

Abstract
Preeclampsia (PE), a hypertensive disorder of pregnancy, is increasing as a major contributor to perinatal and long-term morbidity of mother and offspring. PE is thought to originate from ischemic insults in the placenta driving the release of prohypertensive anti-angiogenic [soluble fms-like tyrosine kinase-1 (sFlt-1)] and proinflammatory [tumor necrosis factor-α (TNF-α)] factors into the maternal circulation. Whereas the increased incidence of PE is hypothesized to be largely due to the obesity pandemic, the mechanisms whereby obesity increases this risk are unknown. The maternal endothelium is targeted by placental and adipose tissue-derived factors like sFlt-1 and TNF-α that promote hypertension during pregnancy, resulting in vascular dysfunction and hypertension. Interestingly, not all obese pregnant women develop PE. Data suggest that obese pregnant women with the greatest metabolic abnormalities have the highest incidence of PE. Identifying obesity-related mechanisms driving hypertension in some obese pregnant women and pathways that protect normotensive obese pregnant women, may uncover novel protocols to treat PE. Metabolic abnormalities, such as increased circulating leptin, glucose, insulin, and lipids, are likely to increase the risk for PE in obese women. It is not only important to understand whether each of these metabolic factors contribute to the increased risk for PE in obesity, but also their cumulative effects. This is particularly relevant to obese pregnant women with gestational diabetes mellitus (GDM) where all of these factors are increased and the risk for PE is highest. It is speculated that these factors potentiate the anti-angiogenic and proinflammatory mechanisms of placental ischemia-induced vascular dysfunction thereby contributing to the increasing incidence of PE.
Keywords: adipose tissue, endothelium, gestational diabetes mellitus, hypertension, women’s health
proper placental and maternal vascular adaptations are important for healthy blood pressure regulation during pregnancy. Hypertensive disorders of pregnancy contribute to a large number of maternal and fetal deaths, not only in developing countries, but also nations like the United States (16). Preeclampsia (PE) is particularly dangerous, as it presents with new-onset hypertension ≥20 wk of gestation along with a number of systemic cardiovascular comorbidities, including endothelial and vascular dysfunction in the kidneys and the brain (3, 54). Human and experimental animal studies support that placental ischemia and hypoxia drive the release of antiangiogenic and proinflammatory factors from the placenta into the maternal circulation (93). There they promote endothelial and vascular dysfunction by reducing the bioavailability of the vasodilator nitric oxide (NO) and resulting in maternal hypertension.
The incidence of PE is on the rise and is thought to be a result of the obesity pandemic (71). A complete understanding of the impact of obesity and its associated metabolic abnormalities on the cascade of events leading from placental ischemia to maternal hypertension is not available. Reciprocally, not all obese pregnant women develop PE. This suggests that: 1) some obese pregnant women are protected against, whereas others are already predisposed to, vascular dysfunction and hypertension and that 2) obesity-related metabolic abnormalities and placental ischemia may be worse in obese preeclamptic patients. Understanding the differences between these two groups is likely to assist in our understanding of obesity’s impact on increasing the risk for developing preeclampsia.
Obesity is a Major Risk Factor for PE
Data overwhelmingly support that obesity is a major risk factor for hypertension during pregnancy (85). In a cohort of primiparous women from Pittsburgh, Pennsylvania having prepregnancy obesity, defined as a body mass index (BMI in kg/m2) >30, the incidence of PE was 14.5% and that of chronic hypertension with superimposed PE was 2.6% (96). These values were both higher than women with a prepregnancy BMI <30 (7.2% and 0.38%, respectively). Furthermore, the greater the degree of obesity, the greater was the incidence and severity for this maternal disorder.
Overweight, obese, and morbidly obese women are at respectively increased risk for developing PE with severe features at ≥34 wk of gestation (21). PE diagnosed at ≥34 wk of gestation is defined as late-onset PE. The American College of Obstetrics and Gynecology defines PE with severe features as new-onset severe hypertension with a systolic blood pressure of >160 mmHg or diastolic blood pressure >110 mmHg, or hypertension requiring antihypertensive therapy exclusive of chronic hypertension, which is concurrent with thrombocytopenia, impaired liver function, persistent right upper quadrant pain or epigastric pain, renal insufficiency, pulmonary edema, or central nervous system disturbances (3). Women with severe PE are more likely to be obese and have small for gestational-age babies (58), and obesity increases the need for inducing preterm birth (66). It may be that placental, adipose tissue, and underlying endothelial dysfunction mediate the impact of obesity on increasing the risk for PE.
Does Obesity Promote Morphological Abnormalities in Placental Vasculature?
Obesity and late-onset PE are associated with normal or larger fetal growth and birth weight with a placenta that is normal weight or even heavier (25, 49, 60). Such placental overgrowth in obesity is associated with fetal hypoxia (49). This increased placental weight may be related to “microvillous crowding” within a limited placental compartment (78). Therefore, the uterine capacity for villous expansion may become constrained at the end of pregnancy, which may be more pronounced in obese gravidas. It is known that the number of capillaries per villous tissue is increased in obesity (55). Future research should focus on the biophysics of uteroplacental constraint, its effects on the placental vascular tree, and the impact of obesity on this. Late PE, especially in obesity, then, may be a manifestation of “villous toxicity,” which is interesting because PE was once called toxemia of pregnancy.
Villous toxicity resulting from crowding and congestion likely cause cell death and hypoxia (78). Placental ischemia and hypoxia induces the release of prohypertensive factors including the proinflammatory cytokine tumor necrosis factor-α (TNF-α) and the anti-angiogenic factor soluble fms-like tyrosine kinase-1 (sFlt-1) from extravillous trophoblast cells. The latter binds and quenches bioavailable angiogenic and vasoprotective factors including vascular endothelial growth factor (VEGF) and placenta growth factor (PlGF), reducing activation of NO synthase (NOS). As the steepest fall in total peripheral vascular resistance and increase in plasma volume occurs late in healthy pregnancies, at ~8 mo of gestation (13, 40), late-onset PE may result from an underlying inability of the maternal cardiovascular system to properly vasodilate in response to placental-derived factors, like PlGF (1, 17, 57, 63, 75, 79), possibly mediated by synergistic actions of obesity-related metabolic factors and placental vascular dysfunction.
Do Maternal Obesity and Related Metabolic Factors Adversely Affect Uteroplacental-Fetal Vascular Function?
Obesity or high-fat diet adversely affect uteroplacental-fetal vascular remodeling and blood flow, which could lead to placental ischemia and hypoxia. In baboons segregated into obese and lean groups while on standard chow diet, the former had significantly greater perirenal fat and fasting venous circulating leptin levels; a slight increase in glucose; and no difference in insulin (24). In the placenta, these obese monkeys had reduced villous surface area but greater villous diameter. It is unclear if the reduced villous surface area was due to the metabolic abnormalities and/or greater numbers of macrophages found in the placental villous tissue and increased CD14 expression [a coreceptor for toll-like receptor (TLR)4 on macrophages] in maternal peripheral blood mononuclear cells and visceral adipose tissue. Blood pressure was not examined. Interestingly, greater numbers of proinflammatory macrophages are also found in placentas from obese pregnant women presenting with insulin resistance and increased leptin, IL-6, and C-reactive peptide along with greater CD68 and CD14 staining in placenta and TNF-α levels in placental macrophages (10).
In high-fat diet-induced obesity in pregnant mice, macrophage activation, but not total cell counts, and their cytokine levels were greater by the end of pregnancy. This was only observed in placentas from male fetuses, which fared worse (44, 67). This is interesting because higher levels of androgens promote insulin resistance in pregnant mice on high-fat diet (61), and inhibition of androgen signaling reduced systolic blood pressure, body fat, triglycerides, leptin, and increased adiponectin and improved glucose tolerance in this high-fat diet pregnant model (62). Placental and maternal vascular function was not examined. As a whole, these data support the need for studies designed to evaluate the combined importance of placental ischemia, sex hormones, obesity-related metabolic factors, and immune cells on blood pressure regulation in obese pregnancies.
Macrophages and T lymphocytes are activated in preeclamptic women (18, 23) and are more prevalent in obese placentas (49). Little is known regarding the role for macrophages in promoting hypertension during pregnancy. It has been shown that adoptive transfer of CD4+ T cells from lean rats with placental ischemia-induced hypertension (reduced uterine perfusion pressure, RUPP, model) elicited hypertension in once normotensive pregnant rats (92). It is unknown if obesity or obesity-related metabolic factors exaggerate the activation and prohypertensive actions of inflammatory cells in response to placental vascular dysfunction and ischemia. Indirect evidence suggests that the TLR4 receptor is a likely pathway whereby obesity could exaggerate these inflammatory mechanisms to increase risk for PE (81, 99). TLR4 is a novel receptor for high mobility group box 1 (HMGB1) (52). Hypoxic trophoblast cells release HMGB1 (43), and this factor is increased in placentas from preeclamptic pregnancies (11). Adipose tissue is also a source of circulating HMGB1, and it is suggested that its release is increased in obesity and insulin resistance, although this has only been studied in men (31). One study did find that circulating HMGB1 levels were greater in overweight pregnant women with gestational diabetes mellitus (GDM) (28). A clear role for this proinflammatory factor in mediating hypertension is not clear, as this cohort of women were only slightly overweight before pregnancy (BMI = 26.9 ± 2.0) vs. controls (BMI = 23.6 ± 2.37), and hypertensive patients were excluded. Placental blood flow was not examined.
Abnormal remodeling and widening of maternal spiral arteries during pregnancy is thought to be a major contributor to placental ischemia/hypoxia and the clinical manifestations of PE, at least the early-onset form. As a result, blood is forced through smaller distal spiral arteries, which would retain their muscular wall, resulting in increased flow turbulence, shear stress, and infarction in the intervillous space (45). Interestingly, endometrial and spiral artery blood flow is reduced in obese women before pregnancy (97). Furthermore, placentas from stillbirths of ≥20 wk of gestation having abnormal spiral artery modifications were associated with a greater incidence of maternal prepregnancy obesity and PE (4). These placentas also had acute atherosis, which is an atherosclerosis-like lesion typically found in the spiral arteries of women with PE (86). Shear stress may be a causative factor in the pathogenesis of such lesions, which could ultimately lead to thrombosis and placental infarcts. It is of utmost importance to understand the role of this in the pathogenesis of PE. This has begun to be examined utilizing atherosclerosis-prone ApoE−/− mice, which have hypertension and proteinuria during pregnancy compared with wild-type controls with greater edema and necrosis of the villous stroma, placental fat deposition and serum sFlt-1 levels (87). The contribution and timing of obesity and acute atherosis in altering placental vascular remodeling, and function and its role in inducing PE, should be determined.
Uteroplacental blood flow has been examined in nonhuman primates under high-fat diet conditions. Monkeys fed a high-fat diet for 4 yr segregated into obese-prone and obese-resistant groups. In the third trimester, the high-fat diet reduced uterine artery blood flow regardless of weight gain, but only reduced placental blood flow in the obese-prone group, as compared with the normal-diet group (26). It could be that those on the high-fat diet without significant weight gain had compensatory mechanisms to protect against placental ischemia, which was lost in those with high-fat diet-induced obesity and metabolic disease. Furthermore, placentas from the obese-prone group had increased infarction, calcification, and syncytial knotting, which are pathologies in ischemic placentas from preeclamptic women. While blood pressure was not examined in this study, there was increased TLR4, monocyte chemoattractant factor (MCP)-1, and interleukin (IL)-1β expression in the placenta. IL-1β has been shown to reduce endothelial NOS (19).
Is NOS/NO System a Target For Obesity-Related Metabolic Abnormalities to Promote Vascular Dysfunction in PE?
The dependency of maternal vascular function and blood pressure regulation on NOS/NO is increased during healthy pregnancy in humans and rodents (84). It has been suggested that NO is a target and reduced in obese hypertensive pregnancies. At delivery, in isolated chorionic plate arteries, there was reduced vasorelaxation response to the NO donor, sodium nitroprusside (SNP). There was no difference in the vasoconstriction response to the thromboxane analog U46619 in obese versus lean pregnant women (34) with uncomplicated pregnancies. At this point, it is not clear if this truly represents vascular dysfunction or an increase in endogenous NOS control of vascular tone, as it was not stated that the SNP responses were generated in the presence of NOS inhibition. This is important information because it has been shown using in vivo and ex vivo approaches that inhibition of NOS increases the response to SNP in isolated arteries (20), which implicates endogenous NOS in blunting the response to an exogenous NO donors. Therefore, it may be that endothelial function was actually increased, especially because they found that acute incubation with leptin at obese levels (100 ng/ml) attenuated U46619-induced vasoconstriction. Leptin blunts vasoconstriction via an endothelial NOS-dependent pathway and could be a potential mechanism whereby some obese pregnant women are protected and do not develop hypertension during pregnancy. It should be determined at which point increasing levels of leptin become vasoconstrictive and hypertensive, as we have found that leptin infusion and increasing circulating levels by five times in once normotensive rats elicited hypertension (69). Chorionic plate arteries from preeclamptic women are more vasoconstrictive (6). Whether underlying endothelial NOS dysfunction in the presence of placental ischemic factors mitigate the vasoprotective effects of leptin should be examined, as NOS inhibition is known to potentiate the prohypertensive actions of leptin (46).
In a separate study, using myometrial artery segments ≤500 µm in diameter isolated from obese pregnant women without pregnancy complications, there was blunted vasoconstriction to U46619 with no difference to arginine vasopressin (AVP). The response to bradykinin and SNP were blunted only in those arteries constricted with AVP, and this was prevented with indomethacin (33). It is not clear to what extent thromboxane and AVP control vascular tone in lean or obese normotensive and preeclamptic pregnancies and how this is regulated by the endothelium or NOS. It is known that during normal pregnancy that there is significantly reduced U46619 responsiveness, and therefore, is seemingly further lowered in uncomplicated obese pregnancies. It should be examined whether this is a compensatory mechanism to protect against placental ischemia in some obese pregnant women, as there are increased myeloperoxidase-producing (myeloperoxidase stimulates production of thromboxane) neutrophils around blood vessels in obese preeclamptic women, and this was positively correlated with blood pressure (80, 83). It is unclear how obesity and these mechanisms affect NOS.
There is evidence of fetoplacental vascular NOS dysfunction in a subset of women with supraphysiological gestational weight gain but having normal weight during before pregnancy (70). In umbilical vein rings, insulin-induced vasorelaxation was significantly impaired, which is suggestive of vascular insulin resistance. The authors proposed that this was due to reduced NOS signaling, as detected by its reduced expression in freshly isolated umbilical vein endothelial cells from these women, but this was not determined functionally in endothelial denudation or NOS inhibition experiments or in the kidney. It should be noted that these women did not have PE or any hypertensive disorder of pregnancy. Again, this suggests that compensatory mechanisms maintained blood pressure regulation. Interestingly, these women did not reach a level of metabolic dysfunction to present with altered oral glucose tolerance tests, suggesting that women may need to have obesity before pregnancy for the development of metabolic dysfunction during pregnancy. It may be necessary that obesity must be present along with underlying endothelial dysfunction for placental ischemic factors and/or obesity-related metabolic abnormalities to exaggerate vasoconstriction and vascular NOS dysfunction to cause hypertension in obese pregnancies. These characteristics, including reduced insulin-induced vasorelaxation, are found in GDM (94), which is thought to be mediated by increased proinflammatory TNF-α and IL-6 levels and altered adipokine levels with lower adiponectin and raised leptin levels (2).
Do Combined Obesity and Metabolic Abnormalities of GDM Promote Even Greater Risk For PE?
In the Hyperglycemia and Adverse Pregnancy Outcome Study (HAPO), a greater risk for PE was observed in obese pregnant women with GDM compared with either morbidity alone (8). Accumulating evidence supports that obesity increases the risk for PE by associating with GDM (12). Indeed, the increased incidence of preeclampsia accompanied by increases in BMI is generally accompanied by diabetes (32). Several studies have documented this risk (22, 37, 68, 89), with GDM even being an independent risk factor for PE (74).
Prepregnancy obesity holds more risk for gestational hypertension and GDM than net maternal gestational weight gain (36, 53). Circulating glucose levels are increased in obese pregnant women but are even greater in those obese pregnancies complicated by GDM (60). Increased fasting glycemia (>5.6 mmol/l) at the first prenatal visit predicted the development of GDM and PE in a Russian cohort (73). Therefore, it is likely that metabolic dysfunction may adversely affect placentation. In vitro studies showed that high glucose concentrations, mimicking those in GDM, inhibit extravillous trophoblast invasion (9). In vivo studies in monkeys emphasized that placental dysfunction in obesity occurred in association with maternal insulin resistance (42). The timing of this insulin resistance was not examined, but at the end of the study, these high-fat diet-fed, obese-prone pregnant monkeys were insulin resistant with elevated fasting insulin and leptin levels. They had reduced placental blood flow and increased placental inflammation. In humans, higher fasting and postload glucose levels were associated with higher cord-blood levels of insulin and leptin (48), but neither placental inflammation nor blood flow were examined. Indirect evidence suggests that uteroplacental blood flow is reduced in GDM, as insulin resistance attenuates the vasodilatory actions of adenosine in GDM in primary human umbilical vein endothelial cells (HUVECs) (94). Interestingly, in HUVECS from patients with GDM, insulin prevented the associated increased transport of l-arginine, which is a cofactor for NOS activity (30). It is unclear if this increased transport allowed uncoupled NOS to produce reactive oxygen species (ROS) with altered insulin signaling, potentially implicating vascular insulin resistance in fetoplacental vascular dysfunction. High-fat diet-fed mice with insulin resistance had increased placental hypoxia inducible factor (HIF)-1α, VEGF-A, and inflammation by the end of pregnancy (50). Eighty six percent of pregnant women with diabetes have some placental abnormality including villous immaturity, villous necrosis, chorangiosis, and increased angiogenesis (39, 42). GDM seems to promote placental ischemia/hypoxia, but its direct role in vascular dysfunction, inflammation, and blood pressure regulation is not known, especially as the circulating metabolic factors are so heterogeneous in this population (38).
To fully understand the role of obesity and metabolic abnormalities in the pathogenesis of PE, experimental animal studies are required to establish cause-and-effect relationships. Sprague-Dawley rats fed a high-fructose diet from the beginning of pregnancy presented with hyperglycemia and hypertension by the end of gestation (82). Induction of Type 1 diabetes mellitus by streptozotocin (STZ) in Wistar rats on gestational day 6 resulted in hypertension, as assessed by tail-cuff and proteinuria by gestational day 19 (41). Intriguingly, this was not found in a spontaneous model of Type 1 diabetes, the nonobese diabetic (NOD) mouse, where blood pressure and heart rate, assessed by telemetry, were reduced by the end of pregnancy but had albuminuria (7). Maternal overweight and obesity increased the risk for PE in patients with Type 1, not Type 2, diabetes (72). Perhaps the timing of obesity and hyperglycemia in pregnancy dictates whether it causes hypertension.
In a systematic review, it was concluded that insufficient glycemic control increased risk for PE (95). Whether hyperglycemia and insulin resistance have a synergistic role together with angiogenic imbalance to elicit exaggeration of hypertension during pregnancy is unknown. With the use of the surrogate marker of insulin sensitivity, sex hormone binding globulin (SHBG), it was found that women with the lowest levels of both SHBG and PlGF had the greatest risk for PE (90). Circulating PlGF levels are reduced in women with GDM (51).
Do Obesity-Related Metabolic Abnormalities Exaggerate Placental Ischemia-Induced Vascular Dysfunction and Hypertension?
Placental ischemia-induced hypertension has a strong dependency on angiogenic imbalance, and this may mediate obesity’s impact to increase the risk for PE. Indeed, PlGF levels are lower in obese versus lean PE patients with no difference in VEGF-A (77). In obese pregnancies examined at each gestational week, BMI was positively related to increased sFlt-1 over time, and the association of BMI versus time was significant in those with placental dysfunction, which was not directly compared with, but seemed greater than, normal pregnancy (98). In a cohort of obese women that went on to develop PE, PlGF levels at 20 to 22 wk of gestation were lower in overweight women compared with those that were underweight or normal weight and were even lower with obesity. These changes were associated with a much stronger correlation for PE in these heavier pregnant women (27). When more specifically examining diabetic preeclamptic pregnancies, those with combined PE and Type 2 diabetes had some of the lowest values of serum PlGF found (91). Moreover, there was a trend for lower cord serum PlGF in diabetic pregnancies with PE compared with those with any hypertensive disorder (56). Furthermore, sFlt-1 levels were increased with reduced PlGF in diabetic pregnancies complicated with PE over diabetic pregnancy alone, although a PE alone or obese groups were not included (14, 15).
It is apparent that prepregnancy obesity and metabolic abnormalities are linked to exaggerated reductions in angiogenic factors found in preeclamptic women. The cause for these reductions, and whether they contribute to the increased risk for PE in obese pregnancies, is unknown. As mentioned earlier, the ischemic placenta is active in promoting the hypertensive phenotype of PE. Placental ischemia and hypoxia-induced release of sFlt-1 quenches and reduces bioavailable proangiogenic and provasodilatory factors, notably VEGF and PlGF. Reduced angiogenic balance is mimicked by infusion of sFlt-1 into once normotensive pregnant rats whereby it drives up maternal blood pressure. Thus placental ischemia/hypoxia and subsequent production of sFlt-1 may be one source for altered angiogenic balance in obese, preeclamptic pregnancies. Interestingly, it is thought that adipose tissue may also contribute to the circulating pool of sFlt-1 (85). The integrative nature of placental ischemia and adipose tissue release of anti-angiogenic factors, for example, by soluble factors derived from the ischemic placenta driving adipose tissue to produce of prohypertensive factors, and vice versa, needs to be examined.
This has begun to be examined by utilizing omental adipose tissue explant cultures. There was no difference in the release of PlGF between pregnant women with preexisting obesity or lean controls with normal glucose tolerance or GDM, but PlGF levels were increased from those with combined obesity and GDM, with no difference in VEGF-A (47). This pattern was also found for sFlt-1 and soluble endoglin (sEng) and the proinflammatory marker soluble intercellular adhesion molecule-1 (sICAM-1). There was no difference for placental explant release of any of these factors between any of the groups, but it is important to note that those women with PE were excluded. Therefore, the interactions between placental ischemia-induced increases in circulating angiogenic imbalance; the ability of adipose tissue to release PlGF; and PlGF signaling in maternal blood vessels and adipose tissue should be examined. Reduced PlGF attenuates the gain of adipose tissue mass in response to high-fat diet during pregnancy in PlGF−/− mice. They were not able to compensate by increasing VEGF-A levels and also had an earlier rise in circulating leptin and hyperinsulinemia compared with wild-type controls (35). It is not known whether these changes are caused by reduced PlGF-mediated vascular dysfunction in the adipose tissue and other metabolic organs. Furthermore, blood pressure was not examined nor was endothelial function or the functional capacity of perivascular adipose tissue (PVAT) to regulate vascular tone examined in the face of reduced PlGF and increased obesity-related metabolic factors.
PVAT is a novel regulator of vasodilator tone with functional ability to buffer vasoconstriction. Adiponectin released from the PVAT induces vasodilation via NOS in human vessels (59), but it is reduced in obesity (29). In combined obesity and GDM, there are greater reductions in circulating adiponectin than obesity alone (76). The high molecular weight form is the only form reduced in PE (65) and GDM (64). High-fat diet-induced obese C57BL/6J mice had reduced circulating high molecular weight adiponectin, hyperinsulinemia and increased fetal weights during pregnancy and adiponectin infusion reversed these findings (5). Blood pressure, endothelial function, or PVAT buffering capacity was not examined.
Perspectives and Significance
The literature suggests that metabolic abnormalities are obligatory for obesity to increase the risk for PE. Understanding the pathogenesis of PE is an important women’s health issue. This is especially true as risk factors for this maternal disorder are increasing, such as prepregnancy obesity and metabolic disease. The individual and combined actions of prepregnancy obesity, high glucose, insulin, leptin, and reduced adiponectin may support the onset of PE by encouraging placental ischemia and exaggerating anti-angiogenic and inflammation-induced maternal vascular dysfunction. Figure 1 illustrates the hypothesis and integrative nature by which obesity and obesity-related metabolic factors reduce cytotrophoblast migration and reduced spiral artery remodeling and widening resulting in placental ischemia. It is also proposed that such obese conditions exaggerate placental ischemia-induced increases in circulating anti-angiogenic factors and proinflammatory pathways that collectively lead to reduced vascular NO levels, increased total peripheral resistance, and reduce renal excretory function culminating in the increased risk for PE in obese pregnancies. Interestingly, not all obese pregnant women develop PE, suggesting upregulation of vasoprotective mechanisms. Such protective pathways may be targeted in the face of obesity-induced GDM and metabolic disease to promote PE. Proper development of therapeutic strategies, such as treatment with PlGF or insulin-sensitizing drugs like metformin (88), may prove beneficial against PE and its long-term consequences. At this time point, the only steadfast treatment for PE is premature delivery of the baby and the ischemic placenta.
Fig. 1.
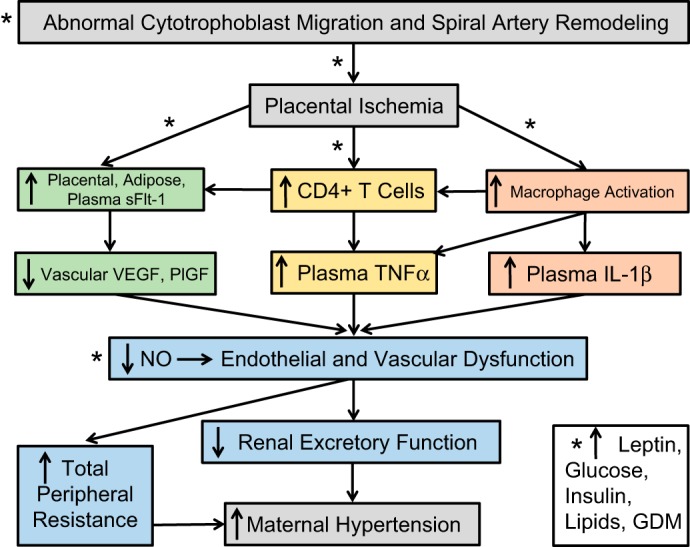
Hypothetical scheme with asterisks denoting where obesity and obesity-related abnormalities, such as increased leptin, glucose, insulin, and lipids, and gestational diabetes mellitus (GDM), may promote and even exaggerate the pathways linking placental ischemia to maternal hypertension.
GRANTS
This paper was funded by National Institute of General Medical Sciences (NIGMS) Grant P20GM104357 and National Heart, Lung, and Blood Institute (NHBLI) Grant K99HL130577.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
F.T.S. performed experiments; F.T.S. analyzed data; F.T.S. interpreted results of experiments; F.T.S. prepared figures; F.T.S. drafted manuscript; F.T.S. edited and revised manuscript; F.T.S. approved final version of manuscript.
REFERENCES
- 1.Aasa KL, Zavan B, Luna RL, Wong PG, Ventura NM, Tse MY, Carmeliet P, Adams MA, Pang SC, Croy BA. Placental growth factor influences maternal cardiovascular adaptation to pregnancy in mice. Biol Reprod 92: 44, 2015. doi: 10.1095/biolreprod.114.124677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Badri MR, Zantout MS, Azar ST. The role of adipokines in gestational diabetes mellitus. Ther Adv Endocrinol Metab 6: 103–108, 2015. doi: 10.1177/2042018815577039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.American College of Obstetricians and Gynecologists; Task Force on Hypertension in Pregnancy . Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet Gynecol 122: 1122–1131, 2013. doi: 10.1097/01.AOG.0000437382.03963.88. [DOI] [PubMed] [Google Scholar]
- 4.Avagliano L, Marconi AM, Romagnoli S, Bulfamante GP. Abnormal spiral arteries modification in stillbirths: the role of maternal prepregnancy body mass index. J Matern Fetal Neonatal Med 25: 2789–2792, 2012. doi: 10.3109/14767058.2012.705395. [DOI] [PubMed] [Google Scholar]
- 5.Aye IL, Rosario FJ, Powell TL, Jansson T. Adiponectin supplementation in pregnant mice prevents the adverse effects of maternal obesity on placental function and fetal growth. Proc Natl Acad Sci U S A 112: 12858–12863, 2015. doi: 10.1073/pnas.1515484112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benoit C, Zavecz J, Wang Y. Vasoreactivity of chorionic plate arteries in response to vasoconstrictors produced by preeclamptic placentas. Placenta 28: 498–504, 2007. doi: 10.1016/j.placenta.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burke SD, Barrette VF, David S, Khankin EV, Adams MA, Croy BA. Circulatory and renal consequences of pregnancy in diabetic NOD mice. Placenta 32: 949–955, 2011. doi: 10.1016/j.placenta.2011.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Catalano PM, McIntyre HD, Cruickshank JK, McCance DR, Dyer AR, Metzger BE, Lowe LP, Trimble ER, Coustan DR, Hadden DR, Persson B, Hod M, Oats JJ; HAPO Study Cooperative Research Group . The hyperglycemia and adverse pregnancy outcome study: associations of GDM and obesity with pregnancy outcomes. Diabetes Care 35: 780–786, 2012. doi: 10.2337/dc11-1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cawyer CR, Horvat D, Leonard D, Allen SR, Jones RO, Zawieja DC, Kuehl TJ, Uddin MN. Hyperglycemia impairs cytotrophoblast function via stress signaling. Am J Obstet Gynecol 211: 541.e1–541.e8, 2014. doi: 10.1016/j.ajog.2014.04.033. [DOI] [PubMed] [Google Scholar]
- 10.Challier JC, Basu S, Bintein T, Minium J, Hotmire K, Catalano PM, Hauguel-de Mouzon S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 29: 274–281, 2008. doi: 10.1016/j.placenta.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Q, Yin YX, Wei J, Tong M, Shen F, Zhao M, Chamley L. Increased expression of high mobility group box 1 (HMGB1) in the cytoplasm of placental syncytiotrophoblast from preeclamptic placentae. Cytokine 85: 30–36, 2016. doi: 10.1016/j.cyto.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Chu SY, Callaghan WM, Kim SY, Schmid CH, Lau J, England LJ, Dietz PM. Maternal obesity and risk of gestational diabetes mellitus. Diabetes Care 30: 2070–2076, 2007. doi: 10.2337/dc06-2559a. [DOI] [PubMed] [Google Scholar]
- 13.Clark SL, Cotton DB, Lee W, Bishop C, Hill T, Southwick J, Pivarnik J, Spillman T, DeVore GR, Phelan J, Hankins GDV, Benedetti TJ, Tolley D. Central hemodynamic assessment of normal term pregnancy. Am J Obstet Gynecol 161: 1439–1442, 1989. doi: 10.1016/0002-9378(89)90900-9. [DOI] [PubMed] [Google Scholar]
- 14.Cohen A, Lim KH, Lee Y, Rana S, Karumanchi SA, Brown F. Circulating levels of the antiangiogenic marker soluble FMS-like tyrosine kinase 1 are elevated in women with pregestational diabetes and preeclampsia: angiogenic markers in preeclampsia and preexisting diabetes. Diabetes Care 30: 375–377, 2007. doi: 10.2337/dc06-1514. [DOI] [PubMed] [Google Scholar]
- 15.Cohen AL, Wenger JB, James-Todd T, Lamparello BM, Halprin E, Serdy S, Fan S, Horowitz GL, Lim KH, Rana S, Takoudes TC, Wyckoff JA, Thadhani R, Karumanchi SA, Brown FM. The association of circulating angiogenic factors and HbA1c with the risk of preeclampsia in women with preexisting diabetes. Hypertens Pregnancy 33: 81–92, 2014. doi: 10.3109/10641955.2013.837175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Creanga AA, Berg CJ, Syverson C, Seed K, Bruce FC, Callaghan WM. Pregnancy-related mortality in the United States, 2006–2010. Obstet Gynecol 125: 5–12, 2015. doi: 10.1097/AOG.0000000000000564. [DOI] [PubMed] [Google Scholar]
- 17.Crovetto F, Figueras F, Triunfo S, Crispi F, Rodriguez-Sureda V, Dominguez C, Llurba E, Gratacós E. First trimester screening for early and late preeclampsia based on maternal characteristics, biophysical parameters, and angiogenic factors. Prenat Diagn 35: 183–191, 2015. doi: 10.1002/pd.4519. [DOI] [PubMed] [Google Scholar]
- 18.Darmochwal-Kolarz D, Saito S, Rolinski J, Tabarkiewicz J, Kolarz B, Leszczynska-Gorzelak B, Oleszczuk J. Activated T lymphocytes in pre-eclampsia. Am J Reprod Immunol 58: 39–45, 2007. doi: 10.1111/j.1600-0897.2007.00489.x. [DOI] [PubMed] [Google Scholar]
- 19.de Frutos T, de Miguel LS, García-Durán M, González-Fernández F, Rodríguez-Feo JA, Montón M, Guerra J, Farré J, Casado S, López-Farré A. NO from smooth muscle cells decreases NOS expression in endothelial cells: role of TNF-alpha. Am J Physiol Heart Circ Physiol 277: H1317–H1325, 1999. [DOI] [PubMed] [Google Scholar]
- 20.Dowell FJ, Henrion D, Duriez M, Michel JB. Vascular reactivity in mesenteric resistance arteries following chronic nitric oxide synthase inhibition in Wistar rats. Br J Pharmacol 117: 341–346, 1996. doi: 10.1111/j.1476-5381.1996.tb15196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Durst JK, Tuuli MG, Stout MJ, Macones GA, Cahill AG. Degree of obesity at delivery and risk of preeclampsia with severe features. Am J Obstet Gynecol 214: 651.e1–651.e5, 2016. doi: 10.1016/j.ajog.2015.11.024. [DOI] [PubMed] [Google Scholar]
- 22.Easmin S, Chowdhury TA, Islam MR, Beg A, Jahan MK, Latif T, Dhar S, Alam MN, Akhter M. Obstetric outcome in early and late onset gestational diabetes mellitus. Mymensingh Med J 24: 450–456, 2015. [PubMed] [Google Scholar]
- 23.Faas MM, Spaans F, De Vos P. Monocytes and macrophages in pregnancy and pre-eclampsia. Front Immunol 5: 298, 2014. doi: 10.3389/fimmu.2014.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farley D, Tejero ME, Comuzzie AG, Higgins PB, Cox L, Werner SL, Jenkins SL, Li C, Choi J, Dick EJ Jr, Hubbard GB, Frost P, Dudley DJ, Ballesteros B, Wu G, Nathanielsz PW, Schlabritz-Loutsevitch NE. Feto-placental adaptations to maternal obesity in the baboon. Placenta 30: 752–760, 2009. doi: 10.1016/j.placenta.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farley DM, Choi J, Dudley DJ, Li C, Jenkins SL, Myatt L, Nathanielsz PW. Placental amino acid transport and placental leptin resistance in pregnancies complicated by maternal obesity. Placenta 31: 718–724, 2010. doi: 10.1016/j.placenta.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 26.Frias AE, Morgan TK, Evans AE, Rasanen J, Oh KY, Thornburg KL, Grove KL. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology 152: 2456–2464, 2011. doi: 10.1210/en.2010-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh SK, Raheja S, Tuli A, Raghunandan C, Agarwal S. Serum placental growth factor as a predictor of early onset preeclampsia in overweight/obese pregnant women. J Am Soc Hypertens 7: 137–148, 2013. doi: 10.1016/j.jash.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 28.Giacobbe A, Granese R, Grasso R, Salpietro V, Corrado F, Giorgianni G, Foti G, Amadore D, Triolo O, Giunta L, Di Benedetto A. Association between maternal serum high mobility group box 1 levels and pregnancy complicated by gestational diabetes mellitus. Nutr Metab Cardiovasc Dis 26: 414–418, 2016. doi: 10.1016/j.numecd.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 29.Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, Laing I, Yates AP, Pemberton PW, Malik RA, Heagerty AM. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation 119: 1661–1670, 2009. doi: 10.1161/CIRCULATIONAHA.108.821181. [DOI] [PubMed] [Google Scholar]
- 30.Guzmán-Gutiérrez E, Armella A, Toledo F, Pardo F, Leiva A, Sobrevia L. Insulin requires A1 adenosine receptors expression to reverse gestational diabetes-increased L-arginine transport in human umbilical vein endothelium. Purinergic Signal 12: 175–190, 2016. doi: 10.1007/s11302-015-9491-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guzmán-Ruiz R, Ortega F, Rodríguez A, Vázquez-Martínez R, Díaz-Ruiz A, Garcia-Navarro S, Giralt M, Garcia-Rios A, Cobo-Padilla D, Tinahones FJ, López-Miranda J, Villarroya F, Frühbeck G, Fernández-Real JM, Malagón MM. Alarmin high-mobility group B1 (HMGB1) is regulated in human adipocytes in insulin resistance and influences insulin secretion in β-cells. Int J Obes (Lond) 38: 1545–1554, 2014. doi: 10.1038/ijo.2014.36. [DOI] [PubMed] [Google Scholar]
- 32.Hauth JC, Clifton RG, Roberts JM, Myatt L, Spong CY, Leveno KJ, Varner MW, Wapner RJ, Thorp JM Jr, Mercer BM, Peaceman AM, Ramin SM, Carpenter MW, Samuels P, Sciscione A, Tolosa JE, Saade G, Sorokin Y, Anderson GD; Eunice Kennedy Shriver National Institute of Child Health and Human Development Maternal-Fetal Medicine Units Network . Maternal insulin resistance and preeclampsia. Am J Obstet Gynecol 204: 327.e1–327.e6, 2011. doi: 10.1016/j.ajog.2011.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayward CE, Cowley EJ, Mills TA, Sibley CP, Wareing M. Maternal obesity impairs specific regulatory pathways in human myometrial arteries. Biol Reprod 90: 65, 2014. doi: 10.1095/biolreprod.113.112623. [DOI] [PubMed] [Google Scholar]
- 34.Hayward CE, Higgins L, Cowley EJ, Greenwood SL, Mills TA, Sibley CP, Wareing M. Chorionic plate arterial function is altered in maternal obesity. Placenta 34: 281–287, 2013. doi: 10.1016/j.placenta.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hemmeryckx B, van Bree R, Van Hoef B, Vercruysse L, Lijnen HR, Verhaeghe J. Adverse adipose phenotype and hyperinsulinemia in gravid mice deficient in placental growth factor. Endocrinology 149: 2176–2183, 2008. doi: 10.1210/en.2007-1272. [DOI] [PubMed] [Google Scholar]
- 36.Heude B, Thiébaugeorges O, Goua V, Forhan A, Kaminski M, Foliguet B, Schweitzer M, Magnin G, Charles MA; EDEN Mother-Child Cohort Study Group . Pre-pregnancy body mass index and weight gain during pregnancy: relations with gestational diabetes and hypertension, and birth outcomes. Matern Child Health J 16: 355–363, 2012. doi: 10.1007/s10995-011-0741-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hildén K, Hanson U, Persson M, Fadl H. Overweight and obesity: a remaining problem in women treated for severe gestational diabetes. Diabet Med 33: 1045–1051, 2016. doi: 10.1111/dme.13156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huynh J, Xiong G, Bentley-Lewis R. A systematic review of metabolite profiling in gestational diabetes mellitus. Diabetologia 57: 2453–2464, 2014. doi: 10.1007/s00125-014-3371-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huynh J, Yamada J, Beauharnais C, Wenger JB, Thadhani RI, Wexler D, Roberts DJ, Bentley-Lewis R. Type 1, type 2 and gestational diabetes mellitus differentially impact placental pathologic characteristics of uteroplacental malperfusion. Placenta 36: 1161–1166, 2015. doi: 10.1016/j.placenta.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hytten FE. Physiological changes in early pregnancy. J Obstet Gynaecol Br Commonw 75: 1193–1197, 1968. doi: 10.1111/j.1471-0528.1968.tb02915.x. [DOI] [PubMed] [Google Scholar]
- 41.Ishihara G, Hiramatsu Y, Masuyama H, Kudo T. Streptozotocin-induced diabetic pregnant rats exhibit signs and symptoms mimicking preeclampsia. Metabolism 49: 853–857, 2000. doi: 10.1053/meta.2000.6750. [DOI] [PubMed] [Google Scholar]
- 42.Jarmuzek P, Wielgos M, Bomba-Opon D. Placental pathologic changes in gestational diabetes mellitus. Neuro Endocrinol Lett 36: 101–105, 2015. [PubMed] [Google Scholar]
- 43.Jiang R, Cai J, Zhu Z, Chen D, Wang J, Wang Q, Teng Y, Huang Y, Tao M, Xia A, Xue M, Zhou S, Chen AF. Hypoxic trophoblast HMGB1 induces endothelial cell hyperpermeability via the TRL-4/caveolin-1 pathway. J Immunol 193: 5000–5012, 2014. doi: 10.4049/jimmunol.1303445. [DOI] [PubMed] [Google Scholar]
- 44.Kim DW, Young SL, Grattan DR, Jasoni CL. Obesity during pregnancy disrupts placental morphology, cell proliferation, and inflammation in a sex-specific manner across gestation in the mouse. Biol Reprod 90: 130, 2014. doi: 10.1095/biolreprod.113.117259. [DOI] [PubMed] [Google Scholar]
- 45.Kingdom JC, Drewlo S. Is heparin a placental anticoagulant in high-risk pregnancies? Blood 118: 4780–4788, 2011. doi: 10.1182/blood-2011-07-319749. [DOI] [PubMed] [Google Scholar]
- 46.Kuo JJ, Jones OB, Hall JE. Inhibition of NO synthesis enhances chronic cardiovascular and renal actions of leptin. Hypertension 37: 670–676, 2001. doi: 10.1161/01.HYP.37.2.670. [DOI] [PubMed] [Google Scholar]
- 47.Lappas M. Markers of endothelial cell dysfunction are increased in human omental adipose tissue from women with pre-existing maternal obesity and gestational diabetes. Metabolism 63: 860–873, 2014. doi: 10.1016/j.metabol.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 48.Lawlor DA, West J, Fairley L, Nelson SM, Bhopal RS, Tuffnell D, Freeman DJ, Wright J, Whitelaw DC, Sattar N. Pregnancy glycaemia and cord-blood levels of insulin and leptin in Pakistani and white British mother-offspring pairs: findings from a prospective pregnancy cohort. Diabetologia 57: 2492–2500, 2014. doi: 10.1007/s00125-014-3386-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leon-Garcia SM, Roeder HA, Nelson KK, Liao X, Pizzo DP, Laurent LC, Parast MM, LaCoursiere DY. Maternal obesity and sex-specific differences in placental pathology. Placenta 38: 33–40, 2016. doi: 10.1016/j.placenta.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 50.Li HP, Chen X, Li MQ. Gestational diabetes induces chronic hypoxia stress and excessive inflammatory response in murine placenta. Int J Clin Exp Pathol 6: 650–659, 2013. [PMC free article] [PubMed] [Google Scholar]
- 51.Li J, Ying H, Cai G, Guo Q, Chen L. Impaired proliferation of pancreatic beta cells, by reduced placental growth factor in pre-eclampsia, as a cause for gestational diabetes mellitus. Cell Prolif 48: 166–174, 2015. doi: 10.1111/cpr.12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li M, Song L, Gao X, Chang W, Qin X. Toll-like receptor 4 on islet β cells senses expression changes in high-mobility group box 1 and contributes to the initiation of type 1 diabetes. Exp Mol Med 44: 260–267, 2012. doi: 10.3858/emm.2012.44.4.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li N, Liu E, Guo J, Pan L, Li B, Wang P, Liu J, Wang Y, Liu G, Baccarelli AA, Hou L, Hu G. Maternal prepregnancy body mass index and gestational weight gain on pregnancy outcomes. PLoS One 8: e82310, 2013. doi: 10.1371/journal.pone.0082310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin LT, Tsui KH, Cheng JT, Cheng JS, Huang WC, Liou WS, Tang PL. Increased risk of intracranial hemorrhage in patients with pregnancy-induced hypertension: a nationwide population-based retrospective cohort study. Medicine (Baltimore) 95: e3732, 2016. doi: 10.1097/MD.0000000000003732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Loardi C, Falchetti M, Prefumo F, Facchetti F, Frusca T. Placental morphology in pregnancies associated with pregravid obesity. J Matern Fetal Neonatal Med 29: 2611–2616, 2016. doi: 10.3109/14767058.2015.1094792. [DOI] [PubMed] [Google Scholar]
- 56.Loukovaara M, Leinonen P, Teramo K, Andersson S. Concentration of cord serum placenta growth factor in normal and diabetic pregnancies. BJOG 112: 75–79, 2005. doi: 10.1111/j.1471-0528.2004.00337.x. [DOI] [PubMed] [Google Scholar]
- 57.Mandalà M, Gokina N, Barron C, Osol G. Endothelial-derived hyperpolarization factor (EDHF) contributes to PlGF-induced dilation of mesenteric resistance arteries from pregnant rats. J Vasc Res 49: 43–49, 2012. doi: 10.1159/000329821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marchetti T, de Moerloose P, Gris JC. Antiphospholipid antibodies and the risk of severe and non-severe pre-eclampsia: the NOHA case-control study. J Thromb Haemost 14: 675–684, 2016. doi: 10.1111/jth.13257. [DOI] [PubMed] [Google Scholar]
- 59.Margaritis M, Antonopoulos AS, Digby J, Lee R, Reilly S, Coutinho P, Shirodaria C, Sayeed R, Petrou M, De Silva R, Jalilzadeh S, Demosthenous M, Bakogiannis C, Tousoulis D, Stefanadis C, Choudhury RP, Casadei B, Channon KM, Antoniades C. Interactions between vascular wall and perivascular adipose tissue reveal novel roles for adiponectin in the regulation of endothelial nitric oxide synthase function in human vessels. Circulation 127: 2209–2221, 2013. doi: 10.1161/CIRCULATIONAHA.112.001133. [DOI] [PubMed] [Google Scholar]
- 60.Martino J, Sebert S, Segura MT, García-Valdés L, Florido J, Padilla MC, Marcos A, Rueda R, McArdle HJ, Budge H, Symonds ME, Campoy C. Maternal body weight and gestational diabetes differentially influence placental and pregnancy outcomes. J Clin Endocrinol Metab 101: 59–68, 2016. doi: 10.1210/jc.2015-2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Masuyama H, Hiramatsu Y. Treatment with constitutive androstane receptor ligand during pregnancy prevents insulin resistance in offspring from high-fat diet-induced obese pregnant mice. Am J Physiol Endocrinol Metab 303: E293–E300, 2012. doi: 10.1152/ajpendo.00167.2012. [DOI] [PubMed] [Google Scholar]
- 62.Masuyama H, Mitsui T, Maki J, Tani K, Nakamura K, Hiramatsu Y. Dimethylesculetin ameliorates maternal glucose intolerance and fetal overgrowth in high-fat diet-fed pregnant mice via constitutive androstane receptor. Mol Cell Biochem 419: 185–192, 2016. doi: 10.1007/s11010-016-2772-4. [DOI] [PubMed] [Google Scholar]
- 63.Masuyama H, Nobumoto E, Segawa T, Hiramatsu Y. Severe superimposed preeclampsia with obesity, diabetes and a mild imbalance of angiogenic factors. Acta Med Okayama 66: 171–175, 2012. [DOI] [PubMed] [Google Scholar]
- 64.Mazaki-Tovi S, Romero R, Vaisbuch E, Erez O, Mittal P, Chaiworapongsa T, Kim SK, Pacora P, Yeo L, Gotsch F, Dong Z, Yoon BH, Hassan SS, Kusanovic JP. Maternal serum adiponectin multimers in gestational diabetes. J Perinat Med 37: 637–650, 2009. doi: 10.1515/JPM.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mazaki-Tovi S, Romero R, Vaisbuch E, Kusanovic JP, Erez O, Gotsch F, Chaiworapongsa T, Than NG, Kim SK, Nhan-Chang CL, Jodicke C, Pacora P, Yeo L, Dong Z, Yoon BH, Hassan SS, Mittal P. Maternal serum adiponectin multimers in preeclampsia. J Perinat Med 37: 349–363, 2009. doi: 10.1515/JPM.2009.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McDonald SD, Han Z, Mulla S, Beyene J; Knowledge Synthesis Group . Overweight and obesity in mothers and risk of preterm birth and low birth weight infants: systematic review and meta-analyses. BMJ 341: c3428, 2010. doi: 10.1136/bmj.c3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Myatt L, Maloyan A. Obesity and placental function. Semin Reprod Med 34: 42–49, 2016. doi: 10.1055/s-0035-1570027. [DOI] [PubMed] [Google Scholar]
- 68.Nerenberg KA, Johnson JA, Leung B, Savu A, Ryan EA, Chik CL, Kaul P. Risks of gestational diabetes and preeclampsia over the last decade in a cohort of Alberta women. J Obstet Gynaecol Can 35: 986–994, 2013. doi: 10.1016/S1701-2163(15)30786-6. [DOI] [PubMed] [Google Scholar]
- 69.Palei AC, Spradley FT, Granger JP. Chronic hyperleptinemia results in the development of hypertension in pregnant rats. Am J Physiol Regul Integr Comp Physiol 308: R855–R861, 2015. doi: 10.1152/ajpregu.00286.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pardo F, Silva L, Sáez T, Salsoso R, Gutiérrez J, Sanhueza C, Leiva A, Sobrevia L. Human supraphysiological gestational weight gain and fetoplacental vascular dysfunction. Int J Obes (Lond) 39: 1264–1273, 2015. doi: 10.1038/ijo.2015.57. [DOI] [PubMed] [Google Scholar]
- 71.Persson M, Cnattingius S, Wikström AK, Johansson S. Maternal overweight and obesity and risk of pre-eclampsia in women with type 1 diabetes or type 2 diabetes. Diabetologia 59: 2099–2105, 2016. doi: 10.1007/s00125-016-4035-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Persson M, Cnattingius S, Wikström AK, Johansson S. Maternal overweight and obesity and risk of pre-eclampsia in women with type 1 diabetes or type 2 diabetes. Diabetologia 59: 2099–2105, 2016. doi: 10.1007/s00125-016-4035-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Popova P, Tkachuk A, Dronova A, Gerasimov A, Kravchuk E, Bolshakova M, Rozdestvenskaya O, Demidova K, Nikolaeva A, Grineva E. Fasting glycemia at the first prenatal visit and pregnancy outcomes in Russian women. Minerva Endocrinol 41: 477–485, 2016. [PubMed] [Google Scholar]
- 74.Powe CE, Allard C, Battista MC, Doyon M, Bouchard L, Ecker JL, Perron P, Florez JC, Thadhani R, Hivert MF. Heterogeneous contribution of insulin sensitivity and secretion defects to gestational diabetes mellitus. Diabetes Care 39: 1052–1055, 2016. doi: 10.2337/dc15-2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Powers RW, Roberts JM, Plymire DA, Pucci D, Datwyler SA, Laird DM, Sogin DC, Jeyabalan A, Hubel CA, Gandley RE. Low placental growth factor across pregnancy identifies a subset of women with preterm preeclampsia: type 1 versus type 2 preeclampsia? Hypertension 60: 239–246, 2012. doi: 10.1161/HYPERTENSIONAHA.112.191213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ramirez VI, Miller E, Meireles CL, Gelfond J, Krummel DA, Powell TL. Adiponectin and IGFBP-1 in the development of gestational diabetes in obese mothers. BMJ Open Diabetes Res Care 2: e000010, 2014. doi: 10.1136/bmjdrc-2013-000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Raymond D, Peterson E. A critical review of early-onset and late-onset preeclampsia. Obstet Gynecol Surv 66: 497–506, 2011. doi: 10.1097/OGX.0b013e3182331028. [DOI] [PubMed] [Google Scholar]
- 78.Redman CW, Sargent IL, Staff AC. IFPA Senior Award Lecture: making sense of pre-eclampsia - two placental causes of preeclampsia? Placenta 35, Suppl: S20–S25, 2014. doi: 10.1016/j.placenta.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 79.Savvidou MD, Noori M, Anderson JM, Hingorani AD, Nicolaides KH. Maternal endothelial function and serum concentrations of placental growth factor and soluble endoglin in women with abnormal placentation. Ultrasound Obstet Gynecol 32: 871–876, 2008. doi: 10.1002/uog.6126. [DOI] [PubMed] [Google Scholar]
- 80.Shah TJ, Leik CE, Walsh SW. Neutrophil infiltration and systemic vascular inflammation in obese women. Reprod Sci 17: 116–124, 2010. doi: 10.1177/1933719109348252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shirasuna K, Seno K, Ohtsu A, Shiratsuki S, Ohkuchi A, Suzuki H, Matsubara S, Nagayama S, Iwata H, Kuwayama T. AGEs and HMGB1 increase inflammatory cytokine production from human placental cells, resulting in an enhancement of monocyte migration. Am J Reprod Immunol 75: 557–568, 2016. doi: 10.1111/aji.12506. [DOI] [PubMed] [Google Scholar]
- 82.Shortliffe LM, Hammam O, Han X, Kouba E, Tsao PS, Wang B. Dietary fructose in pregnancy induces hyperglycemia, hypertension, and pathologic kidney and liver changes in a rodent model. Pregnancy Hypertens 5: 308–314, 2015. [DOI] [PubMed] [Google Scholar]
- 83.Shukla J, Walsh SW. Neutrophil release of myeloperoxidase in systemic vasculature of obese women may put them at risk for preeclampsia. Reprod Sci 22: 300–307, 2015. doi: 10.1177/1933719114557899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sladek SM, Magness RR, Conrad KP. Nitric oxide and pregnancy. Am J Physiol Regul Integr Comp Physiol 272: R441–R463, 1997. [DOI] [PubMed] [Google Scholar]
- 85.Spradley FT, Palei AC, Granger JP. Increased risk for the development of preeclampsia in obese pregnancies: weighing in on the mechanisms. Am J Physiol Regul Integr Comp Physiol 309: R1326–R1343, 2015. doi: 10.1152/ajpregu.00178.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Staff AC, Dechend R, Pijnenborg R. Learning from the placenta: acute atherosis and vascular remodeling in preeclampsia-novel aspects for atherosclerosis and future cardiovascular health. Hypertension 56: 1026–1034, 2010. doi: 10.1161/HYPERTENSIONAHA.110.157743. [DOI] [PubMed] [Google Scholar]
- 87.Sun W, Cui B, Hong F, Xu Y. Establishment of ApoE-knockout mouse model of preeclampsia and relevant mechanisms. Exp Ther Med 12: 2634–2638, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 88.Syngelaki A, Nicolaides KH, Balani J, Hyer S, Akolekar R, Kotecha R, Pastides A, Shehata H. Metformin versus placebo in obese pregnant women without diabetes mellitus. N Engl J Med 374: 434–443, 2016. doi: 10.1056/NEJMoa1509819. [DOI] [PubMed] [Google Scholar]
- 89.Tamayo T, Tamayo M, Rathmann W, Potthoff P. Prevalence of gestational diabetes and risk of complications before and after initiation of a general systematic two-step screening strategy in Germany (2012–2014). Diabetes Res Clin Pract 115: 1–8, 2016. doi: 10.1016/j.diabres.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 90.Thadhani R, Ecker JL, Mutter WP, Wolf M, Smirnakis KV, Sukhatme VP, Levine RJ, Karumanchi SA. Insulin resistance and alterations in angiogenesis: additive insults that may lead to preeclampsia. Hypertension 43: 988–992, 2004. doi: 10.1161/01.HYP.0000124460.67539.1d. [DOI] [PubMed] [Google Scholar]
- 91.Tsiakkas A, Duvdevani N, Wright A, Wright D, Nicolaides KH. Serum placental growth factor in the three trimesters of pregnancy: effects of maternal characteristics and medical history. Ultrasound Obstet Gynecol 45: 591–598, 2015. doi: 10.1002/uog.14811. [DOI] [PubMed] [Google Scholar]
- 92.Wallace K, Novotny S, Heath J, Moseley J, Martin JN Jr, Owens MY, LaMarca B. Hypertension in response to CD4(+) T cells from reduced uterine perfusion pregnant rats is associated with activation of the endothelin-1 system. Am J Physiol Regul Integr Comp Physiol 303: R144–R149, 2012. doi: 10.1152/ajpregu.00049.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Warrington JP, George EM, Palei AC, Spradley FT, Granger JP. Recent advances in the understanding of the pathophysiology of preeclampsia. Hypertension 62: 666–673, 2013. doi: 10.1161/HYPERTENSIONAHA.113.00588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Westermeier F, Salomón C, Farías M, Arroyo P, Fuenzalida B, Sáez T, Salsoso R, Sanhueza C, Guzmán-Gutiérrez E, Pardo F, Leiva A, Sobrevia L. Insulin requires normal expression and signaling of insulin receptor A to reverse gestational diabetes-reduced adenosine transport in human umbilical vein endothelium. FASEB J 29: 37–49, 2015. doi: 10.1096/fj.14-254219. [DOI] [PubMed] [Google Scholar]
- 95.Wotherspoon AC, Young IS, McCance DR, Holmes VA. Evaluation of biomarkers for the prediction of pre-eclampsia in women with type 1 diabetes mellitus: A systematic review. J Diabetes Complications 30: 958–966, 2016. doi: 10.1016/j.jdiacomp.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 96.Young OM, Twedt R, Catov JM. Pre-pregnancy maternal obesity and the risk of preterm preeclampsia in the American primigravida. Obesity (Silver Spring) 24: 1226–1229, 2016. doi: 10.1002/oby.21412. [DOI] [PubMed] [Google Scholar]
- 97.Zeng X, Pang H, Li X, Luo S, Jin S, Li S. Impact of obesity on endometrial blood flow in women without polycystic ovarian syndrome during intracytoplasmic sperm injection. Reprod Biol Endocrinol 11: 57, 2013. doi: 10.1186/1477-7827-11-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zera CA, Seely EW, Wilkins-Haug LE, Lim KH, Parry SI, McElrath TF. The association of body mass index with serum angiogenic markers in normal and abnormal pregnancies. Am J Obstet Gynecol 211: 247.e1–247.e7, 2014. doi: 10.1016/j.ajog.2014.03.020. [DOI] [PubMed] [Google Scholar]
- 99.Zhu L, Zhang Z, Zhang L, Shi Y, Qi J, Chang A, Gao J, Feng Y, Yang X. HMGB1-RAGE signaling pathway in severe preeclampsia. Placenta 36: 1148–1152, 2015. doi: 10.1016/j.placenta.2015.08.006. [DOI] [PubMed] [Google Scholar]