

Abstract
Key points
Rheumatoid arthritis (RA) is a chronic inflammatory condition associated with an increased risk of cardiovascular mortality.
Increased sympathetic nerve activity and reduced cardiac baroreflex sensitivity heighten cardiovascular risk, althogh whether such autonomic dysfunction is present in RA is not known.
In the present study, we observed an increased sympathetic nerve activity and reduced cardiac baroreflex sensitivity in patients with RA compared to matched controls.
Pain was positively correlated with sympathetic nerve activity and negatively correlated with cardiac baroreflex sensitivity.
The pattern of autonomic dysfunction that we describe may help to explain the increased cardiovascular risk in RA, and raises the possibility that optimizing pain management may resolve autonomic dysfunction in RA.
Abstract
Rheumatoid arthritis (RA) is a chronic inflammatory condition associated with increased cardiovascular morbidity/mortality and an incompletely understood pathophysiology. In animal studies, central and blood borne inflammatory cytokines that can be elevated in RA evoke pathogenic increases in sympathetic activity and reductions in baroreflex sensitivity (BRS). We hypothesized that muscle sympathetic nerve activity (MSNA) was increased and BRS decreased in RA. MSNA, blood pressure and heart rate (HR) were recorded in age‐ and sex‐matched RA‐normotensive (n = 13), RA‐hypertensive patients (RA‐HTN; n = 17), normotensive (NC; n = 17) and hypertensive controls (HTN; n = 16). BRS was determined using the modified Oxford technique. Inflammation and pain were determined using serum high sensitivity C‐reactive protein (hs‐CRP) and a visual analogue scale (VAS), respectively. MSNA was elevated similarly in RA, RA‐HTN and HTN patients (32 ± 9, 35 ± 14, 37 ± 8 bursts min–1) compared to NC (22 ± 9 bursts min–1; P = 0.004). Sympathetic BRS was similar between groups (P = 0.927), whereas cardiac BRS (cBRS) was reduced in RA, RA‐HTN and HTN patients [5(3–8), 4 (2–7), 6 (4–9) ms mmHg–1] compared to NC [11 (8–15) ms mmHg–1; P = 0.002]. HR was independently associated with hs‐CRP. Increased MSNA and reduced cBRS were associated with hs‐CRP although confounded in multivariable analysis. VAS was independently associated with MSNA burst frequency, cBRS and HR. We provide the first evidence for heightened sympathetic outflow and reduced cBRS in RA that can be independent of hypertension. In RA patients, reported pain was positively correlated with MSNA and negatively correlated with cBRS. Future studies should assess whether therapies to ameliorate pain and inflammation in RA restores autonomic balance and reduces cardiovascular events.
Keywords: autonomic nervous system, cytokine, inflammation, pain
Key points
Rheumatoid arthritis (RA) is a chronic inflammatory condition associated with an increased risk of cardiovascular mortality.
Increased sympathetic nerve activity and reduced cardiac baroreflex sensitivity heighten cardiovascular risk, althogh whether such autonomic dysfunction is present in RA is not known.
In the present study, we observed an increased sympathetic nerve activity and reduced cardiac baroreflex sensitivity in patients with RA compared to matched controls.
Pain was positively correlated with sympathetic nerve activity and negatively correlated with cardiac baroreflex sensitivity.
The pattern of autonomic dysfunction that we describe may help to explain the increased cardiovascular risk in RA, and raises the possibility that optimizing pain management may resolve autonomic dysfunction in RA.
Abbreviations
- BMI
body mass index
- BP
blood pressure
- BRS
baroreflex sensitivity
- cBRS
cardiac baroreflex sensitivity
- HR
heart rate
- hs‐CRP
high sensitivity C‐reactive protein
- HTN
hypertensive
- IL
interleukin
- MOT
modified Oxford technique
- MSNA
muscle sympathetic nerve activity
- NC
normotensive controls
- RA
rheumatoid arthritis
- RA‐HTN
rheumatoid arthritis hypertensive
- TNF
tumour necrosis factor
- VAS
visual analogue scale
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory condition associated with increased risk of cardiovascular mortality and myocardial infarction, as well as an increased prevalence of hypertension and cardiac arrhythmias (Maradit‐Kremers et al. 2005; Arosio et al. 2007; Levy et al. 2008; Panoulas et al. 2008). Traditional cardiovascular risk factors are present in RA patients but do not wholly account for the increased cardiovascular risk (Maradit‐Kremers et al. 2005) and hence novel mechanisms are sought (Amaya‐Amaya et al. 2013; Panoulas et al. 2014).
A heightened central sympathetic outflow to the heart and/or vasculature has been identified in numerous cardiovascular diseases (Fisher & Paton, 2012) and is associated with the pathogenesis of hypertension (Fisher & Paton, 2012), cardiac arrhythmias (Lown & Verrier, 1976) and increased mortality (Cohn et al. 1984; Barretto et al. 2009). Reciprocal links between inflammation and the sympathetic nervous system have been identified (Niijima et al. 1991; Zhang et al. 2003; Helwig et al. 2008). Central sympatholytic agents have anti‐inflammatory actions in human hypertension (Poyhonen‐Alho et al. 2008), whereas the infusion of inflammatory cytokines known to be elevated in RA patients, such as interleukin (IL)‐6 (Helwig et al. 2008), tumour necrosis factor (TNF)‐α (Zhang et al. 2003) and IL‐1β (Niijima et al. 1991), increases sympathetic nerve activity in rats. Thus, the chronic activation of inflammatory cytokines in RA may elicit deleterious increases in sympathetic neural outflow that further promote activation of pro‐inflammatory cytokines. Furthermore, microinjection of IL‐6 into the nucleus of the solitary tract, a key central regulatory cardiovascular site, is reported to reduce cardiac baroreflex sensitivity (BRS) (Takagishi et al. 2010). Impaired BRS is present in hypertension (Gribbin et al. 1971), heart failure (Ferguson et al. 1992) and coronary artery disease (De Ferrari et al. 2007), and is reported to predict reduced survival following myocardial infarction (La Rovere et al. 1998; De Ferrari et al. 2007) and in patients with heart failure (La Rovere et al. 2009). Depressed BRS may potentially contribute to the recognized increased cardiovascular risk in RA (Maradit‐Kremers et al. 2005; Levy et al. 2008) by precipitating further increases in central sympathetic outflow and reducing cardiac electrical stability.
Whether sympathetic activity is increased and BRS is reduced in RA is presently equivocal, partly as a result of the indirect methods of assessment used to examine this (Adlan et al. 2014). Elevations in plasma noradrenaline have been reported in RA patients (Vlcek et al. 2008), although it is not clear whether this is a consequence of increased central sympathetic outflow, enhanced release from peripheral adrenergic stores or altered local reuptake mechanisms (Esler et al. 1990). Such limitations are overcome by the microneurography technique, which provides a direct assessment of sympathetic nerve activity to the skeletal muscle vasculature (MSNA), although such measures have not been undertaken previously in RA. It is also not known whether baroreflex regulation of MSNA is altered in RA. Although a reduction in cardiac BRS has been suggested in RA patients (Aydemir et al. 2010), baroreflex control of the heart and vasculature do not always function in a parallel fashion (Rudas et al. 1999).
We aimed to test the hypothesis that central sympathetic outflow is elevated in RA patients and is associated with elevated circulating concentrations of inflammatory cytokines. We made the first direct recordings of MSNA in patients with RA. Additionally, we examined whether the sensitivity of baroreflex control of the heart and MSNA are reduced in RA patients and associated with an elevated concentration of circulating inflammatory cytokines. Hypertension control groups (RA and non‐RA) were included to attempt and control for the effect of medications and comorbidities.
Methods
Participants
Thirty patients with a clinical diagnosis of RA based on the 1987 American College of Rheumatology criteria (Arnett et al. 1988) were recruited from the rheumatology clinics at Russells Hall Hospital, Dudley, UK, and Sandwell General Hospital, West Bromwich, UK. The 1987 American College of Rheumatology criteria allows for classification of RA when four of its seven criteria (morning stiffness, arthritis of three or more joint areas, arthritis of hand joints, symmetric arthritis, rheumatoid nodules, positive serum rheumatoid factor and radiographic changes) are present (morning stiffness, arthritis and rheumatoid nodules need to be present for at least 6 weeks). Exclusion criteria included: age <18 or >75 years, atrial fibrillation or other heart rhythm disorder, significant valvular disease, coronary artery disease, diabetes, ischaemic stroke, chronic renal failure, liver impairment, hormone replacement therapy, and those who are pregnant or who might be pregnant. In addition, 33 age‐, sex‐ and body mass index (BMI)‐matched control participants were recruited from the hospitals and surrounding areas. Normotensive control (NC) participants were free from major illnesses, whereas hypertensive controls (HTN) either had a prior diagnosis of hypertension or blood pressure (BP) ≥140/90 mmHg. Ethical approval was obtained by the local Research Ethics Committee (National Research and Ethics Service Committee West Midlands – Edgbaston, 11/WM/0298). Written informed consent was received from participants prior to inclusion in the study, in accordance with the Declaration of Helsinki.
Experimental protocol
Participants were studied according to an observational, case–control study that included four groups: 13 RA patients, 17 RA‐HTN patients, 16 HTN and 17 NC. Participants attended the research laboratory at 09.00 h after an overnight fast (from food, caffeine, alcohol and nicotine). All medications were withheld on the morning of testing. A detailed clinical history was obtained and physical examination was performed in RA patients to count the number of swollen and tender joints aiming to determine the disease activity score C‐reactive protein (DAS28‐CRP) (Wells et al. 2009). A visual analogue scale (VAS) was used as a measure of patient‐reported pain (Huskisson, 1974). Height and weight was measured, and the BMI was determined (weight/height2). All subsequent measurements were performed under uniform conditions in a temperature‐controlled room when the participants were resting quietly in the supine position.
Following instrumentation for heart rate (HR), BP, leg blood flow and MSNA as described below, an i.v. catheter was inserted into a superficial vein in the antecubital fossa for blood sampling and injections. The experimental protocol involved a 10 min baseline period followed by sequential infusion of sodium nitroprusside (100 μg of sodium nitroprusside) and phenylephrine (150 μg of phenylephrine) 1 min later (modified Oxford technique; MOT) (Rudas et al. 1999).
Measurements
HR was continuously recorded using a lead II electrocardiogram (ECG, BioAmp; ADInstruments, Bella Vista, Australia). Beat‐to‐beat BP was recorded using finger photoplethysmography (Portapres; Finapres Medical Systems, Amsterdam, The Netherlands) and was calibrated with brachial BP recordings using an automated sphygmomanometer (Omron 705IT; Omron Corporation, Hoopddorp, The Netherlands). Multi‐unit recordings of MSNA were obtained (FE185 NeuroAmp EX; ADInstruments) from the peroneal nerve using tungsten microelectrodes (200 μm, 1–3 μm uninsulated tip; UNA32F2S; FHC, Bowdoin, ME, USA). Electrical stimulation on the skin surface was used for nerve mapping, and a reference electrode was inserted s.c. 2–3 cm away from the recording electrode, which was inserted into the nerve fascicle. The neural signals were amplified, filtered (100 Hz low pass, 2000 Hz high pass, 60 Hz notch), rectified and integrated (absolute value, time constant decay 0.1 s) to obtain a mean voltage of sympathetic nerve activity. MSNA was confirmed by: listening to the amplified signal on speakers; observing the characteristic bursting pattern on a computer screen; palpating the skin and muscle fibres; and performing a breath hold (Wallin & Fagius, 1988; Grassi & Esler, 1999). Leg blood flow was measured using venous occlusion strain gauge plethysmography in accordance with standard methods (Hokanson EC‐6 plethysmograph; DE Hokanson, Bellevue, WA, USA) (Joyner et al. 2001). A lightweight indium‐in‐silastic strain gauge was positioned around the right calf at the point of greatest circumference. The length of the strain gauge was 2 cm less than the widest girth of the calf. Cuffs were placed around the right ankle and inflated to a pressure of 200 mmHg and maintained for 1 min to achieve arterial occlusion. Sixty seconds later, cuffs placed around the thigh were rapidly inflated to 50 mmHg (Hokanson E20 rapid cuff inflator and AG101 air source; Hokanson, Bellevue, WA, USA) to evoke venous occlusions. Venous occlusion was repeated three times during 1 min, with the thigh cuffs inflated for 5 s and then deflated for 10 s each time.
Data analysis
Data were acquired using the Powerlab 16/35 data acquisition system and Labchart Pro software (ADInstruments) and analysed offline. Sympathetic bursts were identified by a single observer (AMA) using a semi‐automated scoring system created using Spike 2 (Cambridge Electronic Design, Cambridge, UK). MSNA burst frequency (bursts min–1) and incidence [bursts (100 heart beats)–1] was determined. Assessment of cardiac BRS was determined during the phenylephrine induced rise in BP (MOT) (Rudas et al. 1999; Studinger et al. 2007). Analysis was performed from the first concordant change in systolic BP and RR‐interval until they were discordant. To account for baroreflex delays, systolic BP was associated with concurrent (resting RR‐interval >800 ms) or subsequent RR‐intervals (RR‐interval between 500 and 800 ms) (Pickering & Davies, 1973; Eckberg & Eckberg, 1982). Respiratory related variations in RR‐interval were accounted for by averaging RR‐intervals over 3 mmHg pressure bins (Ebert, 1990). Assessment of baroreflex control of MSNA was determined using linear regression [diastolic BP (DBP) vs. MSNA burst activity] during the phenylephrine induced rise in BP (MOT) (Hart et al. 2010). Briefly, each cardiac cycle was scored according to the presence or absence of a sympathetic burst. Data were binned according to the DBP of the previous cardiac cycle (3 mmHg bins). Linear regression analysis was then performed on the relationship between the mean of each DBP bin and MSNA burst incidence (bursts/100 heart beats). All data were weighted for the number of cardiac cycles in each DBP bin (Hart et al. 2010). We used the slope of the linear regression [(bursts/100 heart beats)/mmHg] as an index of sympathetic BRS. Some patients were unsuitable for microneurography (e.g. discomfort laying still for an extended period or declined microneurography) and it was not possible to obtain sufficiently high quality MSNA recordings in all patients; thus, these results were omitted from the MSNA analyses. Participant numbers are stated as appropriate.
Blood sampling
Blood samples for inflammatory markers were centrifuged immediately and the plasma stored at –80°C. Commercially available enzyme‐linked immunosorbent assay kits were used to determine high sensitivity C‐reactive protein (hs‐CRP) (MP Biomedicals, Santa Ana, CA, USA) and cytokines (IL‐6, TNF‐α, IL‐10; BioSupply UK, Bradford, UK). The intra‐ and inter‐assay coefficients of variations were 7.5% and 4.1%, respectively, for hs‐CRP; 4.9% and 6.0% for IL‐6; 8.5% and 9.8% for TNF‐α; and 6.8% and 7.5% for IL‐10. Local routine clinical laboratories were used to analyse full blood count, renal function, fasting lipid profile and fasting glucose.
Statistical analysis
Statistical analysis was performed using SPSS, version 19 (IBM Corp., Armonk, NY, USA). Continuous variables were tested for normality using the Kolmogorov–Smirnov test. Non‐parametric data were (naturally) logarithmically transformed. Group differences were assessed using ANOVA with a least significance difference post hoc test for continuous variables, as well as Pearson's chi‐squared test for categorical data. Differences between the RA normotensive and RA‐HTN groups were assessed using an independent t test. Associations between autonomic parameters and inflammation were assessed before (Pearson's product/Spearman's rank correlation coefficient) and after adjustment for potential confounders. Data are expressed as the mean ± SD for parametric data; geometric mean (95% confidence interval) for non‐parametric data; and frequency (percentages) for categorical variables. P < 0.05 was considered statistically significant.
Results
Baseline participant characteristics are presented in Table 1. Aside from hypertension, there were no significant differences in other cardiovascular risk factors between the groups. Compared to the RA and NC groups, there was a higher prevalence of osteoarthritis (P = 0.003) and statin therapy (P = 0.032) in the RA‐HTN and HTN groups. The RA and RA‐HTN groups had a higher prevalence of proton pump inhibitor (P = 0.016) and folic acid (P < 0.001) therapy, whereas calcium/vitamin D supplementation tended to be highest in the RA‐HTN group (P = 0.06). Similar anti‐hypertensive agent use was noted in the RA‐HTN and HTN groups. A small number of participants were smokers (P = 0.064 between groups), although no use of other nicotine containing products was reported by any participant.
Table 1.
Subject characteristics
| RA | RA‐HTN | NC | HTN | ||
|---|---|---|---|---|---|
| (n = 13) | (n = 17) | (n = 17) | (n = 16) | P | |
| Age (years) | 55.9 ± 11.7 | 60.5 ± 9.6 | 53.6 ± 13.0 | 59.6 ± 10.1 | 0.257 |
| Female, n (%) | 8 (62) | 12 (71) | 10 (59) | 11 (69) | 0.876 |
| Body mass index, (kg m–2) | 27.8 | 29.6 | 26.4 | 25.8 | 0.130 |
| (25.4–30.4) | (26.4–33.3) | (23.8–29.2) | (24.5–27.2) | ||
| Total cholesterol (mmol L–1) | 4.8 ± 1.1 | 5.1 ± 1.0 | 5.1 ± 0.9 | 5.1 ± 1.0 | 0.804 |
| Triglycerides (mmol L–1) | 1.1 (0.9–1.5) | 1.0 (0.8–1.4) | 1.1 (0.8–1.3) | 1.1 (0.9–1.3) | 0.968 |
| HDL (mmol L–1) | 1.3 (1.1–1.5) | 1.5 (1.3–1.8) | 1.4 (1.2–1.6) | 1.4 (1.2–1.7) | 0.498 |
| LDL (mmol L–1) a | 2.9 ± 0.9 | 2.9 ± 0.9 | 3.1 ± 0.8 | 3.0 ± 0.9 | 0.866 |
| eGFR (ml min–1 1.73 m–2) | 100.4 ± 20.3 | 89.9 ± 14.6 | 88.0 ± 19.6 | 83.5 ± 17.9 | 0.099 |
| Haemoglobin (g L–1) | 126 ± 12 * , † | 133 ± 14 | 138 ± 10 | 138 ± 12 | 0.019 |
| Smoking, n (%) | 4 (31) | 1 (6) | 3 (18) | 0 | 0.064 |
| Osteoarthritis, n (%) | 2 (15) | 9 (53) | 0 | 4 (25) | 0.003 |
| Anti‐hypertensive agent, n (%) | – | 7 (41) | – | 11 (69) | 0.112 |
| ACEi or ARB, n (%) | – | 5 (29) | – | 5 (31) | 0.909 |
| Calcium channel blocker, n (%) | – | 4 (24) | – | 7 (44) | 0.218 |
| Thiazide, n (%) | – | 2 (12) | – | 5 (31) | 0.171 |
| β‐blocker, n (%) | – | 2 (12) | – | 1 (6) | 0.582 |
| α‐blocker, n (%) | – | 0 | – | 2 (13) | 0.133 |
| Aspirin, n (%) | 1 (8) | 1 (6) | 0 | 1 (6) | 0.748 |
| Statin, n (%) | 1 (8) | 6 (35) | 0 | 4 (25) | 0.032 |
| Proton pump inhibitor, n (%) | 4 (31) | 6 (35) | 0 | 1 (6) | 0.016 |
| Folic acid, n (%) | 8 (62) | 12 (71) | 0 | 0 | <0.001 |
| Adcal D3, n (%) | 2 (15) | 5 (29) | 1 (6) | 0 | 0.060 |
Values are expressed as the mean ± SD (parametric) for continuous variables and frequency (percentage) for discrete variables. Non‐parametric data were (natural) log transformed and are displayed as the geometric mean (95% confidence intervals). One‐way ANOVA with a least significance difference post hoc test. Pearson's chi‐square test for categorical data. Significance: P ≤ 0.05. Post hoc: P ≤ 0.05.
* P ≤ 0.05 vs. HC
† P ≤ 0.05 vs. RA‐HTN.
aCalculated using the Friedewald formula.
ACEi, angiotensin‐converting enzyme inhibitor; ARB, angiotensin‐renin blocker; CVD, cardiovascular disease; eGFR, estimated glomerular filtration rate; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein. Adcal D3 is a calcium carbonate, vitamin D3 tablet.
RA disease‐related characteristics are presented in Table 2. The RA normotensive and RA‐HTN groups had similar disease duration, sero‐positivity and RA drug therapy. A greater number of swollen joints (P = 0.045) and a trend for higher disease activity (DAS28‐CRP, P = 0.063) were seen in the RA‐HTN group compared to the RA group.
Table 2.
Rheumatoid arthritis‐related characteristics
| RA | RA‐HTN | ||
|---|---|---|---|
| (n = 13) | (n = 17) | P | |
| RA duration (years) | 7.6 (4.6–12.4) | 5.5 (2.9–10.5) | 0.443 |
| RF positive, n (%) | 9 (69) | 10 (59) | 0.768 |
| Swollen joints, n (%) | 5.2 (2.9–9.0) | 2.6 (1.6–4.0) | 0.045 |
| Tender joints, n (%) | 6.7 (2.8–14.7) | 3.5 (1.7–6.7) | 0.194 |
| DAS28‐CRP | 4.8 ± 1.9 | 3.7 ± 1.2 | 0.063 |
| Disease activity, n (%) | |||
| Remission | 4 (31) | 5 (31) | 0.088 |
| Low disease activity | 3 (23) | 9 (56) | |
| High disease activity | 6 (46) | 2 (13) | |
| VAS, % | 36.8 (21.7–62.0) | 12.9 (5.3–30.1) | 0.047 |
| DMARD, n (%) | 11 (85) | 15 (88) | 0.773 |
| Number of DMARDs | 1.8 (1.2–2.4) | 1.8 (1.3–2.2) | 0.850 |
| Methotrexate, n (%) | 8 (62) | 12 (71) | 0.602 |
| Hydroxychloroquine, n (%) | 5 (38) | 10 (59) | 0.269 |
| Sulfasalazine, n (%) | 4 (31) | 3 (18) | 0.400 |
| Leflunomide, n (%) | 2 (15) | 1 (6) | 0.390 |
| Glucocorticoid, n (%) | 1 (8) | 6 (35) | 0.077 |
| Prednisolone, n (%) | 1 (8) | 4 (24) | 0.249 |
| Prednisolone dose (mg) | 3 | 5.3 ± 3 | 0.534 |
| Intramuscular, n (%) | 0 | 1 (6) | 0.374 |
| Intra‐articular, n (%) | 0 | 1 (6) | 0.374 |
| NSAID, n (%) | 5 (38) | 6 (35) | 0.858 |
| Opioid, n (%) | 6 (46) | 7 (41) | 0.785 |
| Weak, n (%) | 5 (38) | 7 (41) | 0.880 |
| Strong, n (%) | 1 (8) | 0 | 0.245 |
| Biological agent, n (%) | 4 (31) | 3 (18) | 0.400 |
| TNF‐α inhibitor, n (%) | 4 (31) | 1 (6) | 0.070 |
| Certolizumab, n (%) | 2 (15) | 0 | 0.094 |
| Etanercept, n (%) | 2 (15) | 0 | 0.094 |
| Golilumab, n (%) | 0 | 1 (6) | 0.374 |
| Rituximab, n (%) | 0 | 2 (12) | 0.201 |
Values expressed as the mean ± SD for continuous variables (parametric) and frequency (percentage) for discrete variables. Non‐parametric data were (natural) log transformed and are displayed as the geometric mean (95% confidence intervals). Statistical differences were tested using an independent t test for continuous variables and Pearson's chi‐square test for categorical data. Significance: P ≤ 0.05.
DAS, disease activity score; DMARD, disease modifying anti‐rheumatic drug; NSAID, non‐steroidal anti‐inflammatory drug; RF, rheumatoid factor.
Haemodynamic parameters
Mean BP was higher in the HTN and RA‐HTN groups compared to the RA and NC groups (P < 0.001), with no significant difference between the RA‐HTN and HTN groups, or between the RA and NC groups (systolic/diastolic BP RA 129 ± 10/79 ± 6, RA‐HTN 154 ± 18/87 ± 10, HTN 147 ± 25/84 ± 11, NC 123 ± 10/75 ± 6 mmHg; P < 0.001; post hoc analysis, P < 0.05 for RA vs. RA‐HTN and HTN, NC vs. RA‐HTN and HTN; diastolic BP RA vs. HTN, P = 0.095) (Fig. 1). Resting HR was higher in the RA and RA‐HTN groups compared to the NC and HTN groups (P = 0.008). MSNA (burst frequency) was higher in the RA, RA‐HTN and HTN groups compared to the NC group (P = 0.004) (Fig. 2). When adjusted for HR, there was a significant difference in MSNA (burst incidence) between the groups (P = 0.029). Post hoc analysis showed significantly higher MSNA burst incidence in the HTN group compared to the NC group; and trends for an elevation in the RA‐HTN vs. NC groups (P = 0.111) and between the RA and HTN groups (P = 0.056) (Fig. 1). Leg blood flow was significantly higher in the RA and RA‐HTN groups compared to the NC group and tended to be higher than in the HTN group (geometric mean, 95% CI, RA 2.0, 1.5–2.6; RA‐HTN 2.0, 1.4–2.8; NC 1.2, 0.9–1.7; HTN 1.4, 1.0–1.8 ml 100–1 ml min–1; P = 0.047; post hoc analysis RA vs. HTN, P = 0.101, RA‐HTN vs. HTN, P = 0.086). However, leg vascular conductance was similar between all groups (RA 21, 15–27; RA‐HTN 18, 12–26; NC 14, 10–19; HTN 13, 10–17 arbitrary units; P = 0.148).
Figure 1. BP and HR.
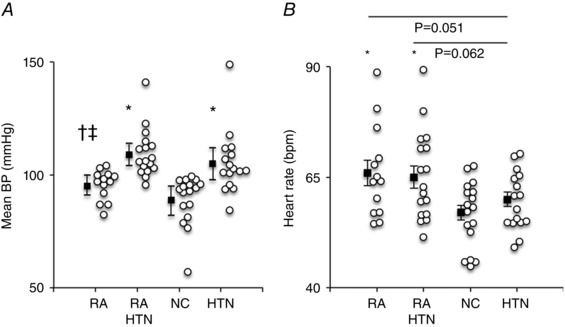
Box and whisker plots showing mean blood pressure (A, geometric mean and 95% confidence intervals) and heart rate (B, mean ± SEM) in the RA, RA‐HTN, NC and HTN groups. Group (black squares) and individual (white circles) data are shown. Overall effect: P < 0.05. Post hoc: * P < 0.05 vs. NC, † P < 0.05 vs. HTN, ‡ P < 0.05 vs. RA‐HTN. A and B, RA, n = 13; RA‐HTN, n = 17; NC, n = 17; HTN, n = 16.
Figure 2. MSNA.
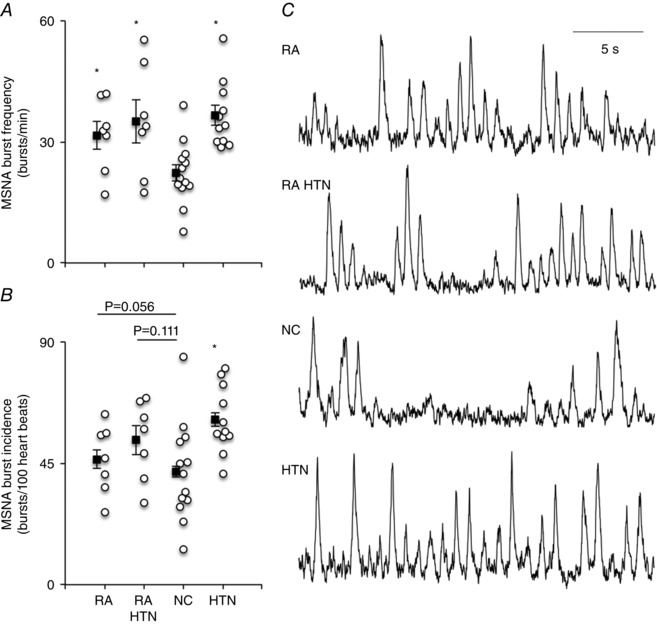
Box and whisker plots showing group mean ± SEM data for MSNA burst frequency (A) and MSNA burst incidence (B), as well as original sympathetic neurograms showing MSNA (C) in representative individuals from the RA, RA‐HTN, NC and HTN groups. Group (black squares) and individual (white circles) data are shown. Overall effect: P < 0.05. Post hoc: * P < 0.05 vs. NC. A and B, RA, n = 7; RA‐HTN, n = 7, NC, n = 13; HTN, n = 11.
Baroreflex sensitivity
Cardiac BRS was lower in the RA, RA‐HTN and HTN groups compared to the NC group (P = 0.002), whereas arterial baroreflex control of MSNA was not different between all groups (P = 0.927) (Fig. 3).
Figure 3. Cardiovagal and arterial sympathetic baroreflex senstivity.
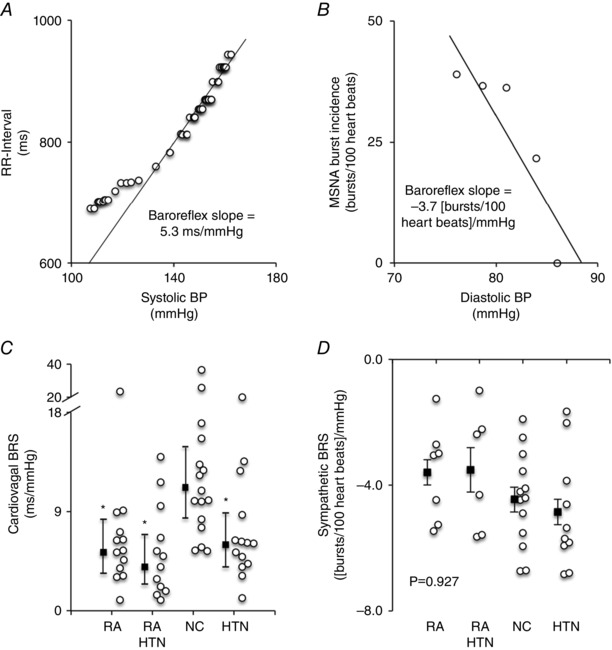
Scatter plots from an original record of an RA patient demonstrating the relationship between RR‐interval and systolic BP (A), as well as between MSNA burst incidence and diastolic BP (B). Box and whisker plots showing cardiovagal baroreflex sensitivity (C), geometric mean and 95% confidence intervals) and baroreflex control of MSNA (D) (mean ± SEM) in the RA, RA‐HTN, NC and HTN groups. Group (black squares) and individual (white circles) data are shown. Overall effect: P < 0.05. Post hoc: * P < 0.05 vs. NC. A, RA, n = 13; RA‐HTN, n = 17; NC, n = 17; HTN, n = 16. B, RA, n = 6; RA‐HTN, n = 5; NC, n = 9; HTN, n = 7.
Inflammation and pain
The RA and RA‐HTN groups had higher hs‐CRP compared to the NC and HTN groups (P < 0.001) (Fig. 4) with no significant difference between the RA and RA‐HTN groups. IL‐6 was higher in the RA group compared to the RA‐HTN, NC and HTN groups, and was higher in the RA‐HTN group compared to the NC group (P < 0.001). The RA group had higher TNF‐α compared to the NC and HTN groups (ANOVA, P = 0.027; post hoc RA vs. NC and RA vs. HTN, P < 0.05) and numerically (but not statistically significantly) higher TNF‐α than the RA‐HTN group (post hoc RA vs. RA‐HTN, P = 0.062). IL‐10 tended to be higher in the RA group compared to the other groups (P = 0.159).
Figure 4. Inflammatory biomarkers.
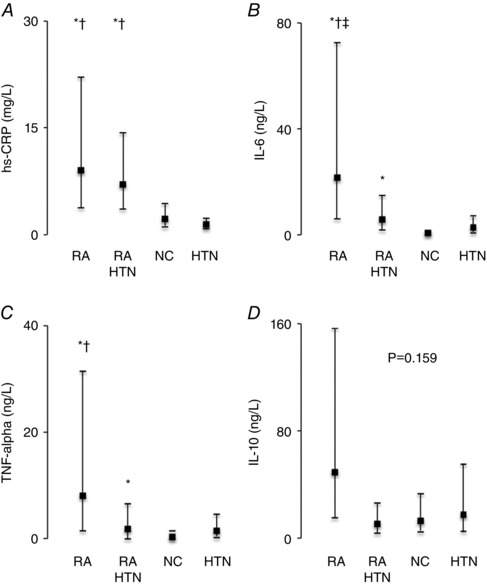
Box and whisker plots showing concentrations (geometric mean and 95% confidence intervals) of hs‐CRP (A), IL‐6 (B), TNF‐α (C) and IL‐10 (D) in the RA, RA‐HTN, NC and HTN groups. Group data are shown. Overall effect: P < 0.05. Post hoc: * P < 0.05 vs. NC, † P < 0.05 vs. HTN, ‡ P < 0.05 vs. RA‐HTN. RA, n = 13; RA‐HTN, n = 17; NC, n = 17; HTN, n = 16.
The RA and RA‐HTN groups had more pain than the NC and HTN groups (as measured by VAS) and the RA group had more pain than the RA‐HTN groups (RA geometric mean 37, 95% CI 22–62; RA‐HTN 13, 5–30; NC 1, 0–2; HTN 1, 0–3; P < 0.001).
Associations between inflammation and autonomic function
Inflammatory cytokines (IL‐6, TNF‐α and IL‐10) were positively associated with each other, whereas hs‐CRP was only associated with IL‐6 (Table 3). Both hs‐CRP and IL‐6 were positively associated with HR, although TNF‐α and IL‐10 were not. MSNA burst frequency was positively associated with hs‐CRP but not with inflammatory cytokines; however, this association was not evident when MSNA was adjusted for HR (MSNA burst incidence). Cardiac BRS was inversely associated with inflammation (hs‐CRP, IL‐6 and TNF‐α), whereas arterial baroreflex control of MSNA was not.
Table 3.
Correlation between inflammation, pain and autonomic function
| VAS | hs‐CRP | IL‐6 | TNF‐α | IL‐10 | |
|---|---|---|---|---|---|
| hs‐CRP | 0.586* | ||||
| <0.001 | – | – | – | – | |
| 57 | |||||
| IL‐6 | 0.519* | 0.339* | |||
| <0.001 | 0.010 | – | – | – | |
| 62 | 57 | ||||
| TNF‐α | 0.306* | 0.010 | 0.753* | ||
| 0.016 | 0.939 | <0.001 | – | – | |
| 62 | 57 | 62 | |||
| IL‐10 | 0.114 | –0.068 | 0.537* | 0.612* | |
| 0.378 | 0.616 | <0.001 | <0.001 | – | |
| 62 | 57 | 62 | 62 | ||
| Heart rate | 0.464* | 0.362* | 0.339* | 0.059 | −0.068 |
| <0.001 | 0.006 | 0.010 | 0.646 | 0.616 | |
| 63 | 57 | 57 | 57 | 57 | |
| Mean blood pressure | 0.224 | 0.253 | 0.073 | 0.025 | −0.138 |
| 0.077 | 0.057 | 0.572 | 0.844 | 0.286 | |
| 63 | 57 | 62 | 62 | 62 | |
| MSNA burst frequency | 0.238 | 0.418* | 0.266 | 0.039 | −0.044 |
| 0.150 | 0.011 | 0.107 | 0.816 | 0.794 | |
| 38 | 36 | 38 | 38 | 38 | |
| MSNA burst incidence | −0.032 | 0.193 | 0.039 | −0.033 | −0.109 |
| 0.849 | 0.260 | 0.815 | 0.844 | 0.516 | |
| 38 | 36 | 38 | 38 | 38 | |
| cBRS | −0.506* | −0.332* | −0.408* | −0.322* | −0.080 |
| <0.001 | 0.019 | 0.002 | 0.016 | 0.561 | |
| 55 | 50 | 55 | 55 | 55 |
Spearman's correlation. Values are expressed as Spearman's rho, P value and n.
*
Significance: P < 0.05.
Table 4 shows the association between inflammation markers and autonomic function before and after multivariable adjustment. Following multivariable adjustment (RA, presence of hypertension, age, sex, BMI, haemoglobin), hs‐CRP remained positively associated with HR (adjusted r 2 = 0.375, P < 0.001), whereas the associations with MSNA burst frequency and cardiac BRS were no longer statistically significant. Similarly, the associations between inflammatory cytokines and autonomic parameters disappeared following multivariable adjustment. In patients with RA, disease activity (DAS28‐CRP) was independently associated with HR (adjusted r 2 = 0.204, P = 0.034) after adjustment for multiple variables (age, sex, BMI, haemoglobin concentration, presence of hypertension and RA duration).
Table 4.
Association between inflammation (hs‐CRP, IL‐6, TNF‐α and IL‐10), pain (VAS) and autonomic function before and after multivariable adjustment
| Univariablea | Multivariablec | |||||
|---|---|---|---|---|---|---|
| N | Rho | P | r 2 | F | P | |
| Dependent variable: MSNA burst frequency | ||||||
| hs‐CRP | 36 | 0.418 | 0.011 | 0.222 | 1.452 | 0.238 |
| VAS | 38 | 0.238 | 0.150 | 0.345 | 7.237 | 0.012* |
| Dependent variable: cBRS | ||||||
| hs‐CRP | 50 | −0.332 | 0.019 | 0.364 | 0.683 | 0.413 |
| IL‐6 | 55 | −0.408 | 0.002 | 0.364 | 0.039 | 0.845 |
| TNF‐α | 55 | −0.322 | 0.016 | 0.364 | 0.055 | 0.815 |
| VAS | 55 | −0.506 | <0.001 | 0.417 | 4.279 | 0.044* |
| Dependent variable: HR | ||||||
| hs‐CRP | 62 | 0.362 | 0.006 | 0.366 | 13.705 | 0.001* |
| IL‐6 | 62 | 0.339 | 0.010 | 0.208 | 0.230 | 0.634 |
| DAS28‐CRPd | 29 | 0.499b | 0.006 | 0.204 | 5.123 | 0.034* |
| VAS | 63 | 0.464 | <0.001 | 0.526 | 36.661 | <0.001* |
Spearman's rank.
Pearson's correlation.
After adjustment for age, sex, BMI, presence of hypertension, RA diagnosis and haemoglobin concentration.
After adjustment for age, sex, BMI, presence of hypertension, haemoglobin concentration and RA duration.
DAS, disease activity score.
* P < 0.05.
Associations between pain and autonomic function
VAS was independently associated with MSNA burst frequency (positively, P = 0.012), cardiac BRS (inversely, P = 0.044), and HR (positively, P < 0.001) after adjustment for multiple variables (RA, presence of hypertension, age, sex, BMI, haemoglobin) (Table 4).
Discussion
In the present study, we provide the first direct evidence for heightened sympathetic outflow and reduced arterial baroreflex control of the heart in RA, whereas baroreflex control of MSNA was preserved. These autonomic alterations could occur independently of hypertension and were associated with increases in both pain and inflammation in RA. We show, in RA patients, that MSNA is heightened, whereas baroreflex control of HR is reduced, and also that this can occur independently of the presence of hypertension. We observed a significant positive but moderate association between inflammation (as measured by hs‐CRP) and MSNA.
Heightened sympathetic outflow to the heart and/or vasculature can have a multitude of deleterious consequences (Fisher & Paton, 2012) and is associated with increased mortality risk (Lown & Verrier, 1976; Cohn et al. 1984; Barretto et al. 2009). Experimental data from animals showing that inflammatory cytokines (IL‐6, TNF‐α, IL‐1β) can increase sympathetic nerve activity (Niijima et al. 1991; Zhang et al. 2003; Helwig et al. 2008) led to our hypothesis that an elevated circulating cytokine concentration is associated with increased MSNA in RA. By using microneurography to provide a direct assessment of sympathetic outflow, we circumvented the limitations associated with the measurement of plasma catecholamines, which reflect tissue clearance and uptake, as well as production (Esler et al. 1990).
Reduced cardiac BRS is present in cardiovascular diseases, such as hypertension and chronic heart failure, and predicts mortality risk following myocardial infarction (De Ferrari et al. 2007). Using the modified Oxford technique, which provides an assessment of BRS control across a wide BP range (Rudas et al. 1999), we observed that cardiac BRS was reduced in patients with RA. The underlying mechanisms may relate to altered central baroreflex modulation, as well as disruptions in afferent or efferent pathways. Nevertheless, this reduced cardiac BRS may contribute to the increased cardiovascular and mortality risk seen in RA (Maradit‐Kremers et al. 2005; Levy et al. 2008). Interestingly, the baroreflex dysfunction was specific to the cardiovagal limb because the control of MSNA was not different between groups. The prognostic significance of alterations in sympathetic BRS has not been studied; however, baroreflex activation therapy has been shown to reduce MSNA, improve symptoms and increase ejection fraction in chronic heart failure patients (Gronda et al. 2014). Studies of elderly individuals (Ebert et al. 1992) and patients with HTN (Grassi et al. 1998) have previously reported a differential effect on baroreflex control of the heart and MSNA similar to that observed in RA in the present study. Such observations may be attributable to distinct baroreflex pathways regulating different end‐organs. This separation may commence at the level of the primary afferent neurones and involve discreet conduits within central reflex circuits that enable the target‐organ specific control of pre‐motor and motor neurones (Polson et al. 2007; Simms et al. 2006). In addition, disparate changes in vagal and sympathetic motoneuronal modulation, and/or cholinergic signalling at the sinoatrial node, may also contribute (Rudas et al. 1999; Dutoit et al. 2010; Shantsila et al. 2015).
In the present study, pain was independently associated with MSNA burst frequency and cardiac BRS. These findings are in agreement with work showing that experimentally evoked chronic pain can heighten sympathetic outflow (Fazalbhoy et al. 2012) and reduce cardiac BRS (Duschek et al. 2007). Although the precise mechanisms are not fully understood, interactions between pain and cardiovascular control may be explained by an overlap in anatomical structures and pathways, including afferent pathways (e.g. baroreceptor and nociceptor projections), central modulation (e.g. nucleus of the solitary tract) and efferent pathways (e.g. descending pain inhibitory, sympathetic and parasympathetic projections) (Bruehl & Chung, 2004). In the present study, RA patients had greater self‐reported pain than RA‐HTN patients despite similar serum concentrations of hs‐CRP. This may reflect the higher pain threshold observed in hypertensive individuals (i.e. hypertensive hypoalgesia) (Ghione et al. 1988) or possibly an effect of the higher concentration of inflammatory cytokines in the RA group. Evidence from animal (Boettger et al. 2008) and human studies shows that inflammatory cytokines have a direct role in the modulation of pain perception (Hess et al. 2011). TNF‐α inhibition acutely blocked both central nociceptive activity and activation of the limbic system in RA patients (Hess et al. 2011). In RA, disease control has been shown to improve survival (Choi et al. 2002; Listing et al. 2015), although further clarification is needed to distinguish between the effects of pain and inflammation control on autonomic function.
Elevated HR is an independent predictor of mortality in the general population (Ho et al. 2014) and in other conditions (e.g. chronic heart failure, coronary artery disease, myocardial infarction, hypertension and diabetes mellitus) (Palatini et al. 2002; Fox et al. 2008; Ho et al. 2010; Hillis et al. 2012; Jabre et al. 2014). Patients with RA were observed to have an elevated HR in the present study, independent of hypertension. Aside from an increase in cardiac sympathetic nerve activity and a decrease in parasympathetic activity, there are several other potential mechanisms: chronic anaemia (Zlateva et al. 2010), increased metabolic rate as a result of chronic inflammation (Straub et al. 2010) or concomitant thyroid dysfunction (Shiroky et al. 1993), medication (Gilani et al. 2012), anxiety or depressive illness (Sheehy et al. 2006) and low physical activity (Sokka et al. 2008). Attempts to lower HR with the use of β‐blockers following myocardial infarction or in chronic heart failure has been shown to improve survival (Lopez‐Sendon et al. 2004). In an experimental model of arthritis, the use of carvedilol (a non‐selective β‐blocker) was associated with a reduction in markers of oxidative stress and the release of inflammatory cytokines (TNF‐α, IL‐6) (Arab & El‐Sawalhi, 2013). Whether such benefits would be manifest in RA patients treated with β‐blockers requires further study. We found that MSNA burst frequency (bursts min–1) was higher in RA patients compared to normotensive controls but MSNA burst incidence [bursts (100 heart beats)–1] was not (RA vs. NC, P = 0.056; RA‐HTN vs. NC, P = 0.111), probably on account of the elevated HR. Nevertheless, the observation that bursts per unit time are elevated in RA and RA‐HTN compared to NC is interpreted as being indicative of heightened sympatho‐excitation. The relatively small sample size is a potential limitation of the present study; however, this did not prevent a significantly elevated MSNA burst frequency being detected in the RA, RA‐HTN and HTN groups compared to controls. A small number of participants were smokers and, although the sympatho‐excitatory effects of smoking cannot be excluded (Hering et al. 2010), smoking was not statistically associated with MSNA in multivariable analysis. Notably, leg blood flow was elevated in RA and RA‐HTN groups compared to controls, possibly as a result of the vasodilatory effects of inflammatory cytokines. In animal models of sepsis, inflammatory cytokines appeared to cause vasodilatation via downregulation of α1‐adrenergic, angiotensin II and vasopressin receptors (Bucher et al. 2001; Bucher et al. 2002; Bucher et al. 2003). We acknowledge that substantial variability was evident in many of the parameters measured (e.g. inflammatory cytokines, HR, cBRS). All data were tested statistically for normality, and non‐normally distributed data were logarithmically transformed and parametric testing was performed. In the multivariable analysis, independent associations between pain VAS, hs‐CRP and autonomic function (HR, MSNA and cBRS) were weak to moderate and only explained between 35% and 53% of the variance. Further studies are required to determine the additional factors that may contribute to autonomic dysfunction in RA.
An important strength of the design of the present study is the inclusion of hypertensive control groups, allowing us to control for the presence of hypertension in RA and to control for the effects of medications. Athough increased MSNA and reduced cardiac BRS are known to be present in hypertension (Carthy, 2014), we have shown that, in RA, these autonomic alterations can occur independently of hypertension, suggesting an alternative mechanism (e.g. cortisol‐induced hypertension). Glucocorticoid use was higher in the RA‐HTN group compared to the RA group and may partly account for the difference in the inflammatory profile and BP seen in the present study. Glucocorticoids have been shown to reduce MSNA (Lenders et al. 1995), which may have attenuated sympatho‐excitation in the RA‐HTN group. Mechanistic links between immunity and hypertension have also been described, where T‐cells, macrophages and monocytes accumulate within the kidney and blood vessel wall, releasing cytokines that deleteriously affect renal and vascular function (McMaster et al. 2015). The cross‐sectional design precludes the establishment of causality; however, we suggest that heightened sympathetic outflow can occur prior to the development of hypertension and may possibly contribute to the development of hypertension in some patients with RA. Autonomic dysfunction may be a causal factor in the pathogenesis of RA or a consequence of the disease, or both. Future randomized controlled interventional studies are needed to clarify whether autonomic dysfunction is a cause or consequence of RA and, in particular, to differentiate between the contributions of inflammation and pain to autonomic dysfunction in RA.
In conclusion, the present study is the first to demonstrate heightened MSNA in RA. Heightened sympathetic outflow and reduced baroreflex control of HR in RA may potentially contribute to the recognized increase in cardiovascular risk. We found that, compared to inflammation, pain was more strongly correlated with MSNA (positively) and cBRS (negatively), suggesting a potential benefit in optimizing pain management to restore autonomic balance in RA. Such findings may mean that therapeutic interventions to reduce inflammation and pain in RA can restore autonomic balance and hence improve morbidity and mortality, whereas therapies that target the autonomic nervous system (e.g. sympatholytic agents, baroreflex activation) may reduce inflammation and/or pain. Interventional studies designed to evaluate these possibilities will help improve our understanding of the pathophysiology of RA and, in particular, will shed light on the interplay between inflammation, pain and the autonomic nervous system.
Additional information
Conflicts of interests/competing interests
The authors declare that they have no competing interests.
Author contributions
JPF, GDK, GYL and JFRP were involved in conception of the work and critical review. AMA and JPF were involved in acquisition, analysis and interpretation of the work. AMA drafted and revised the work. All authors have approved the final manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriate. All persons designated as authors qualify for authorship and all those who qualify for authorship are listed.
Funding
This study was supported by a grant from Arthritis Research UK (grant number 196633). JFRP is funded by the British Heart Foundation.
Acknowledgements
The authors acknowledge the support of the National Institute of Health Research Clinical Research Network (NIHR CRN), Jacqueline Smith for performing the biochemical analysis and the participants for their time.
References
- Adlan AM, Lip GY, Paton JF, Kitas GD & Fisher JP (2014). Autonomic function and rheumatoid arthritis – a systematic review. Semin Arthritis Rheum 44, 283–304. [DOI] [PubMed] [Google Scholar]
- Amaya‐Amaya J, Sarmiento‐Monroy JC, Mantilla RD, Pineda‐Tamayo R, Rojas‐Villarraga A & Anaya JM (2013). Novel risk factors for cardiovascular disease in rheumatoid arthritis. Immunol Res 56, 267–286. [DOI] [PubMed] [Google Scholar]
- Arab HH & El‐Sawalhi MM (2013). Carvedilol alleviates adjuvant‐induced arthritis and subcutaneous air pouch edema: modulation of oxidative stress and inflammatory mediators. Toxicol Appl Pharmacol 268, 241–248. [DOI] [PubMed] [Google Scholar]
- Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS & et al (1988). The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 31, 315–324. [DOI] [PubMed] [Google Scholar]
- Arosio E, De Marchi S, Rigoni A, Prior M, Delva P & Lechi A (2007). Forearm haemodynamics, arterial stiffness and microcirculatory reactivity in rheumatoid arthritis. J Hypertens 25, 1273–1278. [DOI] [PubMed] [Google Scholar]
- Aydemir M, Yazisiz V, Basarici I, Avci AB, Erbasan F, Belgi A & Terzioglu E (2010). Cardiac autonomic profile in rheumatoid arthritis and systemic lupus erythematosus. Lupus 19, 255–261. [DOI] [PubMed] [Google Scholar]
- Barretto AC, Santos AC, Munhoz R, Rondon MU, Franco FG, Trombetta IC, Roveda F, de Matos LN, Braga AM, Middlekauff HR & Negrao CE (2009). Increased muscle sympathetic nerve activity predicts mortality in heart failure patients. Int J Cardiol 135, 302–307. [DOI] [PubMed] [Google Scholar]
- Boettger MK, Hensellek S, Richter F, Gajda M, Stockigt R, von Banchet GS, Brauer R & Schaible HG (2008). Antinociceptive effects of tumor necrosis factor alpha neutralization in a rat model of antigen‐induced arthritis: evidence of a neuronal target. Arthritis Rheum 58, 2368–2378. [DOI] [PubMed] [Google Scholar]
- Bruehl S & Chung OY (2004). Interactions between the cardiovascular and pain regulatory systems: an updated review of mechanisms and possible alterations in chronic pain. Neurosci Biobehav Rev 28, 395–414. [DOI] [PubMed] [Google Scholar]
- Bucher M, Hobbhahn J, Taeger K & Kurtz A (2002). Cytokine‐mediated downregulation of vasopressin V(1A) receptors during acute endotoxemia in rats. Am J Physiol Regul Integr Comp Physiol 282, R979–R984. [DOI] [PubMed] [Google Scholar]
- Bucher M, Ittner KP, Hobbhahn J, Taeger K & Kurtz A (2001). Downregulation of angiotensin II type 1 receptors during sepsis. Hypertension 38, 177–182. [DOI] [PubMed] [Google Scholar]
- Bucher M, Kees F, Taeger K & Kurtz A (2003). Cytokines down‐regulate alpha1‐adrenergic receptor expression during endotoxemia. Crit Care Med 31, 566–571. [DOI] [PubMed] [Google Scholar]
- Carthy ER (2014). Autonomic dysfunction in essential hypertension: A systematic review. Ann Med Surg 3, 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HK, Hernan MA, Seeger JD, Robins JM & Wolfe F (2002). Methotrexate and mortality in patients with rheumatoid arthritis: a prospective study. Lancet 359, 1173–1177. [DOI] [PubMed] [Google Scholar]
- Cohn JN, Levine TB, Olivari MT, Garberg V, Lura D, Francis GS, Simon AB & Rector T (1984). Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med 311, 819–823. [DOI] [PubMed] [Google Scholar]
- De Ferrari GM, Sanzo A, Bertoletti A, Specchia G, Vanoli E & Schwartz PJ (2007). Baroreflex sensitivity predicts long‐term cardiovascular mortality after myocardial infarction even in patients with preserved left ventricular function. J Am Coll Cardiol 50, 2285–2290. [DOI] [PubMed] [Google Scholar]
- Duschek S, Muck I & Reyes Del Paso GA (2007). Relationship between baroreceptor cardiac reflex sensitivity and pain experience in normotensive individuals. Int J Psychophysiol 65, 193–200. [DOI] [PubMed] [Google Scholar]
- Dutoit AP, Hart EC, Charkoudian N, Wallin BG, Curry TB & Joyner MJ (2010). Cardiac baroreflex sensitivity is not correlated to sympathetic baroreflex sensitivity within healthy, young humans. Hypertension 56, 1118–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert TJ ( 1990). Differential effects of nitrous oxide on baroreflex control of heart rate and peripheral sympathetic nerve activity in humans. Anesthesiology 72, 16–22. [DOI] [PubMed] [Google Scholar]
- Ebert TJ, Morgan BJ, Barney JA, Denahan T & Smith JJ (1992). Effects of aging on baroreflex regulation of sympathetic activity in humans. Am J Physiol Heart Circ Physiol 263, H798–H803. [DOI] [PubMed] [Google Scholar]
- Eckberg DL & Eckberg MJ (1982). Human sinus node responses to repetitive, ramped carotid baroreceptor stimuli. Am J Physiol Heart Circ Physiol 242, H638–H644. [DOI] [PubMed] [Google Scholar]
- Esler M, Jennings G, Lambert G, Meredith I, Horne M & Eisenhofer G (1990). Overflow of catecholamine neurotransmitters to the circulation: source, fate, and functions. Physiol Rev 70, 963–985. [DOI] [PubMed] [Google Scholar]
- Fazalbhoy A, Birznieks I & Macefield VG (2012). Individual differences in the cardiovascular responses to tonic muscle pain: parallel increases or decreases in muscle sympathetic nerve activity, blood pressure and heart rate. Exp Physiol 97, 1084–1092. [DOI] [PubMed] [Google Scholar]
- Ferguson DW, Berg WJ, Roach PJ, Oren RM & Mark AL (1992). Effects of heart failure on baroreflex control of sympathetic neural activity. Am J Cardiol 69, 523–531. [DOI] [PubMed] [Google Scholar]
- Fisher JP & Paton JF (2012). The sympathetic nervous system and blood pressure in humans: implications for hypertension. J Hum Hypertens 26, 463–475. [DOI] [PubMed] [Google Scholar]
- Fox K, Ford I, Steg PG, Tendera M, Robertson M, Ferrari R & investigators B (2008). Heart rate as a prognostic risk factor in patients with coronary artery disease and left‐ventricular systolic dysfunction (BEAUTIFUL): a subgroup analysis of a randomised controlled trial. Lancet 372, 817–821. [DOI] [PubMed] [Google Scholar]
- Ghione S, Rosa C, Mezzasalma L & Panattoni E (1988). Arterial hypertension is associated with hypalgesia in humans. Hypertension 12, 491–497. [DOI] [PubMed] [Google Scholar]
- Gilani ST, Khan DA, Khan FA & Ahmed M (2012). Adverse effects of low dose methotrexate in rheumatoid arthritis patients. J Coll Physicians Surg Pak 22, 101–104. [PubMed] [Google Scholar]
- Grassi G, Cattaneo BM, Seravalle G, Lanfranchi A & Mancia G (1998). Baroreflex control of sympathetic nerve activity in essential and secondary hypertension. Hypertension 31, 68–72. [DOI] [PubMed] [Google Scholar]
- Grassi G & Esler M (1999). How to assess sympathetic activity in humans. J Hypertens 17, 719–734. [DOI] [PubMed] [Google Scholar]
- Gribbin B, Pickering TG, Sleight P & Peto R (1971). Effect of age and high blood pressure on baroreflex sensitivity in man. Circ Res 29, 424–431. [DOI] [PubMed] [Google Scholar]
- Gronda E, Seravalle G, Brambilla G, Costantino G, Casini A, Alsheraei A, Lovett EG, Mancia G & Grassi G (2014). Chronic baroreflex activation effects on sympathetic nerve traffic, baroreflex function, and cardiac haemodynamics in heart failure: a proof‐of‐concept study. Eur J Heart Fail 16, 977–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart EC, Joyner MJ, Wallin BG, Karlsson T, Curry TB & Charkoudian N (2010). Baroreflex control of muscle sympathetic nerve activity: a nonpharmacological measure of baroreflex sensitivity. Am J Physiol Regul Integr Comp Physiol 298, H816–H822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helwig BG, Craig RA, Fels RJ, Blecha F & Kenney MJ (2008). Central nervous system administration of interleukin‐6 produces splenic sympathoexcitation. Auton Neurosci 141, 104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering D, Kucharska W, Kara T, Somers VK & Narkiewicz K (2010). Smoking is associated with chronic sympathetic activation in hypertension. Blood Press 19, 152–155. [DOI] [PubMed] [Google Scholar]
- Hess A, Axmann R, Rech J, Finzel S, Heindl C, Kreitz S, Sergeeva M, Saake M, Garcia M, Kollias G, Straub RH, Sporns O, Doerfler A, Brune K & Schett G (2011). Blockade of TNF‐alpha rapidly inhibits pain responses in the central nervous system. Proc Natl Acad Sci USA 108, 3731–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillis GS, Woodward M, Rodgers A, Chow CK, Li Q, Zoungas S, Patel A, Webster R, Batty GD, Ninomiya T, Mancia G, Poulter NR & Chalmers J (2012). Resting heart rate and the risk of death and cardiovascular complications in patients with type 2 diabetes mellitus. Diabetologia 55, 1283–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JE, Bittner V, Demicco DA, Breazna A, Deedwania PC & Waters DD (2010). Usefulness of heart rate at rest as a predictor of mortality, hospitalization for heart failure, myocardial infarction, and stroke in patients with stable coronary heart disease (Data from the Treating to New Targets [TNT] trial). Am J Cardiol 105, 905–911. [DOI] [PubMed] [Google Scholar]
- Ho JE, Larson MG, Ghorbani A, Cheng S, Coglianese EE, Vasan RS & Wang TJ (2014). Long‐term cardiovascular risks associated with an elevated heart rate: the Framingham Heart Study. J Am Heart Assoc 3, e000668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huskisson EC ( 1974). Measurement of pain. Lancet 2, 1127–1131. [DOI] [PubMed] [Google Scholar]
- Jabre P, Roger VL, Weston SA, Adnet F, Jiang R, Vivien B, Empana JP & Jouven X (2014). Resting heart rate in first year survivors of myocardial infarction and long‐term mortality: a community study. Mayo Clin Proc 89, 1655–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner MJ, Dietz NM & Shepherd JT (2001). From Belfast to Mayo and beyond: the use and future of plethysmography to study blood flow in human limbs. J Appl Physiol 91, 2431–2441. [DOI] [PubMed] [Google Scholar]
- La Rovere MT, Bigger JT, Jr. , Marcus FI, Mortara A & Schwartz PJ (1998). Baroreflex sensitivity and heart‐rate variability in prediction of total cardiac mortality after myocardial infarction. ATRAMI (Autonomic Tone and Reflexes After Myocardial Infarction) Investigators. Lancet 351, 478–484. [DOI] [PubMed] [Google Scholar]
- La Rovere MT, Pinna GD, Maestri R, Robbi E, Caporotondi A, Guazzotti G, Sleight P & Febo O (2009). Prognostic implications of baroreflex sensitivity in heart failure patients in the beta‐blocking era. J Am Coll Cardiol 53, 193–199. [DOI] [PubMed] [Google Scholar]
- Lenders JW, Golczynska A & Goldstein DS (1995). Glucocorticoids, sympathetic activity, and presynaptic alpha 2‐adrenoceptor function in humans. J Clin Endocrinol Metab 80, 1804–1808. [DOI] [PubMed] [Google Scholar]
- Levy L, Fautrel B, Barnetche T & Schaeverbeke T (2008). Incidence and risk of fatal myocardial infarction and stroke events in rheumatoid arthritis patients. A systematic review of the literature. Clin Exp Rheumatol 26, 673–679. [PubMed] [Google Scholar]
- Listing J, Kekow J, Manger B, Burmester GR, Pattloch D, Zink A & Strangfeld A (2015). Mortality in rheumatoid arthritis: the impact of disease activity, treatment with glucocorticoids, TNFalpha inhibitors and rituximab. Ann Rheum Dis 74, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Sendon J, Swedberg K, McMurray J, Tamargo J, Maggioni AP, Dargie H, Tendera M, Waagstein F, Kjekshus J, Lechat P, Torp‐Pedersen C & Task ForceOn Beta‐Blockers of the European Society of C (2004). Expert consensus document on beta‐adrenergic receptor blockers. Eur Heart J 25, 1341–1362. [DOI] [PubMed] [Google Scholar]
- Lown B & Verrier RL (1976). Neural activity and ventricular fibrillation. N Engl J Med 294, 1165–1170. [DOI] [PubMed] [Google Scholar]
- Maradit‐Kremers H, Crowson CS, Nicola PJ, Ballman KV, Roger VL, Jacobsen SJ & Gabriel SE (2005). Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population‐based cohort study. Arthritis Rheum 52, 402–411. [DOI] [PubMed] [Google Scholar]
- McMaster WG, Kirabo A, Madhur MS & Harrison DG (2015). Inflammation, immunity, and hypertensive end‐organ damage. Circ Res 116, 1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niijima A, Hori T, Aou S & Oomura Y (1991). The effects of interleukin‐1 beta on the activity of adrenal, splenic and renal sympathetic nerves in the rat. J Auton Nerv Syst 36, 183–192. [DOI] [PubMed] [Google Scholar]
- Palatini P, Thijs L, Staessen JA, Fagard RH, Bulpitt CJ, Clement DL, de Leeuw PW, Jaaskivi M, Leonetti G, Nachev C, O'Brien ET, Parati G, Rodicio JL, Roman E, Sarti C, Tuomilehto J & Systolic Hypertension in Europe Trial I (2002). Predictive value of clinic and ambulatory heart rate for mortality in elderly subjects with systolic hypertension. Arch Intern Med 162, 2313–2321. [DOI] [PubMed] [Google Scholar]
- Panoulas VF, Metsios GS, Pace AV, John H, Treharne GJ, Banks MJ & Kitas GD (2008). Hypertension in rheumatoid arthritis. Rheumatology 47, 1286–1298. [DOI] [PubMed] [Google Scholar]
- Panoulas VF, Toms TE, Douglas KM, Sandoo A, Metsios GS, Stavropoulos‐Kalinoglou A & Kitas GD (2014). Prolonged QTc interval predicts all‐cause mortality in patients with rheumatoid arthritis: an association driven by high inflammatory burden. Rheumatology 53, 131–137. [DOI] [PubMed] [Google Scholar]
- Pickering TG & Davies J (1973). Estimation of the conduction time of the baroreceptor‐cardiac reflex in man. Cardiovasc Res 7, 213–219. [DOI] [PubMed] [Google Scholar]
- Polson JW, Dampney RA, Boscan P, Pickering AE & Paton JF (2007). Differential baroreflex control of sympathetic drive by angiotensin II in the nucleus tracts solitarii. Am J Physiol Regul Integr Comp Physiol 293, R1954–R1960. [DOI] [PubMed] [Google Scholar]
- Poyhonen‐Alho MK, Manhem K, Katzman P, Kibarskis A, Antikainen RL, Erkkola RU, Tuomilehto JO, Ebeling PE & Kaaja RJ (2008). Central sympatholytic therapy has anti‐inflammatory properties in hypertensive postmenopausal women. J Hypertens 26, 2445–2449. [DOI] [PubMed] [Google Scholar]
- Rudas L, Crossman AA, Morillo CA, Halliwill JR, Tahvanainen KU, Kuusela TA & Eckberg DL (1999). Human sympathetic and vagal baroreflex responses to sequential nitroprusside and phenylephrine. Am J Physiol Heart Circ Physiol 276, H1691–H1698. [DOI] [PubMed] [Google Scholar]
- Shantsila A, McIntyre DB, Lip GY, Fadel PJ, Paton JF, Pickering AE & Fisher JP (2015). Influence of age on respiratory modulation of muscle sympathetic nerve activity, blood pressure and baroreflex function in humans. Exp Physiol 100, 1039–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy C, Murphy E & Barry M (2006). Depression in rheumatoid arthritis–underscoring the problem. Rheumatology 45, 1325–1327. [DOI] [PubMed] [Google Scholar]
- Shiroky JB, Cohen M, Ballachey ML & Neville C (1993). Thyroid dysfunction in rheumatoid arthritis: a controlled prospective survey. Ann Rheum Dis 52, 454–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms AE, Paton JF & Pickering AE (2006). Hierarchical recruitment of the sympathetic and parasympathetic limbs of the baroreflex in normotensive and spontaneously hypertensive rats. J Physiol 579, 473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokka T, Hakkinen A, Kautiainen H, Maillefert JF, Toloza S, Mork Hansen T, Calvo‐Alen J, Oding R, Liveborn M, Huisman M, Alten R, Pohl C, Cutolo M, Immonen K, Woolf A, Murphy E, Sheehy C, Quirke E, Celik S, Yazici Y, Tlustochowicz W, Kapolka D, Skakic V, Rojkovich B, Muller R, Stropuviene S, Andersone D, Drosos AA, Lazovskis J, Pincus T & Group Q‐R (2008). Physical inactivity in patients with rheumatoid arthritis: data from twenty‐one countries in a cross‐sectional, international study. Arthritis Rheum 59, 42–50. [DOI] [PubMed] [Google Scholar]
- Straub RH, Cutolo M, Buttgereit F & Pongratz G (2010). Energy regulation and neuroendocrine‐immune control in chronic inflammatory diseases. J Intern Med 267, 543–560. [DOI] [PubMed] [Google Scholar]
- Studinger P, Goldstein R & Taylor JA (2007). Mechanical and neural contributions to hysteresis in the cardiac vagal limb of the arterial baroreflex. J Physiol 583, 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagishi M, Waki H, Bhuiyan ME, Gouraud SS, Kohsaka A, Cui H, Yamazaki T, Paton JF & Maeda M (2010). IL‐6 microinjected in the nucleus tractus solitarii attenuates cardiac baroreceptor reflex function in rats. Am J Physiol Regul Integr Comp Physiol 298, R183–R190. [DOI] [PubMed] [Google Scholar]
- Vlcek M, Rovensky J, Blazicek P, Radikova Z, Penesova A, Kerlik J, Kvetnansky R & Imrich R (2008). Sympathetic nervous system response to orthostatic stress in female patients with rheumatoid arthritis. Ann NY Acad Sci 1148, 556–561. [DOI] [PubMed] [Google Scholar]
- Wallin BG & Fagius J (1988). Peripheral sympathetic neural activity in conscious humans. Annu Rev Physiol 50, 565–576. [DOI] [PubMed] [Google Scholar]
- Wells G, Becker JC, Teng J, Dougados M, Schiff M, Smolen J, Aletaha D & van Riel PL (2009). Validation of the 28‐joint Disease Activity Score (DAS28) and European League Against Rheumatism response criteria based on C‐reactive protein against disease progression in patients with rheumatoid arthritis, and comparison with the DAS28 based on erythrocyte sedimentation rate. Ann Rheum Dis 68, 954–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZH, Wei SG, Francis J & Felder RB (2003). Cardiovascular and renal sympathetic activation by blood‐borne TNF‐alpha in rat: the role of central prostaglandins. Am J Physiol Regul Integr Comp Physiol 284, R916–R927. [DOI] [PubMed] [Google Scholar]
- Zlateva G, Diazaraque R, Viala‐Danten M & Niculescu L (2010). Burden of anemia in patients with osteoarthritis and rheumatoid arthritis in French secondary care. BMC Geriatrics 10, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]