

Age‐related hearing loss (ARHL) affects millions of people, particularly the elderly, whose hearing capacity declines over time and who in severe cases become functionally deaf. Such a deficit negatively impacts the quality of life since speech perception and cognition can be significantly compromised. Numerous studies, in human and animal models, have shown that ageing‐dependent hearing impairments can be largely accounted for by a loss or damage of hair cells in the cochlea. These cells detect sound waves with extraordinary sensitivity and convert mechanical signals to electrical and chemical impulses that can be propagated into the central auditory brain. Being the peripheral detectors of auditory information, hair cells are constantly exposed to various stimuli of noisy environments and are naturally vulnerable to deleterious levels of sound. There is also strong evidence showing morphological and functional deficits in downstream afferent contacts of the auditory nerves derived from spiral ganglion cells, which collect information from hair cells and convey it to the central auditory system. Collectively, it is believed that the peripheral impairments attenuate sound information inflow into the aged brain, and hence manifest as ARHL. However, it is largely unknown whether there are age‐dependent defects along the central auditory pathways, and if so, how such defects at cellular and molecular levels contribute to the compromised capacity of ageing brains to decode the fine and complex temporal structure of sound, such as in speech and music.
In this issue of The Journal of Physiology, Xie & Manis (2017) present an exciting study comparing the properties of auditory neurotransmission in mature and aged CBA/CaJ mice (2–4 vs. 16–20 months old), which did not carry any known genetic mutations of hearing loss but naturally developed ARHL, mimicking the progressive nature of the human condition (Sergeyenko et al. 2013). Indeed, Xie and Manis confirmed that when presented with standard sound clicks, these aged mice had much higher thresholds and lower amplitudes of all waves in their auditory brainstem responses (ABRs) than healthy mature mice. Given flattened waves of ABRs in vivo, one would expect that neurotransmission downstream from the auditory nerve to other central stations would be profoundly affected. Surprisingly, they discovered that in ageing mice synaptic transmission at the endbulb of Held synapse, the first synaptic contact between the auditory nerve and the globular bushy cells of the cochlear nucleus (Fig. 1), was very well preserved when the auditory nerve in brainstem slices in vitro was stimulated at low frequencies (i.e. 100 Hz). In contrast, when stimulated at high frequencies (i.e. 400 Hz), the aged synapses were unable to sustain spike‐ and Ca2+‐dependent synchronized release of neurotransmitters from the auditory nerve as typically seen in mature synapses, leading to numerous asynchronous release events that mismatched the timing of afferent inputs. Interestingly, such a breakdown of synchrony in transmitter release could be readily rescued by either a reduction of Ca2+ entry into the nerve terminal or addition of a membrane‐permeant Ca2+ chelator that buffered intra‐terminal Ca2+ build‐up during high frequency activity. These observations led the authors to conclude that intracellular calcium handing in the aged synapse is deficient during intense activity, being not properly buffered or effectively removed.
Figure 1. Compromised synchrony of transmitter release at the endbulb of Held synapse in the aged brain.
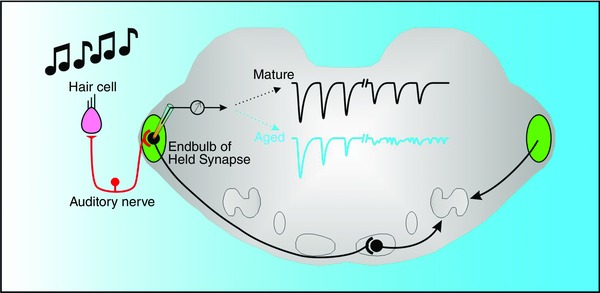
Patch‐clamp recordings from the first central synapse, namely the endbulb of Held–globular bushy cell synapse, in the auditory brainstem slice reveal that neurotransmitter release from the auditory nerve is highly synchronized and phase‐locked to inputs in mature mice, but become asynchronous in aged mice during prolonged high‐frequency activity.
This study has presented compelling evidence that, at a central auditory synapse in the aged mice, deficiency in intra‐terminal Ca2+ homeostasis breaks down the temporal precision of neurotransmitter release. Given auditory processing is critically dependent on decoding the fine temporal structure of sound, one can infer that Ca2+ mishandling may impede the fidelity of information transfer along the auditory pathway for sound perception, and contribute to the development of ARHL. By unravelling frequency‐dependent Ca2+ overload as a novel mechanism for ARHL, this study raises a new set of questions with respect to the cellular and molecular substrates leading to desynchronized transmitter release. For example, is there an age‐dependent down‐regulation of low‐threshold potassium channels that prevent activity‐dependent spike broadening and overload of Ca2+ influx in the nerve terminal (Yang et al. 2014)? Have the topographic distribution and density of voltage‐gated calcium channels in the active zone been remodelled? Do the levels of endogenous Ca2+‐binding proteins, sensors and/or pumps change over time? Are the absorption and release of internal Ca2+ stores altered? With emerging evidence of specific genes associated with the onset of ARHL (Potter et al. 2016), answering these questions will not only provide mechanistic insights into ARHL, but also identify molecular substrates for developing innovative strategies to rectify this pathological condition. The fact that central auditory synapses can remarkably maintain the status quo despite diminishing sensory inputs during ageing raises the promising prospect of reversing or rescuing ARHL by targeting peripheral synapses as well as the intracellular Ca2+ signalling cascade in central synapses (Müller & Barr‐Gillespie 2015), ultimately rejuvenating the capacity of ageing brains to attend a symphony with synchronous neurotransmitter release.
Additional information
Competing interests
None declared.
Author contributions
Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Linked articles This Perspective highlights an article by Xie & Manis. To read this paper, visit http://dx.doi.org/10.1113/JP272683.
References
- Müller U & Barr‐Gillespie PG (2015). New treatment options for hearing loss. Nat Rev Drug Discov 14, 346–365. [DOI] [PubMed] [Google Scholar]
- Potter PK, Bowl MR, Jeyarajan P, et al (2016). Novel gene function revealed by mouse mutagenesis screens for models of age‐related disease. Nat Commun 7, 12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeyenko Y, Lall K, Liberman MC & Kujawa SG (2013). Age‐related cochlear synaptopathy: an early‐onset contributor to auditory functional decline. J Neurosci 33, 13686–13694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie R & Manis PB (2017). Synaptic transmission at the endbulb of Held deteriorates during age‐related hearing loss. J Physiol 595, 919–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YM, Wang W, Fedchyshyn MJ, Zhou Z, Ding JP & Wang LY (2014). Enhancing the fidelity of neurotransmission by activity‐dependent facilitation of presynaptic K+ currents. Nat Commun 5, 4564–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]