

Supplemental Digital Content is available in the text.
Keywords: antiretroviral therapy, clinical trials, HIV-1, pharmacogenomics, phenome-wide association studies
Abstract
Background
High-throughput approaches are increasingly being used to identify genetic associations across multiple phenotypes simultaneously. Here, we describe a pilot analysis that considered multiple on-treatment laboratory phenotypes from antiretroviral therapy-naive patients who were randomized to initiate antiretroviral regimens in a prospective clinical trial, AIDS Clinical Trials Group protocol A5202.
Participants and methods
From among 5 9545 294 polymorphisms imputed genome-wide, we analyzed 2544, including 2124 annotated in the PharmGKB, and 420 previously associated with traits in the GWAS Catalog. We derived 774 phenotypes on the basis of context from six variables: plasma atazanavir (ATV) pharmacokinetics, plasma efavirenz (EFV) pharmacokinetics, change in the CD4+ T-cell count, HIV-1 RNA suppression, fasting low-density lipoprotein-cholesterol, and fasting triglycerides. Permutation testing assessed the likelihood of associations being by chance alone. Pleiotropy was assessed for polymorphisms with the lowest P-values.
Results
This analysis included 1181 patients. At P less than 1.5×10−4, most associations were not by chance alone. Polymorphisms with the lowest P-values for EFV pharmacokinetics (CYPB26 rs3745274), low-density lipoprotein -cholesterol (APOE rs7412), and triglyceride (APOA5 rs651821) phenotypes had been associated previously with those traits in previous studies. The association between triglycerides and rs651821 was present with ATV-containing regimens, but not with EFV-containing regimens. Polymorphisms with the lowest P-values for ATV pharmacokinetics, CD4 T-cell count, and HIV-1 RNA phenotypes had not been reported previously to be associated with that trait.
Conclusion
Using data from a prospective HIV clinical trial, we identified expected genetic associations, potentially novel associations, and at least one context-dependent association. This study supports high-throughput strategies that simultaneously explore multiple phenotypes from clinical trials’ datasets for genetic associations.
Introduction
Access to safe and effective antiretroviral therapy (ART) is critical in the global response to the AIDS pandemic. Genetic polymorphisms in drug absorption, distribution, metabolism, and elimination (ADME) genes and off-target genes have convincingly been shown to be associated with adverse effects and/or pharmacokinetics of antiretroviral drugs including abacavir (ABC) 1, atazanavir (ATV) 2, dolutegravir 3, efavirenz (EFV) 4, etravirine 5, lopinavir 6, and nevirapine 7, and genetic screening to avoid ABC hypersensitivity reaction is now the standard of care in many resource-abundant countries.
Genome-wide association studies (GWAS) explore whether an individual trait (i.e. phenotype) associates with single-nucleotide polymorphisms (SNPs) across the genome. Only one phenotype is typically considered in a GWAS. The term ‘phenome’ describes the aggregate of many phenotypes in a given dataset. Phenome-wide association studies (PheWAS) complement GWAS by testing for genotype–phenotype associations across numerous phenotypes 8–12. A PheWAS may interrogate a single SNP against the phenome or may interrogate numerous SNPs simultaneously. Also unique to PheWAS is the ability to identify pleiotropy, whereby one SNP is found to be associated with multiple seemingly unrelated phenotypes 13,14.
Context-dependent genetic associations with antiretroviral drugs are well described. Failure to consider context may miss or underestimate important genetic associations. For EFV, some individuals with CYP2B6 slow metabolizer genotypes experience extremely high plasma EFV exposure only in the context of concomitant isoniazid 15–17. Among individuals with CYP2B6 slow metabolizer genotypes, the likelihood of EFV discontinuation for central nervous system side effects appears to be greater in the context of European versus African ancestry 18,19. For ATV, among individuals with UGT1A1 low expressor genotypes, the likelihood of bilirubin-related drug discontinuation is considerably greater in the context of European versus African ancestry 20. With nevirapine, among individuals with HLA risk alleles, severe cutaneous reactions occur largely when nevirapine is initiated in the context of higher CD4 T-cell counts 21.
Prospective clinical trials that randomized HIV-infected patients to initiate different antiretroviral regimens, and that involve extensive data collection, offer a special opportunity to apply a multiphenotype analytical approach focused on pharmacogenomics. We previously applied PheWAS to pretreatment (i.e. baseline) laboratory data National Institute of Health-funded AIDS Clinical Trials Group (ACTG) protocols 22. That analysis established that our analysis pipeline for studying multiple phenotypes is robust, with 20 polymorphisms replicating associations with identical or related phenotypes reported in the National Human Genome Research Institute – European Bioinformatics Institute GWAS Catalog 23, including several not reported previously in HIV-positive cohorts.
The present analyses explored associations with multiple on-treatment phenotypes from ACTG protocol A5202 24,25. We considered a total of 774 phenotypes representing ATV pharmacokinetics, CD4 T-cell count, EFV pharmacokinetics, fasting low-density lipoprotein (LDL) cholesterol, HIV-1 RNA, and fasting triglycerides, and that were derived by considering various contexts including sex, race/ethnicity, baseline age, baseline body mass index, baseline CD4+ T-cell count, baseline plasma HIV-1 RNA, randomized antiretroviral regimen, and component antiretroviral drug. These context-dependent phenotypes are useful in interpreting genome–phenome association results and highlight relationships of potential interest between these polymorphisms and phenotypes.
Participants and methods
Study participants
AIDS Clinical Trials Group protocol A5202 (ClinTrials.gov NCT00118898) was a phase IIIb equivalence study of four once-daily regimens for the initial treatment of HIV-1 infection. The primary results of A5202 have been reported previously 24,25. Patients enrolled from 2005 to 2007 were randomized to open-label ATV (300 mg) plus ritonavir (RTV, 100 mg) or EFV (600 mg) with either placebo-controlled ABC/lamivudine (3TC) (600 mg/300 mg) or tenofovir disoproxil fumarate/emtricitabine (TDF/FTC, 300 mg/200 mg). Study evaluations included laboratory testing at entry, at weeks 4, 8, 16, and 24, and every 12 weeks thereafter until the last enrolled patient was followed for 96 weeks. Analyses included A5202 participants who consented to provide DNA for genetic research under ACTG protocol A5128.
Phenotypes
For this analysis, we considered laboratory data from A5202 at entry and subsequent on-study weeks, and representing immunologic, virologic, metabolic, and pharmacologic domains. Immunologic phenotypes were derived from CD4+ T-cell count data, which are known to correlate with mortality on ART 26–30. Virologic phenotypes were derived from data on plasma HIV-1 RNA suppression to less than 200 copies/ml, which decreases transmission 31. Metabolic phenotypes were derived from data on fasting LDL cholesterol and fasting triglyceride levels, which are in the causal pathway to myocardial infarction 32,33. Pharmacologic phenotypes were derived from data on EFV and ATV pharmacokinetics, which relate to drug efficacy and toxicity 34–41.
We define the terms variable, primary phenotype, and subphenotype as follows: variables represent data without regard to study week or context (e.g. among all study patients, all fasting triglyceride data). Primary phenotypes are derived from variables while also considering study week but without regard to context (e.g. among all study patients, fasting triglycerides at baseline, at study weeks 24, 48, and 96, and change in fasting triglycerides from baseline to week 24, to 48, and to 96). Subphenotypes are derived from primary phenotypes while considering context (e.g. the fasting triglyceride primary phenotype noted above, but only among patients randomized to receive ATV/RTV).
Contexts for subphenotypes were defined as follows: categorical context included sex (male or female), self-identified race/ethnicity (White, Black, or Hispanic), randomized antiretroviral regimen (ATV+RTV+ABC/3TC, EFV+ABC/3TC, ATV+RTV+TDF/FTC, or EFV+TDF/FTC), and component antiretroviral drug (ATV+RTV, ABC/3TC, EFV, or TDF/FTC). Because ATV/RTV, ABC/3TC, and TDF/FTC were always prescribed as two-drug combinations, the component drugs could not be analyzed individually. For continuous baseline parameters, continuous context was derived on the basis of percentile cut-offs for age, BMI, CD4+ T-cell count, and plasma HIV-1 RNA (10, 25, 33, 50, 67, 75, and 90 percentile for each). With this approach, we generated a total of 774 primary phenotypes and subphenotypes for analysis, as listed in Supplemental Table 1 (Supplemental digital content 1, http://links.lww.com/FPC/B148).
For each primary phenotype, we examined frequency distribution plots and reviewed summary information, identified phenotypes requiring transformation to approximate normality to fulfill assumptions for linear regression, assured consistent units of measurement, and censored outliers judged to be biologically implausible.
Imputation and QC of genetic data
Patients from A5202 were genotyped with the Illumina 1M duo array as part of a previous immunogenomics project 42. The PLINK program and R statistical programming language were used for QC procedures 43,44. Polymorphisms were censored for call rates below 98%. After excluding 10 samples where genetically inferred sex differed from clinical data, or missing sex status that could not be inferred, 26 samples with overall genotyping call rates below 98%, and one sample with cryptic relatedness on the basis of identity-by-descent estimates of more than 0.3 from ~100 000 pruned SNPs, there were 1221 samples for imputation.
Post-QC data were imputed to 1000 genomes 45 after converting into genome build 37 using liftOver 46 and stratifying by chromosome to parallelize imputation processing. ShapeIt2 47 was used to check strand alignment and to phase data. The IMPUTE2 algorithm 48 was used to impute additional genotypes that were available in the 1000 genomes reference panel, but not directly genotyped. Each chromosome was segmented into 6 Mb regions with at least 3500 reference variants in each region. Imputed genotypes were included if posterior probabilities exceeded 0.9.
The quality of imputed data was assessed following the Electronic Medical Records and Genomics protocol 49. Each chromosome from each phase was checked for 100% concordance with genotyped data. We excluded imputed SNPs with imputation scores less than 0.3, genotyping call rates below 98%, and minor allele frequencies (MAF) less than 0.01.
Candidate polymorphisms for analysis
From the set of imputed SNPs, we included in this analysis only SNPs for which there was some a priori evidence of a pharmacogenetic association with any drug and phenotype on the basis of data from PharmGKB (Pharmacogenomics Knowledgebase 50). There were 2622 such SNPs in 761 genes that were annotated for a possible drug–phenotype association. Of these 2622 SNPs, we included in this analysis only a subset of 2124 SNPs that were also represented in the imputed, post-QC genome-wide data.
In addition to PharmGKB SNPs, from the set of imputed SNPs, we also included SNPs for which previous GWAS had shown evidence of association with any lipid-related trait with a P-value of less than 10−8, as represented in the GWAS Catalog SNPs 23, which includes results from published GWAS fulfilling catalog criteria 51. There were 447 such SNPs, of which we included in this analysis only a subset of 420 SNPs that were also represented in the imputed, post-QC genome-wide data. A total of 2544 SNPs were included in the analysis (listed in Supplemental Table 2, Supplemental digital content 2, http://links.lww.com/FPC/B149).
Statistical analysis
When linked with available laboratory phenotypes, the final analysis dataset included 1181 patients, 2124 PharmGKB SNPs, and 420 GWAS Catalog SNPs. Using the R statistical package, continuous phenotypes were modeled with linear regression and the dichotomous phenotype with logistic regression 44. The first three principal components, calculated using EIGENSOFT 52, were used to adjust for global ancestry. Each analysis was also adjusted for sex and age. Consideration of context resulted in models of varying sample sizes. For models with at least 100 patients, we excluded SNPs with MAF of less than 0.05. For models with fewer than 100 patients, we excluded SNPs with MAF of less than 0.10. We did not infer or impute missing laboratory data.
Permutation testing was used to empirically derive P-value cut-offs (PPT) 53. Briefly, within the analysis dataset, we permuted the connection between genotype and phenotype data. This randomly matches each patient’s genotypes to another patient’s phenotypes, while preserving relationships between genotypes (e.g. linkage disequilibrium) and between phenotypes (e.g. correlations). Permutation was repeated 1000 times, each generating a new dataset. We then carried out the association analysis on each of the 1000 datasets, from which we determined, at various P-value cut-offs, the average number of SNPs per analysis that pass that cut-off in the permuted data (i.e. by chance alone). We compared this average number with the actual number of SNPs that passed that same cut-off in the unpermuted data. This yields probabilities that SNP–phenotype associations at any given P-value threshold in the unpermuted data were by chance alone. Our approach differs from a more traditional permutation approach that would calculate permuted P-values for each association test, the latter permutation approach being computationally prohibitive.
Results
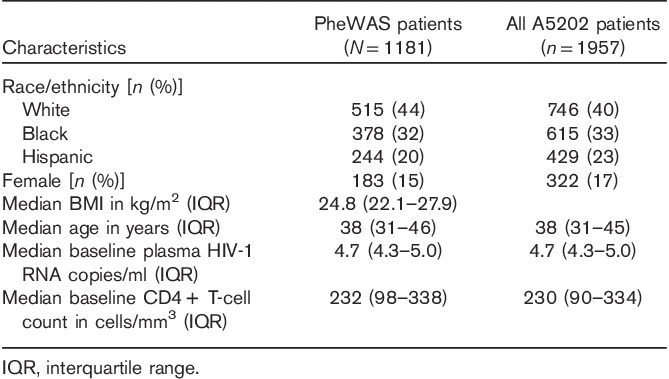
This multiphenotype analysis included data from 1181 patients from A5202, who had consented to provide DNA for genetic research under ACTG protocol A5128. The characteristics of the study patients are presented in Table 1. The characteristics of patients included in the analysis generally reflected the characteristics of all A5202 study patients. From the available baseline and subsequent on-study data, a total of 774 phenotypes were derived for analysis as described under ‘participants and methods’ section. These comprised 19 primary phenotypes as well as 755 subphenotypes that were derived on the basis of baseline age, sex, race/ethnicity, BMI, CD4+ T-cell count, plasma HIV-1 RNA, randomized antiretroviral regimen, and component antiretroviral drug. This generated 68 phenotypes for ATV pharmacokinetics, 84 for CD4 T-cell count, 34 for EFV pharmacokinetics, 252 for fasting LDL cholesterol, 84 for HIV-1 RNA, and 252 for fasting triglycerides. Definitions for each of the 774 phenotypes are provided in Supplemental Table 2 (Supplemental digital content 2, http://links.lww.com/FPC/B149).
Table 1.
Baseline characteristics of study patients included in phenome-wide association studies
From the imputed genome-wide genotype data on these study patients, a total of 2501 SNPs (which were represented in either PharmGKB or the GWAS Catalog) provided at least one association result with at least one phenotype in this analysis. As noted in ‘participants and methods’ section, we excluded SNPs with MAF of less than 0.05 from models with at least 100 patients and SNPs with MAF of less than 0.10 from models with fewer than 100 patients. A total of 1 773 707 SNP–phenotype pairs provided P-values for association.
To assess the likelihood that associations were by chance alone, permutation testing was used to empirically derive PPT to determine the probability that SNP–phenotype associations in the unpermuted data were by chance alone, as described in ‘participants and methods’ section. For example, at PPT less than 1.5×10−4, 50% of SNP–phenotype pairs in this analysis are likely not by chance alone (Fig. 1). Of the 1 773 707 SNP–phenotype pairs noted above, P-values for 737 (0.04%) were less than this PPT threshold. The number of patients included in each model ranged from 18 (e.g. for EFV concentrations in patients younger than 26 years of age) to 1080 (e.g. for HIV-1 RNA response at 48 weeks), with a median of 242 patients per model.
Fig. 1.
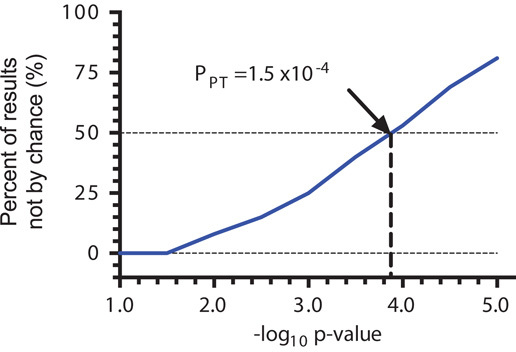
Empirically derived P-values on the basis of permutation testing. Permutation testing was used to empirically derive P-value cut-offs (PPT). Briefly, within the dataset used for association analysis, we permuted the connection between genotype and phenotype data. Permutation was repeated 1000 times, each generating a new dataset. We then carried out analyses on each of the 1000 datasets, from which we determined, at various P-value cut-offs, the average number of single nucleotide polymorphisms (SNPs) per analysis that pass that cut-off in the permuted data. We compared this average number with the actual number of SNPs that passed that same cut-off in the unpermuted data, providing an empiric determination of the probability that SNP–phenotype associations in the unpermuted data were by chance alone.
Within each phenotype domain, association results for the five SNPs with the lowest P-value with at least one phenotype are presented in Table 2. For EFV pharmacokinetics, fasting LDL-cholesterol, and fasting triglyceride phenotypes, the SNP with the lowest P-value had previously been associated with that trait at P less than 5.0×10−8 in at least one GWAS 4,54. For the five SNPs with the lowest P-values in EFV pharmacokinetics, fasting LDL-cholesterol, and fasting triglyceride domains (15 SNPs total), Manhattan plots for associations between each SNP and as many as 774 phenotypes across all six domains are shown in Fig. 2.
Table 2.
Association results for the five lowest P-value single nucleotide polymorphisms within each phenotype domain
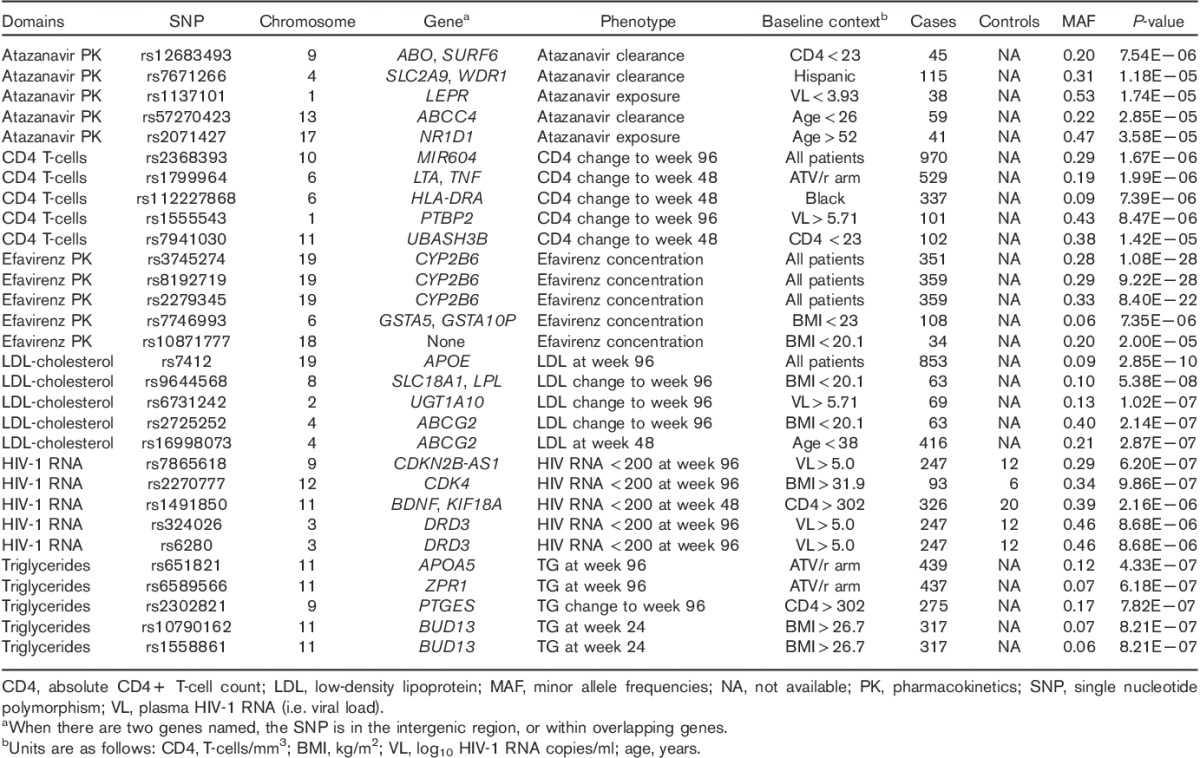
Fig. 2.
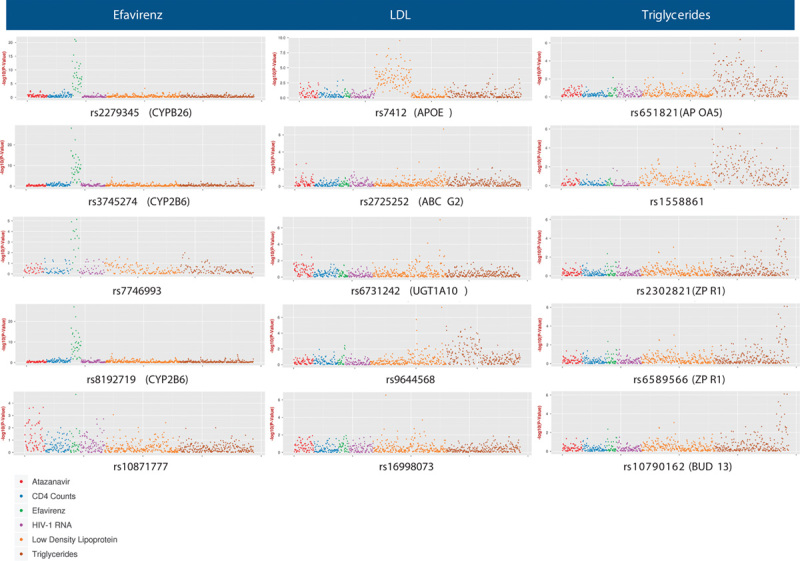
Manhattan plots representing all phenotype associations for the five single nucleotide polymorphisms (SNPs) with the lowest P-values for efavirenz pharmacokinetic, fasting low-density lipoprotein (LDL) cholesterol, and fasting triglyceride phenotypes. We analyzed SNPs that were annotated previously for any drug in the PharmGKB or associated previously with any trait in the GWAS Catalog, and that were also represented in the imputed, post-QC genome-wide data. Each marker represents, for each phenotype, the –log10 P-value for association with the indicated SNP. Color-coded phenotype categories are indicated at bottom left of figure. Note that the scale of the Y-axis differs between plots.
For EFV concentrations, the lowest P-value was with rs3745274 (P=1.1×10−28) among all 351 patients with evaluable data, but rs3745274 was also associated with numerous other context-derived EFV subphenotypes (Fig. 2). Log10 P-values for association between rs3745274 and EFV concentrations correlated very strongly with sample size in the model (Spearman’s ρ=0.95, P<0.0001), suggesting that this genetic association was present irrespective of context (i.e. sex, race/ethnicity, randomized antiretroviral regimen, component antiretroviral drug, baseline age, BMI, CD4+ T-cell count, and plasma HIV-1 RNA). In contrast, an association between rs10871777 and EFV concentration (P=2.0×10−5) was only observed among 34 individuals with baseline BMI in the lowest 10th percentile, but not among 80 individuals with BMI in the lowest 25th percentile (P=0.01), nor among 108 individuals with BMI in the lowest 33rd percentile (P=0.25) (Fig. 2). Furthermore, there was no hint of association between rs10871777 and EFV concentration within any other decile of BMI (i.e. 10th to 20th decile, 20th to 30th decile, etc.), considering both P-values and β coefficients (data not shown).
For fasting LDL-cholesterol, the lowest P-value was between rs7412 in APOE and week 96 LDL-cholesterol among all 853 evaluable patients. As shown in Fig. 2, rs7412 was associated with numerous context-derived LDL-cholesterol phenotypes. Associations were only with absolute values of LDL-cholesterol at individual study weeks, not with LDL-cholesterol change from baseline. Log10 P-values for association between rs7412 and LDL-cholesterol correlated directly with sample size in the model (Spearman’s ρ=0.64, P<0.0001), without strong evidence for context dependence. For example, rs7412 was associated with week 96 LDL-cholesterol among patients randomized to either ATV/RTV-containing ART (n=419, P=2.0×10−7) or to EFV-containing ART (n=435, P=2.7×10−4). In addition, rs9644568 (near LPL) was associated with LDL-cholesterol change to week 96 among 63 individuals with baseline BMI in the lowest 10th percentile, but few other LDL-cholesterol phenotypes, but was more broadly associated with triglyceride phenotypes. In contrast, an association between rs16998073 and LDL-cholesterol at week 48 (P=2.9×10−7) was only at week 48 among 416 individuals in the lower 50th percentile for age (less than 38 years), but less so among individuals in the lowest 33rd percentile for age (n=270; P=3.6×10−3) or in the top 33rd percentile for age (n=277; P=0.042) (Fig. 2).
For fasting triglycerides, the lowest P-value was between rs651821 in APOA5 and week 96 triglycerides among 439 individuals randomized to the ATV/RTV-containing ART. As shown in Fig. 2, rs651821 was associated with numerous context-derived triglyceride subphenotypes, including both absolute values at individual study weeks and change from baseline. Although log10 P-values for associations between rs651821 and triglycerides tended to correlate with phenotype sample size (Spearman’s ρ=0.42, P<0.0001), there was some evidence for context dependence. For example, rs651821 was associated with week 96 triglycerides among patients randomized to ATV/RTV-containing ART (n=439, P=4.3×10−7), but not EFV-containing ART (n=543, P=0.24). Furthermore, among patients randomized to ATV/RTV-containing ART, this association between rs651821 and week 96 triglycerides was also observed with concomitant TDF/FTC (n=219, P=2.3×10−4), with concomitant ABC/3TC (n=221, P=2.5×10−4), and was also observed at week 48 (n=481, P=3.2×10−4). Among patients randomized to EFV-containing ART, an association between rs651821 and week 96 triglycerides was also absent with concomitant TDF/FTC (n=224, P=0.04), with concomitant ABC/3TC (n=230, P=0.86), and at week 48 (n=488, P=0.02). In contrast, among individuals with baseline CD4 count of more than 302, there was an association between rs2302821 and change in triglycerides at week 96 (n=275, P=7.8×10−7), but not at week 48 (n=296, P=0.45).
For ATV pharmacokinetics, CD4 T-cell count, and HIV-1 RNA phenotypes, the SNP with the lowest P-value had not been reported previously to be associated with that trait (Table 2). For the five SNPs with the lowest P-values in ATV pharmacokinetics, CD4 T-cell count, and HIV-1 RNA domains (15 SNPs total), Manhattan plots for associations between each SNP and as many as 774 phenotypes across all six domains are shown in Fig. 3. For ATV pharmacokinetics, the lowest P-value was with ATV clearance among patients with baseline CD4 T-cell count of less than 23 cells/mm3 (n=45), and was with rs12683493, which is intergenic between ABO and SURF6 (P=7.5×10−6). For CD4 T-cells, the lowest P-value was with change in patients CD4 T-cell count from baseline to week 96 among all patients (n=970) and was with rs2368393 in both MIR604 and SVIL (P=1.7×10−6). For HIV-1 RNA, the lowest P-value was with HIV-1 RNA control at week 96 among patients with baseline HIV-1 RNA of more than 5.0 log10 copies/ml (n=247) and was with rs7865618 in CDKN2B-AS1 (P=6.2×10−7).
Fig. 3.
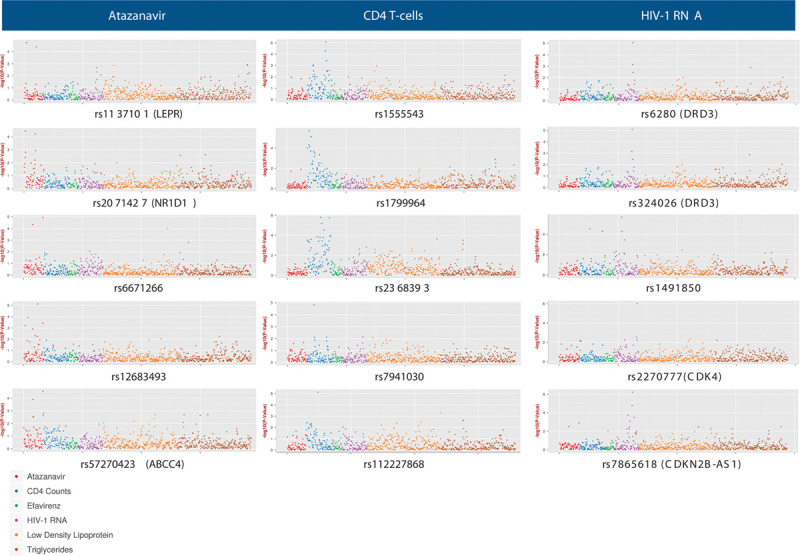
Manhattan plots representing all phenotype associations for the five single nucleotide polymorphisms (SNPs) with the lowest P-values for atazanavir pharmacokinetic, HIV-1 RNA, and CD4 T-cell phenotypes. We analyzed SNPs that were annotated previously for any drug in the PharmGKB or previously associated with any trait in the GWAS Catalog, and that were also represented in the imputed, post-QC genome-wide data. Each marker represents, for each phenotype, the –log10 P-value for association with the indicated SNP. Color-coded phenotype categories are indicated at the bottom left of the figure. Note that the scale of the Y-axis differs between plots.
Discussion
Phenome-wide association studies typically rely on observational data collected from electronic medical records, which may be subject to variability in the timing and completeness of data collection. The present multiphenotype analysis is unique in that it is the first to explore on-treatment data from a prospective clinical trial. The collection of specific data elements at predetermined intervals before and after initiation of therapy makes clinical trials an attractive resource of structured longitudinal data to evaluate pharmacogenomic associations.
The present study characterized associations between 2544 SNPs from the PharmGKB and the GWAS Catalog and 774 context-derived phenotypes among 1181 HIV-infected participants from ACTG protocol A5202. Several associations replicated previous reports. We readily replicated the known association between CYP2B6 variants and plasma EFV concentrations 4,55,56. The lowest P-value was with rs3745274, which was associated with numerous context-derived EFV phenotypes. This genetic association appeared to persist irrespective of context (i.e. sex, race/ethnicity, randomized antiretroviral regimen, component antiretroviral drug, baseline age, BMI, CD4+ T-cell count, and plasma HIV-1 RNA). In contrast, the association between EFV concentration and rs10871777 (an SNP previously associated with obesity 57) is very likely spurious, as this was only observed among individuals with baseline BMI in the lowest 10th percentile.
For LDL-cholesterol, rs7412 in APOE has been associated with LDL-cholesterol levels in previous GWAS 54,58, and was associated with numerous context-derived LDL-cholesterol phenotypes in this analysis. However, it was only associated with absolute values of LDL-cholesterol at individual study weeks, not with LDL-cholesterol change from baseline. In addition, there was no strong evidence for context dependence. An advantage of PheWAS is the ability to detect pleiotropy. In this respect, although rs9644568 (near LPL) was associated with LDL-cholesterol change at week 96 among 63 individuals with baseline BMI in the lowest 10th percentile, but very few other LDL-cholesterol phenotypes, it was associated more with numerous triglyceride phenotypes, consistent with its previously reported association with triglycerides in GWAS 59. In contrast, an association between LDL-cholesterol and rs16998073 (an SNP associated previously with diastolic blood pressure 60) is very likely spurious as an association was observed only at week 48 among individuals in the lower 50th percentile for age.
For triglycerides, rs651821 in APOA5 has been associated with triglycerides in previous GWAS 61, as have three of our other top five SNPs (rs6589566 62; rs10790162 63; and rs1558861 64). The SNP rs651821 was associated with numerous context-derived triglyceride phenotypes, representing both absolute values at individual study weeks and change from baseline. In addition, there was some evidence that this association was context dependent, with rs651821 associated with week 96 LDL-cholesterol among patients randomized to ATV/RTV-containing ART, but not EFV-containing ART. An association between and change in triglycerides and rs2302821 (an SNP associated weakly with cardiovascular toxicity in patients treated with celecoxib 60) is very likely spurious, as this was observed in a phenotype at week 96, but not at week 48.
In contrast to the above findings, SNPs with the lowest P-values for association with ATV pharmacokinetics, CD4 T-cell count, and HIV-1 RNA phenotypes had not been reported previously to be associated with that trait. The validity of multiple other associations, such as those between ATV pharmacokinetics and rs12683493 (intergenic between ABO and SURF6, in a haplotype implicated in cough with enalapril 65), change in CD4 T-cell count from baseline and rs2368393 (in both MIR604 and SVIL, not associated with risk of drug toxicity in children with lymphoblastic leukemia–lymphoma 66), and HIV-1 RNA control and rs7865618 (in CDKN2B-AS1, associated with cardiovascular disease 67 and glaucoma in GWAS 68) may be spurious. Replication for these and other SNP associations (many of which may be spurious) in independent cohorts is warranted.
In this analysis, we used context as a strategy to derive multiple subphenotypes from each primary phenotype. Our rationale is that genetic associations for a given SNP–trait association may differ depending on context; thus, we expect that context-dependent associations may be more readily identified and understood using this approach. One advantage of this approach is that it allows for a very granular exploration of SNP–genotype associations that may be influenced by context. We found such context dependence in the association between rs651821 and triglyceride phenotypes among patients randomized to ATV/RTV-containing ART regimens, but not among patients randomized to EFV-containing ART regimens. The random assignment of A5202 participants to receive either ATV/RTV-containing or EFV-containing ART decreases the likelihood that our finding was because of confounding as unrecognized confounders should be equally distributed across arms. This is an advantage of using clinical trials datasets for genetic association analyses. Another advantage of granular exploration of SNP–genotype associations is the ability to discern associations that are almost certainly spurious by comparing strengths of association between closely related phenotypes. For example, among individuals with baseline CD4 count of more than 302, the association between rs2302821 and change in triglycerides at week 96 (P=7.8×10−7) is almost certainly spurious as there was no such association at week 48 (P=0.45).
Multiple hypothesis testing is inherent in approaches that examine multiple phenotypes such as PheWAS, but Bonferoni correction is not appropriate because the assumption that tests are independent is violated. To address this issue, we performed 1000 permutations of the analysis to create an empirical null distribution. This showed that in the present analysis, the majority of models with P-value less than 1.5×10−4 were unlikely to be by chance alone.
This analysis leveraged extensive a priori knowledge of genes and phenotypes from PharmGKB and the GWAS Catalog. This increases the likelihood of validity, biological plausibility, and supportive publications. As was apparent in both our previous PheWAS 22 and the present study, disease-specific knowledge is useful in interpreting genetic associations and to prioritize associations for further replication and study. Because every phenome is unique, analyses that consider large numbers of phenotypes may benefit more so than GWAS from disease-specific knowledge and understanding, including relationships among phenotypes.
This study had limitations. A larger sample size may have shown additional associations and we have not yet sought to replicate associations in other datasets. We considered a limited number of phenotypes and contexts. We considered ART as intent to treat. We only used available SNPs that were available or imputed from genome-wide genotyping, without additional genotyping. We also focused analyses on individual SNPs, whereas multiple SNPs considered in combination may more strongly associate with some phenotypes. Data from prospective, randomized clinical trials offer distinct advantages (e.g. randomization tends to evenly distribute covariates across study arms), but there are limitations. Although data of ACTG clinical trials are rigorously collected and validated, electronic medical records datasets will likely contain a wider range of variables. In addition, eligibility criteria for clinical trials may exclude some individuals who would otherwise be included in electronic medical records datasets.
In summary, this pilot study supports a multiphenotype analysis strategy to explore clinical trials datasets for genetic associations and to ultimately identify genetic associations with the potential to optimize ART safety and efficacy. This approach complements more established GWAS by performing simultaneous calculations for identifying genotype–phenotype associations across numerous phenotypes. Work is ongoing to further evaluate and optimize multiphenotype analyses for clinical trials datasets. On the basis of results from this pilot study, we plan to extend the PheWAS approach both to a much more extensive set of traits and to multiple other clinical trials datasets. This will include replication of associations identified here.
Supplementary Material
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website (www.pharmacogeneticsandgenomics.com).
Acknowledgements
The authors are grateful to the many patients with HIV infection who volunteered for A5202 and A5128. In addition, they acknowledge the contributions of study teams and site staff for these protocols. The authors thank Paul J. McLaren, PhD (Public Health Agency of Canada, Winnipeg, Canada), for previous involvement and collaborations that used these genome-wide genotype data.
The project described was supported by Award Number U01AI068636 from the National Institute of Allergy and Infectious Diseases (NIAID) and supported by the National Institute of Mental Health (NIMH) and the National Institute of Dental and Craniofacial Research (NIDCR). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. This work was supported by the AIDS Clinical Trials Group funded by the National Institute of Allergy and Infectious Diseases (AI068636, AI038858, AI068634, AI038855). Grant support included AI077505, AI069439, TR000445, AI054999, AI110527 (D.W.H.), BRS-ACURE-06-00140-T001 (G.D.M.), and AI108355 (C.V.).
Clinical Research Sites that participated in ACTG protocol A5202, and collected DNA under protocol A5128, were supported by the following grants from NIAID: AI069477, AI027675, AI073961, AI069474, AI069432, AI069513, AI069423, AI050410, AI069452, AI069450, AI054907, AI069428, AI069439, AI069467, AI045008, AI069495, AI069415, AI069556, AI069484, AI069424, AI069532, AI069419, AI069471, AI025859, AI069418, AI050409, AI069423, AI069501, AI069502, AI069511, AI069434, AI069465, AI069494, AI069472, AI069470, AI046376, AI072626, AI027661, AI034853, AI069447, AI032782, AI027658, AI27666, AI058740, and AI046370, and by the following grants from the National Center for Research Resources (NCRR): RR00051, RR00046, RR025747, RR025777, RR024160, RR024996, RR024156, RR024160, and RR024160. Study drugs were provided by Bristol-Myers Squibb Co., Gilead Sciences, and GlaxoSmithKline, Inc.
Conflicts of interest
Eric S. Daar has been principal investigator on research grants to Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center from Gilead, Merck, and ViiV, and a consultant/advisor to Bristol Myers Squibb, Gilead, Janssen, Merck, Teva, and ViiV. For the remaining authors there are no conflicts of interest.
References
- 1.Mallal S, Phillips E, Carosi G, Molina JM, Workman C, Tomazic J, et al. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med 2008; 358:568–579. [DOI] [PubMed] [Google Scholar]
- 2.Rotger M, Taffe P, Bleiber G, Gunthard HF, Furrer H, Vernazza P, et al. Gilbert syndrome and the development of antiretroviral therapy-associated hyperbilirubinemia. J Infect Dis 2005; 192:1381–1386. [DOI] [PubMed] [Google Scholar]
- 3.Chen S, St, Jean P, Borland J, Song I, Yeo AJ, Piscitelli S, et al. Evaluation of the effect of UGT1A1 polymorphisms on dolutegravir pharmacokinetics. Pharmacogenomics 2014; 15:9–16. [DOI] [PubMed] [Google Scholar]
- 4.Holzinger ER, Grady B, Ritchie MD, Ribaudo HJ, Acosta EP, Morse GD, et al. Genome-wide association study of plasma efavirenz pharmacokinetics in AIDS Clinical Trials Group protocols implicates several CYP2B6 variants. Pharmacogenet Genom 2012; 22:858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kakuda T, Nijs S, van Hoecke G. Pharmacokinetics of etravirine according to CYP2C9 and CYP2C19 metabolizer status: a meta-analysis of phase I trials. Presented at 20th conference on retroviruses and opportunistic infections, Atlanta, GA. February; 2013.
- 6.Lubomirov R, di Iulio J, Fayet A, Colombo S, Martinez R, Marzolini C, et al. ADME pharmacogenetics: investigation of the pharmacokinetics of the antiretroviral agent lopinavir coformulated with ritonavir. Pharmacogenet Genomics 2010; 20:217–230. [DOI] [PubMed] [Google Scholar]
- 7.Yuan J, Guo S, Hall D, Cammett AM, Jayadev S, Distel M, et al. Toxicogenomics of nevirapine-associated cutaneous and hepatic adverse events among populations of African, Asian, and European descent. AIDS 2011; 25:1271–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pendergrass SA, Brown-Gentry K, Dudek S, Frase A, Torstenson ES, Goodloe R, et al. Phenome-wide association study (PheWAS) for detection of pleiotropy within the Population Architecture using Genomics and Epidemiology (PAGE) Network. PLoS Genet 2013; 9:e1003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pendergrass SA, Brown-Gentry K, Dudek SM, Torstenson ES, Ambite JL, Avery CL, et al. The use of phenome-wide association studies (PheWAS) for exploration of novel genotype-phenotype relationships and pleiotropy discovery. Genet Epidemiol 2011; 35:410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pendergrass SA, Verma A, Okula A, Hall MA, Crawford DC, Ritchie MD. Phenome-wide association studies: embracing complexity for discovery. Hum Hered 2015; 79:111–123. [DOI] [PubMed] [Google Scholar]
- 11.Pendergrass SA, Ritchie MD. Phenome-wide association studies: leveraging comprehensive phenotypic and genotypic data for discovery. Curr Genet Med Rep 2015; 3:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bush WS, Oetjens MT, Crawford DC. Unravelling the human genome-phenome relationship using phenome-wide association studies. Nat Rev Genet 2016; 17:129–145. [DOI] [PubMed] [Google Scholar]
- 13.Tyler AL, Crawford DC, Pendergrass SA. The detection and characterization of pleiotropy: discovery, progress, and promise. Brief Bioinform 2016; 17:13–22. [DOI] [PubMed] [Google Scholar]
- 14.Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW. Pleiotropy in complex traits: challenges and strategies. Nat Rev Genet 2013; 14:483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwara A, Lartey M, Sagoe KW, Court MH. Paradoxically elevated efavirenz concentrations in HIV/tuberculosis-coinfected patients with CYP2B6 516TT genotype on rifampin-containing antituberculous therapy. AIDS 2011; 25:388–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McIlleron HM, Schomaker M, Ren Y, Sinxadi P, Nuttall JJ, Gous H, et al. Effects of rifampin-based antituberculosis therapy on plasma efavirenz concentrations in children vary by CYP2B6 genotype. AIDS 2013; 27:1933–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luetkemeyer AF, Rosenkranz SL, Lu D, Grinsztejn B, Sanchez J, Ssemmanda M, et al. Combined effect of CYP2B6 and NAT2 genotype on plasma efavirenz exposure during rifampin-based antituberculosis therapy in the STRIDE study. Clin Infect Dis 2015; 60:1860–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ribaudo HJ, Liu H, Schwab M, Schaeffeler E, Eichelbaum M, Motsinger-Reif AA, et al. Effect of CYP2B6, ABCB1, and CYP3A5 polymorphisms on efavirenz pharmacokinetics and treatment response: an AIDS Clinical Trials Group study. J Infect Dis 2010; 202:717–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leger P, Chirwa S, Turner M, Richardson DM, Baker P, Leonard M, et al. Pharmacogenetics of efavirenz discontinuation for reported central nervous system symptoms appears to differ by race. Pharmacogenet Genomics 2016; 26:473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vardhanabhuti S, Ribaudo HJ, Landovitz RJ, Ofotokun I, Lennox JL, Currier JS, et al. Screening for UGT1A1 genotype in study A5257 would have markedly reduced premature discontinuation of atazanavir for hyperbilirubinemia. Open Forum Infect Dis 2015; 2:ofv085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin AM, Nolan D, James I, Cameron P, Keller J, Moore C, et al. Predisposition to nevirapine hypersensitivity associated with HLA-DRB1*0101 and abrogated by low CD4 T-cell counts. AIDS 2005; 19:97–99. [DOI] [PubMed] [Google Scholar]
- 22.Moore CB, Verma A, Pendergrass S, Setia S, Johnson DH, Daar ES, et al. Phenome-wide associations study (PheWAS) relating pre-treatment laboratory parameters with human genetic variants in AIDS clinical trials group protocols. Open Forum Infect Dis 2014; 2:ofu113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.NHGRI-EBI. GWAS Catalog – The NHGRI-EBI Catalog of published genome-wide association studies. Available at: https://www.ebi.ac.uk/gwas/. [Accessed 1 September 2016].
- 24.Sax PE, Tierney C, Collier AC, Fischl MA, Mollan K, Peeples L, et al. Abacavir–lamivudine versus tenofovir–emtricitabine for initial HIV-1 therapy. N Engl J Med 2009; 361:2230–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daar ES, Tierney C, Fischl MA, Sax PE, Mollan K, Budhathoki C, et al. Atazanavir plus ritonavir or efavirenz as part of a 3-drug regimen for initial treatment of HIV-1. Ann Intern Med 2011; 154:445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baker JV, Peng G, Rapkin J, Abrams DI, Silverberg MJ, MacArthur RD, et al. CD4+ count and risk of non-AIDS diseases following initial treatment for HIV infection. AIDS 2008; 22:841–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Lelyveld SF, Gras L, Kesselring A, Zhang S, De Wolf F, Wensing AM, et al. Long-term complications in patients with poor immunological recovery despite virological successful HAART in Dutch ATHENA cohort. AIDS 2012; 26:465–474. [DOI] [PubMed] [Google Scholar]
- 28.Marin B, Thiebaut R, Bucher HC, Rondeau V, Costagliola D, Dorrucci M, et al. Non-AIDS-defining deaths and immunodeficiency in the era of combination antiretroviral therapy. AIDS 2009; 23:1743–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baker JV, Peng G, Rapkin J, Krason D, Reilly C, Cavert WP, et al. Poor initial CD4+ recovery with antiretroviral therapy prolongs immune depletion and increases risk for AIDS and non-AIDS diseases. JAIDS 2008; 48:541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tenorio AR, Zheng Y, Bosch RJ, Krishnan S, Rodriguez B, Hunt PW, et al. Soluble markers of inflammation and coagulation but not T-cell activation predict non-AIDS-defining morbid events during suppressive antiretroviral treatment. J Infect Dis 2014; 210:1248–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, Kumarasamy N, et al. Prevention of HIV-1 infection with early antiretroviral therapy. N Engl J Med 2011; 365:493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cholesterol Treatment Trialists C, Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet 2012; 380:581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet 2013; 45:1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gutierrez F, Navarro A, Padilla S, Anton R, Masia M, Borras J, et al. Prediction of neuropsychiatric adverse events associated with long-term efavirenz therapy, using plasma drug level monitoring. Clin Infect Dis 2005; 41:1648–1653. [DOI] [PubMed] [Google Scholar]
- 35.Gounden V, van Niekerk C, Snyman T, George JA. Presence of the CYP2B6 516G> T polymorphism, increased plasma efavirenz concentrations and early neuropsychiatric side effects in South African HIV-infected patients. AIDS Res Ther 2010; 7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clifford DB, Evans S, Yang Y, Acosta EP, Goodkin K, Tashima K, et al. Impact of efavirenz on neuropsychological performance and symptoms in HIV-infected individuals. Ann Intern Med 2005; 143:714–721. [DOI] [PubMed] [Google Scholar]
- 37.Rotger M, Colombo S, Furrer H, Bleiber G, Buclin T, Lee BL, et al. Influence of CYP2B6 polymorphism on plasma and intracellular concentrations and toxicity of efavirenz and nevirapine in HIV-infected patients. Pharmacogenet Genom 2005; 15:1–5. [DOI] [PubMed] [Google Scholar]
- 38.Marzolini C, Telenti A, Decosterd LA, Greub G, Biollaz J, Buclin T. Efavirenz plasma levels can predict treatment failure and central nervous system side effects in HIV-1-infected patients. AIDS 2001; 15:71–75. [DOI] [PubMed] [Google Scholar]
- 39.Gallego L, Barreiro P, del Rio R, Gonzalez de Requena D, Rodriguez-Albarino A, Gonzalez-Lahoz J, et al. Analyzing sleep abnormalities in HIV-infected patients treated with Efavirenz. Clin Infect Dis 2004; 38:430–432. [DOI] [PubMed] [Google Scholar]
- 40.Gandhi M, Ameli N, Bacchetti P, Anastos K, Gange SJ, Minkoff H, et al. Atazanavir concentration in hair is the strongest predictor of outcomes on antiretroviral therapy. Clin Infect Dis 2011; 52:1267–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson DH, Venuto C, Ritchie MD, Morse GD, Daar ES, McLaren PJ, et al. Genomewide association study of atazanavir pharmacokinetics and hyperbilirubinemia in AIDS Clinical Trials Group protocol A5202. Pharmacogenet Genom 2014; 24:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 2010; 330:1551–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Team RDC. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2011. [Google Scholar]
- 45.1000 Genomes Project Consortium, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature 2010; 467:1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.liftOver. Lift genome annotations. Available at: http://genome.ucsc.edu/cgi-bin/hgLiftOver. [Accessed 1 September 2016].
- 47.Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods 2013; 10:5–6. [DOI] [PubMed] [Google Scholar]
- 48.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009; 5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verma SS, de Andrade M, Tromp G, Kuivaniemi H, Pugh E, Namjou B, et al. Imputation and quality control steps for combining multiple genome-wide datasets. Front Genet 2014; 5:370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.PharmGKB – The Pharmacogenomics Knowledgebase. Available at: https://www.pharmgkb.org/; 2016. [Acecessed 1 September 2016].
- 51.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA 2009; 106:9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Price A. EIGENSOFT. Available at: https://www.hsph.harvard.edu/alkes-price/software/. [Accessed 14 December 2016].
- 53.Good PI. Permutation, parametric and bootstrap tests of hypotheses: a practical guide to resampling methods for testing hypotheses 2004. 3rd ed New York, NY: Springer. [Google Scholar]
- 54.Kettunen J, Tukiainen T, Sarin AP, Ortega-Alonso A, Tikkanen E, Lyytikainen LP, et al. Genome-wide association study identifies multiple loci influencing human serum metabolite levels. Nat Genet 2012; 44:269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rotger M, Tegude H, Colombo S, Cavassini M, Furrer H, Decosterd L, et al. Predictive value of known and novel alleles of CYP2B6 for efavirenz plasma concentrations in HIV-infected individuals. Clin Pharmacol Ther 2007; 81:557–566. [DOI] [PubMed] [Google Scholar]
- 56.Haas DW, Ribaudo HJ, Kim RB, Tierney C, Wilkinson GR, Gulick RM, et al. Pharmacogenetics of efavirenz and central nervous system side effects: an adult AIDS Clinical Trials Group study. AIDS 2004; 18:2391–2400. [PubMed] [Google Scholar]
- 57.Berndt SI, Gustafsson S, Magi R, Ganna A, Wheeler E, Feitosa MF, et al. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat Genet 2013; 45:501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rasmussen-Torvik LJ, Pacheco JA, Wilke RA, Thompson WK, Ritchie MD, Kho AN, et al. High density GWAS for LDL cholesterol in African Americans using electronic medical records reveals a strong protective variant in APOE. Clin Transl Sci 2012; 5:394–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weissglas-Volkov D, Aguilar-Salinas CA, Nikkola E, Deere KA, Cruz-Bautista I, Arellano-Campos O, et al. Genomic study in Mexicans identifies a new locus for triglycerides and refines European lipid loci. J Med Genet 2013; 50:298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet 2009; 41:666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tan A, Sun J, Xia N, Qin X, Hu Y, Zhang S, et al. A genome-wide association and gene–environment interaction study for serum triglycerides levels in a healthy Chinese male population. Hum Mol Genet 2012; 21:1658–1664. [DOI] [PubMed] [Google Scholar]
- 62.Coram MA, Duan Q, Hoffmann TJ, Thornton T, Knowles JW, Johnson NA, et al. Genome-wide characterization of shared and distinct genetic components that influence blood lipid levels in ethnically diverse human populations. Am J Hum Genet 2013; 92:904–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kraja AT, Vaidya D, Pankow JS, Goodarzi MO, Assimes TL, Kullo IJ, et al. A bivariate genome-wide approach to metabolic syndrome: STAMPEED consortium. Diabetes 2011; 60:1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kooner JS, Chambers JC, Aguilar-Salinas CA, Hinds DA, Hyde CL, Warnes GR, et al. Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nat Genet 2008; 40:149–151. [DOI] [PubMed] [Google Scholar]
- 65.Luo JQ, He FZ, Luo ZY, Wen JG, Wang LY, Sun NL, et al. Rs495828 polymorphism of the ABO gene is a predictor of enalapril-induced cough in Chinese patients with essential hypertension. Pharmacogenet Genomics 2014; 24:306–313. [DOI] [PubMed] [Google Scholar]
- 66.Lopez-Lopez E, Gutierrez-Camino A, Pinan MA, Sanchez-Toledo J, Uriz JJ, Ballesteros J, et al. Pharmacogenetics of microRNAs and microRNAs biogenesis machinery in pediatric acute lymphoblastic leukemia. PLoS One 2014; 9:e91261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wild PS, Zeller T, Schillert A, Szymczak S, Sinning CR, Deiseroth A, et al. A genome-wide association study identifies LIPA as a susceptibility gene for coronary artery disease. Circ Cardiovasc Genet 2011; 4:403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nakano M, Ikeda Y, Tokuda Y, Fuwa M, Omi N, Ueno M, et al. Common variants in CDKN2B-AS1 associated with optic-nerve vulnerability of glaucoma identified by genome-wide association studies in Japanese. PLoS One 2012; 7:e33389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website (www.pharmacogeneticsandgenomics.com).