

Abstract
Key points
Retigabine is a KCNQ voltage‐gated potassium channel opener that was recently approved as an add‐on therapeutic for patients with drug‐resistant epilepsy.
Retigabine exhibits very little specificity between most KCNQ channel subtypes, and there is interest in generating more potent and specific KCNQ channel openers.
The present study describes the marked specificity of ICA069673 for KCNQ2 vs. KCNQ3, and exploits this property to investigate determinants of KCNQ subtype specificity.
ICA069673 acts on a binding site in the voltage‐sensing domain that is distinct from the putative retigabine site in the channel pore.
ICA069673 has two separable effects on KCNQ channel activity. We identify two channel residues required for subtype specificity of KCNQ channel openers and show that these are sufficient to generate ICA069673 sensitivity in KCNQ3.
Abstract
Retigabine (RTG) is the first approved anti‐epileptic drug that acts via activation of voltage‐gated potassium channels, targeting KCNQ channels that underlie the neuronal M‐current. RTG exhibits little specificity between KCNQ2–5 as a result of conservation of a Trp residue in the pore domain that binds to the drug. The RTG analogue ICA‐069673 (‘ICA73’) exhibits much stronger effects on KCNQ2 channels, including a large hyperpolarizing shift of the voltage‐dependence of activation, an ∼2‐fold enhancement of peak current and pronounced subtype specificity for KCNQ2 over KCNQ3. Based on ICA73 sensitivity of chimeric constructs of the transmembrane segments of KCNQ2 and KCNQ3, this drug appears to interact with the KCNQ2 voltage sensor (S1–S4) rather than the pore region targeted by RTG. KCNQ2 point mutants in the voltage sensor were generated based on KCNQ2/KCNQ3 sequence differences, and screened for ICA73 sensitivity. These experiments reveal that KCNQ2 residues F168 and A181 in the S3 segment are essential determinants of ICA73 subtype specificity. Mutations at either position in KCNQ2 abolish the ICA73‐mediated gating shift, but preserve RTG sensitivity. Interestingly, A181P mutant channels show little ICA73‐mediated gating shift but retain current potentiation by the drug. Mutations (L198F and P211A), which introduce these critical KCNQ2 residues at corresponding positions in KCNQ3, transplant partial ICA73 sensitivity. These findings demonstrate that RTG and ICA73 act via distinct mechanisms, and also reveal specific residues that underlie subtype specificity of KCNQ channel openers.
Keywords: epilepsy, KCNQ activator, KCNQ channel, M‐current, potassium channel, retigabine
Key points
Retigabine is a KCNQ voltage‐gated potassium channel opener that was recently approved as an add‐on therapeutic for patients with drug‐resistant epilepsy.
Retigabine exhibits very little specificity between most KCNQ channel subtypes, and there is interest in generating more potent and specific KCNQ channel openers.
The present study describes the marked specificity of ICA069673 for KCNQ2 vs. KCNQ3, and exploits this property to investigate determinants of KCNQ subtype specificity.
ICA069673 acts on a binding site in the voltage‐sensing domain that is distinct from the putative retigabine site in the channel pore.
ICA069673 has two separable effects on KCNQ channel activity. We identify two channel residues required for subtype specificity of KCNQ channel openers and show that these are sufficient to generate ICA069673 sensitivity in KCNQ3.
Abbreviations
- RTG
retigabine
- VSD
voltage‐sensing domain
- WT
wild‐type
Introduction
Retigabine (RTG) is a recently approved anti‐epileptic drug currently used as an adjunct add‐on treatment for pharmaco‐resistant epilepsy (Miceli et al. 2008). RTG and its closely‐related analogue flupirtine are currently the only voltage‐gated potassium channel openers approved for human use. Their molecular mechanism of action is well described; they are considered to interact with a Trp side chain located in the pore‐forming S5 segment of KCNQ2–5 channel subunits, and cause a pronounced hyperpolarizing shift of the voltage‐dependence of channel activation (Schenzer et al. 2005; Lange et al. 2009). Importantly, RTG exhibits little specificity between KCNQ2–5 channel subunits, and the effects arising from KCNQ channels in peripheral tissues, including the bladder, are a recurring problem in patients (Brickel et al. 2012; Martyn‐St et al. 2012). However, RTG has served as an important chemical tool for investigating the therapeutic potential of KCNQ channel modulation, and also as a template for the development of further analogues that may exhibit better subunit selectivity and centrally‐restricted actions (Xiong et al. 2008; Kalappa et al. 2015; Kumar et al. 2016).
There is growing recognition of small molecules with KCNQ channel opener activity (Miceli et al. 2011). Certain oxindole analogues (e.g. BMS‐204352) act as potent KCNQ2–5 activators, whereas others, such as R‐L3, activate KCNQ1 (Wu & Dworetzky, 2005; Dalby‐Brown et al. 2006; Xiong et al. 2007). The widely‐used non‐steroidal anti‐inflammatory drugs diclofenac and meclofenamic acid, usually categorized as non‐selective cyclooxygenase inhibitors, are also relatively effective activators of KCNQ2 and KCNQ2/3 heteromers (Munster et al. 2002; Peretz et al. 2007; Peretz et al. 2010). Zinc pyrithione, used for controlling dandruff and treating psoriasis, is reported to be a strong KCNQ activator, causing potentiation of all KCNQ channel currents except KCNQ3 (Xiong et al. 2007). Some commercially available compounds are suggested to exhibit subtype selectivity, including ML‐213, as well as a family of pyridinyl benzamide compounds, including ICA‐27243, ICA‐110381 and ICA‐069673 (Wickenden et al. 2008; Blom et al. 2010; Gao et al. 2010).
With this progress in discovery of KCNQ openers, fundamental principles underlying their actions are being revealed. For example, our group recently demonstrated the requirement for a hydrogen bond donor in the KCNQ3 Trp265 side chain that is assumed to interact with RTG (Kim et al. 2015). This chemical property correlated with the polarity of a hydrogen bond acceptor carbonyl found in many KCNQ channel openers. Drugs with very weak polarity at this position, such as ICA‐069673, exhibit weak effects on KCNQ3. Also, an analogue with much stronger bond polarity (ML‐213) exhibited a more potent activation of KCNQ3 (Yu et al. 2013). This detailed description of the chemical basis of drug action at the putative RTG binding site, as well as the weak effects of ICA‐069673 in KCNQ3, were inconsistent with reports of the activating effects of ICA‐069673 (and other close analogues) on KCNQ channels (Boehlen et al. 2013; Brueggemann et al. 2014). Several studies have suggested the possibility that certain KCNQ activators may act on a distinct site present in certain KCNQ subtypes. For example, a family of diclofenac derivatives were shown to be insensitive to mutation of the S5 Trp residue that is essential for RTG actions, and they may interact with the voltage‐sensing domain (VSD) rather than the pore region of KCNQ (Padilla et al. 2009; Gao et al. 2010; Peretz et al. 2010).
In the present study, we describe strong subunit specificity of ICA‐069673, and exploit this property to investigate sequence determinants underlying subunit specificity. The present study identifies two residues in the voltage‐sensing domain of KCNQ2 that are required for the actions of ICA‐069673, and whose mutations do not dramatically alter channel gating or RTG sensitivity. Moreover, the introduction of these residues into KCNQ3 transplants significant ICA‐069673 sensitivity. These results demonstrate unambiguously that KCNQ channel openers can act independently of the RTG binding site with a distinct mechanism. The present study also highlights the multi‐pronged effects of ICA‐069673, which causes a marked hyperpolarizing shift of KCNQ2, and a dramatic potentiation of current. These two effects can be separated by point mutations in the KCNQ2 voltage‐sensing domain.
Methods
KCNQ2 and KCNQ3 channel constructs
Mutant channels were derived from human KCNQ2 or KCNQ3 genes (originally in pTLN vector; gifts from Dr M. Taglialatela, University of Molise, Campobasso, Italy; and Dr T. Jentsch, Leibniz‐institut fuer Molekulare Pharmakologie, Berlin, Germany), expressed in pcDNA3.1(–) plasmid (Invitrogen, Carlsbad, CA, USA). KCNQ3* channels refer to KCNQ3[A315T], carrying a point mutation that allows homomeric expression of KCNQ3 (Gomez‐Posada et al. 2010). Chimeras between KCNQ2 and KCNQ3 were constructed using an overlapping PCR method. Flanking primers were used to amplify respective segments of KCNQ2 and KCNQ3. PCR approaches were then used to sequentially combine overlapping fragments until all necessary components segments of the chimera were incorporated. Break points for the chimeras were generated. For Q2+Q3[S1–S2] channels, KCNQ2 residues 89–148 were replaced by KCNQ3 residues 117–178. For Q2+Q3[S3–S4], KCNQ2 residues 153–207 were replaced by KCNQ3 residues 183–236. For Q2+Q3[S5–S6] channels, KCNQ2 residues 239–324 were replaced by KCNQ3 residues 268–363. For Q2+Q3[S1] channels, KCNQ2 residues 89–115 were replaced by KCNQ3 residues 117–145. For Q2+Q3[S2] channels, KCNQ2 residues 115–149 were replaced by KCNQ3 residues 145–178. Point mutants in KCNQ2 and KCNQ3 were constructed using a two‐step overlapping PCR method. All constructs were subcloned into pcDNA3.1(–) using NheI and EcoRI (KCNQ2) or NheI and XhoI (KCNQ3) restriction enzymes and verified by Sanger sequencing approaches (Genewiz, South Plainfield, NJ, USA; or Applied Genomics Core, University of Alberta, Edmonton, AB, Canada).
Cell culture and whole‐cell patch clamp recordings
HEK293 cells were cultured in 50 ml polystyrene tissue culture flasks (Falcon; Corning Inc., Tewksbury, MA, USA) in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin. Cells were grown in an incubator at 5% CO2 and 37°C. Cells were plated into six‐well plates and co‐transfected with plasmids encoding the channel of interest and green fluorescent protein using jetPRIME DNA transfection reagent (Polyplus, New York, NY, USA). After 24 h of incubation with transfection reagent, cells were split onto sterile glass coverslips at low density to allow recordings from individual cells, and electrophysiological experiments were conducted 1 day later.
Whole‐cell patch clamp recordings were performed using extracellular solution consisting (in mm): 135 NaCl, 5 KCl, 2.8 sodium acetate, 1 CaCl2(2H2O), 1 MgCl2(6H2O) and 10 Hepes (pH 7.4). Intracellular solution contained (in mm): 135 KCl, 5 EGTA and 10 Hepes (pH 7.3). RTG and ICA‐069673 were stored as 100 mm stocks in DMSO, and were diluted to working concentrations in extracellular solution on each experimental day. Glass pipette tips were manufactured using soda lime glass (Fisher Scientific Co., Pittsburgh, PA, USA) and had open tip resistances of 1–3 MΩ using the standard experimental solutions. Series resistance compensation of 75–85% was used in all recordings. Recordings were filtered at 5 kHz and sampled at 10 kHz using a Digidata 1440A (Molecular Devices, Sunnyvale, CA, USA) controlled by pClamp, version 10 (Molecular Devices). Experimental compounds were purchased from Toronto Research Chemicals (Toronto, ON, Canada) (RTG) or Tocris (Avonmouth, Bristol, UK) (ICA‐069673).
Two‐electrode voltage clamp recordings
For experiments in Xenopus laevis oocytes, complementary RNA was transcribed from the cDNA of several constructs using the mMessage mMachine Kit (Ambion, Austin, TX, USA). Stage V–VI X. laevis oocytes were prepared as described previously and were injected with cRNA (Kim et al. 2015). We used female X. laevis frogs that were 100 g or greater in size and the oocytes were prepared using protocols approved by the University of British Columbia Animal Care Committee, or the University of Alberta Animal Care Committee, in accordance with the Canadian Council for Animal Care guidelines. Frogs were anaesthetized by bathing in 1.5% tricaine methanesulphonate sulphonate solution buffered with bicarbonate, prior to the surgery for harvesting of oocytes. Oocytes were incubated post injection for 12–96 h at 18°C before recording. We recorded voltage‐clamped potassium currents in standard Ringer solution (in mm): 116 NaCl, 2 KCl, 1 MgCl2, 0.5 CaCl2 and 5 Hepes (pH 7.4) using an OC‐725C voltage clamp (Warner, Hamden, CT, USA). Glass microelectrodes were backfilled with 3 m KCl and had resistances of 0.1–1 MΩ. Data were filtered at 5 kHz and digitized at 10 kHz using a Digidata 1440A (Molecular Devices) controlled by pClamp, version 10 (Molecular Devices).
Non‐radioactive Rb+ efflux assay
HEK293 cells were plated into 24‐well plates and co‐transfected with the KCNQ mutant channel of interest, IRK1 and green fluorescent protein, using jetPRIME DNA transfection reagent (Polyplus). The IRK1 construct was included to ensure reproducible control of membrane voltage by changes to extracellular K+ concentration. Cells were incubated in the transfection mixture for 24 h, followed by 24 h in regular media. Two days post transfection, cells were incubated with Rb+ loading solution (1 mm RbCl in Dulbecco's modified Eagle's medium with 10% FBS and 1% penicillin–streptomycin) for 2 h. This was followed by washing twice with 0 K+ buffer (140 mm NaCl, 1.2 mm MgSO4, 2.5 mm CaCl2 and 10 mm Hepes, pH 7.4). The reversal potential of this nominally 0 K+ solution was measured experimentally as being more negative than –90 mV (with our standard intracellular recording solution). The use of a hyperpolarizing solution during washes dramatically reduces the extent of Rb+ efflux during wash steps (because the KCNQ channel‐mediated efflux pathway is held closed) and therefore enhances the detectable readout of the assay. Cells were then incubated in 600 μl of 0 K+ (‘hyperpolarizing’) buffer, 100 K+ activating buffer (40 mm NaCl, 100 mm KCl, 1.2 mm MgSO4, 2.5 CaCl2 and 10 mm Hepes at pH 7.4) or 0 K+ buffer supplemented with ICA 069673. Assay aliquots (200 μl) were removed at multiple time points (5, 10 and 20 min) and cells were lysed with 1% SDS RIPA lysis buffer (25 mM Tris, 150 mM NaCl, 1% SDS, 0.5% Na‐deoxycholate, 1% NP‐40) upon completion of the assay. Rb+ concentrations were measured by flame atomic absorption spectroscopy using ICR8000 instrument (Aurora Biomed, Vancouver, BC, Canada). The amount of Rb+ efflux was calculated as a fraction of total Rb+ loaded (sum of Rb+ remaining in lysed cells and Rb+ extruded from cells). Normalized Rb+ efflux in drug solution was calculated as (EffluxICA069673 – Efflux0 K +)/(Efflux100 K + – Efflux0 K +). This calculation can be thought of as the ‘drug‐stimulated efflux’ normalized to the ‘maximum voltage‐activation evoked efflux’ (i.e. both include a subtraction of background efflux in nominally 0 K+ conditions). For simplicity, only the 20 min time point data (mean ± SEM) are reported in the present study.
Molecular models of KCNQ2
Molecular models of KCNQ2 were kindly shared with us by Dr M. Taglialatela (University of Molise, Campobasso, Italy) (Miceli et al. 2013) and Dr B. Attali (Tel Aviv University, Israel) (Gourgy‐Hacohen et al. 2014) with detailed methods for model generation being described in the respective studies.
Statistical analysis
Voltage dependence of channel activation was fitted with a standard single component Boltzmann equation of the form G/G max = 1/(1 + /k, where V 1/2 is the voltage where channels exhibit half‐maximal activation and k is a slope factor. Tail current deactivation kinetics were fit with a single exponential equation in the form of I(t) = I(0)* e −t/τ.
In most cases, the ICA‐069673‐responsive channels exhibited such a dramatic drug‐mediated hyperpolarizing gating shift that we could not confidently fit their conductance–voltage relationships or tail current kinetics. Thus, most conductance–voltage relationships in the presence of ICA 069673 are fit by a non‐descriptive sigmoidal relationship and we have not focused on specific gating parameters in these cases. For statistical comparisons of drug effects on KCNQ2 mutants, we used fit independent parameters describing either the potentiation (conductance at +20 mV relative to peak conductance in control) or the shift (conductance at –80 mV relative to peak conductance in control). For KCNQ3 channel mutants, conventional gating parameters (V 1/2, slope factor k) could be derived in all experimental conditions and were used for statistical comparisons. Specific statistical tests were carried out using SigmaPlot, version 10.0 (Systat Software Inc., Chicago, IL, USA) and are described in as appropriate. Data are reported as the mean ± SEM.
Results
Subtype specific effects of ICA73
Certain KCNQ activators, including ML‐213, ICA‐069673 (referred to as ICA73) and Zn‐pyrithione, have been reported to show some subtype specificity, although some reports are unclear and the determinants of subtype specificity remain unknown (Xiong et al. 2007; Boehlen et al. 2013). For example, we recently observed that ML‐213, previously described as a KCNQ2‐specific activator, could also activate KCNQ3* (KCNQ3[A315T]) channels with greater potency than RTG (Yu et al. 2013; Kim et al. 2015).
Through ongoing investigation of commercially available KCNQ channel openers, we recognized that ICA73 and RTG (Fig. 1 A) have dramatically different effects on KCNQ2 vs. KCNQ3* channels (Fig. 1 B and C). RTG caused a leftward shift of the V 1/2 of activation in both KCNQ2 and KCNQ3* (Schenzer et al. 2005). Although the effects of RTG are more pronounced in KCNQ3*, these findings reflect that RTG is a relatively non‐selective KCNQ channel activator. We have recently reported a larger effect of RTG in KCNQ2 than observed in the present study (Kim et al. 2015), which may be a result of differences in drug sensitivity in different batches of oocytes. Nevertheless, RTG consistently causes a clear hyperpolarizing shift of both KCNQ2 and KCNQ3*. By contrast, 30 μm ICA73 induced a dramatic hyperpolarizing shift of KCNQ2 channel activation (Fig. 1 B), whereas even higher concentrations (100 μm) elicited much smaller effects on KCNQ3* (Fig. 1 C). Because of challenges with confidently fitting conductance–voltage relationships generated in the presence of ICA73, we used a fit independent comparison of conductance at −80 mV for KCNQ2(Fig. 1 B, inset). In the absence of drug, channels are predominantly closed at −80 mV, with increased conductance in the presence of ICA73 or RTG reflecting the magnitude of the shift in voltage‐dependent gating (Fig. 1 B). Based on these unique effects of ICA73, we set out to investigate differences between KCNQ2 and KCNQ3* that underlie subtype‐specific drug effects. Some previous studies have highlighted an alternative binding site in the voltage‐sensing domain of KCNQ channels for certain KCNQ openers. However, potential residues reported to influence drug effects tend to be highly conserved and essential for normal activation gating of Kv channels (e.g. S2 positions E130 and F137, as well as S4 residues L197, R198 and R201) and do not provide a rationale for understanding subtype specific effects (Tao et al. 2010; Pless et al. 2011; Li et al. 2013 b; Brueggemann et al. 2014).
Figure 1. ICA069673 exhibits subtype‐specificity for KCNQ2 over KCNQ3 channels.
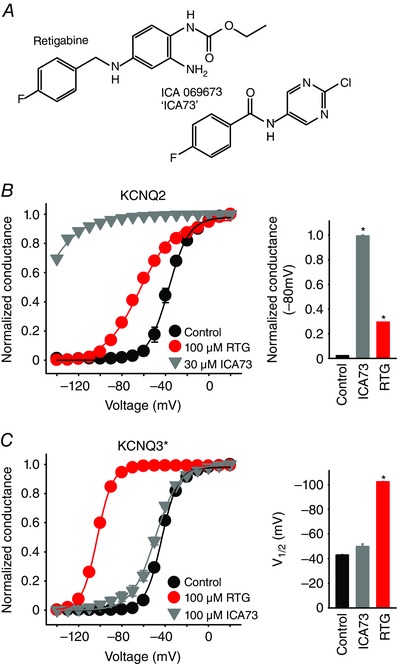
A, chemical structures of RTG and ICA069673 (‘ICA73’). B and C, conductance–voltage relationships for KCNQ2 and KCNQ3* were generated using recordings from X. laevis oocytes expressing each respective channel. KCNQ2 parameters of activation were: control V 1/2 = –37 ± 2 mV, k = 10 ± 1 mV; RTG V 1/2 = –61 ± 1 mV, k = 17 ± 1 mV, n = 5. KCNQ3 parameters of activation were: control V 1/2 = –43 ± 1 mV, k = 8 ± 1 mV; ICA73 V 1/2 = –50 ± 2 mV, k = 11 ± 2 mV; RTG V 1/2 = –103 ± 1 mV, k = 7 ± 1 mV, n = 5. (B and C, insets) Statistical comparisons between control and ICA73 or RTG (ANOVA, Dunnett's post hoc test. * P < 0.05 relative to control condition).
ICA73 causes current potentiation and a shift in voltage‐dependent gating of KCNQ2
ICA73 exhibits two very prominent but distinct effects on KCNQ2 channels: a shift in the voltage‐dependence of activation, and current potentiation at voltages where channel activation is saturated (Fig. 2). These effects are sometimes not obvious in published work because of a common practice of normalizing conductance–voltage relationships. Although we have observed current potentiation by ICA73 in channels expressed in both oocytes and mammalian cell lines, we have found these responses to be more consistent in mammalian cell lines. Also, several of the mutants that we have generated proved to be surprisingly challenging to express in oocytes, and so the remainder of the experiments in the present study were carried out using HEK293 cells as an expression system. The multiple effects of ICA73 on KCNQ2 are apparent in exemplar traces taken from the same cell (Fig. 2 A and B) and in conductance–voltage relationships normalized to the peak conductance in control conditions (Fig. 2 C). These data highlight both a shift in voltage‐dependence of activation, and an approximate doubling of peak channel current by ICA73. As noted above, there are challenges fitting conductance–voltage relationships in the presence of ICA73 because of the large drug‐mediated gating shift. We have addressed this by comparing the conductance (normalized to peak control conductance) at either +20 mV (as an index of current potentiation) or –80 mV (as an index of the drug‐mediated gating shift, Fig. 2 C). The maximal ICA73‐mediated gating shift is much larger than RTG, such that we are unable to close channels completely even with very negative voltage pulses (illustrated by sample tail currents at –130 mV) (Fig. 2 D). Also, the substantial KCNQ2 open probability at the holding potential of –80 mV in the presence of 30 μm ICA73 causes a pronounced instantaneous current upon subsequent membrane depolarization (Fig. 2 B). This generation of a prominent potassium conductance around the resting membrane potential is an important feature of KCNQ channel openers that probably underlies their therapeutic benefit.
Figure 2. ICA73 potentiates KCNQ2 currents and induces a large hyperpolarizing shift of the conductance–voltage relationship.

A and B, exemplar patch clamp current traces of WT KCNQ2 (expressed in HEK cells) in control conditions or 30 μM ICA73. Cells were held at –80 mV, and pulsed between –140 mV and +20 mV for 2 s, with tail currents measured at –20 mV. C, conductance–voltage relationships for WT KCNQ2, normalized to peak conductance in control for each cell (mean ± SEM) (control V 1/2 = –40 ± 5 mV, k = 12 ± 1 mV; n = 4). Student's t test was used to compare conductance (normalized to peak conductance in control conditions) at –80 mV or +20 mV. * P < 0.05. D, exemplar sweeps to elicit tail currents in WT KCNQ2 pulsed to –130 mV in control or ICA73 (from a holding voltage of +20 mv).
ICA73 acts within the VSD
We investigated the channel elements responsible for ICA73‐selective effects on KCNQ2 over KCNQ3, first by using a chimeric approach of replacing transmembrane domains in KCNQ2 (ICA73‐sensitive) with the corresponding (ICA73‐insensitive) KCNQ3 sequence (Fig. 3 A, D and G). Whole‐cell patch clamp experiments revealed that substitution of the KCNQ3 pore (S5–S6) for KCNQ2 largely preserved ICA73 sensitivity, with persistence of both a large hyperpolarizing gating shift and current potentiation by ICA73 (Fig. 3 H and I). This finding suggests that determinants of ICA73‐sensitivity lie within the S1–S4 segments of KCNQ2. Consistent with this, substitution of the KCNQ3 S3–S4 sequence for KCNQ2 rendered channels completely insensitive to ICA73, abolishing both the gating shift and current potentiation effects (Fig. 3 E and F). Finally, introduction of the KCNQ3 S1–S2 sequence caused a reduced ICA73 effect that appeared to preserve current potentiation but abolish most of the drug‐mediated gating shift (Fig. 3 B and C). The S1–S2 chimeric channels also exhibited notably slower gating kinetics relative to wild‐type (WT) KCNQ2 channels (Fig. 3 C). These results highlight the voltage‐sensing domain (S1–S4) as a probable site for ICA73 subtype selectivity, with a particular importance of the S3–S4 segments. These findings also highlight that the gating shift and current potentiation effects can occur separately, as demonstrated by the Q2+Q3[S1,S2] chimera (Fig. 3 A–C).
Figure 3. Substitution of KCNQ3 VSD into KCNQ2 alters channel sensitivity to ICA73.

A, D and G, cartoon representations of chimeric channels of KCNQ2 (grey) and KCNQ3 (red). B, E and H, conductance–voltage relationships for (B) Q2+ Q3[S1,S2] (control V 1/2 = –9 ± 1 mV, k = 8 ± 1 mV; ICA73 V 1/2 = –22 mV ± 5 mV, k = 5 ± 1 mV, n = 5); (E) Q2+Q3[S3–S4] (control V 1/2 = –40 ± 3 mV, k = 11 ± 1 mV; ICA73 V 1/2 = –39 ± 2 mV, k = 10 ± 1 mV, n = 4); and (H) Q2+Q3[S5–S6] (control V 1/2 = –25 ± 7 mV, k = 11 ± 2 mV; n = 5) normalized to peak conductance in control. C, F and I, sample current traces for (C) Q2+Q3[S1,S2]; (F) Q2+Q3[S3–S4]; and (I) Q2+Q3[S5–S6] illustrating the lack of ICA73 effect when S3–S4 of KCNQ3 is substituted into KCNQ2. J, relative conductance at –80 mV was used as an index of the gating shift effect of ICA73 (ANOVA, Dunnett's post hoc test. * P < 0.05). K, relative conductance at +20 mV for WT KCNQ2 and chimeric constructs in the presence of ICA73 to illustrate current potentiation effect (statistical tests as in J).
Identification of amino acid determinants of ICA73 subtype specificity
To identify residues in the KCNQ2 voltage‐sensing domain that confer ICA73 specificity, we used sequence alignments of KCNQ2 and KCNQ3 (Fig. 4 A) to guide mutagenesis of KCNQ2 (to corresponding KCNQ3 residues) at positions throughout S3 and S4. In addition, because of a large number of residue differences in S1 and S2, we generated two additional chimeras with either S1 or S2 from KCNQ3 substituted for KCNQ2. We employed a non‐radioactive Rb+ efflux assay to screen these mutants for ICA73 sensitivity (Li et al. 2013 a). Because ICA73 effectively activates KCNQ2 channels even at very hyperpolarized potentials (Fig. 2 C), ICA73‐mediated Rb+ efflux in hyperpolarizing solutions (nominally external K+‐free) was used to identify mutants that retain ICA73 sensitivity. Cells were also co‐transfected with IRK1 to ensure that manipulations of extracellular K+ would strongly influence the membrane voltage (see Methods). Using this approach, we identified the KCNQ2[F168L] and KCNQ2[A181P] mutations in S3 as highly perturbative to ICA73 sensitivity (Fig. 4 B and C). It is also noteworthy that the S1 chimera and S2 chimera both retained significant ICA73 sensitivity. This was unexpected because the S1–S2 chimera described in Fig. 3 A–C is predominantly closed in the voltage range targeted by the Rb+ efflux assay (more negative than –90 mV as a result of the very low external K+ concentration). This finding suggested that there may be important interactions between S1 and S2 playing a role in ICA73 sensitivity (Fig. 3 B and C), although swapping individual KCNQ2 and KCNQ3 residues may not be a powerful approach for investigating this question. We focused the remainder of the present study on the S3–S4 residues identified in this initial scan because these are previously unrecognized determinants of subtype specificity of KCNQ activators.
Figure 4. Rubidium efflux screen identifies two S3 mutations that diminish ICA73 response.

A, alignment of KCNQ2–5 sequences. Differences between KCNQ2 and KCNQ3 were used to identify candidate non‐conserved residues in the VSD that may be essential for ICA73 sensitivity. B, a rubidium efflux assay was used to scan ICA73 responses from mutants as indicated. Normalized ICA73 response was calculated as the ratio of the response in ICA73 (in hyperpolarizing solution) to the maximal rubidium efflux observed in depolarizing solution (ANOVA, Dunnett's post hoc test. * P < 0.05). Data are presented as the mean ± SEM (n = 3 per construct). C, structural representation of a molecular model of a single KCNQ2 subunit highlighting all candidate residues screened (red), F168L and A181P (blue), and Trp236 (yellow).
Aromatic residues at KCNQ2 position 168 preserve ICA73 sensitivity
KCNQ2[F168L] channels generated functional currents with activation kinetics and voltage‐dependence generally similar to WT KCNQ2 but with some quantitative differences (Fig. 5 A and B and Table 1). However, ICA73 had very small effects on these mutant channels (Fig. 5 A and B), which is consistent with data from the Rb+ efflux experiments. The F168L mutation abolished both the ICA73‐mediated hyperpolarizing gating shift, and current potentiation (Fig. 5 A, B and I). Also, ICA73 had almost no effect on kinetics of F168L channel closure (Fig. 5 G). Gating effects of the KCNQ2 F168L mutation were previously investigated in detail (Miceli et al. 2013), reporting a modest (∼7 mV) depolarizing gating shift in the F168L mutant, along with slower deactivation kinetics, comparable to our findings (Table 1). We tested additional aromatic mutations at position 168 (Fig. 5 C–F). Conductance–voltage relationships indicate that aromatic mutations preserve potentiation and gating effects of ICA73, although the magnitude of the effects of ICA73 was not as pronounced in the F168Y mutant (Fig. 5 I); this mutant also caused a large intrinsic gating shift in the absence of drug (Fig. 5 D). Residue F168 is located in an interesting region at the cytoplasmic side of the S3 transmembrane helix (Fig. 5 H). Previously published models of the KCNQ2 open state (Miceli et al. 2011; Gourgy‐Hacohen et al. 2014) place F168 in close proximity with a conserved lysine (KCNQ2 K219) in the S4–S5 linker, and a conserved arginine (KCNQ2 R213) at the cytoplasmic side of the S4 segment. It is also possible that F168 could come into close contact with other S4 charged side chains in alternative voltage sensor configurations.
Figure 5. Functional characterization of KCNQ2[F168L] mutant channels.

A, C and E, exemplar patch clamp current recordings of (A) KCNQ2[F168L], (C) KCNQ2[F168Y] and (E) KCNQ2[F168W]. Cells were held at –80 mV, and pulsed between –140 mV and +40 mV for 2 s, with tail currents measured at –20 mV. B, D and F, conductance–voltage relationships of (B) KCNQ2[F168L] (control V 1/2 = –23 ± 2 mV, k = 8.9 ± 0.6 mV; ICA73 V 1/2 = –20 ± 7 mV, k = 15 ± 4 mV, n = 4); (D) KCNQ2[F168Y] (control V 1/2 = –7 ± 5 mV, k = 19 ± 3, n = 4); and (F) KCNQ2[F168W] (control V 1/2 = –29 ± 9 mV, k = 17 ± 2 mV, n = 3). The conductance–voltage relationship in ICA73 is normalized to the peak conductance in control conditions for each individual cell. G, time constants (τ) of channel closure were measured by pulsing to a range of negative voltages after a depolarizing prepulse to +20 mV; inset, sample tail currents of KCNQ2[F168L] at –130 mV. H, structural model of the KCNQ2 open state, depicted as a ‘bottom view’ of the intracellular side of the channel transmembrane domain. KCNQ2 residue F168 is in close proximity with R213 and K219, suggesting possible interactions that may underlie the role of F168. I, relative conductance at –80 mV and +20 mV of WT KCNQ2 and F168 mutants in the presence of ICA73 to illustrate ICA73‐mediated gating shift and current potentiation effects, respectively (ANOVA, Dunett's post hoc test. * P < 0.05 relative to WT KCNQ2).
Table 1.
Gating parameters for various KCNQ2 channel mutants under control conditions
| Construct | V 1/2 (mV) | k (mV) | N |
|---|---|---|---|
| WT KCNQ2 | –35 ± 4 | 12 ± 1 | 8 |
| KCNQ2 [F168L] | –22 ± 2 * | 10.7 ± 0.9 | 8 |
| KCNQ2 [A181P] | –20 ± 1 * | 11.9 ± 0.8 | 8 |
| Q2+Q3(S1,S2) | –8 ± 1 | 8.4 ± 0.4 | 5 |
| Q2+Q3(S3,S4) | –40 ± 3 | 11 ± 1 | 4 |
| Q2+Q3(S5,S6) | –25 ± 9 | 11 ± 2 | 5 |
| KCNQ2 [F168W] | –29 ± 9 | 16 ± 2 | 4 |
| KCNQ2 [F168Y] | –6 ± 5 | 19 ± 3 | 4 |
| KCNQ2 [A181G] | –18 ± 8 | 11 ± 2 | 3 |
| KCNQ2 [A181L] | –10 ± 6 | 12.0 ± 0.9 | 3 |
Conductance–voltage relationships were fit with a Boltzman equation (see Methods). Data are presented as the mean ± SEM. The study was not powered for large numbers of multiple comparisons, and so statistical tests were restricted to the italicized constructs (WT KCNQ2, KCNQ2[F168L], KCNQ2[A181P], cumulative data from Figs 2, 5, 6 and 7) compared using ANOVA and Dunnett's post hoc test (* P < 0.05 relative to WT KCNQ2).
Proline at position 181 in KCNQ2 disrupts the effects of ICA73
Unexpectedly, upon detailed characterization, we observed that the KCNQ2[A181P] mutant retained pronounced potentiation of peak current but exhibited a much smaller shift of voltage‐dependent activation relative to WT KCNQ2 (Fig. 6 A, B and I). This behaviour was similar to the Q2+Q3[S1,S2] chimera (Fig. 3 A–C), once again indicating that the gating shift and current potentiation effects do not necessarily coincide, and thus may not be mediated by the same molecular determinants. Tail current kinetics also illustrate channel susceptibility to the drug, although ICA73‐mediated deceleration of channel closure is not as dramatic as that observed in WT KCNQ2 (compare Fig. 6 G, inset and Fig. 2 D). Because prolines can perturb α‐helical structures, we tested additional mutations of either a bulky residue (Leu, high helical propensity) or a small and flexible residue (Gly, low helical propensity but still amenable to α‐helical configuration) at position 181. ICA73 had a large effect on both mutants (Fig. 6 C–F), suggesting that A181 is not essential for drug binding or a helical configuration of S3, although the proline present in KCNQ3 may deform the top of the S3 helix (Fig. 6 H) and alter the channel response to ICA73.
Figure 6. Functional characterization of KCNQ2[A181P] mutant channels illustrates the separable nature of current potentiation and gating shift.

A, C and E, exemplar patch clamp current recordings of (A) KCNQ2[A181P], (C) KCNQ2[A181L] and (E) KCNQ2[A181G]. B, D and F, conductance–voltage relationships of (B) KCNQ2[A181P] (control V 1/2 = –20 ± 2 mV, k = 10.5 ± 0.7 mV; ICA73 V 1/2 = –32 ± 2 mV, k = 12 ± 1 mV, n = 5); (D) KCNQ2[A181L] (control V 1/2 = –10 ± 6 mV, k = 11 ± 2 mV, n = 3); and (F) KCNQ2[A181G] (control V 1/2 = –18 ± 8 mV, k = 12 ± 1 mV, n = 3). The conductance–voltage relationship in ICA73 is normalized to the peak conductance in control conditions for each individual cell. (G) Time constants (τ) of channel closure were measured by pulsing to a range of negative voltages from a holding potential of +20 mV; inset, sample tail currents of KCNQ2[A181P] at –130 mV. (H) Molecular model of the voltage‐sensing domain of KCNQ2 channels in the open state. Residue A181 is located on the extracellular side of the S3 segment. (I) Relative conductance at –80 mV and +20 mV of WT KCNQ2 and A181 mutants in the presence of ICA73 to illustrate gating shift and current potentiation effects, respectively (ANOVA, Dunnett's post hoc test. * P < 0.05 relative to WT KCNQ2).
ICA73‐insensitive mutations preserve RTG sensitivity
The experiments conducted thus far suggest that ICA73 and RTG probably act via different binding sites and different mechanisms. To further investigate the effects of the A181P and F168L mutations, and the effects of RTG vs. ICA73, we compared the effects of RTG on KCNQ2[F168L] and KCNQ2[A181P] with WT KCNQ2. KCNQ2[F168L] (Fig. 7 C and D) or [A181P] (Fig. 7 E and F) mutant channels, shown in the present study to exhibit disrupted ICA73 sensitivity (Figs 5 and 6), retain sensitivity to RTG that is comparable to WT KCNQ2 (Fig. 7 A, B and E, inset). These data suggest that ICA73 and RTG act on distinct channel sites.
Figure 7. ICA73‐insensitive mutants do not perturb RTG sensitivity.

A, C and E, conductance voltage relationships for (A) KCNQ2 WT (control V 1/2 = –26 ± 3 mV, k = 13 ± 2 mV; RTG V 1/2 = –66 ± 4 mV, k = 16 ± 1 mV, n = 4) and the ICA73‐insensitive mutants (C) KCNQ2[F168L] (control V 1/2 = –24 ± 3 mV, k = 13 ± 1 mV; RTG V 1/2 = –60 ± 5 mV, k = 16 ± 2 mV, n = 4) and (E) KCNQ2[A181P] (control V 1/2 = –24 ± 4 mV, k = 13 ± 1 mV; RTG V 1/2 = –60 ± 4 mV, k = 17 ± 2 mV, n = 4) in control conditions or 100 μm RTG as indicated. Inset: change in V 1/2 in the presence of RTG for the indicated constructs (no statistical difference between any of the mutants and WT KCNQ2). Note that the KCNQ2[F168L] and [A181P] mutations exhibited variable channel rundown in the presence of RTG (no potentiation as observed with ICA73), so their conductance–voltage relationships have been normalized to 1 for clarity. B, D and F, exemplar patch clamp current recordings of (B) KCNQ2 WT (D) KCNQ2[F168L] and (F) KCNQ2[A181P].
Transfer of ICA73 sensitivity with point mutations in the KCNQ3 voltage sensor
We also tested the effects of point mutations in the KCNQ3 voltage sensor, at positions corresponding to KCNQ2 F168 and A181, on ICA73 sensitivity. Each individual point mutant (L198F and P211A) generated modestly enhanced ICA73 sensitivity relative to KCNQ3* channels (Fig. 8 C and D). The combination of both mutations in KCNQ3*[L198F][P211A] resulted in even stronger effects of ICA73 (Fig. 8 B). Although this effect was not nearly as pronounced as the effects of ICA73 on KCNQ2, it is quite a dramatic increase in ICA73 sensitivity relative to KCNQ3* (Fig. 8 A) and demonstrates that local changes in the voltage‐sensing domain can influence ICA73 sensitivity. None of these point mutations caused any appreciable effect on KCNQ3* gating (Table 2).
Figure 8. ICA73‐sensitivity can be transferred into KCNQ3 via point mutations in the voltage sensor.

A–D, conductance voltage relationships for (A) KCNQ3* (control V 1/2 = –52 ± 4 mV, k = 6 ± 2 mV, ICA73 V 1/2 = –61 ± 3 mV, k = 7 ± 2 mV, n = 6); (B) KCNQ3*[L198F][P211A] (control V 1/2 = –58 ± 2 mV, k = 6 ± 1 mV, ICA73 V 1/2 = –91 ± 6 mV, k = 15 ± 2 mV, n = 11); (C) KCNQ3*[L198F] (control V 1/2 = –56 ± 3 mV, k = 5 ± 1 mV, ICA73 V 1/2 = –77 ± 5 mV, k = 10 ± 2 mV, n = 7); and (D) KCNQ3*[P211A] (control V 1/2 = –55 ± 3 mV, k = 5 ± 1 mV, ICA73 V 1/2 = –75 ± 3 mV, k = 8 ± 1 mV, n = 7). E, change in V 1/2 in the presence of ICA73 for indicated channels (ANOVA, Dunnett's post hoc test. * P < 0.05 relative to KCNQ3* control).
Table 2.
Gating parameters under control conditions for various KCNQ3* channel mutants
| Construct | V 1/2 (mV) | k (mV) | N |
|---|---|---|---|
| KCNQ3* | –52 ± 4 | 5.9 ± 1.4 | 6 |
| KCNQ3*[L198F] | –56 ± 3 | 5.0 ± 1.2 | 7 |
| KCNQ3*[P211A] | –55 ± 2 | 5.0 ± 0.7 | 7 |
| KCNQ3*[L198F][P211A] | –58 ± 2 | 6.1 ± 0.6 | 11 |
Conductance–voltage relationships were fit with a Boltzmann equation (see Methods). Data are presented as the mean ± SEM No statistical differences were detected (one‐way ANOVA).
Discussion
The development of RTG and flupirtine as ‘first‐in‐class’ voltage‐gated potassium channel openers has led to efforts to further develop and understand the pharmacophore of this emerging drug class (Bentzen et al. 2006; Xiong et al. 2008; Blom et al. 2010; Gao et al. 2010; Peretz et al. 2010; Kim et al. 2015). RTG exhibits little subtype specificity between KCNQ2–5, and so a more detailed understanding of the mechanism of action of KCNQ openers may contribute to the development of more specific or potent analogues. The lack of subtype specificity of RTG arises because of its essential interaction with a S5 Trp residue that is conserved in KCNQ2–5 subunits (Lange et al. 2009). Several KCNQ openers have been reported to exhibit some subtype selectivity, although the mechanism and sequence determinants underlying this specificity have been unclear (Padilla et al. 2009; Yu et al. 2011; Brueggemann et al. 2014). In addition, the effects of KCNQ openers are often studied in heteromeric KCNQ channel mixtures, making it difficult to investigate how different KCNQ subtypes may differentially contribute to drug sensitivity.
In the present study, we exploited dramatic differences in the effects of ICA73 in KCNQ2 vs. KCNQ3 to identify residues that are essential for ICA73 subtype specificity. ICA73 induces a very large hyperpolarizing shift of the KCNQ2 (but not KCNQ3) conductance–voltage relationship, together with a large potentiation of current magnitude, with both effects far exceeding what has been reported for RTG. Using a chimeric approach, we identified S3 residues A181 and F168 in KCNQ2 that are essential for normal ICA73 sensitivity (Figs 4, 5, 6). Moreover, mutation of the corresponding positions in KCNQ3 (L198F and P211A) enhances the effects of ICA73, although these mutations do not fully transfer the sensitivity observed in KCNQ2 (Fig. 8). Interestingly, our findings also show that certain mutations (KCNQ2[A181P] or the Q2+Q3[S1,S2] chimera) dissociate gating shift effects from effects on current potentiation.
Previous investigations have reported voltage sensor‐mediated effects of certain KCNQ openers (Padilla et al. 2009; Gao et al. 2010; Peretz et al. 2010). For example, a class of diclofenac‐derived compounds was reported to interact with conserved voltage‐sensing residues in KCNQ2 (Peretz et al. 2010). Also, a chimeric study suggested that the voltage sensor plays an important role in mediating the effects of ICA73 and other related compounds (Padilla et al. 2009). Lastly, recent work has demonstrated that mutation of KCNQ2 residues E130 and F137 influence sensitivity to ICA73 and other closely‐related compounds (Li et al. 2013 b; Brueggemann et al. 2014). However, these residues are highly conserved among voltage‐gated potassium channels, serving important roles as voltage sensor counter‐charges (E130) or as the ‘gating charge transfer center’ (F137) (Papazian et al. 1995; Tao et al. 2010; Pless et al. 2011; Pless et al. 2014). Mutations at these positions often have significant effects on channel function, and cannot account for the ICA73 subtype specificity for KCNQ2 over KCNQ3 (because both channels have identical residues at these positions).
The present study identifies KCNQ2 F168 as an essential determinant of ICA73 sensitivity. KCNQ2[F168L] mutant channels were completely unresponsive to ICA73, exhibiting no hyperpolarizing shift or current potentiation in the presence of the drug (Fig. 5). Based on our findings thus far, an aromatic side chain at this position appears to be required for ICA73 sensitivity. In molecular models of KCNQ2 (Miceli et al. 2013; Gourgy‐Hacohen et al. 2014), the F168L position is located at a convergence of many positively charged side chains including a voltage‐sensing arginine (R213), as well as a lysine (K219) in the S4‐S5 linker. The requirement for an aromatic side chain may involve this proximity to positively charged side chains, or may be related to positioning of S3 near the membrane interface. In any case, both Lys219 and Arg213 are almost certainly conformationally mobile during channel gating and we are pursuing a more detailed understanding of the interactions between these residues with F168 in different channel states.
The KCNQ2 A181P mutation had a very unexpected outcome on ICA73 response: the drug‐mediated hyperpolarizing gating shift was almost abolished, although the current potentiation effect was preserved (Fig. 6). This observation highlights the importance of monitoring both of these distinct parameters when characterizing KCNQ openers; as noted earlier, conductance–voltage relationships are often normalized, and such data transformation may mask the current potentiating effect of ICA73 or other openers. Because the current potentiation effect of ICA73 persists in KCNQ2[A181P], we speculate that this mutation does not abolish drug binding but, instead, alters how drug interactions influence the gating process. In support of this suggestion, ICA73 interaction clearly decelerates channel closure despite not significantly changing the voltage‐dependence of channel opening. We also observed that ICA73 sensitivity was preserved with a variety of mutations at position A181, suggesting that the native alanine may not directly contribute to binding. Rather, the presence of a proline (as in KCNQ3) may alter the nature of the response to the drug, rather than altering binding.
Importantly, there may be additional residues/mechanisms generating subtype specificity. There appears to be strong evidence that the voltage‐sensing domain controls sensitivity to ICA73‐like compounds; however KCNQ2 residues F168 and A181 are almost on opposite sides of the voltage sensor, and it is difficult to rationalize how both might be directly involved. Also, KCNQ5 has been reported to be insensitive to ICA73 (Brueggemann et al. 2014) but shares the essential alanine and phenylalanine residues identified in the present study. This observation suggests that other residues may impede drug binding in KCNQ5 and that, potentially, a similar chimeric study of KCNQ2 and KCNQ5 may be a good approach for identifying additional positions that influence drug binding and specificity.
An additional consideration is the probable importance of state‐dependent binding of ICA73 and similar drugs. A generally true description of a channel opener that causes a hyperpolarizing gating shift is that it must have an energetically favourable interaction with open/activated states compared to closed states. Thus, mutations that influence voltage sensor equilibria during channel activation could alter the accessibility of a binding site in the VSD, without necessarily directly altering binding. The absence of a gating shift in KCNQ3* does not necessarily imply that ICA73 does not bind the channel but, instead, could indicate that the drug binds comparably well to closed and open/activated states of the VSD (and therefore causes no gating shift). Thus, the observation of a modest ‘transfer’ of ICA73‐sensitivity into KCNQ3 (Fig. 8) could also be consistent with the P211A and L198F mutations altering the state‐dependent accessibility of the ICA73 binding site, rather than reconstituting a binding site.
Taken together, our findings demonstrate that despite sharing structural similarities with RTG, ICA73 acts via an entirely different mechanism of action. Specific residues distant from the putative RTG binding site are able to influence the effects of ICA73 on KCNQ2, and underlie subtype specificity of the drug for KCNQ2 over KCNQ3. Moreover, mutations of these residues in the voltage sensor domain have multi‐pronged effects on ICA73 actions because some are able to specifically abolish the effects of ICA73 on voltage‐dependence at the same time as preserving current potentiation. These findings demonstrate unambiguously that KCNQ openers should be classified into at least two subgroups based on their primary site of action (which can be determined based on differential effects of the KCNQ2[W236L/F] or [F168L] mutations). Ongoing investigation of the multi‐pronged effects of KCNQ openers acting on the voltage sensor will hopefully lead to a deeper understanding of the general principles that underlie the actions of these drugs.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
Research described in this manuscript was carried out in the laboratories of Dr H. T. Kurata at the University of British Columbia (Deprtment of Anesthsiology, Pharmacology and Therapeutics) and at the University of Alberta (Department of Pharmacology, Alberta Diabetes Institute, from August 2015 onwards). HTK, AWW and RY conceived and designed the experiments. AWW, RY and HTK collected, assembled, analysed and interpretated data. HTK, AWW and RY drafted the article or revised it critically for important intellectual content. All authors were involved in the interpretation of final results and the writing of the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This research project was supported by CIHR operating grant MOP 142482 to HTK, and Human Frontiers in Science Program Young Investigator Award 0082 to HTK. HTK was also supported by a CIHR New Investigator salary award and a Michael Smith Foundation for Health Research Scholar award.
Acknowledgements
We thank Drs Thomas Jentsch and Maurizio Taglialatela for kindly sharing plasmids encoding KCNQ2 and KCNQ3. Drs Maurizio Taglialatela and Bernard Attali kindly shared the molecular models of KCNQ2 that were used to interpret our data.
Linked articles This article is highlighted by a Perspective by Barro‐Soria et al. To read this Perspective, visit http://dx.doi.org/10.1113/JP273250.
References
- Bentzen BH, Schmitt N, Calloe K, Dalby BW, Grunnet M & Olesen SP (2006). The acrylamide (S)‐1 differentially affects Kv7 (KCNQ) potassium channels. Neuropharmacology 51, 1068–1077. [DOI] [PubMed] [Google Scholar]
- Blom SM, Schmitt N & Jensen HS (2010). Differential effects of ICA‐27243 on cloned K(V)7 channels. Pharmacology 86, 174–181. [DOI] [PubMed] [Google Scholar]
- Boehlen A, Schwake M, Dost R, Kunert A, Fidzinski P, Heinemann U & Gebhardt C (2013). The new KCNQ2 activator 4‐chlor‐N‐(6‐chlor‐pyridin‐3‐yl)‐benzamid displays anticonvulsant potential. Br J Pharmacol 168, 1182–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickel N, Gandhi P, VanLandingham K, Hammond J & DeRossett S (2012). The urinary safety profile and secondary renal effects of retigabine (ezogabine): a first‐in‐class antiepileptic drug that targets KCNQ (K(v)7) potassium channels. Epilepsia 53, 606–612. [DOI] [PubMed] [Google Scholar]
- Brueggemann LI, Haick JM, Cribbs LL & Byron KL (2014). Differential activation of vascular smooth muscle Kv7.4, Kv7.5, and Kv7.4/7.5 channels by ML213 and ICA‐069673. Mol Pharmacol 86, 330–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalby‐Brown W, Hansen HH, Korsgaard MP, Mirza N & Olesen SP (2006). K(v)7 channels: function, pharmacology and channel modulators. Curr Top Med Chem 6, 999–1023. [DOI] [PubMed] [Google Scholar]
- Gao Z, Zhang T, Wu M, Xiong Q, Sun H, Zhang Y, Zu L, Wang W & Li M (2010). Isoform‐specific prolongation of Kv7 (KCNQ) potassium channel opening mediated by new molecular determinants for drug‐channel interactions. J Biol Chem 285, 28322–28332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Posada JC, Etxeberria A, Roura‐Ferrer M, Areso P, Masin M, Murrell‐Lagnado RD & Villarroel A (2010). A pore residue of the KCNQ3 potassium M‐channel subunit controls surface expression. J Neurosci 30, 9316–9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourgy‐Hacohen O, Kornilov P, Pittel I, Peretz A, Attali B & Paas Y (2014). Capturing distinct KCNQ2 channel resting states by metal ion bridges in the voltage‐sensor domain. J Gen Physiol 144, 513–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalappa BI, Soh H, Duignan KM, Furuya T, Edwards S, Tzingounis AV & Tzounopoulos T (2015). Potent KCNQ2/3‐specific channel activator suppresses in vivo epileptic activity and prevents the development of tinnitus. J Neurosci 35, 8829–8842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim RY, Yau MC, Galpin JD, Seebohm G, Ahern CA, Pless SA & Kurata HT (2015). Atomic basis for therapeutic activation of neuronal potassium channels. Nat Commun 6, 8116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M, Reed N, Liu R, Aizenman E, Wipf P & Tzounopoulos T (2016). Synthesis and evaluation of potent KCNQ2/3‐specific channel activators. Mol Pharmacol 89, 667–677. [DOI] [PubMed] [Google Scholar]
- Lange W, Geissendorfer J, Schenzer A, Grotzinger J, Seebohm G, Friedrich T & Schwake M (2009). Refinement of the binding site and mode of action of the anticonvulsant retigabine on KCNQ K+ channels. Mol Pharmacol 75, 272–280. [DOI] [PubMed] [Google Scholar]
- Li JB, Huang X, Zhang RS, Kim RY, Yang R & Kurata HT (2013. a). Decomposition of slide helix contributions to ATP‐dependent inhibition of Kir6.2 channels. J Biol Chem 288, 23038–23049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Chen Z, Xu H, Sun H, Li H, Liu H, Yang H, Gao Z, Jiang H & Li M (2013. b). The gating charge pathway of an epilepsy‐associated potassium channel accommodates chemical ligands. Cell Res 23, 1106–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martyn‐St JM, Glanville J, McCool R, Duffy S, Cooper J, Hugel P & Lane PW (2012). The efficacy and safety of retigabine and other adjunctive treatments for refractory partial epilepsy: a systematic review and indirect comparison. Seizure 21, 665–678. [DOI] [PubMed] [Google Scholar]
- Miceli F, Soldovieri MV, Ambrosino P, Barrese V, Migliore M, Cilio MR & Taglialatela M (2013). Genotype‐phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of K(v)7.2 potassium channel subunits. Proc Natl Acad Sci USA 110, 4386–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miceli F, Soldovieri MV, Iannotti FA, Barrese V, Ambrosino P, Martire M, Cilio MR & Taglialatela M (2011). The voltage‐sensing domain of K(v)7.2 channels as a molecular target for epilepsy‐causing mutations and anticonvulsants. Front Pharmacol 2, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miceli F, Soldovieri MV, Martire M & Taglialatela M (2008). Molecular pharmacology and therapeutic potential of neuronal Kv7‐modulating drugs. Curr Opin Pharmacol 8, 65–74. [DOI] [PubMed] [Google Scholar]
- Munster T, Gibbs JP, Shen D, Baethge BA, Botstein GR, Caldwell J, Dietz F, Ettlinger R, Golden HE, Lindsley H, McLaughlin GE, Moreland LW, Roberts WN, Rooney TW, Rothschild B, Sack M, Sebba AI, Weisman M, Welch KE, Yocum D & Furst DE (2002). Hydroxychloroquine concentration–response relationships in patients with rheumatoid arthritis. Arthritis Rheum 46, 1460–1469. [DOI] [PubMed] [Google Scholar]
- Padilla K, Wickenden AD, Gerlach AC & McCormack K (2009). The KCNQ2/3 selective channel opener ICA‐27243 binds to a novel voltage‐sensor domain site. Neurosci Lett 465, 138–142. [DOI] [PubMed] [Google Scholar]
- Papazian DM, Shao XM, Seoh SA, Mock AF, Huang Y & Wainstock DH (1995). Electrostatic interactions of S4 voltage sensor in Shaker K+ channel. Neuron 14, 1293–1301. [DOI] [PubMed] [Google Scholar]
- Peretz A, Degani‐Katzav N, Talmon M, Danieli E, Gopin A, Malka E, Nachman R, Raz A, Shabat D & Attali B (2007). A tale of switched functions: from cyclooxygenase inhibition to M‐channel modulation in new diphenylamine derivatives. PLoS ONE 2, e1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz A, Pell L, Gofman Y, Haitin Y, Shamgar L, Patrich E, Kornilov P, Gourgy‐Hacohen O, Ben‐Tal N & Attali B (2010). Targeting the voltage sensor of Kv7.2 voltage‐gated K+ channels with a new gating‐modifier. Proc Natl Acad Sci USA 107, 15637–15642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pless SA, Elstone FD, Niciforovic AP, Galpin JD, Yang R, Kurata HT & Ahern CA (2014). Asymmetric functional contributions of acidic and aromatic side chains in sodium channel voltage‐sensor domains. J Gen Physiol 143, 645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pless SA, Galpin JD, Niciforovic AP & Ahern CA (2011). Contributions of counter‐charge in a potassium channel voltage‐sensor domain. Nat Chem Biol 7, 617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenzer A, Friedrich T, Pusch M, Saftig P, Jentsch TJ, Grotzinger J & Schwake M (2005). Molecular determinants of KCNQ (Kv7) K+ channel sensitivity to the anticonvulsant retigabine. J Neurosci 25, 5051–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Lee A, Limapichat W, Dougherty DA & MacKinnon R (2010). A gating charge transfer center in voltage sensors. Science 328, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickenden AD, Krajewski JL, London B, Wagoner PK, Wilson WA, Clark S, Roeloffs R, McNaughton‐Smith G & Rigdon GC (2008). N‐(6‐chloro‐pyridin‐3‐yl)‐3,4‐difluoro‐benzamide (ICA‐27243): a novel, selective KCNQ2/Q3 potassium channel activator. Mol Pharmacol 73, 977–986. [DOI] [PubMed] [Google Scholar]
- Wu YJ & Dworetzky SI (2005). Recent developments on KCNQ potassium channel openers. Curr Med Chem 12, 453–460. [DOI] [PubMed] [Google Scholar]
- Xiong Q, Gao Z, Wang W & Li M (2008). Activation of Kv7 (KCNQ) voltage‐gated potassium channels by synthetic compounds. Trends Pharmacol Sci 29, 99–107. [DOI] [PubMed] [Google Scholar]
- Xiong Q, Sun H & Li M (2007). Zinc pyrithione‐mediated activation of voltage‐gated KCNQ potassium channels rescues epileptogenic mutants. Nat Chem Biol 3, 287–296. [DOI] [PubMed] [Google Scholar]
- Yu H, Wu M, Townsend SD, Zou B, Long S, Daniels JS, McManus OB, Li M, Lindsley CW & Hopkins CR (2011). Discovery, synthesis, and structure activity relationship of a series of N‐aryl‐ bicyclo[2.2.1]heptane‐2‐carboxamides: characterization of ML213 as a novel KCNQ2 and KCNQ4 potassium channel opener. ACS Chem Neurosci 2, 572–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Wu M, Hopkins C, Engers J, Townsend S, Lindsley C, McManus OB & Li M (2013). A small molecule activator of KCNQ2 and KCNQ4 channels 2010–2011, Mar 29 [updated 2013 Feb 28]. [PubMed] [Google Scholar]