

ABSTRACT
Compared to other fungal pathogens, Cryptococcus neoformans is particularly adept at avoiding detection by innate immune cells. To explore fungal cellular features involved in immune avoidance, we characterized cell surface changes of the C. neoformans rim101Δ mutant, a strain that fails to organize and shield immunogenic epitopes from host detection. These cell surface changes are associated with an exaggerated, detrimental inflammatory response in mouse models of infection. We determined that the disorganized strain rim101Δ cell wall increases macrophage detection in a contact-dependent manner. Using biochemical and microscopy methods, we demonstrated that the rim101Δ strain shows a modest increase in the levels of both cell wall chitin and chitosan but that it shows a more dramatic increase in chito-oligomer exposure, as measured by wheat germ agglutinin staining. We also created a series of mutants with various levels of cell wall wheat germ agglutinin staining, and we demonstrated that the staining intensity correlates with the degree of macrophage activation in response to each strain. To explore the host receptors responsible for recognizing the rim101Δ mutant, we determined that both the MyD88 and CARD9 innate immune signaling proteins are involved. Finally, we characterized the immune response to the rim101Δ mutant in vivo, documenting a dramatic and sustained increase in Th1 and Th17 cytokine responses. These results suggest that the Rim101 transcription factor actively regulates the C. neoformans cell wall to prevent the exposure of immune stimulatory molecules within the host. These studies further explored the ways in which immune cells detect C. neoformans and other fungal pathogens by mechanisms that include sensing N-acetylglucosamine-containing structures, such as chitin and chitosan.
IMPORTANCE
Infectious microorganisms have developed many ways to avoid recognition by the host immune system. For example, pathogenic fungi alter their cell surfaces to mask immunogenic epitopes. We have created a fungal strain with a targeted mutation in a pH response pathway that is unable to properly organize its cell wall, resulting in a dramatic immune reaction during infection. This mutant cell wall is defective in hiding important cell wall components, such as the chito-oligomers chitin and chitosan. By creating a series of cell wall mutants, we demonstrated that the degree of chito-oligomer exposure correlates with the intensity of innate immune cell activation. This activation requires a combination of host receptors to recognize and respond to these infecting microorganisms. Therefore, these experiments explored host-pathogen interactions that determine the degree of the subsequent inflammatory response and the likely outcome of infection.
INTRODUCTION
Over the last several decades, the increased use of immunosuppressive drugs and the HIV/AIDS pandemic have greatly expanded the population of people who are susceptible to disseminated fungal infections. The opportunistic fungal pathogen Cryptococcus neoformans has emerged as a particularly deadly pathogen, causing over 300,000 deaths each year, primarily among those suffering from HIV/AIDS (1, 2). C. neoformans first colonizes the lungs, where it can disseminate to the central nervous system to cause life-threatening fungal meningitis, which is universally fatal without treatment (1). The initial interactions between C. neoformans and the innate immune cells in the lung elicit either a robust, protective immune response or a weak, nonprotective response. This infection can also lead to an overexuberant pattern of immune activation resulting in excessive host damage that can be fatal (3). Understanding this initial host-microbe interaction will allow us to better define what constitutes a beneficial immune response to this pathogen.
C. neoformans has a highly dynamic cell surface that changes in composition and architecture during infection. Some of these changes include alterations in the cell wall carbohydrate composition and the attachment of a polysaccharide capsule (4–6). Alterations in the cell wall influence the interaction of C. neoformans with immune cells. The capsule, which is primarily composed of the polysaccharide glucuronoxylomannan (GXM), shields potentially immune-stimulatory molecules in the cell wall from detection. GXM also actively inhibits proinflammatory receptors and signaling in innate immune cells (7–11). While no detailed cell wall analysis has been performed during C. neoformans infection, increased levels of chitin and α-1,3-glucan in C. neoformans cells recovered from infected mice or from cells cultured in host-mimicking tissue culture media have been noted (4, 12). Additionally, the C. neoformans cell wall has been shown to thicken during infection (13). Within the host, during infection, C. neoformans produces Titan cells, representing a morphological state with a very thick cell wall and dense capsule (14, 15). The surface properties of Titan cells have recently been shown to guide the immune response to achieve a more favorable environment for C. neoformans survival (16). Compared to other fungal pathogens, C. neoformans has an increased amount of chitin and chitosan present in its cell wall and a corresponding decrease in the amounts of many α- and β-glucans and mannoproteins (17). Interestingly, relatively little is known about how the innate immune system recognizes distinct components of the C. neoformans cell surface and especially its cell wall.
Previously, members of our laboratory identified the C. neoformans Rim101 transcription factor as an important regulator of the adaptive cell surface changes that occur during infection. The RIM101 gene is one of the most highly induced genes during human cryptococcosis, suggesting that it plays a central role in microbial survival in vivo (18). The rim101Δ mutant has a defect in capsule attachment, as well as an aberrant cell wall morphology (4, 19). Despite these cell surface defects, this mutant was unexpectedly hypervirulent in a mouse inhalation model of infection. Further investigation demonstrated that the rim101Δ strain induces a dramatic inflammatory response in the lungs of infected mice, leading to excessive host damage (4, 19).
The particular cell surface changes in the rim101Δ mutant that drive this increased inflammatory response were not immediately evident. During infection, and when cultured in host-mimicking tissue culture media, this mutant displays increased staining by wheat germ agglutinin (WGA), a reagent commonly used to detect and quantify chitin-derived fungal cell wall carbohydrates (4, 16, 20, 21). In previous studies, these have been defined in C. neoformans as chito-oligomers consisting of 3 to 20 residues of N-acetylglucosamine, which can include chitin and chitosan molecules (20). Chitin is a polymer composed of repeating subunits of β-1,4-N-acetyl glucosamine, serving as an important structural component of most fungal cell walls (22). Chitin makes up a particularly high proportion of the C. neoformans cell wall, and it is significantly more abundant in C. neoformans than in other important fungal pathogens such as Candida albicans (17, 23). C. neoformans cell walls also contain significantly more of the chitin-related carbohydrate chitosan than those of many other fungi (17, 24). Chitosan is a polymer of β-1,4-N-glucosamine and is synthesized by the deacetylation of chitin. A polymer that is at least 60% deacetylated is generally considered to be chitosan; however, these polymers can be heterogeneous, containing both N-acetylglucosamine and N-glucosamine residues (25, 26). Previous work has shown that immune recognition of chitin is a complex process and can result in both increased and decreased inflammatory responses, depending on the source and size of the chitin molecule (16, 24, 27). C. neoformans provides an excellent model to better understand the immune response to fungal cell wall chitin and chitosan.
In this study, we explored the Rim101-regulated cell surface properties that control innate immune detection. In addition to a previously described capsule attachment defect, we find that the rim101Δ mutant has a modest increase in total cell wall chitin and chitosan levels. However, the rim101Δ mutant also displays a previously unappreciated increase in chito-oligomer exposure, as demonstrated by staining with WGA. We show that these cell wall defects result in increased recognition by macrophages in vitro and that this response likely involves members of the Toll-like receptor (TLR) and C-type lectin receptor (CLR) families. We also demonstrate that the rim101Δ mutant induces a dramatic inflammatory response in the lungs of infected mice that persists throughout a prolonged infection. Our results demonstrate that a properly organized C. neoformans cell wall is not readily recognized by macrophages and that it is much less stimulatory than the cell walls from other common fungal pathogens (28–30). Active regulation of the C. neoformans cell wall architecture is important for evading immune detection. In particular, increased WGA staining is associated with an increase in C. neoformans recognition by the immune system, altering the balance between pathogen recognition/clearance and host recovery from immune activation.
RESULTS
The rim101Δ mutation increases macrophage activation.
We previously noted that the rim101Δ mutant induces a dramatic inflammatory response in the lungs of infected mice (4). During a C. neoformans infection, macrophages serve in the initial identification of and response to the pathogen. To determine whether macrophages would respond differently to the rim101Δ mutant, we quantified tumor necrosis factor alpha (TNF-α) secreted by bone marrow-derived macrophages (BMMs) that were cocultured with the mutant strain and compared the results to the levels seen with the wild-type (WT) and rim101Δ + RIM101 reconstituted strains (Fig. 1). While this in vitro coculture system does not include all opsonins and other factors that may be present during a true infection, it does measure direct fungal activation of these immune cells. The rim101Δ strain was previously shown to induce significantly more TNF-α in the lungs of infected mice (4). This cytokine is also one marker of macrophage activation in vitro. Overall, we observed a TNF-α response to all C. neoformans strains that was much lower than previously published macrophage responses to other prominent fungal pathogens (30–33). In this assay, the rim101Δ mutant induced BMMs to secrete significantly more TNF-α than the WT. We observed a similar pattern of TNF-α secretion when the fungal cells were heat killed prior to coincubation with BMMs (Fig. 1). UV-killed fungi produced a similar response (data not shown). Therefore, the enhanced immune cell response to the rim101Δ strain does not appear to require fungal cell viability or metabolism.
FIG 1 .
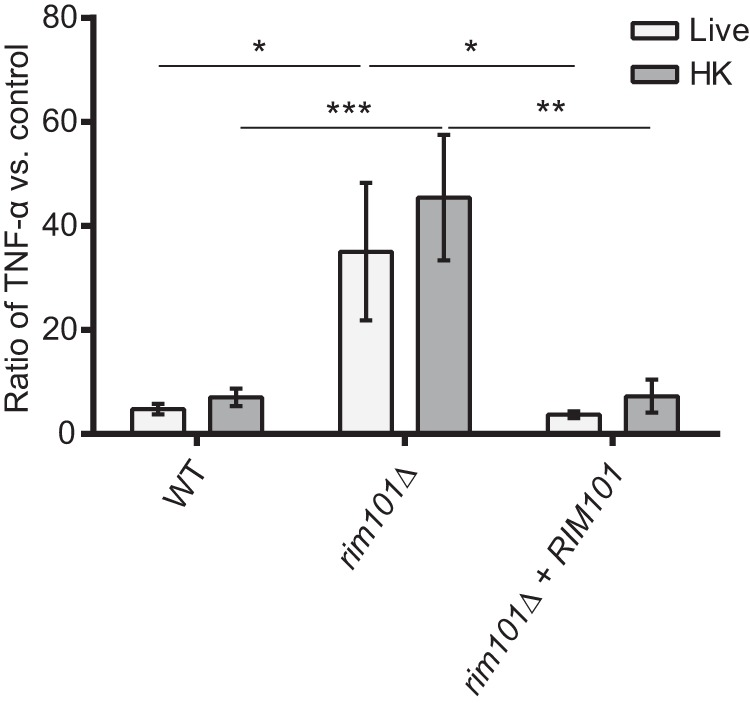
The rim101Δ mutant induces increased TNF-α secretion from macrophages. BMMs were coincubated with the indicated live or heat-killed (HK) strains at a multiplicity of infection (MOI) of 10:1 C. neoformans/BMM cells. Secreted TNF-α levels were quantified after 6 h of coincubation, and data are represented as a ratio of TNF-α levels (in picograms per milliliter) normalized to a control incubation with no added fungal cells. Primary data for TNF-α levels are listed in Table S1. Two-way analysis of variance (ANOVA) and Tukey’s multiple-comparison test were used to compare the means of results from three replicates for three independent experiments (n = 9). ***, P < 0.001; **, P < 0.01; *, P < 0.05. Error bars represent standard errors of the means.
These data demonstrate that certain static properties of the rim101Δ mutant cells are immune stimulatory. We utilized this in vitro measure of macrophage stimulation to further analyze innate immune cell interaction with the rim101Δ mutant. To determine whether the response to this mutant was contact dependent, we used a 3-µm-pore-size Transwell system to separate fungal and BMM cells in a coculture assay (Fig. 2A). Without Transwell separation, we again observed an increased TNF-α response to the rim101Δ mutant compared to the response to the WT strain. With Transwell separation, however, we observed no significant difference in the TNF-α responses to the various strains. This result suggests that the observed enhanced macrophage response requires direct contact with the rim101Δ strain and also suggests that the response to this mutant is not driven by a secreted immune stimulatory molecule.
FIG 2 .

The rim101Δ mutant cell wall induces TNF-α secretion from BMMs in a contact-dependent and capsule-independent manner. (A) WT and rim101Δ cells were coincubated with BMMs at an MOI of 10:1 C. neoformans/BMM cells, either “in direct contact” or “separated” by a Transwell system. (n = 3 for each strain/condition.) (B) BMM TNF-α levels (in picograms per milliliter) after 6 h of incubation with 10 µg/ml purified cell wall material from the indicated fungal strains. Data represent means of results from three replicate samples for three independent cell wall preparations (n = 9 for each strain). (C) BMMs were coincubated for 6 h with UV-killed fungal cells that had been precultured in either YPD (rich) medium or tissue culture (TC), CO2-independent medium. TNF-α levels in the culture medium were normalized to a control incubation with no added fungal cells. Primary data for TNF-α levels are listed in Table S2. Data represent means of results from three replicate samples for two independent experiments (n = 6 for each strain/condition). (D) BMMs were coincubated with the indicated strains for 6 h. The gray line separates strains normalized by cell number (WT and rim101Δ strains) and by weight (cap59Δ strain or cap59Δ rim101Δ strain). Data represent means of results from three replicate samples (n = 9). Two-way (A) and one-way (B, C, and D) ANOVA and Tukey’s multiple-comparison test were used to compare means. ***, P < 0.001; **, P < 0.01; *, P < 0.05. Error bars represent standard errors of the means.
Rim101 is a central regulator of the cell wall during infection, and mutations disrupting Rim101 function result in an altered cell surface (4). To determine whether the rim101Δ mutant cell wall could independently induce increased BMM activation, we purified cell wall material from the C. neoformans strains after growth in tissue culture medium was performed and we treated BMMs with 10 µg/ml of the cell wall extracts. Similarly to whole cells, cell wall material isolated from the rim101Δ mutant induced significantly more TNF-α from BMMs than the cell wall material isolated from either the WT or the reconstituted strain (Fig. 2B). Examined microscopically, the isolated cell wall material was composed of empty cell-shaped particles (data not shown), indicating that architecture of the cell wall may be largely maintained in the cell wall extracts. Together, these data suggest that macrophages recognize rim101Δ C. neoformans more readily than WT C. neoformans, which is likely due to changes in the composition or architecture of the rim101Δ cell wall.
The rim101Δ mutant cell surface defects manifest only when the strain is cultured under host-mimicking conditions (tissue culture medium), and the mutant has a normal-appearing cell wall when cultured in rich medium (4). We determined whether the BMM response to the rim101Δ mutant would also be dependent on the growth medium. First, we precultured WT and rim101Δ mutant cells in rich medium (yeast extract-peptone-dextrose [YPD]) or tissue culture medium. In order to prevent active cell wall remodeling during coincubation with BMMs, we killed the fungal cells using UV irradiation prior to coculture. Cultured under tissue culture conditions, the rim101Δ mutant again induced more TNF-α secretion than the WT cells (Fig. 2C). In contrast, these strains induced similar levels of TNF-α after the fungi were precultured in rich medium. This demonstrates that the rim101Δ mutant is stimulatory only when cultured under conditions that induce cell surface alterations.
The rim101Δ mutation increases immune recognition of the acapsular cap59Δ mutant.
C. neoformans polysaccharide capsule components actively suppress innate immune cell activation, and they also serve to shield the more immunogenic cell wall from recognition (7, 32). As such, we considered that the previously reported rim101Δ capsule defect could potentially explain the increased immune recognition of these mutant strains. To determine the role of the capsule in the recognition of the rim101Δ strain, we compared the BMM responses to strain cap59Δ, the acapsular mutant, and the rim101Δ cap59Δ double mutant. Due to cell aggregation in the cap59Δ background, we normalized the cell concentrations based on weight instead of cell number. Overall, we observed an increased TNF-α response to both acapsular strains (the cap59Δ and rim101Δ cap59Δ mutants) compared to the WT strain (Fig. 2D), though the precision of the comparisons was limited due to differing cell normalization methods. We also noted that the cap59Δ rim101Δ double mutant induced significantly more TNF-α production than the cap59Δ single mutant (Fig. 2D). These data indicate that the rim101Δ mutant cell wall changes increase immune recognition by another mechanism in addition to the effect due to loss of surface capsule.
Analysis of cell wall carbohydrate content and organization in the rim101Δ mutant cell wall.
We previously demonstrated that host-mimicking conditions in vitro induced the rim101Δ mutant to produce a thick, disordered cell wall, a phenotype that was absent when cells were cultured in rich media (4). The rim101Δ mutant cell wall demonstrated a dramatic increase in cell surface staining with wheat germ agglutinin (WGA), a lectin that binds chito-oligomers (4). This result suggested the presence of a very different cell wall structure in this strain.
To better define the actual composition of the rim101Δ mutant cell wall compared to that of the WT, we biochemically quantified the levels of three major cell wall carbohydrates that comprise a typical fungal cell wall: glucosamine, glucan, and mannose. After culturing the strains in host-like tissue culture media, the cell walls were extracted and hydrolyzed into their individual sugar components. This technique deacetylates the chitin to chitosan and therefore measures chitin and chitosan together as glucosamine. The levels of glucosamine, glucan, and mannose in these cell wall fractions were quantified using high-performance liquid chromatography (HPLC) (Fig. 3A). We found that the levels of mannose, which is found on cell wall mannoproteins, were not significantly different between the WT, rim101Δ, and rim101Δ + RIM101 strains. Changes in total glucose/glucan content were also not observed between the rim101Δ and rim101Δ + RIM101 strains. The WT strain had slightly less glucose/glucan than the rim101Δ and reconstituted strains. As the WT and rim101Δ + RIM101 strains induce similar macrophage responses, the slightly altered glucan content likely does not contribute to the increased immune response to the rim101Δ mutant. The mannose-rich capsule did not appear to contaminate our analysis, as the acapsular cap59Δ strain had mannose levels comparable to those seen with the WT cells (data not shown). Despite the relatively low level of glucosamine, we observed a trending though statistically insignificant increase in the level of glucosamine in the rim101Δ mutant compared to both the WT strain (P = 0.998) and the rim101Δ + RIM101 reconstituted strain (P = 0.999). Overall, by this method, the total relative levels of glucosamine, glucan, and mannose in the rim101Δ mutant cell wall were not dramatically different.
FIG 3 .

The rim101Δ mutant has increased chito-oligomer exposure. (A) The cell walls were isolated from the WT, rim101Δ mutant, and rim101Δ + RIM101 reconstituted strains after incubation for 18 h in CO2-independent medium. Glucosamine, glucan, and mannose levels were quantified in the isolated cell walls using high-performance anion-exchange chromatography with pulse amperometric detection (HPAEC-PAD). Data represent means of results from three independent cell wall preparations (n = 3 for each strain). (B) The WT and rim101Δ mutant were incubated for 18 h in CO2-independent medium, and cell walls were isolated. Chitin and chitosan levels were quantified for each strain using a modified 3-methyl-2-benzothiazolinone hydrazone hydrochloride (MBTH) colorimetric assay. Data represent means of results from three independent cell wall preparations (n = 3 for each strain). (C) C. neoformans strains were incubated for 18 h in CO2-independent medium and stained with fluorescein isothiocyanate (FITC)-conjugated wheat germ agglutinin (WGA) or calcofluor white (CFW). Staining intensity was assessed using epifluorescence microscopy with identical exposures for all images. (D) Fluorescence levels for 100 individual cells (represented in panel C) were quantified using ImageJ (Fiji). One-way ANOVA and Tukey’s multiple-comparison test were used to compare means to the WT values. Means represent the fluorescent (Fluor.) intensity levels from three independent experiments (n = 3). ***, P < 0.001; **, P < 0.01; *, P < 0.05; ns, not significant. Error bars represent standard errors of the means.
To differentiate between alterations in the chitin and chitosan levels in the rim101Δ mutant, we utilized a recently developed biochemical assay to quantify these components separately (reference 23 and Jennifer Lodge, personal communication). We found no significant difference in chitin levels between the rim101Δ mutant and WT strains, but we did observe a statistically significant increase in chitosan levels in the mutant strain (Fig. 3B). Our results also demonstrate that the growth medium likely alters chitin and chitosan levels in C. neoformans, as cells cultured in a rich medium have been shown to have chitosan/chitin ratios that are higher than what we observed here from cells cultured in tissue culture medium (23).
We previously observed a dramatic increase in the staining intensity of the rim101Δ mutant cell wall with the chitin-binding lectin WGA when this strain was incubated under host-mimicking conditions such as those using tissue culture medium. This observation suggested that the rim101Δ mutant cell wall might contain significantly higher levels of chito-oligomers or other N-acetylglucosamine-containing molecules (4, 34). However, this result is not consistent with the relatively small increases in relative chitin/chitosan levels observed in the HPLC analysis and biochemical chitin/chitosan quantification. To explore the discrepancy between the WGA staining and HPLC analysis, we analyzed rim101Δ mutant cell wall chitin levels using a different fluorescent stain, calcofluor white (CFW). CFW is a small (917-Da) fluorescent molecule that also binds chitin and chitosan, in addition to cellulose (35). We compared the mean CFW fluorescent levels of WT, rim101Δ, and rim101Δ + RIM101 reconstituted strains that were cultured in tissue culture medium, the condition under which we observed increased WGA binding (Fig. 3C). We also stained the cells with WGA. Similarly to our previous study, we found that WGA cell wall fluorescence was 6-fold higher for the rim101Δ mutant than for the WT and rim101Δ + RIM101 reconstituted strains. For the same cells, the CFW fluorescence was only 1.5-fold higher in the rim101Δ mutant cells than in the WT and rim101Δ + RIM101 strains. This observation is more consistent with the biochemical measurement of total chitin and chitosan levels in these strains (Fig. 3B). CFW also displayed a staining pattern different from that of WGA, with CFW staining the entire cell wall whereas WGA brightly stained only the bud necks and bud scars on the WT and rim101Δ + RIM101 strains, as well as showing punctate staining throughout the rim101Δ cell surface. Chitin and chitosan make up the basal, structural layer of the fungal cell wall, and these carbohydrates are present in higher levels at bud necks and bud scars of yeast cells (22). This suggests that WGA, which is a 38-kDa protein dimer (34), primarily binds exposed chito-oligomers, while the much smaller CFW molecule has access to total chito-oligomers present in the fungal cell wall (36, 37). Together, these data suggest that the rim101Δ cell wall has a modest increase in total chitin/chitosan content but displays an altered pattern of organization resulting in a more dramatic increase in chito-oligomer exposure.
Alteration in chitin and chitosan synthesis and exposure increases recognition by BMMs.
C. neoformans possesses eight chitin synthase genes (CHS1 to CHS8 [CHS1-8]) that may be involved in distinct temporal, morphological, or site-specific aspects of chitin biosynthesis. Similarly to the case in other fungi, this apparent gene family expansion results in potential functional redundancy for synthesizing the fundamental components of such an important biological structure as the cell wall. Possession of multiple chitin-modifying genes also suggests the potential for precise, condition-specific modification of the molecular structure of chitin and chitosan (23, 38, 39). However, despite some degree of functional overlap, previous studies demonstrated that mutations disrupting single CHS genes result in measurable alterations in chitin and chitosan levels. For example, the chs3Δ mutant cannot produce chitosan, and the chs5Δ mutant has a marked decrease in total levels of cell wall chitin (23).
We hypothesized that increased chitin and chitosan levels in the rim101Δ mutant cell wall were responsible, at least partially, for its increased immune recognition. Therefore, we sought to decrease both chitin and chitosan levels by disrupting the CHS3 (chitosan) and CHS5 (chitin) biosynthetic genes in the rim101Δ mutant background. In previously published data from experiments using biochemical assays and a rich culture medium, the same chs mutants displayed alterations in either chitin or chitosan content even though the total chitin/chitosan levels were not dramatically changed. However, our culturing of the resulting chs mutant strains in tissue culture medium induced unexpected effects on cell wall staining of these chito-oligomers. Interestingly, neither the chs3Δ nor the chs5Δ mutation decreased CFW or WGA staining in the rim101Δ strain. Additionally, the chs3Δ and chs5Δ mutations in the WT background resulted in increased CFW and WGA staining (Fig. 4A and B). Together, these data suggest that chs3Δ and chs5Δ mutations have differing effects on cell wall chito-oligomer levels (CFW staining) and chito-oligomer exposure (WGA staining) that depend on the background of the mutant strain and the growth conditions.
FIG 4 .
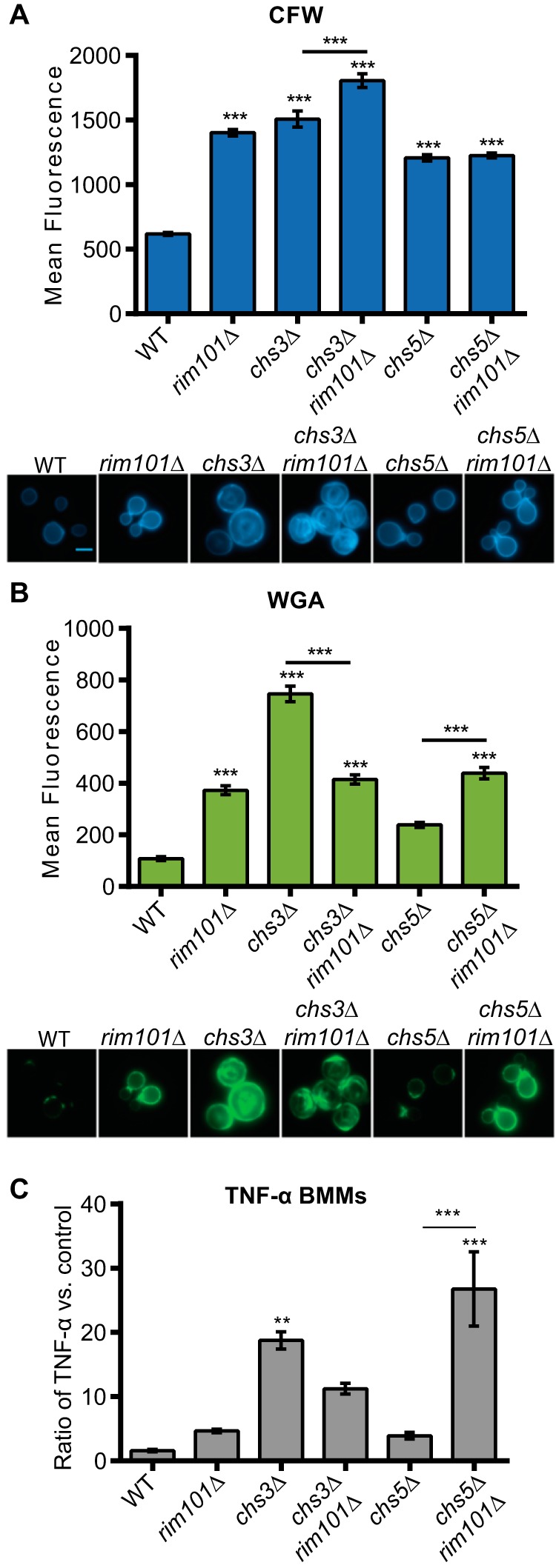
Fungal cell WGA staining intensity correlates with levels of induced TNF-α secretion from BMMs. (A and B) C. neoformans strains were stained with CFW (A) or FITC-WGA (B) after incubation in tissue culture medium at 37°C for 18 h. Fluorescence levels for 100 individual cells were quantified using ImageJ (Fiji) (upper panels) (n = 100). Representative fluorescent microscopic images for each strain (at a magnification of ×600) are demonstrated in the lower panels. (C) C. neoformans strains were coincubated with BMMs at a multiplicity of infection (MOI) of 10:1 C. neoformans/BMM cells. TNF-α levels in the culture medium were normalized to a control incubation with no added fungal cells. One-way ANOVA and Tukey’s multiple-comparison test were used to compare the means of results from three independent experimental samples. ***, P < 0.001; **, P < 0.01; *, P < 0.05. Error bars represent standard errors of the means.
By disrupting CHS3 and CHS5 in the rim101Δ mutant, we inadvertently created a series of strains with various levels of CFW and WGA staining. We used these strains as tools to further probe the association between macrophage activation and CFW/WGA staining intensities. Consistent with our previous experiments, we observed that the rim101Δ strain induced more TNF-α secretion by BMMs than the WT strain, although this increase did not reach statistical significance in this experiment. We also noted that the chs3Δ, chs3Δ rim101Δ, and chs5Δ rim101Δ mutants induced even more TNF-α secretion than the rim101Δ strain, while the chs5Δ mutation alone had no significant effect of the TNF-α levels (Fig. 4C). Importantly, the level of WGA staining of each strain correlated with the level of BMM TNF-α secretion.
The chs3 mutation did result in an alteration of cell size after incubation in this tissue culture medium, with increases in average cell area compared to the wild type of 3.8-fold for the chs3Δ mutant and 2.6-fold for the chs3Δ rim101Δ double mutant. However, this cellular change is insufficient to account for the larger measured increases in WGA intensity for these cells (Fig. 4C). In contrast, increased CFW staining was not associated with increased BMM TNF-α secretion in each case (Fig. 4). The chs5Δ rim101Δ double mutant was somewhat of an exception to this pattern, as it induced significantly more TNF-α secretion than the other tested strains. Together, these data suggest that increased chito-oligomer exposure, but not necessarily the total cell wall chitin or chitosan level, is closely associated with increased macrophage activation.
Macrophage activation in vitro does not require the inflammasome.
Previous studies have demonstrated that purified chitosan activates the inflammasome and induces inflammatory cytokine secretion (40, 41). As such, we considered that elevated chitosan levels in the rim101Δ mutant cell wall might increase inflammasome activation. Two signals are required for inflammasome activation: an initial Toll-like receptor (TLR)- or C-type lectin receptor (CLR)-activating signal and a subsequent stimulus that activates one of the NLRP inflammasomes. Activation induces the production, processing, and secretion of the inflammatory cytokines interleukin-33 (IL-33), IL-18, and IL-1β. We treated both lipopolysaccharide (LPS)-primed (TLR-stimulated) and unprimed BMMs with the WT, rim101Δ, rim101Δ + RIM101, cap59Δ, and rim101Δ cap59Δ strains. We then quantified the level of secreted IL-1β. IL-1β levels were not detected above baseline for any strain (Fig. S1A and B). As a positive control, we also treated BMMs with purified chitosan, and we observed elevated IL-1β secretion from LPS-primed BMMs (Fig. S1C). Therefore, in this in vitro assay, the rim101Δ mutation does not appear to have induced macrophage activation through inflammasome-mediated signaling recognizing its increased cell wall chitosan content or exposure.
The rim101Δ mutation does not induce measurable IL-1β secretion (A and B). BMMs were primed with or without 100 ng/ml UltraPure LPS for 3 h. BMMs were coincubated with the indicated C. neoformans strains at an MOI of 10 C. neoformans fungal cells to 1 BMM. cap59Δ and rim101Δ cap59Δ cells were normalized to 2 mg cells/ml of media (C) LPS-primed and unprimed BMMs were treated with 0.1 mg/ml chitosan for 6 h. Secreted IL-1β levels (in picograms per milliliter) were quantified by ELISA (BioLegend) after 6 h of coincubation. Two-way or one-way ANOVA and Tukey’s multiple-comparison test were used to compare means of results of three replicates for each strain (n = 3). ***, P < 0.001; **, P < 0.01; *, P < 0.05. Error bars represent standard errors of the means. Download FIG S1, EPS file, 0.8 MB (848.1KB, eps) .
Copyright © 2017 Ost et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In vitro response to the rim101Δ strain involves MyD88 and CARD9.
Macrophages recognize and respond to fungi through the activation of multiple pattern recognition receptors, or PRRs. Of these receptors, members of the Toll-like receptor (TLR) and the C-type lectin receptor (CLR) families have been shown to be responsible for recognizing fungal cell wall carbohydrates and mannoproteins (42). To determine whether members of the TLR or CLR families are responsible for the increased recognition of the rim101Δ mutant strain, we utilized BMMs from mice lacking key downstream signaling adaptor proteins required for TLR and CLR activity. Most TLRs require the MyD88 adaptor protein to activate cytokine production, while the CLRs require CARD9 to propagate their downstream stimulatory signals (42).
We coincubated C. neoformans strains with BMMs derived from MyD88−/− or CARD9−/− mutant mice, and we compared their TNF-α responses to those seen with WT BMMs. To better visualize subtle alterations in the TNF-α response to the rim101Δ mutant, we utilized the acapsular cap59Δ mutant background to amplify the TNF-α response (Fig. 5). Although the responses to cap59Δ C. neoformans cells were not significantly altered in the MyD88−/− and CARD9−/− BMMs, these mutant macrophages failed to induce TNF-α production in response to the rim101Δ cap59Δ strain (Fig. 5). These results demonstrate that the increased recognition of the rim101Δ cap59Δ strain by macrophages requires both MyD88 and CARD9, suggesting that one or more members of the TLR and CLR families are involved.
FIG 5 .
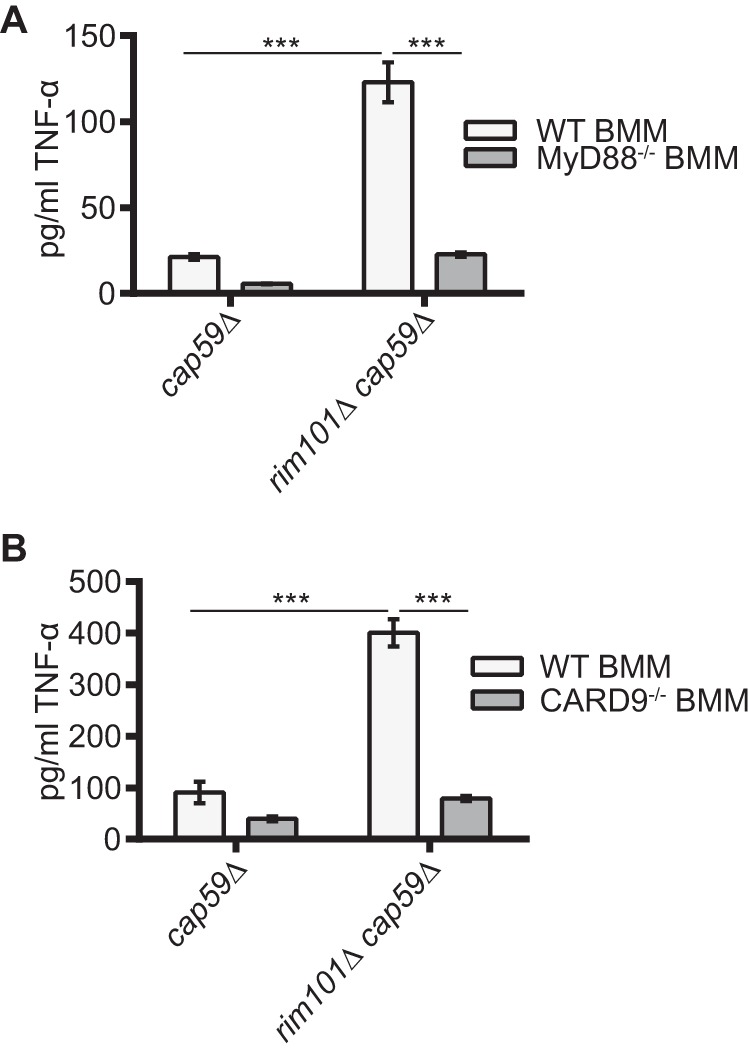
MyD88 and CARD9 contribute to the TNF-α response to the rim101Δ strain. BMMs were harvested from WT (A and B) and MyD88−/− mutant (A) or CARD9−/− mutant (B) mice in the C57BL-6 background. These cells were coincubated with 2 mg/ml of the cap59Δ strain or the rim101Δ cap59Δ strain. TNF-α levels (in picograms per milliliter) in the culture medium were quantified after 6 h coincubation. Two-way ANOVA and Tukey’s multiple-comparison test were used to compare the means of results from three replicate samples (n = 3). ***, P < 0.001; **, P < 0.01; *, P < 0.05. Error bars represent standard errors of the means.
The rim101Δ mutant induces a global increase in inflammatory cytokine production in the lungs of infected mice.
To better characterize the immune response to the rim101Δ mutant, we intranasally inoculated BALB/c mice with 104 cells of each strain and analyzed the levels of 18 cytokines in infected lungs at day 7 and day 14 postinfection (Fig. 6; Table S3). In the rim101Δ mutant-infected lungs, there were increased levels of several inflammatory cytokines measured at day 7 and a global increase in the levels of almost all cytokines measured at day 14 compared to the levels seen with the WT and rim101Δ + RIM101 mutant infections. Several of the most highly upregulated cytokines, including the neutrophil chemoattractant CXCL1 (Fig. 6D) and the inflammatory cytokines IL-1α, IL-1β, TNF-α, IL-6, monocyte chemoattractant protein 1 (MCP-1), macrophage inflammatory protein 1 alpha (MIP-1α), and MIP-1β (Fig. 6), are known to induce inflammatory cell infiltration. Several Th1-, Th2-, and Th17 (inflammatory)-driving cytokines were upregulated by the rim101Δ mutant, making it difficult to determine the specific type of adaptive immune response induced by this strain. Notably, the only cytokine whose level was significantly reduced in the rim101Δ-infected lungs at day 14 was the Th2-associated cytokine IL-4 (Fig. 6Q). In contrast, the levels of key Th1 and Th17 (inflammatory) polarizing cytokines, such as IFN-γ and IL-17A, were significantly higher or trended higher at day 14 (Fig. 6). Altered cytokine profiles were not due to increased proliferation of the rim101Δ mutant, as similar numbers of viable rim101Δ mutant and WT cells were recovered from the lung and the brain at day 7 (lung only) and day 14 (lung and brain) postinfection (Fig. S2). Overall, the rim101Δ mutant induced a dramatic immune response in the lungs of infected mice, characterized by a global increase in cytokine secretion.
FIG 6 .

The rim101Δ strain elicits an inflammatory and Th1 cytokine response in infected lungs. The WT, rim101Δ mutant, and rim101Δ + RIM101 reconstituted strains were inoculated (104 fungal cells) into BALB/c mice by inhalation. At day 7 and day 14 postinfection, the indicated cytokines were quantified from whole-lung digestions. 4 mice were inoculated for each strain. Data represent results of 2 independent experiments (8 mice in total). Primary data for cytokine levels are listed in Table S3. One-way ANOVA and Tukey’s multiple comparison tests were used to compare means of results to the WT infection data for each day. G-CSF, granulocyte colony-stimulating factor. ***, P < 0.001; **, P < 0.01; *, P < 0.05. Error bars represent standard errors of the means.
TNF-α levels (quantified in picograms per milliliter) for Fig. 1. Download TABLE S1, DOCX file, 0.01 MB (14.5KB, docx) .
Copyright © 2017 Ost et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
TNF-α levels (quantified in picograms per milliliter) for Fig. 2C. Data represent the BMM TNF-α response to C. neoformans cultured in rich media versus tissue culture media. Download TABLE S2, DOCX file, 0.01 MB (14.4KB, docx) .
Copyright © 2017 Ost et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
All cytokines quantified. Download TABLE S3, DOCX file, 0.02 MB (20.8KB, docx) .
Copyright © 2017 Ost et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The rim101Δ mutant proliferates at a rate similar to that of the WT strain during infection. Fungal CFU counts were determined from whole-lung (A and B) or whole-brain (C) homogenates. 4 BALB/c mice were intranasally inoculated with 104 cells for each strain. Data represent results of 2 independent experiments (8 mice in total). One-way ANOVA and Tukey’s multiple-comparison test were used to compare means. ***, P < 0.001; **, P < 0.01; *, P < 0.05. Error bars represent standard errors of the means. Download FIG S2, EPS file, 1 MB (1MB, eps) .
Copyright © 2017 Ost et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
Like many fungal pathogens, C. neoformans has a highly dynamic cell surface that it actively modifies during infection, thereby shaping its interaction with the immune system. Resulting immune responses can range from nonprotective and weak, in the case of cryptococcal meningitis, to overexuberant and damaging. Damaging immune responses to C. neoformans infection have become significant issues for people recovering from immune suppression, especially patients with late-stage HIV infection as they initiate antiretroviral therapy. This condition is known as immune reconstitution inflammatory syndrome, or IRIS, and it results in mortality rates similar to those encountered in cases of untreated C. neoformans meningitis (3). Typical infection models of cryptococcosis explore infection in highly susceptible hosts with nonprotective, weak immune responses. Few models explore the damaging immune responses that can also occur during these infections in humans. Therefore, little is known about the C. neoformans cellular attributes that drive highly active inflammatory responses, including microbial features that are typically hidden during infection with natural fungal isolates. C. neoformans cells induce a much lower innate immune response than other prominent fungal pathogens. Therefore, C. neoformans-driven inflammatory responses are difficult to analyze in vitro. The rim101Δ mutant was previously shown to induce a detrimental inflammatory response and an increased death rate in two separate mouse strains (4, 19). Alterations in the cell wall structure and composition, and the resulting defect in surface capsule, were hypothesized to increase immune recognition and drive the inflammatory response. In this study, as modeled in Fig. 7, we demonstrated that the rim101Δ mutant has increased chito-oligomer exposure on its cell surface in response to host-like conditions, without a major change in total cell wall carbohydrate content. This cell wall alteration, in addition to its decreased capsule, leads to increased recognition by macrophages in vitro and increased inflammation in vivo (Fig. 7).
FIG 7 .

Model for the immune stimulatory mechanism of the rim101Δ mutant. When C. neoformans enters the host, it senses host environmental cues (elevated pH, increased CO2, high temperature) and uses the Rim101 signaling pathway to adaptively regulate various cellular structures. These adaptations include the organization of its cell wall and the production/attachment of its polysaccharide capsule. Proper cell surface regulation prevents recognition by innate immune receptors. Without Rim101 regulation, the cell wall becomes highly disorganized, resulting in increased chitosan levels, a dramatic increase in chito-oligomer exposure, and impaired capsule attachment. These cell surface defects allow enhanced detection by CARD9- and MyD88-dependent innate immune receptors, leading to increased inflammatory cytokine secretion by host immune cells.
Rim101 limits C. neoformans chitin exposure and chitosan levels under host-mimicking conditions and correspondingly suppresses recognition by innate immune cells. While other rim101Δ cell surface attributes undoubtedly contribute to immune recognition, our data strongly associate chito-oligomer exposure with immune stimulation by C. neoformans. Exposure of these carbohydrate molecules also appeared to be more important than total chitin or chitosan levels, which suggests that cell wall components are typically detected by immune cells primarily at the outer, exposed cell surface, perhaps emphasizing why fungi tend to place more immunogenic molecules deeply within the cell wall structure.
Cell surface alterations that contribute to rim101Δ mutant immune activation.
Recent studies have demonstrated that chitin-derived structures are important in C. neoformans capsule formation and immune recognition. When WGA was added to cryptococcal cultures, the capsule polysaccharide failed to form, likely due to altered capsule gene expression and trafficking of capsular polysaccharide to the cell surface (21). Additionally, blocking surface chito-oligomers with WGA resulted in defective phagocytosis by macrophages (21). These results are consistent with our findings in which chito-oligomer exposure enhanced macrophage activation in vitro and in vivo.
The role of chitin in the immune response to fungi is complex. Unlike β-1,3-glucan, which is usually proinflammatory, chitin has been shown to both stimulate and inhibit immune responses, depending on the source and size of the chitin molecule. The reported type of immune response to chitin can range from an allergic Th2 response (24, 43) to a proinflammatory Th1 response (44–46) to an anti-inflammatory response (24, 47, 48). Several recent studies have begun to reveal the mechanisms behind the observed differing immune responses to fungal chitin. Wagener et al. found that initial interactions between BMMs and purified chitin from several fungal pathogens, including C. neoformans, resulted in proinflammatory cytokine secretion. Once the BMMs were activated, chitin was digested by secreted mammalian chitinases and subsequently phagocytosed by the macrophage. The ingested chitin then stimulated the secretion of an immunoregulatory cytokine, IL-10, that acted to inhibit the inflammatory response (24). Our in vitro experiments explored the initial interaction between macrophages and fungal strains with various degrees of chitin exposure. The observed association between increased chitin exposure and enhanced secretion of the proinflammatory cytokine TNF-α is therefore consistent with the previously described immune responses to chitin in vitro.
Alterations specifically in cell wall chitosan levels may also play a role in the detection of the rim101Δ mutant. Our biochemical analysis showed that the rim101Δ mutant has a modest increase in levels of cell wall chitosan. C. neoformans is among the few prominent fungal pathogens that have significant levels of chitosan in their cell walls (17, 24). Chitosan alone has been shown to activate the inflammasome in macrophages, inducing the secretion of inflammatory cytokines such as IL-18 and IL-1β. Notably, IL-1β was one of the most highly upregulated cytokines in the mouse lungs infected with the rim101Δ mutant (40, 41). However, the rim101Δ mutant does not appear to induce IL-1β secretion in vitro. Furthermore, the chs3Δ mutation did not reduce the in vitro TNF-α response to the rim101Δ mutant. The chs3Δ mutation disrupts almost all chitosan production (23) and would be predicted to significantly reduce chitosan levels in the chs3Δ rim101Δ strain. As we have observed with chitin, perhaps whether chitosan exposure occurs is more important for immune recognition than its total levels. C. neoformans chitosan may play an important role in organizing the cell wall and shielding cells from excessive immune activation; mutants with significantly decreased chitosan levels were both avirulent and capable of inducing robust and protective immune responses in multiple mouse backgrounds (49). Similarly to the response to chitin, the response to chitosan is clearly complex and likely dependent on the precise chemical form in which it is presented to the immune cells. Further experimentation is needed to more precisely determine the role of chitosan in the immune response to the rim101Δ mutant.
It is likely that increased chito-oligomer exposure is not the only rim101Δ mutant cell surface alteration that stimulates macrophages. The rim101Δ mutant also has a significant capsule defect which would further expose cell surface pathogen-associated molecular patterns (PAMPs) to the immune system. There is an extensive body of research demonstrating the anti-immune recognition properties of the polysaccharide capsule, showing that it both acts as a barrier to direct detection of cell wall PAMPs and actively suppresses immune signaling pathways (7–11). While direct comparisons were difficult in our in vitro assays, we consistently observed an increase in TNF-α secretion by macrophages under conditions of stimulation with cap59Δ single and double mutants compared to WT or single rim101Δ mutant strains. This finding also suggests that the GXM secreted by the rim101Δ mutant is still able to suppress immune responses.
Mechanisms of immune recognition of fungal cell surfaces.
The innate immune receptors responding to increased C. neoformans chito-oligomer exposure are still unknown; however, we demonstrated here that multiple pattern recognition receptors are likely involved. The adaptor proteins required for most TLR and CLR signaling, MyD88 and CARD9, respectively, were both required for a complete response to the rim101Δ cap59Δ strain in vitro. Members of the TLR and CLR families have been previously implicated in chitin sensing. MyD88 and TLR2 were shown to be required for the in vitro proinflammatory response to chitin (46). TLR9, an intracellular receptor, was more recently shown to detect chitin that had been phagocytosed, although this interaction induced an anti-inflammatory response (24). Mannose receptor and dectin-1 have also been implicated in fungal chitin detection (24, 37). Future studies will identify the specific receptors, including non-TLR MyD88-dependent receptors such as IL-1R, required for the macrophage recognition of the rim101Δ mutant.
Several known chitin-sensing host proteins were not accounted for in our in vitro assays. Host chitinases also play an important role in the detection and killing of invading fungal pathogens. Chitotriosidase 1 (Chit1) has been associated with the induction of the nonprotective Th2 immune response during C. neoformans infections (50), and the acidic mammalian chitinase (AMCase) suppresses the Th2 response to fungi and other chitin-containing organisms (43, 51). The rim101Δ mutant and other strains with increased chito-oligomer exposure are likely more prone to attack by Chit1 and AMCase. A recent study demonstrated that IgG and the Fcγ receptor detect purified chitin particles and, when combined with several TLR agonists, induce inflammatory cytokine production from human peripheral blood mononuclear cells (48). Further analysis will determine whether chitinases and IgG are involved in the immune response to C. neoformans strains with various degrees of chito-oligomer exposure.
Characterization of the immune response to the rim101Δ mutant in vivo.
The immune response to the rim101Δ mutant in a murine lung infection model was characterized by an increase in the levels of many proinflammatory cytokines. Levels of certain cytokines, including several favoring Th1- and IL-17-weighted responses, were particularly elevated during this infection. This response is consistent with previous studies demonstrating a strong Th1 and Th17 response to chitin or to fungal mutants producing high levels of chitin (44–46). While levels of several Th2 cytokines were elevated during the entire course of the rim101Δ mutant infection, IL-4 was the only cytokine whose level was significantly decreased during the rim101Δ mutant infection at day 14 postinfection. IL-4 secretion is an important driver of the Th2 response, as it induces the production of other Th2-associated cytokines while suppressing Th1 and Th17 responses (52). This observation demonstrates that alteration of the rim101Δ cell wall primarily induced a proinflammatory Th1/Th17 immune response in this infection model and is largely consistent with our previous analysis of the rim101Δ mutant infection (4). Here, however, we analyzed the response at later time points, and in a different mouse background (BALB/c versus C57B/6), demonstrating a consistent proinflammatory response to the rim101Δ mutant throughout the infection that was independent of the mouse genotype.
Many studies have demonstrated that a strong Th1 immune response is essential for clearing C. neoformans infections and that a Th2 response is associated with a worse disease outcome (53). However, the inflammatory condition IRIS results primarily from an overexuberant Th1 immune response (3). Our findings, in combination with findings revealing the extensive fungal cell death that occurs under this condition, suggest that increased chito-oligomer exposure may drive, or worsen, the inflammatory response in Cryptococcus-associated IRIS. C. neoformans cells have a wide range of morphologies, sizes, and cell surface properties during infection. These include the production of Titan cells, which have recently been shown to possess increased chitin levels compared to smaller yeast forms (50). In addition, dead or dying fungal cells often have aberrant cell wall compositions and arrangements (37). The results determined with the chitin synthase mutants analyzed in this study demonstrated that mutations disrupting cell wall composition can increase chito-oligomer exposure, even when these mutations are expected to decrease total chitin/chitosan levels (23). C. neoformans cells with altered cell wall architecture may be present in patients with recurrent or latent C. neoformans infections, who are particularly susceptible to developing IRIS. We previously found that the rim101Δ cap59Δ double mutant was also avirulent and cleared from infected mice, despite being highly immunostimulatory in vitro (4). In addition, the chs3Δ mutant, shown here to have increased chito-oligomer exposure, is more readily recognized by BMMs and yet is avirulent in mice (12). These examples demonstrate that the particular immune response to various levels of fungal chito-oligomer exposure can be both beneficial and detrimental to the host. Additional phenotypes, such as temperature sensitivity, would affect the fitness of certain strains during an infection. Therefore, the duration of fungal cell persistence is likely a contributor whose importance is equal to that of the cell wall organization with respect to the ultimate outcome of the infection.
Conclusion.
We have demonstrated that the combination of increased chito-oligomer exposure and decreased capsule attachment in C. neoformans is strongly associated with immune stimulation in vitro and in vivo. Future experiments are needed to determine whether immune cells directly detect chitin and/or chitosan exposure or if these molecules serve as a binding site for unknown immune stimulatory molecules. Furthermore, while the rim101Δ mutant would rarely be encountered in the environment, understanding C. neoformans cell surface patterns that stimulate immune recognition is vitally important for vaccine development and prevention of damaging inflammatory responses. These data also increase our understanding of how C. neoformans utilizes Rim pathway signaling to actively avoid immune recognition.
MATERIALS AND METHODS
Strains, media, and growth conditions.
The C. neoformans strains used in this study are listed in Table 1. All strains were generated in the C. neoformans var. grubii strain H99 and were maintained on YPD medium (yeast extract [2%], peptone [1%], dextrose [2%]). Unless otherwise stated, C. neoformans strains were cultured in CO2-independent medium (Gibco) at 37°C for cell wall analysis and cell wall staining and prior to immune cell coculture experiments.
TABLE 1 .
Strains used in this study
Molecular biology.
Gene disruptions were made as previously described (54, 55) using nourseothricin (NAT) or neomycin (NEO) genes as the dominant drug resistance genes. Disruption cassettes were transformed using biolistic transformation (55). All disruptions were confirmed by Southern blotting.
Cell wall isolation and HPLC.
C. neoformans strains were inoculated from an overnight YPD culture into 25 ml CO2-independent medium and incubated for 18 h at 37°C. Cell wall isolation and high-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD) analysis were performed as previously described (56).
A modified MBTH (3-methyl–benzothiazolinone hydrazone hydrochloride) method was used to quantify chitin and chitosan levels (Jennifer Lodge, personal communication) (57, 58). Strains were cultured overnight in 50 ml YPD at 30°C and diluted to an optical density at 600 nm (OD600) of 0.5 in 25 ml of CO2-independent media. Each strain was cultured in triplicate for 18 to 20 h at 37°C with shaking. In preweighed tubes, cells were harvested, washed with H2O, freeze-dried, and weighed. Dried cells were then resuspended in 10 ml 6% KOH and incubated in an 80°C water bath for 90 min. Cells were spun and washed twice with phosphate-buffered saline (PBS) (pH 7.0) and then twice with H2O. Samples were then resuspended in H2O to a concentration of 10 mg/ml (using the dried cell weight) and sonicated to homogenize the samples. For each sample, two 2-ml tubes were prepared (one for chitin plus chitosan and one for chitosan only) with 100 µl (1 mg dry weight material) of the cell suspension mixed with 100 µl of 1 M HCl and subjected to vortex mixing. Two sets of hexosamine standards (d-glucosamine) were also prepared that consisted of 6 2-fold serial dilutions in 100 µl of H2O starting from 5 µM. One hundred microliters of 1 M HCl was also added to each standard sample. The chitin-plus-chitosan tubes were incubated for 2 h at 110°C, while the chitosan tubes were left at room temperature (RT). Heating in HCl deacetylates the chitin to chitosan, allowing total chitin/chitosan levels to be quantified. Samples were then cooled to room temperature. Next, all samples (boiled and unboiled) were deaminated as follows: in a fume hood, 400 µl of 2.5% sodium nitrite was then added to both sets of tubes, and the tubes were subjected to vortex mixing and left at room temperature for 15 min. A 200-µl volume of 12.5% ammonium sulfamate was slowly added, and the mixture was subjected to vortex mixing and incubated at room temperature for 5 min. Nitrogen oxide fumes (brown fumes) were allowed to dissipate before being removed from the fume hood. To initiate MBTH complex formation, 200 µl 0.25% MBTH (0.25% [wt/vol] 3-methyl–2-benzothiazolinone hydrazone hydrochloride monohydrate [Sigma]) was added to each sample, and the samples were subjected to vortex mixing and incubated for 30 min at 37°C. Finally, for blue color formation, 200 µl of 0.5% ferric chloride was added and the samples were mixed by pipetting and incubated for 5 min at 37°C. The samples were cooled to room temperature and spun in a microcentrifuge at maximum speed for 2 min. A 200-µl volume of the supernatant was added to a 96-well plate, and absorbance was measured at 650 nm with a spectrophotometer blanked with MBTH alone. Two standard curves corresponding to the level of absorbance versus that of hexosamine (in nanomoles) (average values) were prepared: one for chitin plus chitosan (boiled standards) and one for chitosan only (unboiled standards). Chitin levels were determined by finding the difference between chitin plus chitosan and chitosan for each sample.
Cell wall staining and microscopy.
Cells from overnight cultures in either CO2-independent medium or YPD were pelleted, washed, and resuspended in phosphate-buffered saline (pH 7.4). Cells were stained either with 100 µg/ml WGA conjugated to Alexa Fluor 488 (Molecular Probes) for 45 min in the dark or with 25 µg/ml CFW for 10 min. Cells were washed 2 times with PBS and imaged on a Zeiss Axio Imager A1 fluorescence microscope equipped with an AxioCam MRm digital camera. The same exposure time was used to image all strains. The mean gray value (sum of gray values for all the pixels in a cell divided by the number of pixels that make up the cell) was calculated for at least 100 individual cells using ImageJ (Fiji) (59), and the results are reported as average fluorescence values ± the standard errors of the means.
In vitro macrophage experiments.
Bone marrow-derived macrophages were isolated and prepared as previously described (60, 61). The harvested cells were plated in 96-well plates in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) (non-heat inactivated) and 1 U/ml penicillin/streptomycin at a concentration of 5 × 104 cells/well. A total of 5 × 105 C. neoformans cells (10:1 C. neoformans cells/macrophages) were added to each well, and the coculture was incubated at 37°C and 5% CO2. Each fungal strain was tested with a minimum of three biological replicates per experiment. After 6 h of coincubation, the medium from these experiments was harvested, and secreted TNF-α was quantified by enzyme-linked immunosorbent assay (ELISA; BioLegend). Data are represented as the average TNF-α values (in picograms per milliliter) for these biological triplicates or as these values normalized to macrophage-only control wells in which fresh tissue culture medium alone, rather than fungal cells, was added to each well. This type of normalization allowed comparisons between experiments performed on different days with different macrophage cultures and different ELISA plates.
All strains with the cap59Δ mutation aggregated in large, adherent clumps that made accurate quantification by hemocytometer or optical density impossible. Instead, these were normalized to 2 mg wet cell pellet/ml of medium. This cell concentration was used since it approximated the milligram-per-milliliter concentration of the rim101Δ mutant inoculum for the standard multiplicity of infection (MOI) of 10 fungal cells to 1 BMM used for the other coculture assays performed in this study (the rim101Δ strain was used as a comparator because it has a significant capsule defect and therefore has a mass/cell ratio similar to that of the acapsular cap59Δ strains). A concentration of 2 milligrams/ml of the rim101Δ mutant was approximately 5 × 106 cells/ml.
Cell separation experiments were performed using Transwell (Corning) tissue culture plates. Bone marrow-derived macrophages were harvested and plated (as described above) in the lower Transwell chamber. The various C. neoformans strains were incubated in the upper chamber and separated from the macrophages by a 3-µm-pore-size membrane to prevent cell migration while allowing free transit of diffusible molecules between the two chambers.
To test the effect of isolated cell walls on macrophage activation, cell wall material (isolated as described above) was added at a concentration of 10 mg/ml to BMMs and incubated at 37°C and 5% CO2 for 6 h. After coincubation, the medium from these experiments was harvested, and secreted TNF-α was quantified by ELISA as described above.
Animal experiments.
We used the murine inhalation model of cryptococcosis to assess organ fungal burden. BALB/c mice were intranasally infected with 104 fungal cells for each strain. At specified time points, the animals were sacrificed, and lungs and brains were dissected and homogenized in 2 ml PBS. Viable cells were calculated as CFU counts per gram of tissue by quantitative culture. Pulmonary leukocyte isolation, staining, and flow cytometry were assessed as described previously (62). Lung cytokine levels were quantified from lung homogenates as described previously (62). All animal experimentation was performed in accordance with institutional guidelines established at Duke University and the University of Texas at San Antonio.
ACKNOWLEDGMENTS
We acknowledge Jennifer Lodge, Woei Lam, and Rajendra Upadhya for developing and sharing the chitin and chitosan MTBH assay. We thank Todd Brennan of Duke University for providing MyD88-deficient mice. We acknowledge Neil Gow for providing access to the Dionex HPAEC-PAD instrumentation. We also acknowledge Connie Nichols for critical reading of the manuscript.
These experiments were supported by an NIH grant to J.A.A. and F.L.W., Jr. (R01 AI074677). C.M.L.W. was supported by a fellowship provided through the Army Research Office of the Department of Defense (no. W911NF-11-1-0136 f) (F.L.W., Jr.). J.W., L.W., and C.M. were supported by the Wellcome Trust Strategic Award in Medical Mycology and Fungal Immunology (097377) and the MRC, Centre for Medical Mycology (MR/N006364/1).
Footnotes
Citation Ost KS, Esher SK, Leopold Wager CM, Walker L, Wagener J, Munro C, Wormley FL, Jr., Alspaugh JA. 2017. Rim pathway-mediated alterations in the fungal cell wall influence immune recognition and inflammation. mBio 8:e02290-16. https://doi.org/10.1128/mBio.02290-16.
REFERENCES
- 1.Maziarz EK, Perfect JR. 2016. Cryptococcosis. Infect Dis Clin North Am 30:179–206. doi: 10.1016/j.idc.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM. 2009. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23:525–530. doi: 10.1097/QAD.0b013e328322ffac. [DOI] [PubMed] [Google Scholar]
- 3.Perfect JR. 2012. The impact of the host on fungal infections. Am J Med 125:S39–S51. doi: 10.1016/j.amjmed.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 4.O’Meara TR, Holmer SM, Selvig K, Dietrich F, Alspaugh JA. 2013. Cryptococcus neoformans Rim101 is associated with cell wall remodeling and evasion of the host immune responses. mBio 4:e0052-12. doi: 10.1128/mBio.00522-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Meara TR, Alspaugh JA. 2012. The Cryptococcus neoformans capsule: a sword and a shield. Clin Microbiol Rev 25:387–408. doi: 10.1128/CMR.00001-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doering TL. 2009. How sweet it is! Cell wall biogenesis and polysaccharide capsule formation in Cryptococcus neoformans. Annu Rev Microbiol 63:223–247. doi: 10.1146/annurev.micro.62.081307.162753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Retini C, Vecchiarelli A, Monari C, Bistoni F, Kozel TR. 1998. Encapsulation of Cryptococcus neoformans with glucuronoxylomannan inhibits the antigen-presenting capacity of monocytes. Infect Immun 66:664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vecchiarelli A, Pietrella D, Lupo P, Bistoni F, McFadden DC, Casadevall A. 2003. The polysaccharide capsule of Cryptococcus neoformans interferes with human dendritic cell maturation and activation. J Leukoc Biol 74:370–378. doi: 10.1189/jlb.1002476. [DOI] [PubMed] [Google Scholar]
- 9.Piccioni M, Monari C, Kenno S, Pericolini E, Gabrielli E, Pietrella D, Perito S, Bistoni F, Kozel TR, Vecchiarelli A. 2013. A purified capsular polysaccharide markedly inhibits inflammatory response during endotoxic shock. Infect Immun 81:90–98. doi: 10.1128/IAI.00553-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kozel TR, Gotschlich EC. 1982. The capsule of Cryptococcus neoformans passively inhibits phagocytosis of the yeast by macrophages. J Immunol 129:1675–1680. [PubMed] [Google Scholar]
- 11.Guo C, Chen M, Fa Z, Lu A, Fang W, Sun B, Chen C, Liao W, Meng G. 2014. Acapsular Cryptococcus neoformans activates the NLRP3 inflammasome. Microbes Infect 16:845–854. doi: 10.1016/j.micinf.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 12.Baker LG, Specht CA, Lodge JK. 2011. Cell wall chitosan is necessary for virulence in the opportunistic pathogen Cryptococcus neoformans. Eukaryot Cell 10:1264–1268 doi: 10.1128/EC.05138-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feldmesser M, Kress Y, Casadevall A. 2001. Dynamic changes in the morphology of Cryptococcus neoformans during murine pulmonary infection. Microbiology 147:2355–2365. doi: 10.1099/00221287-147-8-2355. [DOI] [PubMed] [Google Scholar]
- 14.Okagaki LH, Strain AK, Nielsen JN, Charlier C, Baltes NJ, Chrétien F, Heitman J, Dromer F, Nielsen K. 2010. Cryptococcal cell morphology affects host cell interactions and pathogenicity. PLoS Pathog 6:e1000953. doi: 10.1371/journal.ppat.1000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaragoza O, García-Rodas R, Nosanchuk JD, Cuenca-Estrella M, Rodríguez-Tudela JL, Casadevall A. 2010. Fungal cell gigantism during mammalian infection. PLoS Pathog 6:e1000945. doi: 10.1371/journal.ppat.1000945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiesner DL, Specht CA, Lee CK, Smith KD, Mukaremera L, Lee ST, Lee CG, Elias JA, Nielsen JN, Boulware DR, Bohjanen PR, Jenkins MK, Levitz SM, Nielsen K. 2015. Chitin recognition via chitotriosidase promotes pathologic type-2 helper T cell responses to cryptococcal infection. PLoS Pathog 11:e1004701. doi: 10.1371/journal.ppat.1004701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilbert NG, Lodge JK, Specht CA. 2011. The cell wall of Cryptococcus, p 67–79. In Heitman J, Kozel TR, Kwon-Chung KJ, Perfect JR, Casadevall A (ed), Cryptococcus: from human pathogen to model yeast. ASM Press, Washington, DC. [Google Scholar]
- 18.Chen Y, Toffaletti DL, Tenor JL, Litvintseva AP, Fang C, Mitchell TG, McDonald TR, Nielsen K, Boulware DR, Bicanic T, Perfect JR. 2014. The Cryptococcus neoformans transcriptome at the site of human meningitis. mBio 5:e01087-13. doi: 10.1128/mBio.01087-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Meara TR, Norton D, Price MS, Hay C, Clements MF, Nichols CB, Alspaugh JA. 2010. Interaction of Cryptococcus neoformans Rim101 and protein kinase A regulates capsule. PLoS Pathog 6:e1000776. doi: 10.1371/journal.ppat.1000776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodrigues ML, Alvarez M, Fonseca FL, Casadevall A. 2008. Binding of the wheat germ lectin to Cryptococcus neoformans suggests an association of chitinlike structures with yeast budding and capsular glucuronoxylomannan. Eukaryot Cell 7:602–609. doi: 10.1128/EC.00307-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fonseca FL, Guimarães AJ, Kmetzsch L, Dutra FF, Silva FD, Taborda CP, Araujo GDS, Frases S, Staats CC, Bozza MT, Schrank A, Vainstein MH, Nimrichter L, Casadevall A, Rodrigues ML. 2013. Binding of the wheat germ lectin to Cryptococcus neoformans chitooligomers affects multiple mechanisms required for fungal pathogenesis. Fungal Genet Biol 60:64–73. doi: 10.1016/j.fgb.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Free SJ. 2013. Fungal cell wall organization and biosynthesis. Adv Genet 81:33–82. doi: 10.1016/B978-0-12-407677-8.00002-6. [DOI] [PubMed] [Google Scholar]
- 23.Banks IR, Specht CA, Donlin MJ, Gerik KJ, Levitz SM, Lodge JK. 2005. A chitin synthase and its regulator protein are critical for chitosan production and growth of the fungal pathogen Cryptococcus neoformans. Eukaryot Cell 4:1902–1912. doi: 10.1128/EC.4.11.1902-1912.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wagener J, Malireddi RKS, Lenardon MD, Köberle M, Vautier S, MacCallum DM, Biedermann T, Schaller M, Netea MG, Kanneganti TD, Brown GD, Brown AJP, Gow NAR. 2014. Fungal chitin dampens inflammation through IL-10 induction mediated by NOD2 and TLR9 activation. PLoS Pathog 10:e1004050. doi: 10.1371/journal.ppat.1004050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aranaz I, Mengibar M, Harris R, Panos I, Miralles B, Acosta N, Galed G, Heras A. 2009. Functional characterization of chitin and chitosan. Curr Chem Biol 3:203–230. [Google Scholar]
- 26.Tsigos I, Martinou A, Kafetzopoulos D, Bouriotis V. 2000. Chitin deacetylases: new, versatile tools in biotechnology. Trends Biotechnol 18:305–312. doi: 10.1016/S0167-7799(00)01462-1. [DOI] [PubMed] [Google Scholar]
- 27.Bueter CL, Specht CA, Levitz SM. 2013. Innate sensing of chitin and chitosan. PLoS Pathog 9:e1003080. doi: 10.1371/journal.ppat.1003080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis SE, Hopke A, Minkin SC, Montedonico AE, Wheeler RT, Reynolds TB. 2014. Masking of β(1-3)-glucan in the cell wall of Candida albicans from detection by innate immune cells depends on phosphatidylserine. Infect Immun 82:4405–4413. doi: 10.1128/IAI.01612-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bom VLP, de Castro PA, Winkelströter LK, Marine M, Hori JI, Ramalho LNZ, dos Reis TF, Goldman MHS, Brown NA, Rajendran R, Ramage G, Walker LA, Munro CA, Rocha MC, Malavazi I, Hagiwara D, Goldman GH. 2015. The Aspergillus fumigatus sitA phosphatase homologue is important for adhesion, cell wall integrity, biofilm formation, and virulence. Eukaryot Cell 14:728–744. doi: 10.1128/EC.00008-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rappleye CA, Eissenberg LG, Goldman WE. 2007. Histoplasma capsulatum alpha-(1,3)-glucan blocks innate immune recognition by the beta-glucan receptor. Proc Natl Acad Sci U S A 104:1366–1370. doi: 10.1073/pnas.0609848104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shepardson KM, Ngo LY, Aimanianda V, Latgé JP, Barker BM, Blosser SJ, Iwakura Y, Hohl TM, Cramer RA. 2013. Hypoxia enhances innate immune activation to Aspergillus fumigatus through cell wall modulation. Microbes Infect 15:259–269. doi: 10.1016/j.micinf.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vecchiarelli A, Pericolini E, Gabrielli E, Kenno S, Perito S, Cenci E, Monari C. 2013. Elucidating the immunological function of the Cryptococcus neoformans capsule. Future Microbiol 8:1107–1116. doi: 10.2217/fmb.13.84. [DOI] [PubMed] [Google Scholar]
- 33.Zhang SQ, Zou Z, Shen H, Shen SS, Miao Q, Huang X, Liu W, Li LP, Chen SM, Yan L, Zhang JD, Zhao JJ, Xu GT, An MM, Jiang YY. 2016. Mnn10 maintains pathogenicity in Candida albicans by extending α-1,6-mannose backbone to evade host dectin-1 mediated antifungal immunity. PLoS Pathog 12:e1005617. doi: 10.1371/journal.ppat.1005617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allen AK, Neuberger A, Sharon N. 1973. The purification, composition and specificity of wheat-germ agglutinin. Biochem J 131:155–162. doi: 10.1042/bj1310155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Somashekar D, Joseph R. 1997. A new spectrophotometric method of assay for chitosanase based on calcofluor white dye binding. Carbohydr Polym 34:343–346. doi: 10.1016/S0144-8617(97)00052-0. [DOI] [Google Scholar]
- 36.Roncero C, Valdivieso MH, Ribas JC, Durán A. 1988. Isolation and characterization of Saccharomyces cerevisiae mutants resistant to calcofluor white. J Bacteriol 170:1950–1954. doi: 10.1128/jb.170.4.1950-1954.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mora-Montes HMHM, Netea MG, Ferwerda G, Lenardon MD, Brown GD, Mistry AR, Kullberg BJ, O’Callaghan CA, Sheth CC, Odds FC, Brown AJP, Munro CA, Gow NAR. 2011. Recognition and blocking of innate immunity cells by Candida albicans chitin. Infect Immun 79:1961–1970. doi: 10.1128/IAI.01282-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rogg LE, Fortwendel JR, Juvvadi PR, Lilley A, Steinbach WJ. 2011. The chitin synthase genes chsA and chsC are not required for cell wall stress responses in the human pathogen Aspergillus fumigatus. Biochem Biophys Res Commun 411:549–554. doi: 10.1016/j.bbrc.2011.06.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rogg LE, Fortwendel JR, Juvvadi PR, Steinbach WJ. 2012. Regulation of expression, activity and localization of fungal chitin synthases. Med Mycol 50:2–17. doi: 10.3109/13693786.2011.577104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bueter CL, Lee CK, Rathinam VAK, Healy GJ, Taron CH, Specht CA, Levitz SM. 2011. Chitosan but not chitin activates the inflammasome by a mechanism dependent upon phagocytosis. J Biol Chem 286:35447–35455. doi: 10.1074/jbc.M111.274936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bueter CL, Lee CK, Wang JP, Ostroff GR, Specht CA, Levitz SM. 2014. Spectrum and mechanisms of inflammasome activation by chitosan. J Immunol 192:5943–5951. doi: 10.4049/jimmunol.1301695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plato A, Hardison SE, Brown GD. 2015. Pattern recognition receptors in antifungal immunity. Semin Immunopathol 37:97–106. doi: 10.1007/s00281-014-0462-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, Locksley RM. 2007. Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature 447:92–96. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Strong P, Clark H, Reid K. 2002. Intranasal application of chitin microparticles down-regulates symptoms of allergic hypersensitivity to Dermatophagoides pteronyssinus and Aspergillus fumigatus in murine models of allergy. Clin Exp Allergy 32:1794–1800. doi: 10.1046/j.1365-2222.2002.01551.x. [DOI] [PubMed] [Google Scholar]
- 45.Shibata Y, Foster LA, Bradfield JF, Myrvik QN. 2000. Oral administration of chitin down-regulates serum IgE levels and lung eosinophilia in the allergic mouse. J Immunol 164:1314–1321. doi: 10.4049/jimmunol.164.3.1314. [DOI] [PubMed] [Google Scholar]
- 46.Da Silva CA, Hartl D, Liu W, Lee CG, Elias JA. 2008. TLR-2 and IL-17A in chitin-induced macrophage activation and acute inflammation. J Immunol 181:4279–4286. doi: 10.4049/jimmunol.181.6.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leão CA, Ferreira-Paim K, Andrade-Silva L, Mora DJ, da Silva PR, Machado AS, Neves PF, Pena GS, Teixeira LS, Silva-Vergara ML. 2011. Primary cutaneous cryptococcosis caused by Cryptococcus gattii in an immunocompetent host. Med Mycol 49:352–355. doi: 10.3109/13693786.2010.530697. [DOI] [PubMed] [Google Scholar]
- 48.Becker KL, Aimanianda V, Wang X, Gresnigt MS, Ammerdorffer A, Jacobs CW, Gazendam RP, Joosten LAB, Netea MG, Latgé JP, van de Veerdonk FL. 2016. Aspergillus cell wall chitin induces anti- and proinflammatory cytokines in human PBMCs via the Fc-γ receptor/Syk/PI3K pathway. mBio 7:e01823-15. doi: 10.1128/mBio.01823-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Upadhya R, Lam WC, Maybruck B, Specht CA, Levitz SM, Lodge JK. 2016. Induction of protective immunity to cryptococcal infection in mice by a heat-killed, chitosan-deficient strain of Cryptococcus neoformans. mBio 7:e00547-16. doi: 10.1128/mBio.00547-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wiesner DL, Boulware DR. 2011. Cryptococcus-related immune reconstitution inflammatory syndrome(IRIS): pathogenesis and its clinical implications. Curr Fungal Infect Rep 5:252–261. doi: 10.1007/s12281-011-0064-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim LK, Morita R, Kobayashi Y, Eisenbarth SC, Lee CG, Elias J, Eynon EE, Flavell RA, Gow NAR, Sohn MH. 2015. AMCase is a crucial regulator of type 2 immune responses to inhaled house dust mites. Proc Natl Acad Sci U S A 112:E2891–E2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Na H, Cho M, Chung Y. 2016. Regulation of Th2 Cell immunity by dendritic cells. Immune Netw 16:1–12. doi: 10.4110/in.2016.16.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kwon-Chung KJ, Fraser JA, Doering TL, Wang Z, Janbon G, Idnurm A, Bahn YS. 2014. Cryptococcus neoformans and Cryptococcus gattii, the etiologic agents of cryptococcosis. Cold Spring Harb Perspect Med 4:a019760. doi: 10.1101/cshperspect.a019760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fraser JA, Subaran RL, Nichols CB, Heitman J. 2003. Recapitulation of the sexual cycle of the primary fungal pathogen Cryptococcus neoformans var. gattii: implications for an outbreak on Vancouver Island, Canada. Eukaryot Cell 2:1036–1045. doi: 10.1128/EC.2.5.1036-1045.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Toffaletti DL, Rude TH, Johnston SA, Durack DT, Perfect JR. 1993. Gene transfer in Cryptococcus neoformans by use of biolistic delivery of DNA. J Bacteriol 175:1405–1411. doi: 10.1128/jb.175.5.1405-1411.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Plaine A, Walker L, Da Costa G, Mora-Montes HM, McKinnon A, Gow NAR, Gaillardin C, Munro CA, Richard ML. 2008. Functional analysis of Candida albicans GPI-anchored proteins: roles in cell wall integrity and caspofungin sensitivity. Fungal Genet Biol 45:1404–1414. doi: 10.1016/j.fgb.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsuji A, Kinoshita T, Hoshino M. 1969. Microdetermination of hexosamines. Chem Pharm Bull (Tokyo) 17:217–218. doi: 10.1248/cpb.17.217. [DOI] [PubMed] [Google Scholar]
- 58.Tsuji A, Kinoshita T, Hoshino M. 1969. Analytical chemical studies on amino sugars. II. Determination of hexosamines using 3-methyl-2-benzothiazolone hydrazone hydrochloride. Chem Pharm Bull (Tokyo) 17:1505–1510. doi: 10.1248/cpb.17.1505. [DOI] [PubMed] [Google Scholar]
- 59.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen M, Xing Y, Lu A, Fang W, Sun B, Chen C, Liao W, Meng G. 2015. Internalized Cryptococcus neoformans activates the canonical caspase-1 and the noncanonical caspase-8 inflammasomes. J Immunol 195:4962–4972. doi: 10.4049/jimmunol.1500865. [DOI] [PubMed] [Google Scholar]
- 61.Johnson CR, Kitz D, Little JR. 1983. A method for the derivation and continuous propagation of cloned murine bone marrow macrophages. J Immunol Methods 65:319–332. doi: 10.1016/0022-1759(83)90127-8. [DOI] [PubMed] [Google Scholar]
- 62.Zhai B, Wozniak KL, Masso-Silva J, Upadhyay S, Hole C, Rivera A, Wormley FL, Lin X. 2015. Development of protective inflammation and cell-mediated immunity against Cryptococcus neoformans after exposure to hyphal mutants. mBio 6:e01433-15. doi: 10.1128/mBio.01433-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The rim101Δ mutation does not induce measurable IL-1β secretion (A and B). BMMs were primed with or without 100 ng/ml UltraPure LPS for 3 h. BMMs were coincubated with the indicated C. neoformans strains at an MOI of 10 C. neoformans fungal cells to 1 BMM. cap59Δ and rim101Δ cap59Δ cells were normalized to 2 mg cells/ml of media (C) LPS-primed and unprimed BMMs were treated with 0.1 mg/ml chitosan for 6 h. Secreted IL-1β levels (in picograms per milliliter) were quantified by ELISA (BioLegend) after 6 h of coincubation. Two-way or one-way ANOVA and Tukey’s multiple-comparison test were used to compare means of results of three replicates for each strain (n = 3). ***, P < 0.001; **, P < 0.01; *, P < 0.05. Error bars represent standard errors of the means. Download FIG S1, EPS file, 0.8 MB (848.1KB, eps) .
Copyright © 2017 Ost et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
TNF-α levels (quantified in picograms per milliliter) for Fig. 1. Download TABLE S1, DOCX file, 0.01 MB (14.5KB, docx) .
Copyright © 2017 Ost et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
TNF-α levels (quantified in picograms per milliliter) for Fig. 2C. Data represent the BMM TNF-α response to C. neoformans cultured in rich media versus tissue culture media. Download TABLE S2, DOCX file, 0.01 MB (14.4KB, docx) .
Copyright © 2017 Ost et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
All cytokines quantified. Download TABLE S3, DOCX file, 0.02 MB (20.8KB, docx) .
Copyright © 2017 Ost et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The rim101Δ mutant proliferates at a rate similar to that of the WT strain during infection. Fungal CFU counts were determined from whole-lung (A and B) or whole-brain (C) homogenates. 4 BALB/c mice were intranasally inoculated with 104 cells for each strain. Data represent results of 2 independent experiments (8 mice in total). One-way ANOVA and Tukey’s multiple-comparison test were used to compare means. ***, P < 0.001; **, P < 0.01; *, P < 0.05. Error bars represent standard errors of the means. Download FIG S2, EPS file, 1 MB (1MB, eps) .
Copyright © 2017 Ost et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.