between WT and CD36−/− MLMVEC (2). However, further experiments using confocal microscopy will be needed to determine whether any Fyn protein actually moves into the nucleus or if it remains solely perinuclear.
Loss of membrane tethering has been previously shown to affect Fyn kinase activity (2). It is likely that loss of CD36 not only changes Fyn localization, but also phosphorylation at the Y419 residue, which regulates the Fyn activity. We previously showed that phosphorylation at the Y419 residue was significantly decreased in CD36−/− mice compared with WT mice (2). Another feature specific to Fyn is colocalization with focal adhesion kinase under stimulated conditions (19), which has also been shown to be abnormal in CD36−/− MLMVEC (2). Upon stimulation with H2O2, Fyn remained perinuclear and failed to localize to actin stress fibers, focal adhesions, or other cell-cell interfaces (2). These data, together with the findings presented here, suggest a critical role for tethering of Fyn to the cell membrane in regulating its participation in cell signaling events at baseline and under stimulated conditions. Together, the restricted distribution of Fyn to the perinuclear region and attenuation of H2O2-induced Ca2+ influx in CD36−/− MLMVEC, but not by inhibition of CD36 fatty acid transport, suggest that physical association of Fyn with surface-associated CD36 may be necessary for H2O2-induced Ca2+ influx.
The mechanism by which CD36 and Fyn alter TRPV4 responsiveness to ROS appears to involve changes in TRPV4 phosphorylation. Our immunoprecipitation data suggest that Fyn associates with TRPV4 under normal conditions, but with loss of CD36 and redistribution of Fyn, both TRPV4 phosphorylation and the amount of TRPV4 bound to Fyn decrease. In lysates where Fyn was immunoprecipitated, the total amount of Fyn was similar in WT and CD36−/− lysates, suggesting that the difference in TRPV4 phosphorylation is not due simply to decreased expression of Fyn in CD36−/− MLMVEC. We previously showed that inhibition of Fyn was associated with decreased TRPV4 phosphorylation and attenuated H2O2-induced Ca2+ influx in LMVEC. Taken as a whole, our data suggest that decreased TRPV4 phosphorylation reduced sensitivity of TRPV4 to activation by ROS, but not chemical agonists. This possibility is supported by prior work showing that mutation of a SFK tyrosine residue in TRPV4 leads to decreased channel responsiveness to certain stimuli, such as heat, but the ability of the external chemical agonist 4α-PDD to activate the channel remained intact (40). Similarly, we recently showed that inhibition of Fyn prevented H2O2-induced Ca2+ influx via TRPV4, but not TRPV4 responsiveness to GSK (36). Although our data suggests a link between CD36, Fyn, and phosphorylated TRPV4, further studies including proteomic approaches to more definitely show changes in specific tyrosine residues and site-specific mutagenesis of these residues on TRPV4 will be required to more definitely establish the site of phosphorylation.
It should be noted that the band observed for TRPV4 in our immunoprecipitation experiments (~75 kDa) was at a lower molecular weight than observed in human LMVEC (100 kDa) (36). This might be explained by lower molecular weight splice variants of TRPV4 that have been reported in mice and rats (24, 44), including a splice variant at ~75 kDa (24). Since no additional bands at 100 kDa were observed in our immunoprecipitate or input lysates, and we confirmed that our band of interest was absent in TRPV4−/− lysates, we conclude that the band observed in MLMVECs is in fact TRPV4. We probed immunoprecipitate lysates with a different TRPV4 antibody and did not observe additional bands at 100 kDa. Since multiple splice variants of this protein have been reported, it is possible that a larger molecular variant not recognized by our antibodies during our IP experiments may be phosphorylated differently. As mentioned above, additional studies using mass spectroscopy will be needed to definitively assess TRPV4 phosphorylation in the absence of CD36 and Fyn.
Although our present data suggest that activation of CD36 is not necessary for H2O2-induced Ca2+ influx in the lung, binding of fatty acids to CD36 has been shown to increase [Ca2+]i in other cell types by activating the release of endoplasmic reticulum stores of calcium (20). However, it is likely that changes in [Ca2+]i due to CD36 fatty acid binding are distinct from H2O2-induced Ca2+ influx since binding of CD36 by fatty acids in taste bud cells has been shown to increase [Ca2+]i by activating release of internal Ca2+ stores, which we have shown was not required for initiation of H2O2-induced Ca2+ influx in LMVEC (15, 36). Thus the mechanism by which CD36 mediates H2O2-induced Ca2+ influx appears distinct from other mechanisms of CD36-mediated Ca2+ influx that have been described.
ECIS experiments revealed that, although agonism of TRPV4 with GSK induced similar peak barrier dysfunction in WT, CD36−/−, and Fyn−/− MLMVEC, recovery to baseline in CD36−/− and Fyn−/− MLMVEC was slowed. We speculate that this difference in barrier recovery observed in both CD36−/− and Fyn−/− MLMVEC may be related to the role of Fyn in promoting barrier recovery by phosphorylating focal adhesion kinase (19), with loss of membrane-bound Fyn, due to either loss of CD36−/− or deletion of Fyn, leading to loss of the barrier-protective effects of Fyn that mediate barrier recovery. Another possibility is that loss of Fyn alters TRPV4 responsiveness to GSK, although, based on our Ca2+ influx data, we suspect that this possibility is less likely.
The finding that CD36 plays a protective role in IR lung injury in vivo is consistent with prior findings noting a protective role for CD36 in IR models in other vascular beds (32, 34). The model of IR used in this study has several advantages. First, since our model involved selective occlusion and subsequent release of the left pulmonary artery, the right lungs of individual mice served as internal controls for measurement of barrier dysfunction. That EBD ratio did not increase in our sham control groups confirmed that the surgical procedure alone did not cause significant trauma to the pulmonary circulation. Also, we have previously shown that this model of IR injury reliably produced lung injury and that measurement of lung edema using EBD ratio mirrored changes in lung weight and changes in lung Kf (14). This model additionally offers the advantage of allowing reperfusion with blood rather than bloodless perfusate typically used in isolated perfused lung models of IR. One limitation of our in vivo data is that our mice were global CD36−/− knockouts; thus a contribution to IR injury protection due to absence of CD36 in inflammatory cells cannot be entirely ruled out, although given the short time points used in assessment of lung injury (3 h after reperfusion), this seems less likely. However, future experiments with additional measures of lung edema (e.g., Kf, wet/dry weights, BAL fluid analysis) are still needed to solidify the barrier-protective role of CD36 in IR and other forms of lung injury.
While our in vitro data show that fatty acid transport by CD36 was not required for H2O-induced Ca2+ influx, another caveat of this model is that it is possible that the observed effects of CD36 on IR lung injury are due to Ca2+-independent effects. We believe this possibility is unlikely, however, given that 1) our prior in vitro data suggesting inhibition of Ca2+ influx are insufficient to attenuate H2O2-induced barrier dysfunction and 2) others have shown inhibition of Ca2+ channels is sufficient to attenuate IR injury in vivo (41). Nonetheless, future in vivo IR experiments in mice treated with SSO or transgenic mice with a mutant CD36 construct that lacks fatty acid transport functionality will be needed to determine whether fatty acid transport by CD36 may be important for non-Ca2+-signaling events related to IR lung injury.
Based on earlier data implicating TRPV4 as the primary initiator of H2O2-induced Ca2+ influx, the experiments in this study focused only on this channel. However, other studies have reported a critical role for other TRP channels, including TRPC6, in lung IR injury (41). Our experiments to date have only examined the initial Ca2+ influx that occurs following introduction of exogenous ROS, and it is likely that there is some degree of spatial and temporal heterogeneity regarding the activation of channels involved in LMVEC Ca2+ flux during IR injury. Thus it is likely that while TRPV4 activation may serve as an initial source of Ca2+, activation of other calcium channels likely occurs with ongoing injury. Our present data raise the interesting possibility that CD36, acting as a scaffold for regulatory kinases, may modulate endothelial Ca2+ channel activity. Further studies examining the exact role of CD36 in mediating Src kinase localization to the cell membrane and the effect of disrupting this mechanism on LMVEC TRP channel function will be instructive in this regard.
In conclusion, our data suggest that loss of CD36 protects against lung IR injury, perhaps due to CD36-mediated changes in H2O2-induced Ca2+ influx. Our data further indicate that the mechanism by which CD36 participates in this process may be by acting as a membrane scaffold for Fyn, allowing for interactions between Fyn and TRPV4 that are required for activation of TRPV4 in response to oxidant stress. Further studies are needed to more fully delineate the role of TRPV4 phosphorylation in lung injury and to determine whether removal of CD36 from the cell membrane could be achieved through pharmacological means.
GRANTS
Support for this study was provided by the National Heart, Lung and Blood Institute Grants F32-HL124930 (K. Suresh), R01HL-73859 (L. A. Shimoda), R25-HL084762 (L. A. Shimoda), and T32HL-007534.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
K.S., L.S., J.R., C.U., J.Z., O.R., S.M., and J.M.D.-o. performed experiments; K.S., J.R., and J.M.D.-o. analyzed data; K.S., J.M.D.-o., and D.B.P. interpreted results of experiments; K.S. prepared figures; K.S. drafted manuscript; A.L.S., D.B.P., and L.A.S. edited and revised manuscript; A.L.S., D.B.P., and L.A.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Michael Caterina for technical expertise regarding TRPV4 and providing TRPV4−/− mice.
REFERENCES
- 1.Aleshin A, Finn RS. SRC: a century of science brought to the clinic. Neoplasia 12: 599–607, 2010. doi: 10.1593/neo.10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anidi IU, Servinsky LE, Rentsendorj O, Stephens RS, Scott AL, Pearse DB. CD36 and Fyn kinase mediate malaria-induced lung endothelial barrier dysfunction in mice infected with Plasmodium berghei. PLoS One 8: e71010, 2013. doi: 10.1371/journal.pone.0071010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balakrishna S, Song W, Achanta S, Doran SF, Liu B, Kaelberer MM, Yu Z, Sui A, Cheung M, Leishman E, Eidam HS, Ye G, Willette RN, Thorneloe KS, Bradshaw HB, Matalon S, Jordt SE. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 307: L158–L172, 2014. doi: 10.1152/ajplung..2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bocharov AV, Wu T, Baranova IN, Birukova AA, Sviridov D, Vishnyakova TG, Remaley AT, Eggerman TL, Patterson AP, Birukov KG. Synthetic amphipathic helical peptides targeting cd36 attenuate lipopolysaccharide-induced inflammation and acute lung injury. J Immunol 197: 611–619, 2016. doi: 10.4049/jimmunol.1401028. [DOI] [PubMed] [Google Scholar]
- 5.Bogeski I, Kappl R, Kummerow C, Gulaboski R, Hoth M, Niemeyer BA. Redox regulation of calcium ion channels: chemical and physiological aspects. Cell Calcium 50: 407–423, 2011. doi: 10.1016/j.ceca.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 6.Bubolz AH, Mendoza SA, Zheng X, Zinkevich NS, Li R, Gutterman DD, Zhang DX. Activation of endothelial TRPV4 channels mediates flow-induced dilation in human coronary arterioles: role of Ca2+ entry and mitochondrial ROS signaling. Am J Physiol Heart Circ Physiol 302: H634–H642, 2012. doi: 10.1152/ajpheart.00717.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chabot F, Mitchell JA, Gutteridge JM, Evans TW. Reactive oxygen species in acute lung injury. Eur Respir J 11: 745–757, 1998. [PubMed] [Google Scholar]
- 8.Chan B, Lanyi A, Song HK, Griesbach J, Simarro-Grande M, Poy F, Howie D, Sumegi J, Terhorst C, Eck MJ. SAP couples Fyn to SLAM immune receptors. Nat Cell Biol 5: 155–160, 2003. doi: 10.1038/ncb920. [DOI] [PubMed] [Google Scholar]
- 9.Cho S, Park EM, Febbraio M, Anrather J, Park L, Racchumi G, Silverstein RL, Iadecola C. The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J Neurosci 25: 2504–2512, 2005. doi: 10.1523/JNEUROSCI.0035-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu LY, Ramakrishnan DP, Silverstein RL. Thrombospondin-1 modulates VEGF signaling via CD36 by recruiting SHP-1 to VEGFR2 complex in microvascular endothelial cells. Blood 122: 1822–1832, 2013. doi: 10.1182/blood-2013-01-482315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cioffi DL, Lowe K, Alvarez DF, Barry C, Stevens T. TRPing on the lung endothelium: calcium channels that regulate barrier function. Antioxid Redox Signal 11: 765–776, 2009. doi: 10.1089/ars.2008.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cioffi DL, Stevens T. Regulation of endothelial cell barrier function by store-operated calcium entry. Microcirculation 13: 709-723, 2006. doi: 10.1080/10739680600930354. [DOI] [PubMed] [Google Scholar]
- 13.Dodd-o JM, Hristopoulos ML, Faraday N, Pearse DB. Effect of ischemia and reperfusion without airway occlusion on vascular barrier function in the in vivo mouse lung. J Appl Physiol 95: 1971-1978, 2003. doi: 10.1152/japplphysiol.00456.2003. [DOI] [PubMed] [Google Scholar]
- 14.Dodd-o JM, Hristopoulos ML, Kibler K, Gutkowska J, Mukaddam-Daher S, Gonzalez A, Welsh-Servinsky LE, Pearse DB. The role of natriuretic peptide receptor-A signaling in unilateral lung ischemia-reperfusion injury in the intact mouse. Am J Physiol Lung Cell Mol Physiol 294: L714–L723, 2008. doi: 10.1152/ajplung.00185.2007. [DOI] [PubMed] [Google Scholar]
- 15.El-Yassimi A, Hichami A, Besnard P, Khan NA. Linoleic acid induces calcium signaling, Src kinase phosphorylation, and neurotransmitter release in mouse CD36-positive gustatory cells. J Biol Chem 283: 12949–12959, 2008. doi: 10.1074/jbc.M707478200. [DOI] [PubMed] [Google Scholar]
- 16.Gandhirajan RK, Meng S, Chandramoorthy HC, Mallilankaraman K, Mancarella S, Gao H, Razmpour R, Yang XF, Houser SR, Chen J, Koch WJ, Wang H, Soboloff J, Gill DL, Madesh M. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J Clin Invest 123: 887–902, 2013. doi: 10.1172/JCI65647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamanaka K, Jian MY, Weber DS, Alvarez DF, Townsley MI, Al-Mehdi AB, King JA, Liedtke W, Parker JC. TRPV4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. Am J Physiol Lung Cell Mol Physiol 293: L923–L932, 2007. doi: 10.1152/ajplung.00221.2007. [DOI] [PubMed] [Google Scholar]
- 18.Huang MM, Bolen JB, Barnwell JW, Shattil SJ, Brugge JS. Membrane glycoprotein IV (CD36) is physically associated with the Fyn, Lyn, and Yes protein-tyrosine kinases in human platelets. Proc Natl Acad Sci USA 88: 7844–7848, 1991. doi: 10.1073/pnas.88.17.7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knezevic N, Tauseef M, Thennes T, Mehta D. The G protein betagamma subunit mediates reannealing of adherens junctions to reverse endothelial permeability increase by thrombin. J Exp Med 206: 2761–2777, 2009. doi: 10.1084/jem.20090652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuda O, Jenkins CM, Skinner JR, Moon SH, Su X, Gross RW, Abumrad NA. CD36 protein is involved in store-operated calcium flux, phospholipase A2 activation, and production of prostaglandin E2. J Biol Chem 286: 17785–17795, 2011. doi: 10.1074/jbc.M111.232975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuda O, Pietka TA, Demianova Z, Kudova E, Cvacka J, Kopecky J, Abumrad NA. Sulfo-N-succinimidyl oleate (SSO) inhibits fatty acid uptake and signaling for intracellular calcium via binding CD36 lysine 164: SSO also inhibits oxidized low density lipoprotein uptake by macrophages. J Biol Chem 288: 15547–15555, 2013. doi: 10.1074/jbc.M113.473298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li K, Yang L, Zhang C, Niu Y, Li W, Liu JJ. HPS6 interacts with dynactin p150Glued to mediate retrograde trafficking and maturation of lysosomes. J Cell Sci 127: 4574–4588, 2014. doi: 10.1242/jcs.141978. [DOI] [PubMed] [Google Scholar]
- 23.Li W, Febbraio M, Reddy SP, Yu DY, Yamamoto M, Silverstein RL. CD36 participates in a signaling pathway that regulates ROS formation in murine VSMCs. J Clin Invest 120: 3996–4006, 2010. doi: 10.1172/JCI42823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4-/- mice. Proc Natl Acad Sci USA 100: 13698–13703, 2003. doi: 10.1073/pnas.1735416100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martínez J, Moreno JJ. Role of Ca2+-independent phospholipase A2 on arachidonic acid release induced by reactive oxygen species. Arch Biochem Biophys 392: 257–262, 2001. doi: 10.1006/abbi.2001.2439. [DOI] [PubMed] [Google Scholar]
- 26.Nagendran J, Pulinilkunnil T, Kienesberger PC, Sung MM, Fung D, Febbraio M, Dyck JR. Cardiomyocyte-specific ablation of CD36 improves post-ischemic functional recovery. J Mol Cell Cardiol 63: 180–188, 2013. doi: 10.1016/j.yjmcc.2013.07.020. [DOI] [PubMed] [Google Scholar]
- 27.Resh MD. Fyn, a Src family tyrosine kinase. Int J Biochem Cell Biol 30: 1159–1162, 1998. doi: 10.1016/S1357-2725(98)00089-2. [DOI] [PubMed] [Google Scholar]
- 28.Sarmiento D, Montorfano I, Cerda O, Cáceres M, Becerra A, Cabello-Verrugio C, Elorza AA, Riedel C, Tapia P, Velásquez LA, Varela D, Simon F. Increases in reactive oxygen species enhance vascular endothelial cell migration through a mechanism dependent on the transient receptor potential melastatin 4 ion channel. Microvasc Res 98: 187–196, 2014. doi: 10.1016/j.mvr.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Seeley EJ, Rosenberg P, Matthay MA. Calcium flux and endothelial dysfunction during acute lung injury: a STIMulating target for therapy. J Clin Invest 123: 1015–1018, 2013. doi: 10.1172/JCI68093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sengupta D, Truschel S, Bachert C, Linstedt AD. Organelle tethering by a homotypic PDZ interaction underlies formation of the Golgi membrane network. J Cell Biol 186: 41–55, 2009. doi: 10.1083/jcb.200902110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shenoy-Scaria AM, Dietzen DJ, Kwong J, Link DC, Lublin DM. Cysteine3 of Src family protein tyrosine kinase determines palmitoylation and localization in caveolae. J Cell Biol 126: 353–363, 1994. doi: 10.1083/jcb.126.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silverstein RL. Inflammation, atherosclerosis, and arterial thrombosis: role of the scavenger receptor CD36. Cleve Clin J Med 76, Suppl 2: S27–S30, 2009. doi: 10.3949/ccjm.76.s2.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal 2: re3, 2009. doi: 10.1126/scisignal.272re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steinbusch LK, Luiken JJ, Vlasblom R, Chabowski A, Hoebers NT, Coumans WA, Vroegrijk IO, Voshol PJ, Ouwens DM, Glatz JF, Diamant M. Absence of fatty acid transporter CD36 protects against Western-type diet-related cardiac dysfunction following pressure overload in mice. Am J Physiol Endocrinol Metab 301: E618–E627, 2011. doi: 10.1152/ajpendo.00106.2011. [DOI] [PubMed] [Google Scholar]
- 35.Stuart LM, Bell SA, Stewart CR, Silver JM, Richard J, Goss JL, Tseng AA, Zhang A, Khoury JBE, Moore KJ. CD36 signals to the actin cytoskeleton and regulates microglial migration via a p130Cas complex. J Biol Chem 282: 27392–27401, 2007. doi: 10.1074/jbc.M702887200. [DOI] [PubMed] [Google Scholar]
- 36.Suresh K, Servinsky L, Reyes J, Baksh S, Undem C, Caterina M, Pearse DB, Shimoda LA. Hydrogen peroxide-induced calcium influx in lung microvascular endothelial cells involves TRPV4. Am J Physiol Lung Cell Mol Physiol 309: L1467–L1477, 2015. doi: 10.1152/ajplung.00275.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tauseef M, Knezevic N, Chava KR, Smith M, Sukriti S, Gianaris N, Obukhov AG, Vogel SM, Schraufnagel DE, Dietrich A, Birnbaumer L, Malik AB, Mehta D. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J Exp Med 209: 1953–1968, 2012. doi: 10.1084/jem.20111355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian MY, Costell M, Maniscalco-Hauk K, Krawiec JA, Olzinski A, Gordon E, Lozinskaya I, Elefante L, Qin P, Matasic DS, James C, Tunstead J, Donovan B, Kallal L, Waszkiewicz A, Vaidya K, Davenport EA, Larkin J, Burgert M, Casillas LN, Marquis RW, Ye G, Eidam HS, Goodman KB, Toomey JR, Roethke TJ, Jucker BM, Schnackenberg CG, Townsley MI, Lepore JJ, Willette RN. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci Transl Med 4: 159ra148, 2012. doi: 10.1126/scitranslmed.3004276. [DOI] [PubMed] [Google Scholar]
- 39.Villalta PC, Townsley MI. Transient receptor potential channels and regulation of lung endothelial permeability. Pulm Circ 3: 802–815, 2013. doi: 10.1086/674765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wegierski T, Lewandrowski U, Müller B, Sickmann A, Walz G. Tyrosine phosphorylation modulates the activity of TRPV4 in response to defined stimuli. J Biol Chem 284: 2923–2933, 2009. doi: 10.1074/jbc.M805357200. [DOI] [PubMed] [Google Scholar]
- 41.Weissmann N, Sydykov A, Kalwa H, Storch U, Fuchs B, Mederos y Schnitzler M, Brandes RP, Grimminger F, Meissner M, Freichel M, Offermanns S, Veit F, Pak O, Krause KH, Schermuly RT, Brewer AC, Schmidt HH, Seeger W, Shah AM, Gudermann T, Ghofrani HA, Dietrich A. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat Commun 3: 649, 2012. doi: 10.1038/ncomms1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolfram Kuhlmann CR, Wiebke Lüdders D, Schaefer CA, Kerstin Most A, Backenköhler U, Neumann T, Tillmanns H, Erdogan A. Lysophosphatidylcholine-induced modulation of Ca2+-activated K+ channels contributes to ROS-dependent proliferation of cultured human endothelial cells. J Mol Cell Cardiol 36: 675–682, 2004. doi: 10.1016/j.yjmcc.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 43.Xu Y, Wang J, Bao Y, Jiang W, Zuo L, Song D, Hong B, Si S. Identification of two antagonists of the scavenger receptor CD36 using a high-throughput screening model. Anal Biochem 400: 207–212, 2010. doi: 10.1016/j.ab.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 44.Zhao Y, Huang H, Jiang Y, Wei H, Liu P, Wang W, Niu W. Unusual localization and translocation of TRPV4 protein in cultured ventricular myocytes of the neonatal rat. Eur J Histochem 56: e32, 2012. doi: 10.4081/ejh.2012.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]