

Abstract
Key points
Neuroinflammation associated with CNS insults leads to neuronal hyperexcitability, which may culminate in epileptiform discharges.
Application of the endotoxin lipopolysaccharide (LPS) to brain tissue initiates a neuroinflammatory cascade, providing an experimental model to study the mechanisms of neuroinflammatory neuronal hyperexcitability.
Here we show that LPS application to hippocampal slices markedly enhances the excitability of CA1 pyramidal cells by inhibiting a specific potassium current, the M‐current, generated by KV7/M channels, which controls the excitability of almost every neuron in the CNS.
The LPS‐induced M‐current inhibition is triggered by sequential activation of microglia, astrocytes and pyramidal cells, mediated by metabotropic purinergic and glutamatergic transmission, leading to blockade of KV7/M channels by calcium released from intracellular stores.
The identification of the downstream molecular target of neuroinflammation, namely the KV7/M channel, potentially has far reaching implications for the understanding and treatment of many acute and chronic brain disorders.
Abstract
Acute brain insults and many chronic brain diseases manifest an innate inflammatory response. The hallmark of this response is glia activation, which promotes repair of damaged tissue, but also induces structural and functional changes that may lead to an increase in neuronal excitability. We have investigated the mechanisms involved in the modulation of neuronal activity by acute inflammation. Initiating inflammatory responses in hippocampal tissue rapidly led to neuronal depolarization and repetitive firing even in the absence of active synaptic transmission. This action was mediated by a complex metabotropic purinergic and glutamatergic glia‐to‐neuron signalling cascade, leading to the blockade of neuronal KV7/M channels by Ca2+ released from internal stores. These channels generate the low voltage‐activating, non‐inactivating M‐type K+ current (M‐current) that controls intrinsic neuronal excitability, and its inhibition was the predominant cause of the inflammation‐induced hyperexcitability. Our discovery that the ubiquitous KV7/M channels are the downstream target of the inflammation‐induced cascade, has far reaching implications for the understanding and treatment of many acute and chronic brain disorders.
Keywords: CA1, hippocampus, intrinsic excitability, M‐current, neuroinflammation
Key points
Neuroinflammation associated with CNS insults leads to neuronal hyperexcitability, which may culminate in epileptiform discharges.
Application of the endotoxin lipopolysaccharide (LPS) to brain tissue initiates a neuroinflammatory cascade, providing an experimental model to study the mechanisms of neuroinflammatory neuronal hyperexcitability.
Here we show that LPS application to hippocampal slices markedly enhances the excitability of CA1 pyramidal cells by inhibiting a specific potassium current, the M‐current, generated by KV7/M channels, which controls the excitability of almost every neuron in the CNS.
The LPS‐induced M‐current inhibition is triggered by sequential activation of microglia, astrocytes and pyramidal cells, mediated by metabotropic purinergic and glutamatergic transmission, leading to blockade of KV7/M channels by calcium released from intracellular stores.
The identification of the downstream molecular target of neuroinflammation, namely the KV7/M channel, potentially has far reaching implications for the understanding and treatment of many acute and chronic brain disorders.
Abbreviations
- aCSF
artificial cerebrospinal fluid
- ADPβS
adenosine 5′‐O‐2‐thiodiphosphate
- AP‐5
2‐amino‐5‐phosphono‐valeric acid
- BAPTA
1,2‐bis(o‐aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid
- [Ca2+]i
intracellular calcium concentration
- CNQX
6‐cyano‐7‐nitro‐quinoxaline‐2,3‐dione
- EPSC
excitatory postsynaptic current
- f–I
frequency–current intensity
- l‐AAA
l‐α‐aminoadipic acid
- IL‐1β
interleukin 1β
- IM
M‐current
- IPSP
inhibitory postsynaptic potentials
- LPS
lipopolysaccharide
- LY341495
(2S)‐2‐amino‐2‐[(1S,2S)‐2‐carboxycycloprop‐1‐yl]‐3‐(xanth‐9‐yl) propanoic acid
- MCPG
(RS)‐α‐methyl‐4‐carboxyphenylglycine
- mEPSC
miniature excitatory postsynaptic current
- mGluR
metabotropic glutamate receptor
- MPEP
2‐methyl‐6‐(phenylethynyl)pyridine hydrochloride
- mGlu5
metabotropic glutamate receptors group I subtype 5
- MRS2179
2′‐deoxy‐N 6‐methyladenosine 3′,5′‐bisphosphate tetrasodium salt
- Rin
input resistance
- SR‐101
sulforhodamine 101
- t‐ACPD
((±)‐1‐aminocyclopentane‐trans‐1,3‐dicarboxylic acid
- TLR‐4
toll‐like receptor 4
- TNFα
tumour necrosis factor α
- TTX
tetrodotoxin
- Vm
membrane potential
- XE991
10,10‐bis(4‐pyridinylmethyl)‐9(10H)‐anthracenone
- ZD7288
4‐ethylphenylamino‐1,2‐dimethyl‐6‐methylaminopyrimidinium chloride
Introduction
Acute insults to the brain (e.g. stroke, haemorrhage, trauma, encephalitis and status epilepticus) and many chronic brain diseases (e.g. epilepsy, Alzheimer's disease and amyotrophic lateral sclerosis) manifest an innate inflammatory response (Tian et al. 2012). The hallmark of this response is activation of microglia and astrocytes, acting to promote repair of damaged tissue (Amor & Woodroofe, 2014). However, activation of glia may also induce structural and functional changes in neuronal networks that lead to brain hyperexcitability, predisposing it to acute or even chronic seizures (Vezzani et al. 2011; Devinsky et al. 2013; Seifert & Steinhauser, 2013). Multiple mechanisms are likely to be involved in inflammation‐induced hyperexcitability. Prevailing hypotheses revolve around activation of microglia and astrocytes, leading to release of glial inflammatory mediators, such as interleukin 1β (IL‐1β) and tumour necrosis factor α (TNFα), as well as a variety of gliotransmitters, which reportedly augment neuronal excitability (Vezzani & Viviani, 2015). Innate brain inflammation can be produced experimentally by applying lipopolysaccharide (LPS; Lund et al. 2006), an endotoxin derived from the outer membrane of gram‐negative bacteria (Freudenberg & Galanos, 1990), to brain tissue. LPS induces inflammation predominately by activating toll‐like receptor 4 (TLR‐4) in microglia (Poltorak et al. 1998; Chow et al. 1999; Olson & Miller, 2004; Lu et al. 2008). It has been shown recently that the acute inflammatory response to LPS leads to enhanced neuronal excitability. Thus, when applied on the cortex of sedated rats, LPS induces epileptiform discharges within minutes, sometimes culminating in full‐blown seizures (Rodgers et al. 2009). Likewise, when applied acutely to hippocampal slices, LPS facilitates the generation of epileptiform discharges (Pascual et al. 2012; Gao et al. 2014). At the cellular level, acute application of LPS to hippocampal slices was reported to enhance the frequency of excitatory postsynaptic currents (EPSCs; Pascual et al. 2012) and to increase neuronal excitability (Gao et al. 2014). These two actions, cooperatively, may underlie LPS‐induced brain hyperexcitability. However, the underlying molecular and biophysical mechanisms of LPS‐induced enhancement of neuronal excitability have not been clarified.
Here, using electrophysiological, pharmacological and imaging techniques in rat hippocampal slices, we show that LPS triggers a complex glia‐to‐neuron signalling cascade that targets a particular class of potassium (K+) channels, namely, the neuronal KV7/M channels. These channels underlie the ubiquitous muscarine‐sensitive, low voltage‐activating, non‐inactivating K+ current (M‐current, or I M; Brown & Passmore, 2009). As I M strongly dampens intrinsic neuronal excitability (Yue & Yaari, 2004; Gu et al. 2005), its inhibition by LPS leads inevitably to a marked enhancement in excitability.
Methods
Ethical approval
All animal experiments were conducted in accordance with the guidelines of the Animal Care Committee of the Hebrew University.
Hippocampal slice preparation
Male Sprague–Dawley rats (125–150 g) were decapitated under ketamine (90 mg kg−1, i.p.) and xylazine (90 mg kg−1, i.p.) anaesthesia. Transverse hippocampal slices (300 μm) were prepared as previously described (Chen et al. 2014) using a VT1200S (Leica, Mannheim, Germany) vibrating blade microtome and transferred to a storage chamber perfused with oxygenated (95% O2–5% CO2) artificial cerebrospinal fluid (aCSF). After at least 30 min of incubation, slices were transferred to a recording bath attached to the stage of an upright fixed stage epifluorescence microscope (BX51WI, Olympus, Tokyo, Japan), with Nomarski DIC optics and an Exi Aqua monochromator camera (QImaging, Surrey, BC, Canada). Slices were transilluminated, maintained by a platinum grid and continuously perfused (2–3 ml min−1) with oxygenated aCSF (95% O2–5% CO2) at room temperature (22–24°C). The standard aCSF contained (in mm): 125 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 26 NaHCO3, 25 d‐glucose and 1 NaH2PO4.H2O. Single neurons were viewed with a ×60 LUMPlanFL objective, with 1.0 numerical aperture (Olympus). Recordings were performed under infrared DIC microscopic transillumination.
Electrophysiological recordings
Patch pipettes (4–5 MΩ) were pulled from borosilicate glass capillaries (Sutter Instrument Co., Novato, CA, USA) on a P‐1000 puller (Sutter Instrument Co.). Whole‐cell membrane currents and voltages were recorded in voltage‐clamp and fast current‐clamp modes, respectively, using a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA). Only CA1 pyramidal cells showing less than 10% change in access resistance during the entire recording period were included in the study.
Series resistance was maintained at 6–8 MΩ. For voltage‐clamp recordings, capacitive currents were minimized and series resistance was compensated by 80–90%. Command voltage protocols were generated with a Digidata 1440A A/D interface (Molecular Devices). Data were sampled at 50 kHz and low‐pass filtered at 10 kHz (–3 dB, 8 pole Bessel filter). Data were digitized using pCLAMP 10.3 (Molecular Devices). Data averaging and peak detection were performed using Clampfit 10.3 software. Data were fitted using Origin 8 (OriginLab Corp., Northampton, MA, USA).
Measurements of changes in passive and active membrane properties of pyramidal cells were made at the original (control) resting V m (membrane potential), maintained constant by injecting an appropriate steady current. Single spikes were evoked by 10 ms depolarizing current pulses of 0.01 nA increments and used for analysing action potential parameters. Spike duration was measured at half‐maximal amplitude. Spike thresholds were obtained by analysing the phase plots (dV/dt) when plotted versus time or versus membrane voltage. The voltage of the threshold was determined as the first minimal value of dV/dt after the peak. The rheobase current and apparent R in were measured using a series of 500 ms hyperpolarizing and depolarizing square current pulses of 0.01 nA increments. R in was provided by the slope of the relationship of voltage deviation versus current intensity in the linear part of the hyperpolarizing range. Spike amplitude was measured from threshold to peak voltage. The relationships between action potential frequency and injected current intensity (f–I curves) were fitted best by a linear function, y = mx, where m is a slope of the function, known as gain.
Current‐clamp recordings
In these experiments the aCSF contained (in mm): 125 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 26 NaHCO3, 25 d‐glucose and 1.25 NaH2PO4.H2O. Unless stated otherwise, the glutamate receptor antagonists 6‐cyano‐7‐nitro‐quinoxaline‐2,3‐dione (CNQX; 15 μm), 2‐amino‐5‐phosphono‐valeric acid (AP‐5; 50 μm) and kynurenic acid (2.5 mm) were added to block fast EPSPs, and the GABAA receptor antagonist picrotoxin (100 μm) was added to block fast IPSPs. Nominally Ca2+‐free aCSF was prepared by replacing CaCl2 with 2 mm MgCl2. The intracellular solution contained (in mm): 130 potassium gluconate, 6 KCl, 2 Mg‐ATP, 8 NaCl and 10 Hepes (pH 7.2, adjusted with KOH). In some experiments, when indicated, 1,2‐bis(o‐aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid (BAPTA; 13 mm) was added to this solution. Pipette potential was zeroed before seal formation and V m was corrected for a liquid junction potential of –14.6 mV. Only neurons having stable resting V m > –55 mV and overshooting spikes were used.
Voltage‐clamp recordings
In these experiments, designed to isolate I M, the aCSF contained (in mm): 123 NaCl, 3.5 KCl, 2 MgCl2, 2 CaCl2, 26 NaHCO3, 25 d‐glucose and 1.25 NaH2PO4.H2O. Tetrodotoxin (TTX; 1 μm), CdCl2 (50 μm) and 4‐ethylphenylamino‐1,2‐dimethyl‐6‐methylaminopyrimidinium chloride (ZD7288; 20 μm) were added to block voltage‐gated Na+ and Ca2+ channels, as well as Ca2+‐gated K+ channels and h‐current. In some experiments, the selective I M blocker 10,10‐bis(4‐pyridinylmethyl)‐9(10H)‐anthracenone (XE991; 20 μm) was applied in the aCSF. Intracellular solutions contained (in mm): 140 KCl, 3 Mg‐ATP, 9 NaCl, 1 MgCl2, 0.3 GTP‐Tris and 10 Hepes (pH 7.2, adjusted with KOH). In some experiments, when indicated, BAPTA (13 mm) was added to this solution. Pipette potential was zeroed before seal formation and V m was not corrected for the small liquid junction potential (–3.4 mV).
The following voltage‐command protocol was used to evoke I M (Halliwell & Adams, 1982; Caspi et al. 2009): from a holding potential of approximately –20 mV, 11 hyperpolarizing voltage steps (duration 500 ms) incrementing by –8 mV were applied. These steps induced slow current ‘relaxations’ after the instantaneous inward current drops, representing the slow I M deactivation. Current relaxations were fitted by exponential curves (starting after the capacitance artefact) and were extrapolated back to the beginning of the hyperpolarizing command pulses. I M amplitudes were assessed as the differences between the instantaneous peak currents at command onset and the steady‐state currents just before command offset.
Imaging
Two complementary techniques were used to measure [Ca2+]i changes in neurons and astrocytes. First, single cells were patched in the whole‐cell configuration and dialysed with intracellular solution containing the Ca2+ indicator Fluo‐4 pentapotassium salt (25 μm; Life Technologies, Carlsbad, CA, USA). The patch pipette was either maintained attached to the cell for parallel electrophysiological recordings or slowly withdrawn after several minutes of dialysis. Imaging was performed using wide field epifluorescence microscopy. Astrocytes were labelled by incubating slices for 1 min in aCSF containing 10 μm sulforhodamine 101 (SR‐101) at room temperature. SR‐101 was excited at 543 nm and detected using a 605 ± 75 nm filter (red channel). Fluo‐4 was excited at 488 nm and detected with a 515 ± 15 nm filter (green channel). Cells showing staining in two channels were regarded as astrocytes. Triangular cells showing staining only in the ‘green’ channel were considered as neurons. Cells that showed staining only in the ‘red’ channel were discarded from the analysis. Second, astrocytes and neurons were unselectively filled with fluorophore by preincubating them in the dark at room temperature with oxygenated aCSF containing 1 mm Fluo‐4 AM (Life Technologies) for 35 min, after which the slices were transferred to normal aCSF for at least 20 min before imaging. Imaging was performed using two‐photon excitation microscopy, to avoid collection of photons from cells below or above the focal plane. Both SR‐101 and Fluo‐4 were excited using an 800 nm laser. The SR‐101 fluorescence was separated from the Fluo‐4 fluorescence using a dichroic mirror (562 nm, Semrock Inc., Rochester, NY, USA). Fluorescence emissions were detected simultaneously by two non‐descanned photomultiplier tubes with a 542/50 nm filter (Semrock) for ‘green’ fluorescence emission and a 617/73 nm filter (Semrock) for ‘red’ fluorescence emission. The regions of interest were scanned at 1 Hz.
Wide‐field epifluorescence microscopy fluorescence was performed using an Olympus BX51WI microscope equipped with a CoolLED illuminator (CoolLED, Andover, UK) and an Exi Aqua monochromator camera (QImaging). The imaging was performed using a ×60 LUMPlanFL objective with an NA of 1 (Olympus). The data were sampled at 3 Hz.
Two‐photon excitation microscopy was performed using a LSM 7 MP (Zeiss, Oberkochen, Germany) equipped with a ×20 water‐immersion objective (NA 1.0; Zeiss). Laser excitation was performed with a Ti:Sapphire laser system (Chameleon vision II; Coherent, Ely, UK).
Fluorescence signals were quantified by measuring the mean pixel intensities of the regions of interest, using NIS elements software (Nikon) or ZEN 2009 software (Zeiss). Changes in [Ca2+]i were expressed as F/F 0 values, where F 0 was the averaged baseline fluorescence collected for at least 1 min before drug application. The value of 0.02 F/F 0 was considered as optical noise. In all Ca2+ imaging experiments, fluorescence intensity decreased with time in an exponential manner regardless of treatment (photo‐bleaching). For each experiment this decrease was fitted by a monoexponential curve, which then was subtracted from the data.
Chemicals and drugs
CNQX was purchased from Tocris Bioscience (Bristol, United Kingdom), TTX from Alomone Labs (Jerusalem, Israel), XE991 and ZD7288 from Tocris Bioscience, and Fluo‐4 AM and Fluo‐4 pentapotassium salt from Life Technologies. All other chemicals and drugs, including LPS, were purchased from Sigma‐Aldrich (St Louise, MO, USA). Stock solutions of Fluo‐4 AM were prepared in dimethylsulfoxide, and diluted 1:1000 when added to the aCSF. Also, t‐ACPD was titrated with NaOH (1 m) for solubility. All other drugs were added to the aCSF solution from aqueous stock solutions.
The effects of 2′‐deoxy‐N 6‐methyladenosine 3′,5′‐bisphosphate tetrasodium salt (MRS2179), (RS)‐α‐methyl‐4‐carboxyphenylglycine (MCPG), (2S)‐2‐amino‐2‐[(1S,2S)‐2‐carboxycycloprop‐1‐yl]‐3‐(xanth‐9‐yl) propanoic acid (LY341495), 2‐methyl‐6‐(phenylethynyl)pyridine hydrochloride (MPEP), suramin, thapsigargin, l‐α‐aminoadipic acid (l‐AAA) and minocycline were usually assessed 45 min after bath perfusion with aCSF containing the drug, as well as throughout the entire recording session. In some experiments retigabine (10 μm) was added to LPS‐treated slices shortly (∼5 min) after LPS application.
Focal drug application
In some experiments, drugs were focally applied onto somata of single CA1 pyramidal cells or of nearby astrocytes using pressure puffs supplied by a pneumatic picopump PV820 (World Precision Instruments, Sarasota, FL, USA), connected to a fine pipette (5 MΩ). The tip of the puff pipette was positioned near (∼5 μm) the cell soma. Single or multiple short pulses (2 s) were applied. The applied pressure was calibrated so that the puff pipette solution would target the cell soma by measuring the dispersion of a fluorescent solution.
Data analysis
Offline analyses were performed with pCLAMP 10.3 software (Molecular Devices). All averaged data are presented as the mean ± SEM. Assessment of statistical significance of differences between means was performed with Student's paired t test or repeated‐measures ANOVA, as appropriate.
Results
The proinflammatory agent LPS increases intrinsic neuronal excitability of CA1 pyramidal cells
To test the effects of the proinflammatory agent LPS on intrinsic properties of CA1 pyramidal cells, we perfused acute hippocampal slices with artificial cerebrospinal fluid (aCSF) containing 2.5 mm kynurenic acid, 15 μm CNQX, 50 μm AP‐5 and 100 μm picrotoxin to block fast excitatory and inhibitory synaptic transmission, and performed whole‐cell patch‐clamp recordings in current‐clamp mode. At resting potential (–58.1 ± 1.5 mV), all neurons were quiescent (n = 56). LPS (10 μg ml–1; bath‐applied 7–10 min after the onset of recording) caused within 1–5 min a 7–10 mV depolarization in all neurons (Table 1), leading to repetitive firing in 75% of the neurons (30/40; Fig. 1 A). These excitatory effects persisted throughout drug application (5–8 min) and in many cases were sustained also during its washout (Fig. 1 A; up to 40 min). Precisely similar effects of LPS on CA1 pyramidal cells’ excitability were observed when the synaptic blockers were omitted from the aCSF (n = 6; data not shown). In LPS‐untreated slices, no increase in neuronal excitability was seen during sustained recordings (up to 15 min), and the neurons retained their sensitivity to LPS throughout this period (n = 4; Fig. 1 A, inset).
Table 1.
Values and statistical analysis of differences in membrane excitability parameters before (Control) and following application of LPS, ADPβS, t‐ACPD and XE991 at the indicated conditions
| Peak amp. | Half‐width | |||||
|---|---|---|---|---|---|---|
| Treatment | Resting V m (mV) | R in (MΩ) | Threshold (mV) | m (f–I curves) | (mV) | (ms) |
| LPS, aCSF with synaptic blockers (n = 30) | ||||||
| Control (aCSF) | –60.9 ± 1. 7 | 415.6 ± 25.9 | –46.6 ± 0.9 | 0.12 ± 0.01 | 103.6 ± 2.9 | 2.2 ± 0.09 |
| LPS (10 μg ml–1) | –55.5 ± 2.7* | 484.3 ± 26* | –51.4 ± 1.3*** | 0.16 ± 0.01* | 100 ± 3 (ns) | 2.4 ± 0.1 (ns) |
| XE991 (n = 10) | ||||||
| Control (aCSF) | –53.2 ± 1.5 | 572.5 ± 61.6 | –38.6 ± 2.7 | 0.1 ± 0.02 | ||
| XE991 (20 μm) | –47.5 ± 1.8*** | 655 ± 170.2*** | –43 ± 3.4** | 0.15 ± 0.03* | ||
| LPS, aCSF with XE991 (n = 6) | ||||||
| Control (aCSF) | –49.8 ± 3.6 | 444.4 ± 80.1 | –41.3 ± 2.7 | 0.21 ± 0.01 | ||
| LPS (10 μg ml–1) | –48.9 ± 3.7 (ns) | 472.2 ± 68.1 (ns) | –39.7 ± 3.1 (ns) | 0.22 ± 0.01 (ns) | ||
| LPS, aCSF with thapsigargin (n = 6) | ||||||
| Control (aCSF) | –58.3 ± 1.2 | 410 ± 35.6 | –44.3 ± 2.5 | 0.12 ± 0.02 | ||
| LPS (10 μg ml–1) | –57 ± 1.1 (ns) | 438.2 ± 63.8 (ns) | –42.4 ± 2.3 (ns) | 0.12 ± 0.03 (ns) | ||
| LPS, BAPTA in intracellular solution (n = 6) | ||||||
| Control (aCSF) | –57.2 ± 1.1 | 371.1 ± 31.6 | –42.2 ± 1 | 0.1 ± 0.02 | ||
| LPS (10 μg ml–1) | –57.3 ± 1.2 (ns) | 339.1 ± 27.4 (ns) | –41.8 ± 1 (ns) | 0.1 ± 0.02 (ns) | ||
| LPS, aCSF with l‐AAA (n = 6) | ||||||
| Control (aCSF) | –60.0 ± 1.1 | 415.7 ± 46.16 | –45.6 ± 1.4 | 0.11 ± 0.02 | ||
| LPS (10 μg ml–1) | –58.7 ± 1.1 (ns) | 409.4 ± 53.9 (ns) | –45 ± 1.5 (ns) | 0.12 ± 0.02 (ns) | ||
| LPS, aCSF with MRS2179 (n = 5) | ||||||
| Control (aCSF) | –57.8 ± 3.7 | 311.2 ± 33.9 | –42.2 ± 3.6 | 0.11 ± 0.02 | ||
| LPS (10 μg ml–1) | –55.8 ± 32.1 (ns) | 302 ± 19.1 (ns) | –41.4 ± 3.4 (ns) | 0.13 ± 0.01 (ns) | ||
| LPS, aCSF with MCPG (n = 5) | ||||||
| Control (aCSF) | –58.696 ± 1.8 | 451.666 ± 41.8 | –45.663 ± 2.5 | 0.11 ± 0.008 | ||
| LPS (10 μg ml–1) | –55.829 ± 1.8 (ns) | 504.666 ± 39.9 (ns) | –45.312 ± 2.5 (ns) | 0.13 ± 0.007 (ns) | ||
| Puff of ADPβS on astrocytes (n = 8) | ||||||
| Control (aCSF) | –56.6 ± 1.2 | 375.3 ± 22.5 | –44.7 ± 2.1 | 0.12 ± 0.01 | ||
| ADPβS (100 μm) | –50.4 ± 2.1*** | 515.5 ± 20.2* | –52.8 ± 0.7*** | 0.15 ± 0.01* | ||
| t‐ACPD, aCSF with l‐AAA and minocycline (n = 6) | ||||||
| Control (aCSF) | –57.2 ± 0.7 | 384.5 ± 11.3 | –32.9 ± 3.1 | 0. 12 ± 0.02 | ||
| t‐ACPD (50 μm) | –47.7 ± 2.9** | 488.2 ± 41.7* | –41.5 ± 2.7*** | 0.16 ± 0.02* | ||
ns, not significant, * P < 0.05, ** P < 0.01, *** P < 0.001.
Figure 1. LPS increases intrinsic excitability of CA1 pyramidal cells.
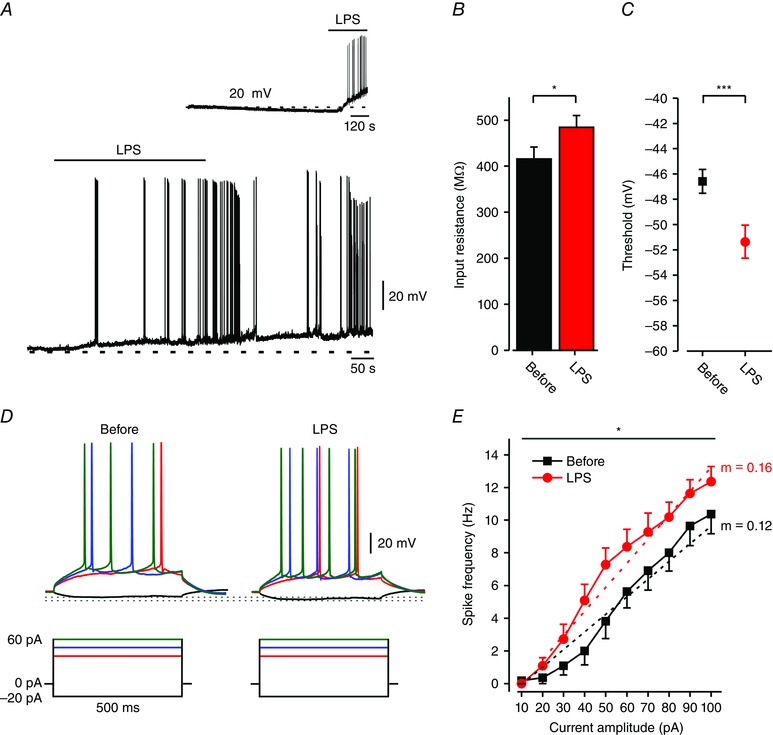
A, typical responses of CA1 pyramidal cells to bath application (marked by horizontal bar) of 10 μg ml–1 LPS (representative of 30/40 experiments). Current‐clamp patch clamp recordings performed in slices perfused with aCSF containing blockers of fast synaptic transmission. Note, membrane depolarization and onset of spike discharges shortly after application of LPS. Dashed lines indicate resting potentials before drug application (–63 mV). Inset: control experiment. A 15 min long current‐clamp recording from a CA1 pyramidal cell in a slice perfused with aCSF. Note, no spontaneous depolarization developed during this period. Application of LPS produced robust depolarization and spike discharges (representative of 4/4 neurons). B, bar graph depicting R in (see Methods) of all responding neurons before (black) and after 5 min exposure to LPS (red); * P < 0.05, paired t test, n = 16. C, spike thresholds (assessed from phase plot analyses of single spike; see Methods) before (black square) and after 5 min exposure to LPS (red circle); *** P < 0.001, paired t test, n = 16. D, typical voltage responses (top) to current steps (bottom) in a pyramidal cell recorded before and after 5 min exposure to LPS (representative of 13/16 neurons). Dotted lines indicate peak voltage deflections in the two conditions. E, mean f–I curves of 13 pyramidal cells recorded before (black squares) and after 5 min exposure to LPS (red circles). Note, LPS induced a significant increase in spike frequency (* P < 0.05, two‐way ANOVA) and gain (m, dotted lines; *** P < 0.001, paired t test; see also Table 1). All measurements described in panels D–G were performed at the native resting potential, adjusted after LPS application by injecting appropriate repolarizing currents. [Colour figure can be viewed at wileyonlinelibrary.com]
We also measured passive and active membrane properties of pyramidal cells before and during application of LPS. Membrane potential (V m) was kept constant by applying steady negative current to counteract its depolarizing action. LPS significantly increased input resistance (R in; Fig. 1 B) and decreased spike threshold (Fig. 1 C) without modifying spike amplitude and width (Table 1). LPS also increased the frequency of spikes evoked by depolarizing current steps (Fig. 1 D), causing a leftward shift in the spike frequency versus current intensity relationship (frequency–current intensity (f–I) curve), and a significant increase in gain (i.e. slope of the f–I curve; Fig. 1 E).
LPS inhibits I M in pyramidal cells
The excitatory effects of LPS in CA1 pyramidal cells, namely, the mild depolarization, the increase in R in, and the increase in spike threshold without concomitant changes in spike amplitude and width (Table 1), are reminiscent of the effects of pharmacologically blocking I M (Table 1; Yue & Yaari, 2004; Gu et al. 2005). Therefore, we tested whether the LPS‐induced increase in neuronal excitability is due to I M inhibition. We used a well‐established voltage‐clamp protocol to isolate I M from other currents (see Methods; Fig. 2 A, left panel). Bath‐applied LPS completely (5/7) or almost completely (2/7) inhibited I M within a few minutes (Fig. 2 A (middle panel), B and C). In the latter two neurons, residual I M was fully abolished by subsequent application of 20 μm 10,10‐bis(4‐pyridinylmethyl)‐9(10H)‐anthracenone (XE991), a selective I M blocker (Wang et al. 1998).
Figure 2. LPS inhibits I M .
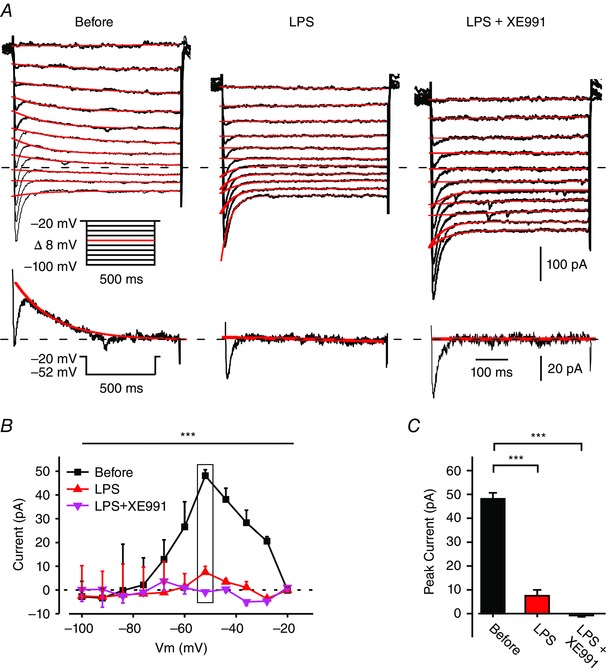
A, voltage‐clamp recordings from a pyramidal cell in a slice perfused with modified aCSF designed to isolate I M (see Methods). Each of the three subpanels illustrates a family of currents evoked by a series of 500 ms, 8 mV hyperpolarizing voltage steps from a holding potential of –20 mV (voltage protocol is shown in inset). The current response obtained by stepping to –52 mV is shown at the bottom of each subpanel. The I M relaxation was fitted with a monoexponential line (red), which was extrapolated to the beginning of the voltage step. I M amplitudes were measured as the differences between the instantaneous peak currents at command onset and the steady‐state currents just before command offset. The dotted line indicates zero current level. The three subpanels depict the current responses before (left), 5 min after bath application of 10 μg ml–1 LPS (middle), and 5 min after additional bath application of 20 μm XE991 (right). Note I M inhibition by LPS (representative of 5/7 experiments). B, plots of I M amplitudes versus hyperpolarizing command potentials in the three conditions shown in A: before (black squares), after 5 min of exposure to LPS (red triangles), and after 5 min of exposure to XE991 (pink inverted triangles). The rectangle outlines peak I M values. The first (Before) and second (LPS, 5 min) plots differ significantly (*** P < 0.001, two‐way ANOVA, n = 7). C, bar graph comparing peak I M amplitudes measured at –52 mV (shown in A, inset) in the three conditions as in B (*** P < 0.001, Student's t test, n = 7). [Colour figure can be viewed at wileyonlinelibrary.com]
In slices pretreated with XE991 to completely block I M, LPS did not further affect membrane properties and excitability of the pyramidal cells (Table 1). This XE991‐mediated occlusion of LPS action indicates that I M inhibition is the main, if not the sole, mechanism of LPS‐induced increase in intrinsic neuronal excitability.
LPS‐induced I M inhibition and increased excitability are mediated by an increase in [Ca2+]i in the pyramidal cells
It is well established that I M is inhibited by multiple transmitters released from neurons and glia (Brown & Passmore, 2009). In some cases this inhibition was shown to be mediated by an increase in [Ca2+]i (Cruzblanca et al. 1998; Gamper & Shapiro, 2003; Caspi et al. 2009). Indeed, KV7/M channels are dose‐dependently blocked by Ca2+ applied to their cytoplasmic surface (Selyanko & Brown, 1996; Chen & Yaari, 2008). We therefore tested whether LPS‐induced I M inhibition and the associated increase in pyramidal cell excitability are caused by an increase in [Ca2+]i. To that end, we combined patch‐clamp recordings with Ca2+ imaging in single neurons filled with the membrane non‐permeant dye Fluo‐4 via the recording pipette. In 80% of the neurons (8/10), bath‐applied LPS induced a substantial, albeit transient, increase in global [Ca2+]i, preceding the neuronal depolarization by 30–60 s (Fig. 3 A). This early increase in [Ca2+]i was followed by additional rapid intracellular Ca2+ transients synchronous with spike discharges (Fig. 3 A). In search of the source of the elevated [Ca2+]i, we found that LPS induces an early rise in [Ca2+]i, as well as an increase in excitability, also in slices bathed in nominally Ca2+‐free aCSF, though spike‐associated Ca2+ transients were entirely absent in this condition (Fig. 3 B). In contrast, in slices preincubated in aCSF containing 1 μm thapsigargin to deplete endoplasmic reticular Ca2+ stores (Thastrup et al. 1990), LPS failed to affect neuronal [Ca2+]i (Fig. 3 C). In the latter condition the neurons displayed normal firing behaviour, which also was unaffected by LPS (Fig. 3 C (inset) and D and Table 1).
Figure 3. LPS‐induced release of Ca2+ from internal stores in pyramidal cells is essential for its excitatory action.
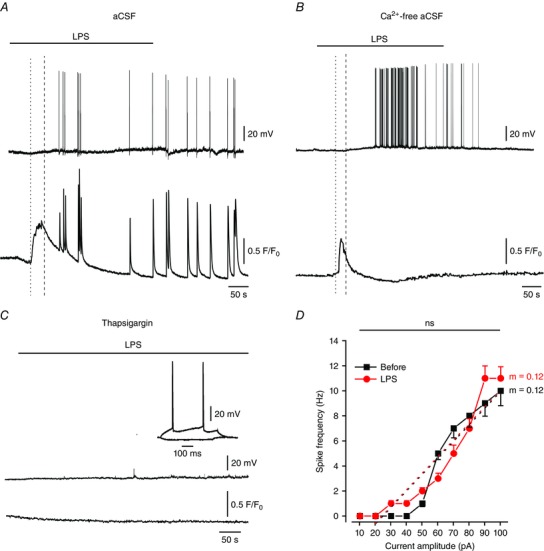
A, LPS induces a two‐phase increase in [Ca2+]i in pyramidal cells. Simultaneous current‐clamp (upper trace) and [Ca2+]i (lower trace) recordings from single pyramidal cells filled with 1μm Fluo‐4 during bath application of 10 μg ml–1 LPS (representative of 8/10 experiments). Note the early increase in [Ca2+]i (onset marked with the dotted line), preceding the cellular depolarization (onset marked with the dashed line) and lasting a few minutes, followed by repetitive transient [Ca2+]i increases coincident with spikes. B, same as in A, but the slices were perfused with nominally Ca2+‐free aCSF. Note the early LPS‐induced increase in [Ca2+]i, but the absence of spike‐associated transient [Ca2+]i increases (representative of 5/7 experiments). C, same as in A, but the slices were pretreated with 1 μm thapsigargin for 45 min prior to LPS application. Note the lack of any cellular response to LPS, though the cell fired normally upon current injection (inset; representative of 5/7 experiments). D, mean f–I curves of 6 pyramidal cells obtained before (black squares) and after 5 min of exposure to LPS (red circles), recorded in slices treated with thapsigargin; ns, not significant, paired t test for gain and two‐way ANOVA for spike frequency. [Colour figure can be viewed at wileyonlinelibrary.com]
To further test for a causal relationship between the LPS‐induced increases in [Ca2+]i and in excitability, we filled neurons with the fast Ca2+ chelator BAPTA. In this condition LPS application did not alter neuronal excitability (Fig. 4 A and B and Table 1), suggesting that the neuronal [Ca2+]i increase is obligatory for the excitability increase.
Figure 4. LPS inhibits I M in an intracellular Ca2+‐dependent manner.
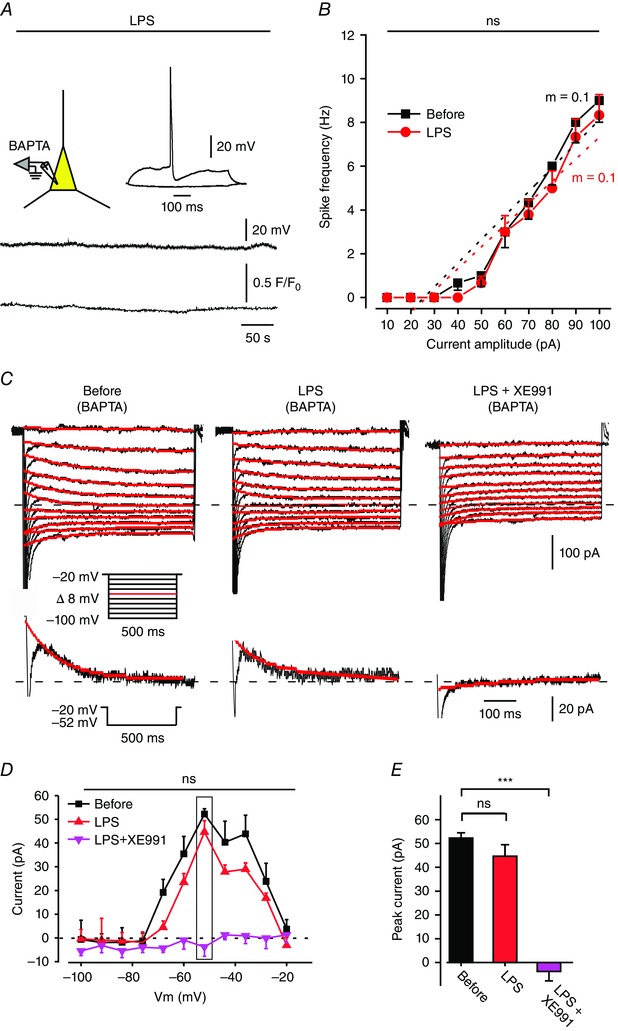
A, simultaneous current‐clamp (upper trace) and [Ca2+]i (lower trace) recordings from single pyramidal cells filled with 1 μm Fluo‐4 and 13 mm BAPTA during bath application of 10 μg ml–1 LPS. Note that BAPTA prevented the LPS‐induced spike discharge, yet the cell fired normally upon injection of depolarizing current pulses (inset; representative of 6/7 experiments). B, mean f–I curves of 6 pyramidal cells filled with BAPTA, obtained before (black squares) and after 5 min of exposure to LPS (red circles). ns, not significant; paired t test for gain and two‐way ANOVA for spike frequency. Note, that BAPTA prevented LPS‐induced increase in gain. C, voltage‐clamp recordings of I M (see Methods) from a pyramidal cell filled with 13 mm BAPTA. Each of the three subpanels illustrates a family of currents evoked by a series of 500 ms, 8 mV hyperpolarizing voltage steps from a holding potential of –20 mV (voltage protocol is shown in inset). The dotted line indicates zero current level. The three subpanels depict the current responses before (left), 5 min after bath application of 10 μg ml–1 LPS (middle), and 5 min after additional bath application of 20 μm XE991 (right). Note no I M reduction in LPS but complete inhibition in XE991 (representative of 6/6 experiments). D, plots of I M amplitudes versus hyperpolarizing command potentials in the three conditions shown in A: before (black squares), after 5 min of exposure to LPS (red triangles), and after 5 min of exposure to XE991 (pink inverted triangles). The rectangle outlines peak I M values. The first (Before) and second (LPS, 5 min) plots did not differ significantly (ns, not significant (** P > 0.05), two‐way ANOVA, n = 6). E, bar graph comparing peak I M amplitudes measured at –52 mV (shown in A, inset) in the three conditions as in B. Note lack of significant I M inhibition by LPS (ns, not significant (** P > 0.05), Student's t test, n = 6) and complete inhibition by XE991 (*** P < 0.001, Student's t test, n = 6). [Colour figure can be viewed at wileyonlinelibrary.com]
Finally, we examined the effects of LPS on I M in BAPTA‐filled neurons. Bath‐applied LPS consistently (6/6) failed to modify I M in this condition (Fig. 4 C (middle panel), D and E), whereas subsequent XE991 application fully inhibited I M (Fig. 4 C (right panel), D and E).
Altogether, these data indicate that in the pyramidal cells, LPS‐induced Ca2+ release from internal stores mediates I M inhibition, which, in turn, enhances pyramidal cell excitability.
LPS effects on pyramidal cells are mediated by astrocytes
It is widely accepted that LPS exerts its effects predominately by activating TLR‐4 receptors in microglial cells (Poltorak et al. 1998; Chow et al. 1999; Lu et al. 2008). These, in turn, activate astrocytes via the release of gliotransmitters, such as ATP (Imura et al. 2013). Activated astrocytes can then further convey the LPS signal to neurons by releasing their own gliotransmitters, such as ATP or glutamate (Parpura & Zorec, 2010; Gundersen et al. 2015). To characterize the involvement of astrocytes and their gliotransmitters in the LPS‐induced increase in neuronal excitability, we first examined the effects of LPS on astrocytic [Ca2+]i in slices labelled with SR‐101 and with Fluo‐4 AM using two‐photon microscopy (see Methods). Bath‐applied LPS induced a substantial [Ca2+]i increase in 14/19 astrocytes, concomitant with the neuronal [Ca2+]i increase (13/16; Fig. 5 A). The increase in astrocytic [Ca2+]i was due to Ca2+ release from intracellular stores, as it occurred also in slices bathed in nominally Ca2+‐free aCSF (Fig. 5 B), but not in slices pretreated with thapsigargin (Fig. 5 C).
Figure 5. Astrocytes mediate neuronal effects of LPS.
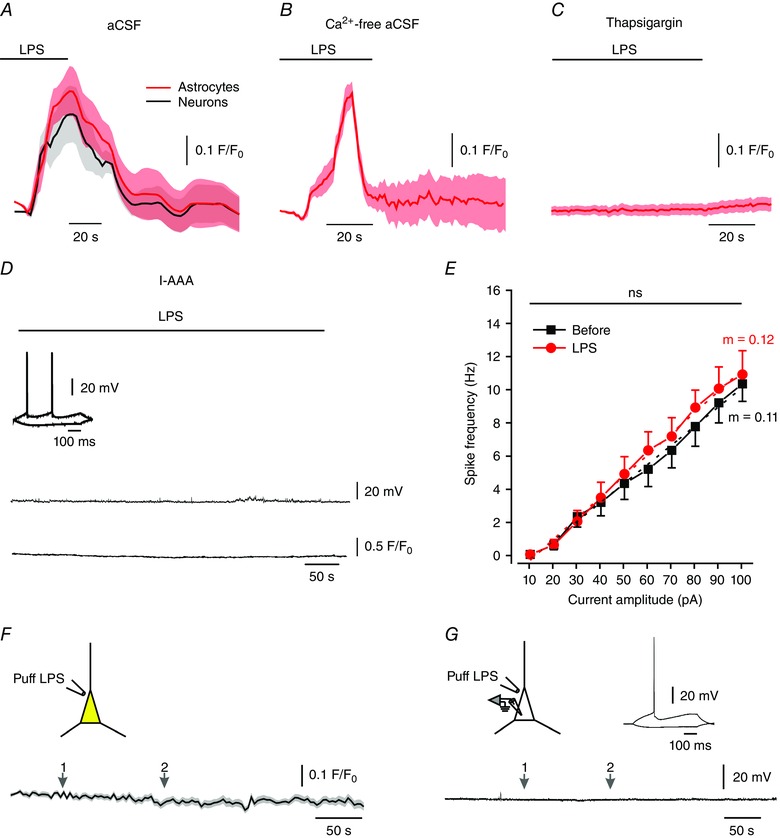
A, LPS (bath‐applied; 10 μg ml–1) causes a marked [Ca2+]i increase in astrocytes and in pyramidal cells. The slices were preloaded with SR‐101 and Fluo‐4 AM. Shown are averaged responses of 14 astrocytes (red circles) and 13 neurons (black circles) in 7 slices. B, LPS (bath‐applied; 10 μg ml–1) caused a marked [Ca2+]i increase in astrocytes in slices perfused with nominally Ca2+‐free aCSF. Averaged responses of 22 astrocytes in 7 slices. C, LPS (bath‐applied; 10 μg ml–1) had no effect on [Ca2+]i in astrocytes in slices pretreated with 1 μm thapsigargin for 45 min prior to LPS application. Averaged responses of 18 astrocytes in 7 slices. D, bath‐applied LPS (10 μg ml–1) has no effect on pyramidal cell excitability and [Ca2+]i in a slice pretreated with the gliotoxin l‐AAA (1 mm). Current‐clamp recordings in pyramidal cells filled with 1μm Fluo‐4. Note that the neuron fired normally upon current injection (inset). Representative of 6/6 experiments. E, mean f–I curves of 6 pyramidal cells obtained before (black squares) and after 5 min of exposure to LPS (red circles) recorded from slices treated with l‐AAA. ns, not significant, paired t test for gain and two‐way ANOVA for spike frequency. F, effects of LPS focally applied by a puff pipette onto a pyramidal cell on neuronal [Ca2+]i in slices preloaded with SR‐101 and Fluo‐4 AM. Puffing LPS (10 μg ml–1) twice (arrows 1, 2) did not affect [Ca2+]i in the neuron. Shown are averaged responses of 24 neurons. G, in current‐clamp recordings, puffing LPS on the pyramidal cell twice (arrows 1, 2) did not affect neuronal excitability. The neuron displayed normal excitability when tested with current pulses (inset). Representative of 7/7 experiments. [Colour figure can be viewed at wileyonlinelibrary.com]
To examine whether astrocytic activation by LPS is required for inducing its neuronal effects, we pretreated slices with the gliotoxin l‐α‐aminoadipic acid (l‐AAA) to inhibit astrocytic functions (Huck et al. 1984; McBean, 1994; Brown & Kretzschmar, 1998). In this condition, bath‐applied LPS exerted no neuronal effects (n = 6, Fig. 5 D–E and Table 1). It is noteworthy that l‐AAA pretreatment by itself neither enhanced nor compromised pyramidal cell excitability (Fig. 5 D, inset). Likewise, it did not abolish its ability to release Ca2+ from intracellular stores, as indicated by marked [Ca2+]i increases in response to bath‐applied t‐ACPD, a metabotropic glutamate receptor (mGluR) agonist (see below, Fig. 9 A), or to 40 mm caffeine (Garaschuk et al. 1997; data not shown). Additionally, in a separate set of experiments we found that acutely applied l‐AAA for up to 1 h affected neither CA1 pyramidal cells’ passive membrane properties nor their excitability (data not shown).
Figure 9. Activation of neuronal group I mGluRs mimics the neuronal effects of LPS.
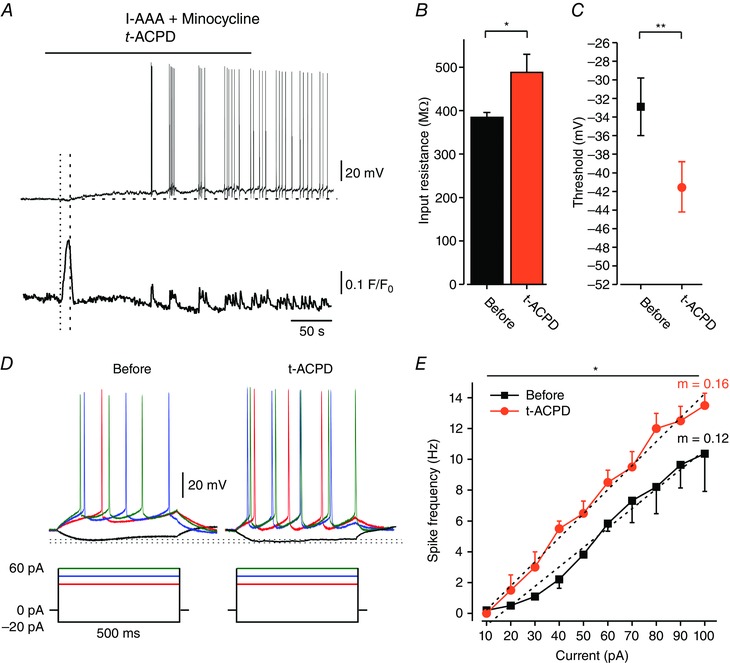
All slices were pretreated with 1 mm l‐AAA and 50 nm minocycline to induce glia dysfunction. A, effects of t‐ACPD (50 μm; bath‐applied) on a Fluo‐4‐filled pyramidal cell in simultaneous current‐clamp (upper trace) and [Ca2+]i (lower trace) recordings. The horizontal dashed line indicates the resting potential of the neuron before drug application. Note the early increase in [Ca2+]i (onset marked with the dotted line), preceding the cellular depolarization (onset marked with the dashed line), followed by repetitive transient [Ca2+]i increases coincident with spikes (representative of 5/5 experiments). B, bar graph depicting R in (see Methods) of all responding neurons before (black) and after 5 min exposure to t‐ACPD (orange); * P < 0.05, paired t test, n = 6. C, spike thresholds (assessed from phase plot analyses of single spike; see Methods) before (black square) and after 5 min of exposure to t‐ACPD (orange circle); ** P < 0.01, paired t test, n = 6. D, typical voltage responses to current steps (shown below) in a pyramidal cell recorded before and after 5 min exposure to t‐ACPD (representative of 6/6 neurons). Dotted lines indicate peak voltage deflections in the two conditions. E, mean f–I curves of 6 pyramidal cells recorded before (black squares) and after 5 min exposure to t‐ACPD (orange circles). Note, t‐ACPD induced a significant increase in spike frequency (* P < 0.05, two‐way ANOVA) and gain (m, dotted lines; * P < 0.05, paired t test; see also Table 1). [Colour figure can be viewed at wileyonlinelibrary.com]
Together, these findings indicate that astrocytic activation is a critical step in LPS‐induced neuronal excitation. They also suggest that LPS does not excite the pyramidal cells directly, i.e. by acting on neuronal TLR‐4 receptors. However, it has been shown that some neurons express TLR‐4 receptors and respond to LPS (Leow‐Dyke et al. 2012; Zhao et al. 2014). We could not support this notion in CA1 pyramidal cells, because direct puff application of LPS onto these neurons in Fluo‐4 AM preloaded slices affected neither [Ca2+]i (n = 24; Fig. 5 F) nor excitability (n = 7; Fig. 5 G).
LPS‐induced neuronal effects require activation of astrocytic P2Y1 receptors
It is well established that microglial cells, astrocytes and neurons can release ATP and respond to it via its multiple purinergic type 2 (P2) receptors (Butt, 2011). Therefore, we explored whether P2 receptors play a role in the neuronal effects of LPS. In slices treated with the broad‐spectrum P2 receptor blocker suramin (Dunn & Blakeley, 1988; Lambrecht et al. 2002), bath‐applied LPS did not produce any change in [Ca2+]i in astrocytes (n = 33; Fig. 6 A, red), as well as in pyramidal cells (n = 28; Fig. 6 A, black). Similar results were obtained in slices treated with 2′‐deoxy‐N 6‐methyladenosine 3′,5′‐bisphosphate tetrasodium salt (MRS2179, Fig. 6 B; astrocytes in red, n = 20; neurons in black, n = 28), a selective P2Y1 receptor blocker (Boyer et al. 1998; Weisman et al. 2012). Likewise, treatment with MRS2179 prevented LPS‐induced increase in neuronal excitability (Fig. 6 C; n = 8/8). Congruently, we found that bath application of the selective P2Y1 receptor agonist adenosine 5′‐O‐2‐thiodiphosphate (ADPβS; von Kugelgen, 2006) caused a substantial [Ca2+]i increase in all astrocytes (n = 13) and neurons (n = 17; Fig. 6 D). This effect was observed also in slices bathed in nominally Ca2+‐free aCSF, but not in slices preincubated with thapsigargin, indicating that ADPβS causes Ca2+ release from intracellular stores (36, data not shown). ADPβS also produced a prominent depolarization and firing in most (6/8) pyramidal cells (Fig. 6 E), strongly resembling the effects of LPS (Table 1). However, ADPβS did not exert any neuronal effects in slices pretreated with l‐AAA to cause astrocytic dysfunction (n = 38 neurons; Fig. 6 F).
Figure 6. LPS‐induced neuronal effects critically depend on activation of astrocytic P2Y1 receptors.
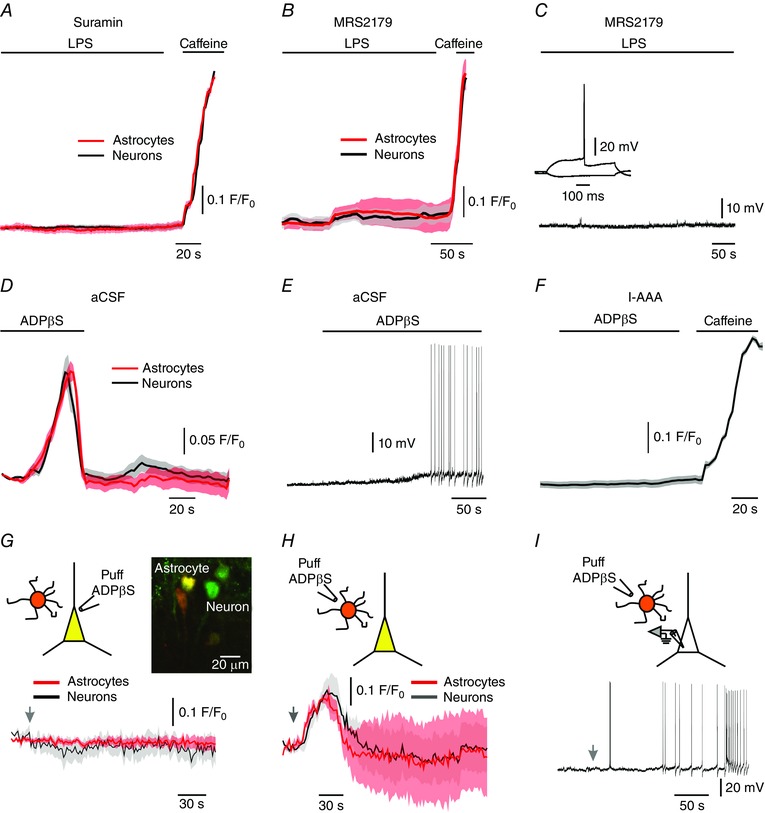
A, LPS (bath‐applied; 10 μg ml–1) had no effect on [Ca2+]i in both astrocytes (red circles) and pyramidal cells (black circles) in slices pretreated with the P2 receptor blocker suramin (100 μm). Note substantial [Ca2+]i increases evoked in both cell types by caffeine (bath applied; 40 mm) in the presence of suramin. Shown are averaged responses of 33 astrocytes and 28 neurons in 5 slices preloaded with SR‐101 and Fluo‐4 AM. B, same as in A, but for slices treated with the P2Y1 receptor antagonist MRS2179 (30 μm). Averaged responses of 20 astrocytes and 28 neurons in 15 slices. Note substantial [Ca2+]i increases evoked in both cell types by caffeine (bath applied; 40 mm) in the presence of MRS2179. C, in slices treated with MRS2179, LPS (bath‐applied; 10 μg ml–1) had no effect on pyramidal cells’ excitability (current‐clamp recordings; representative of 8/8 neurons). D, the P2Y1 receptor agonist ADPβS (bath‐applied; 100 μm) induced [Ca2+]i increases in both astrocytes (red circles) and pyramidal cells (black circles). Averaged responses of 13 astrocytes and 17 neurons in 15 slices preloaded with SR‐101 and Fluo‐4 AM. E, ADPβS (bath‐applied; 100 μm) caused neuronal depolarization and firing (current clamp recordings; representative, 6/8 neurons). F, in slices pretreated with the gliotoxin l‐AAA (1 mm) ADPβS (bath‐applied; 100 μm) had no effect on [Ca2+]i in pyramidal cells. Averaged responses of 38 neurons from 11 slices preloaded with Fluo‐4 AM. Note substantial [Ca2+]i increases evoked by caffeine (bath applied; 40 mm). G, effects of focally applied ADPβS on [Ca2+]i in neighbouring pyramidal cell (black trace) and astrocyte (red trace). The slices were preloaded with SR‐101 and Fluo‐4 AM. Inset: example photomicrograph showing the spatial relationship between a pyramidal cell labelled with Fluo‐4 AM (green) and a nearby astrocyte labelled with SR‐101 and Fluo‐4 AM (yellow). ADPβS was puffed onto the pyramidal cell from a puff pipette containing 250 μm ADPβS at the time indicated by an arrow. Note lack of any [Ca2+]i responses to ADPβS in both pyramidal cell and astrocyte. Shown are averaged responses of 7 astrocytes (red) and 14 neurons (black) in 7 slices. H, same as in G, but ADPβS was puffed onto the astrocyte. Note, [Ca2+]i increases in both astrocytes and neurons in response to ADPβS. Averaged responses of 6 astrocytes and 10 neurons in 7 slices. I, effects of ADPβS puffed (at the time marked by an arrow) onto an astrocyte on the excitability of a nearby pyramidal cell (current‐clamp recordings; representative of 8/11 experiments). [Colour figure can be viewed at wileyonlinelibrary.com]
These data indicate that P2Y1 receptor activation is required for the LPS‐induced astrocytic [Ca2+]i response, which then leads to increased neuronal [Ca2+]i and excitability. They also imply that unlike astrocytes, pyramidal cells do not express functional P2Y1 receptors coupled to intracellular Ca2+ stores. To examine the latter notion more directly, we puffed ADPβS focally onto pyramidal cells or nearby astrocytes (Fig. 6 G, inset, see Methods). Puffing ADPβS onto single neurons did not produce any detectable effects in these neurons (n = 14) or in nearby astrocytes (within a perimeter of 20 μm; n = 7; Fig. 6 G). In contrast, puffing ADPβS onto single astrocytes produced a substantial [Ca2+]i increase in astrocytes (n = 6) and in nearby neurons (n = 10, Fig. 6 H), as well as neuronal depolarization and firing (8/11; Fig. 6 I). Altogether, these results indicate that activation of astrocytic P2Y1 receptors is essential for the downstream increases in neuronal [Ca2+]i and excitability induced by LPS.
LPS‐induced neuronal effects require activation of neuronal group 1 mGluRs
Activation of astrocytic P2Y1 receptors in response to LPS is likely to be mediated by ATP released by microglial cells and/or by the astrocytes themselves (Davalos et al. 2005; Pascual et al. 2012). The question arises whether ATP released by astrocytes also mediates the LPS‐induced neuronal effects. Although our data suggest that pyramidal cells do not express functional P2Y1 receptors, they might still be activated by ATP via other P2 receptors (Koles et al. 2011), such as the Ca2+‐permeable P2X4 receptors (Rubio & Soto, 2001; Sim et al. 2006). We tested this notion by continuously (at 0.5 Hz) puffing suramin onto a single pyramidal cell and simultaneously onto a single astrocyte (via two separate puff pipettes; see scheme in Fig. 7 A) in order to block P2 receptors mostly in these two cells. In this situation, bath‐applied LPS still induced the archetypal increases in [Ca2+]i and excitability in the pyramidal cell (Fig. 7 B (neuron) and C–E), but failed to affect [Ca2+]i in the corresponding astrocyte (Fig. 7 B, astrocyte 1). Yet nearby astrocytes, not exposed to suramin, responded to LPS with a distinctive [Ca2+]i increase (Fig. 7 B, astrocytes 2–4). These results were replicated in 5/7 similar experiments. It is noteworthy that after applying LPS, [Ca2+]i increased simultaneously in the responding three astrocytes, whereas the onset of the [Ca2+]i response in the pyramidal cell was delayed by an additional 2 s (1.9 ± 1.2; n = 5 neurons and 10 astrocytes).
Figure 7. LPS‐induced neuronal excitation is not mediated by neuronal P2 receptors.
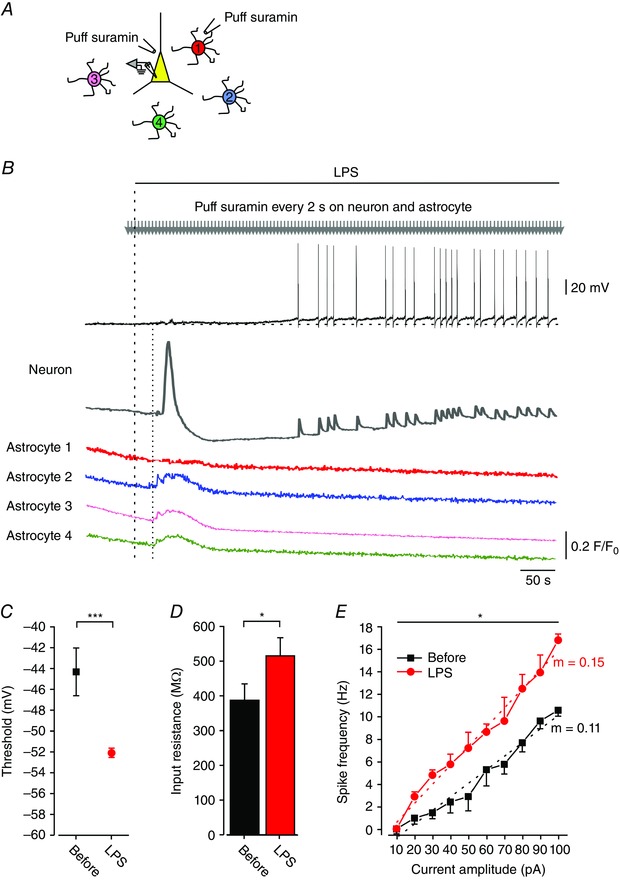
A, scheme of the experimental arrangement. The slice was preloaded with SR‐101 and four astrocytes (1–4), adjacent to a selected pyramidal cell, were individually filled with Fluo‐4 (via a patch‐pipette) for Ca2+ imaging. The pyramidal cell was similarly filled and the patch pipette was maintained for simultaneous Ca2+ imaging and current‐clamp recording. Two puff pipettes filled with suramin (250 μm) were positioned, one on the pyramidal cell and one on astrocyte 1. Suramin was then focally applied onto the two cells at 0.5 Hz. B, LPS (10 μg ml–1; bath‐applied at the time indicated by the dashed line) induced neuronal excitation (Neuron, black trace) preceded by a [Ca2+]i increase (Neuron, dark grey trace), but did not induce any [Ca2+]i response in astrocyte 1 (red trace). All other astrocytes (astrocyte 2, blue trace; astrocyte 3, orange trace; and astrocyte 4, green trace) responded to LPS with a simultaneous [Ca2+]i rise, whose onset (dotted line) preceded the neuronal [Ca2+]i response by ∼3 s (dotted line). Representative of 5/7 experiments. C–E, recordings were performed in CA1 pyramidal cells before and after bath application of LPS (10 μg ml−1) and during puff application of suramin (250 μm) onto a recorded neuron and a nearby astrocyte. C, spike thresholds measured before (black square) and after 5 min of exposure to LPS (red circle); *** P < 0.001, paired t test, n = 7. D, bar graph depicting R in measured before (black) and after 5 min of exposure to LPS (red); * P < 0.05, paired t test, n = 7. E, mean f–I curves of 7 pyramidal cells obtained before (black squares) and after 5 min of exposure to LPS (red circles). For spike frequency: * P < 0.05, two‐way ANOVA; and for gain (m, dotted lines): ** P < 0.01, paired t test. All measurements were performed at the native resting potential, adjusted by injecting appropriate repolarizing currents to counteract the ∼10 mV depolarization induced by ADPβS (resting V m,before = –57.3 ± 1.9 mV; resting V m,LPS = –47.5 ± 2.2 mV; *** P < 0.001, paired t test, n = 7). [Colour figure can be viewed at wileyonlinelibrary.com]
Together, these results indicate that ATP is not the gliotransmitter directly exciting the pyramidal cells within the inflammatory cascade triggered by LPS. An alternative candidate is glutamate, known to be released from active astrocytes (Parpura et al. 1994; Araque et al. 1998; Pasti et al. 2001; Fiacco & McCarthy, 2004), also in response to LPS (Pascual et al. 2012), and to affect nearby neurons. In our conditions of blocked ionotropic glutamate receptors, astrocytic glutamate could excite pyramidal cells via metabotropic glutamate receptors (mGluRs) (Perea & Araque, 2007; Pascual et al. 2012). The most abundant postsynaptic mGluRs in CA1 pyramidal cells are group I mGluRs (Fotuhi et al. 1994). Activating these receptors stimulates PLC and the subsequent IP3 formation leads to Ca2+ release from intracellular stores (Abe et al. 1992; Aramori & Nakanishi, 1992). This effect is associated with pyramidal cell depolarization, increase in R in and enhanced firing (Gereau & Conn, 1995), resembling LPS effects.
We first tested this notion using (RS)‐α‐methyl‐4‐carboxyphenylglycine (MCPG), a group I and II mGluR antagonist (Hayashi et al. 1994; Davies et al. 1995). Application of LPS to slices treated with 2 mm MCPG caused a prominent increase in astrocytic [Ca2+]i (9/9 astrocytes) without any change in neuronal [Ca2+]i (17/17 pyramidal cells; Fig. 8 A), indicating that transmission from astrocytes to pyramidal cells was blocked. To further differentiate between the two groups of mGluRs, we applied LPS to slices treated with 100 μm LY341495, a selective group II mGluR antagonist (Fitzjohn et al. 1998; Kingston et al. 1998). In this condition 16/20 astrocytes and 21/26 pyramidal cells responded to LPS application with a [Ca2+]i increase (Fig. 8 B). Similar results were obtained in five slices treated with 200 μm LY341495 (18/22 astrocytes and 26/31 neurons, data not shown).
Figure 8. LPS‐induced neuronal effects depend on activation of neuronal group I mGluRs.
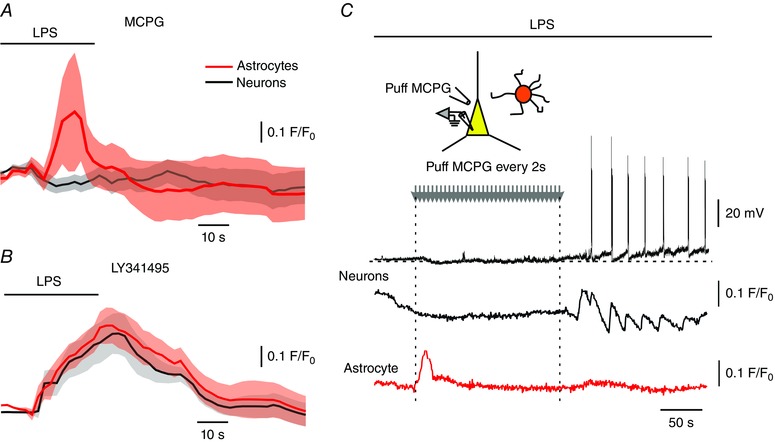
A, LPS (bath‐applied; 10 μg ml–1) induced a [Ca2+]i rise in astrocytes (red circles) but not in pyramidal cells (black circles) in slices treated with the group I/II mGluR antagonist MCPG (2 mm). Shown are averaged responses of 9 astrocytes and 17 neurons in 13 slices preloaded with SR‐101 and Fluo‐4 AM. B, in slices treated with the group II mGluR selective antagonist LY341495 (100 μm), bath‐applied LPS (10 μg ml–1) led to increase in [Ca2+]i in astrocytes (red) and pyramidal cells (black). Shown are averaged responses of 16 astrocytes and 21 neurons in 4 slices. C, group I/II mGluRs were blocked in single pyramidal cells. An astrocyte and a nearby pyramidal cell were filled with a Fluo‐4 via the patch pipette, which was also used for current‐clamp recording from the neuron. A puff pipette filled with MCPG (5 mm) was positioned on the neuron and MCPG was focally applied at 0.5 Hz at the indicated time window (grey arrows; starting 60 s before LPS wash‐in). Bath‐applied LPS induced a [Ca2+]i in the astrocyte (lower red trace), but did not affect the pyramidal cell as long as MCPG was applied (black and grey traces). Representative of 5/5 experiments. [Colour figure can be viewed at wileyonlinelibrary.com]
The latter results suggest that neurons are excited by glutamate released from activated astrocytes and acting at neuronal group I mGluRs (Pascual et al. 2012). However, they do not refute the possibility that astrocytic glutamate activates autocrine group I mGluRs (Shelton & McCarthy, 1999), causing the release of yet another gliotransmitter that excites the pyramidal cells. To differentiate between the two options, we puffed MCPG continuously (at 0.5 Hz) onto single neurons before and during bath application of LPS. In this situation bath‐applied LPS induced astrocytic [Ca2+]i increases but failed to affect the neuron until MCPG puffing was halted (Fig. 8 C). These results confirm that the neuronal effects of LPS are mediated downstream by direct activation of neuronal group I mGluRs.
Next, we examined whether direct activation of neuronal group I mGluRs mimics the LPS‐induced neuronal effects. To that end, we bath‐applied (±)‐1‐aminocyclopentane‐trans‐1,3‐dicarboxylic acid (t‐ACPD, 50 μm), a group I mGluR agonist (Palmer et al. 1989; Davies et al. 1995). To avoid inadvertent indirect neuronal effects of t‐ACPD due to activation of mGluRs in glia, we minimized glia involvement by pretreating slices with l‐AAA, as well as with minocycline (50 nm), a microglia inhibitor (Yrjanheikki et al. 1999; Kim & Suh, 2009). Bath application of t‐ACPD led to an increase in [Ca2+]i (n = 5; Fig. 9 A) followed by depolarization and increase in excitability in all tested pyramidal cells (n = 5; Fig. 9 A–E, Table 1), thus mimicking the neuronal effects of LPS, as well as of ADPβS (Table 1).
Interestingly, the time‐to‐onset of the t‐ACPD‐induced [Ca2+]i increase was significantly shorter compared to that induced by LPS (onsett ‐ACPD = 23.1 ± 3.9 s vs. onsetLPS = 47.9 ± 5.8 s, P < 0.05, n = 5; unpaired t test), suggesting that t‐ACPD acts directly on the pyramidal cells. Altogether, these results suggest that glutamate released by astrocytes, acting on neuronal group I mGluRs and causing a [Ca2+]i increase, mediates LPS‐induced I M inhibition and enhancement of intrinsic neuronal excitability.
LPS‐induced I M inhibition is mediated by mGlu5 receptors
Group I mGluRs comprise mGlu1 and mGlu5 receptors (Luscher & Huber, 2010). In CA1 pyramidal cells the expression of mGlu5 receptors predominates (Baude et al. 1993; Romano et al. 1995). In congruence with the above results (Fig. 8 A and C), blocking both mGlu1 and mGlu5 receptors with MCPG prevented LPS‐induced I M inhibition (Fig. 10 A (middle panel), B and C). A similar preventive effect was exerted by MPEP (10 μm), a selective antagonist of mGlu5 receptors (Salt et al. 1999); Fig. 10 D (middle panel), E and F). Expectedly, application of XE991 to MCPG‐ or MPEP‐treated slices fully blocked I M (Fig. 10 A (right panel), B and C and Fig. 10 D (right panel), E and F, respectively).
Figure 10. Blockade of neuronal mGlu5 receptor prevents LPS‐induced inhibition of I M .
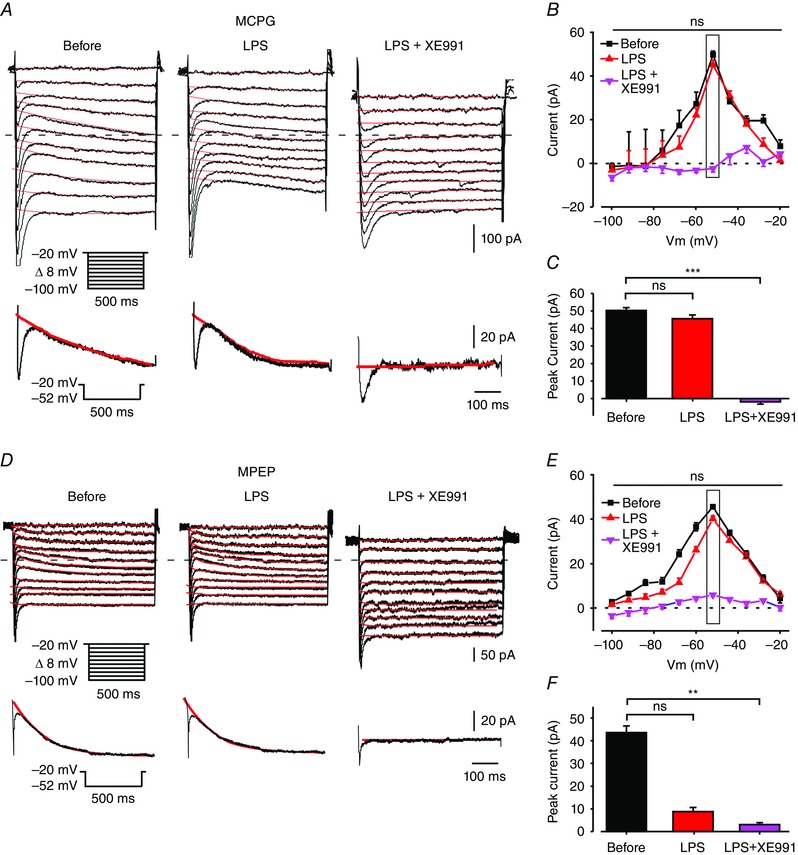
A, voltage‐clamp recordings from a pyramidal cell in a slice perfused with modified aCSF designed to isolate I M (see Methods) and containing MCPG (2 mm). Each subpanel illustrates a family of currents evoked by a series of 500 ms, 8 mV hyperpolarizing voltage steps from a holding potential of –20 mV (voltage protocol is shown in inset). The dotted line indicates zero current level. The current response obtained by stepping to –52 mV is shown at the bottom of each subpanel. The three subpanels depict the current responses before (left), 5 min after bath application of 10 μg ml–1 LPS (middle) and 5 min after additional bath application of 20 μm XE991 (right). Note absence of I M inhibition by LPS (representative of 7/7 experiments). B, plots of I M amplitudes versus hyperpolarizing command potentials in the three conditions shown in A: before (black squares), after 5 min of exposure to LPS (red triangles) and after 5 min of exposure to XE991 (pink inverted triangles) in MCPG‐containing aCSF. The rectangle outlines peak I M values. The first (Before) and second (LPS) plots do not differ significantly (ns, not significant, * P > 0.05, two‐way ANOVA, n = 7). C, bar graph comparing peak I M amplitudes measured at –52 mV (shown in C, inset) in the three conditions as in B. Note lack of significant I M inhibition by LPS and complete inhibition by XE991 (ns, not significant (* P > 0.05), *** P < 0.001, Student's t test, n = 7). D, same as in A, but in a slice treated with the selective mGlu5 receptor antagonist MPEP (10 μm). Note absence of I M inhibition by LPS (representative of 8/10 experiments). E, same as in B, but for slices treated with MPEP. Note lack of significant I M inhibition by LPS (* P > 0.05, two‐way ANOVA, n = 10) and complete inhibition by XE991 (*** P < 0.001, two‐way ANOVA, n = 10). F, same as in C, but for slices treated with MPEP. ns, not significant, *** P < 0.001. [Colour figure can be viewed at wileyonlinelibrary.com]
Additionally, we found that t‐ACPD application to l‐AAA and minocycline‐treated slices inhibited I M in all recorded neurons (n = 6; Fig. 11 A–C). The extent of I M inhibition by t‐ACPD was similar to that exerted by LPS (measured at –52 mV; P = 0.09, n = 7 for LPS; n = 6 for t‐ACPD). As in the case of LPS, filling pyramidal cells with BAPTA barred I M inhibition by t‐ACPD (Fig. 11 D–F), but not by subsequent addition of XE991 to the aCSF (Fig. 11 D–F; n = 7/7). Finally, we found that blockade of mGlu5 receptors with MPEP completely prevented t‐ACPD‐induced inhibition of I M (Fig. 11 G–I; n = 6/6). Again, subsequent addition of XE991 completely blocked I M (Fig. 11 G–I; n = 6/6).
Figure 11. Activation of neuronal mGlu5 receptors inhibits I M in an intracellular Ca2+‐dependent manner.
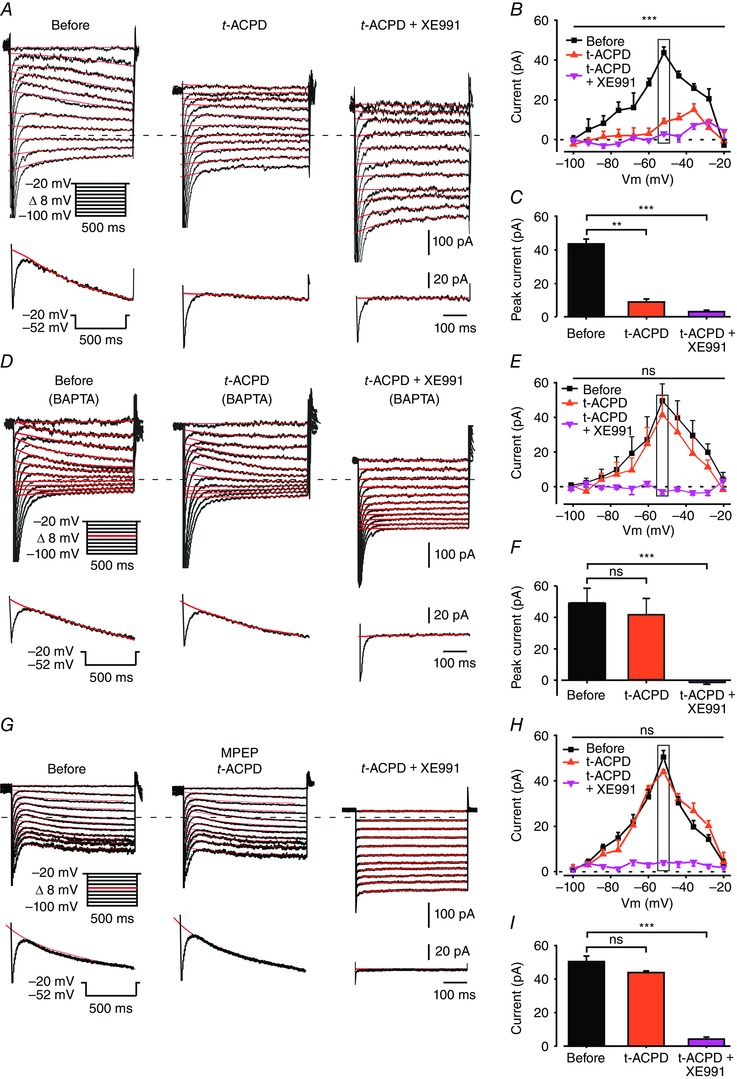
All slices were pretreated with 1 mm l‐AAA and 50 nm minocycline to induce glia dysfunction. A, voltage‐clamp recordings from a pyramidal cell in a slice perfused with modified aCSF designed to isolate I M (see Methods). Each subpanel illustrates a family of currents evoked by a series of 500 ms, 8 mV hyperpolarizing voltage steps from a holding potential of –20 mV (voltage protocol is shown in inset). The current response obtained by stepping to –52 mV is shown at the bottom of each subpanel. The I M relaxation was fitted with a monoexponential line (red), which was extrapolated to the beginning of the voltage step. I M amplitudes were measured as described in Methods. The dotted line indicates zero current level. The three subpanels depict the current responses before (left), 5 min after bath application of 50 μm t‐ACPD (middle) and 5 min after additional bath application of 20 μm XE991 (right). Note, almost complete I M inhibition by t‐ACPD (representative of 6/6 experiments). B, plots of I M amplitudes versus hyperpolarizing command potentials in the three conditions shown in A: before (black squares), after 5 min of exposure to t‐ACPD (red triangles) and after 5 min of exposure to XE991 (pink inverted triangles). Rectangle outlines peak I M values. The first (Before) and second (t‐ACPD, 5 min) plots differ significantly (*** P < 0.001, two‐way ANOVA, n = 6). C, bar graph comparing peak I M amplitudes measured at –52 mV (shown in A, inset) in the three conditions as in B. t‐ACPD significantly inhibits I M (** P < 0.01, *** P < 0.001, Student's t test, n = 6). D, same as in A, but pyramidal cells were filled with 13 mm BAPTA. Note no I M reduction by t‐ACPD in BAPTA‐filled neurons, but complete inhibition by XE991 (representative of 7/7 experiments). E, same as in B, but for the BAPTA‐filled pyramidal cells. Note lack of significant I M inhibition by t‐ACPD (P > 0.05, two‐way ANOVA, n = 7) and complete inhibition by XE991 (*** P < 0.001, two‐way ANOVA, n = 7). F, same as in C, but for the BAPTA‐filled pyramidal cells. ns, not significant, *** P < 0.001. G, same as in A, but in a slice treated with MPEP (10 μm). Note no I M reduction by t‐ACPD, but complete inhibition by XE991 (representative of 6/6 experiments). E, same as in B, but in slices treated with MPEP. Note lack of significant I M inhibition by t‐ACPD (* P > 0.05, two‐way ANOVA, n = 6) and complete inhibition by XE991 (*** P < 0.001, two‐way ANOVA, n = 6). F, same as in C, but in slices treated with MPEP. ns, not significant, *** P < 0.001. [Colour figure can be viewed at wileyonlinelibrary.com]
Altogether, these findings implicate mGlu5 receptors as the predominant downstream receptor mediating I M inhibition.
Retigabine reverses LPS‐induced neuronal hyperexcitability
Several drugs were shown to enhance KV7/M channel activity thereby augmenting I M (Miceli et al. 2008). The foremost of these drugs is retigabine (Main et al. 2000; Wickenden et al. 2000), currently used as an antiepileptic medication (Amabile & Vasudevan, 2013). In CA1 pyramidal cells, retigabine was shown to cause neuronal hyperpolarization, reduction in input resistance, and reduction in spike firing (Yue and Yaari, 2004, 2006). We tested whether retigabine can reverse LPS‐induced increase in neuronal excitability. To that end, slices were perfused with aCSF containing LPS while recording the activity of single CA1 pyramidal cells. One minute after the onset of LPS‐induced spike discharge, retigabine (10 μm) was co‐applied to the aCSF. Within 40 s of its application, retigabine repolarized and silenced the neurons (Fig. 12 A). It also reversed R in (Fig. 12 B), spike threshold (Fig. 12 C) and firing response gain (Fig. 12 D and E) to pre‐LPS values (9/10 neurons). These results clearly show that retigabine maintains its enhancing action even at KV7/M channels subjected to LPS‐induced blockade.
Figure 12. Retigabine reverses LPS‐induced increase in neuronal excitability.
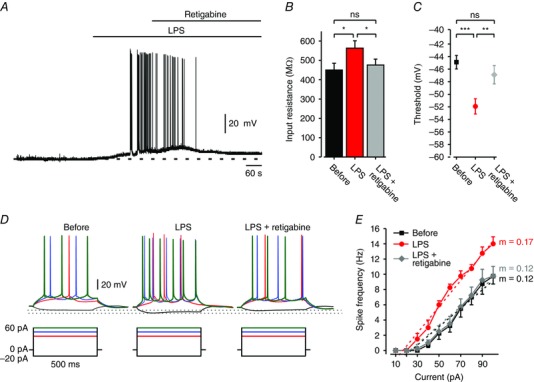
A, representative current‐clamp recording from a CA1 pyramidal cell exposed to LPS (10 μg ml–1). Coapplication of retigabine (10 μm) 1 min after the onset of repetitive firing repolarized and silenced the neuron. (representative of 9/10 neurons responding to LPS). Dashed lines indicate resting potentials before LPS application (–65 mV). B, bar graph depicting R in of all neurons before (black), after 5 min of exposure to LPS (red), and after 5 min of exposure to retigabine coapplied with LPS (overall 10 min exposure to LPS, grey). ns, not significant, * P < 0.05, paired t test, n = 10. C, spike thresholds measured at the same points before (black square), under LPS (red circle), and under LPS and retigabine (grey diamond); ns, not significant, *** P < 0.001, ** P < 0.01, paired t test, n = 10. D, voltage responses (top) to current steps (bottom) in a pyramidal cell recorded at the same points before, under LPS, and under LPS and retigabine (representative of 9/10 neurons). Dotted lines indicate peak voltage deflections in the three conditions. E, mean f–I curves of 10 pyramidal cells obtained at the same points before (black squares), under LPS (red circles), and under LPS and retigabine (grey diamonds). Note, LPS‐induced increase in spike frequency and gain (m, dotted lines) was reversed by retigabine. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
Here we show that the proinflammatory agent LPS acutely produces a marked increase in the intrinsic excitability of CA1 pyramidal cells. The inflammatory cascade by which LPS exerts this action requires ATP‐mediated astrocytic P2Y1 receptor activation, leading to glutamatergic activation of neuronal group I mGluRs, which in turn causes the release of Ca2+ from internal stores and subsequent inhibition of neuronal I M, thereby enhancing intrinsic neuronal excitability (Fig. 13).
Figure 13. Key steps in the LPS‐induced neuroinflammation‐hyperexcitability cascade.
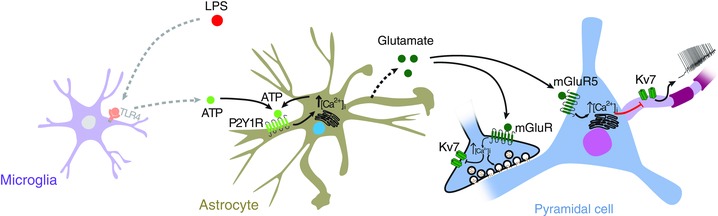
In the scheme, LPS activates TLR‐4 on microglia, leading to ATP release. ATP, in turn, acts on astrocytic P2Y1 receptors, increasing astrocytic [Ca2+]i. This signal may be further amplified by ATP release from astrocytes and activation of autocrine P2Y1 receptors. Consequently, activated astrocytes release glutamate, which acts postsynaptically on mGlu5 receptors (mGluR5) to block neuronal KV7/M channels by releasing Ca2+ from internal stores. The resultant inhibition of I M leads to an increase in intrinsic neuronal excitability. The scheme also depicts LPS‐induced inhibition of putative presynaptic KV7/M channels, leading to enhanced excitatory neurotransmission. [Colour figure can be viewed at wileyonlinelibrary.com]
LPS increases intrinsic excitability via Ca2+‐mediated I M inhibition
I M is a slow, low voltage‐activating, non‐inactivating neuronal K+ current, widely expressed in the nervous system and strongly modulated by multiple factors (Brown & Passmore, 2009; Caspi et al. 2009; Jones et al. 2014). In CA1 pyramidal cells, the predominant K+ channel subunits underlying I M are KV7.2 and KV7.3 (Shah et al. 2002). These subunits are highly expressed in the axon initial segment (Pan et al. 2006), the site of spike generation in these neurons (Spruston et al. 1995). Due to its unique biophysical properties and subcellular localization, I M controls resting V m and R in, affecting spike threshold and firing frequency. Therefore, blocking I M pharmacologically causes neuronal depolarization, increase in R in, and enhanced spike discharge and bursting (Yue & Yaari, 2004; Gu et al. 2005; Yue & Yaari, 2006). Here we show that LPS application induces similar effects. Furthermore, in XE991‐treated slices, subsequent application of LPS exerted no additional effects. These findings strongly implicate I M as the predominant, if not the sole, downstream neuronal target for the LPS‐induced increase in pyramidal cell excitability.
We found that the earliest observed neuronal effect of LPS is a marked increase in [Ca2+]i. Previous studies have shown that an increase in [Ca2+]i inhibits I M (Selyanko & Brown, 1996) and that this inhibition is conferred predominantly by the intracellular Ca2+ sensor calmodulin (Gamper & Shapiro, 2003), which is tethered to the KV7/M channel (Wen & Levitan, 2002). Here we showed that I M inhibition was prevented by filling the neurons with BAPTA, consistent with the notion that also in CA1 PCs a rise in [Ca2+]i inhibits I M (Chen & Yaari, 2008). Intriguingly, the LPS‐induced increase in [Ca2+]i appeared to be transient, lasting about a minute, whereas the increase in neuronal excitability persisted for tens of minutes thereafter. A similar discrepancy in time course was previously described in rat sympathetic neurons for bradykinin‐induced I M inhibition, which also depends on a rise in [Ca2+]i (Cruzblanca et al. 1998). As we measured global somatic changes in [Ca2+]i, we cannot exclude a more persistent [Ca2+]i increase at the axon initial segment, where KV7/M channels are preferentially localized (Devaux et al. 2004; Pan et al. 2006) or at submembrane domains. Alternatively, the initial increase in [Ca2+]i may produce a slow I M inhibition that surpasses the duration of the [Ca2+]i increase (Cruzblanca et al. 1998). In either case, the I M inhibition could be further sustained by spike Ca2+ entry during the resultant repetitive discharge (Chen & Yaari, 2008).
LPS‐induced increase in neuronal excitability is mediated by astrocytic P2Y1 receptors
Our results strongly indicate that LPS‐induced neuronal excitation is critically dependent on recruitment of astrocytes. First, astrocytes responded to bath‐applied LPS with substantial increases in [Ca2+]i. Second, LPS was ineffective in causing neuronal excitation when astrocytic functions were impeded by l‐AAA. The LPS‐induced recruitment of astrocytes is likely to be mediated by activated microglial cells, as TLR‐4 receptors are predominantly expressed by microglia (Pascual et al. 2012), and these cells are known to release multiple factors that modulate the activity of astrocytes (Raetz & Whitfield, 2002; Liao et al. 2010). The expression of TLR‐4 receptors by astrocytes is a controversial issue (Bowman et al. 2003; Pascual et al. 2012; Rannikko et al. 2015). Therefore, we cannot entirely dismiss the possibility that LPS activates astrocytes also directly to produce neuronal excitation.
How does LPS‐induced activation of astrocytes lead to excitation of the pyramidal cells? It has been shown recently that LPS evokes ATP release from microglia and astrocytes (Pascual et al. 2012). The released ATP undoubtedly mediates the astrocytic and neuronal effects of LPS, as they were completely abrogated by the broad spectrum P2 receptor antagonist suramin. Further pharmacological analysis implicated P2Y1 receptors in the LPS‐induced cascade, as the selective P2Y1 receptor antagonist MRS2179 prevented, whereas the selective P2Y1 receptor agonist ADPβS mimicked, all LPS‐induced astrocytic and neuronal effects.
Both hippocampal microglia (Boucsein et al. 2003) and astrocytes (Zhu & Kimelberg, 2001; Bowser & Khakh, 2004; Domercq et al. 2006; Zeng et al. 2008) express P2Y1 receptors, but there have been conflicting reports regarding their expression by pyramidal cells (Moran‐Jimenez & Matute, 2000; Bowser & Khakh, 2004). Hippocampal pyramidal cells grown in culture were shown by immunostaining to express P2Y1 receptors. Furthermore, application of ADPβS to these neurons caused a partial Ca2+‐independent inhibition of I M (Filippov et al. 2006). In contrast, immunostaining for P2Y1 receptors in acute hippocampal slices failed to detect their expression in CA1 pyramidal cells (Bowser & Khakh, 2004). Our results using focal applications of ADPβS are consistent with the latter finding and suggest differences in neuronal P2Y1 receptor expression between the two preparations. Further studies are necessary to resolve this discrepancy. However, as focal ADPβS application onto astrocytes, but not onto pyramidal cells, fully replicated the astrocytic and neuronal effects of LPS, we posit that in hippocampal slices, ATP released from LPS‐activated microglia acts predominantly on astrocytic P2Y1 receptors to increase astrocytic [Ca2+]i (Anderson et al. 2004). This, in turn, may cause astrocytic ATP release, boosting the activation of astrocytes in a regenerative manner (Bowser & Khakh, 2007).
LPS‐induced increase in neuronal excitability is mediated by neuronal mGlu5 receptors
While playing a critical role in the LPS‐induced cascade, ATP is unlikely to be the astrocytic gliotransmitter directly exciting the neurons. This conclusion was ascertained by experiments showing that puffing suramin onto pyramidal cells does not block the neuronal effects of LPS. Rather, several lines of evidence led us to conclude that astrocytic glutamate excites the pyramidal cells via neuronal mGlu5 receptors. First, the group I and II mGluR antagonist MCPG, but not the selective group II mGluR antagonist LY341495, blocked the neuronal, but not the astrocytic, effects of LPS. Second, direct activation of mGluRs with t‐ACPD, in the absence of functional glia, mimicked all neuronal effects of LPS, including [Ca2+]i‐dependent I M inhibition. Finally, the mGlu5 receptor antagonist MPEP prevented I M inhibition by both LPS and t‐ACPD.
Previous studies have shown that glutamate released by astrocytes can mediate astrocytic–neuronal interactions (e.g. Parpura et al. 1994; Angulo et al. 2004; Fiacco & McCarthy, 2004; Volterra et al. 2014). Although the exact mechanism by which astrocytes release glutamate in response to P2Y1 receptor activation remains controversial (Wang et al. 2013), the sheer existence of this release has been demonstrated convincingly (Pascual et al. 2012). Altogether, these data indicate that glutamate is the downstream gliotransmitter, linking between astrocytes and pyramidal cells in the LPS‐induced inflammatory cascade.
Reversibility of LPS‐induced neuronal effects
We found that application of retigabine, a KV7/M channel enhancer (Main et al. 2000; Wickenden et al. 2000), readily reverses the excitatory effects of LPS. This result is congruent with a previous study showing that retigabine and related KV7/M channel enhancers reverse I M inhibition and associated neuronal excitation induced by the proinflammatory agent bradykinin (Linley et al. 2012). Interestingly, bradykinin was shown to block KV7/M channels by transiently raising [Ca2+]i (Cruzblanca et al. 1998), similar to the mechanism attributed to mGlu5 receptors in our present study. Altogether, these studies suggest that retigabine can reverse KV7/M channel blockade exerted via G‐protein‐coupled receptors, highlighting its potential therapeutic role in neuroinflammatory hyperexcitability.
Physiological and pathophysiological implications
There is a large body of evidence from human and animal model studies showing that inflammation of brain tissue increases neuronal network excitability and seizure susceptibility by activating microglia and astrocytes (Vezzani et al. 2011; Devinsky et al. 2013; Seifert & Steinhauser, 2013). These, in turn, release multiple factors that modulate synaptic transmission towards enhanced excitability (Vezzani & Viviani, 2015). Given that excitatory synaptic transmission is shaped foremost by neuronal spike discharge causing synaptic glutamate release, our findings showing that inflammatory factors increase the intrinsic excitability of neurons provide a mechanism for some of the reported synaptic changes in inflammation. Thus, in a recent study in murine hippocampal slices, LPS application enhanced the frequency of excitatory postsynaptic currents (EPSCs) in CA1 pyramidal cells, including that of miniature EPSCs (mEPSCs), via a microglia–astrocytes–neurons cascade, leading to activation of neuronal group I mGluRs (Pascual et al. 2012). However, the mechanism linking mGluR activation to enhanced EPSC frequency was not specified. We posit that LPS induces I M inhibition and enhanced discharge in CA3 pyramidal cells as well. This effect would be reflected in increased EPSC frequency in CA1 pyramidal cells, as the CA3 neurons provide the main excitatory input to CA1 and viable CA3–CA1 connections persist in the hippocampal slice preparation (Schwartzkroin, 1975).
It is also conceivable that the LPS‐induced cascade causes I M inhibition not only at the axon initial segment, where it leads to enhanced discharge, but also at or near the axon terminals (Fig. 11). This action would depolarize the terminals and facilitate spontaneous glutamate release, reflected in enhanced frequency of mEPSCs. Indeed, pharmacological I M inhibition was shown to enhance mEPSC frequency in hippocampal neurons (Peretz et al. 2007), suggesting that Kv7/M channels are expressed in axon terminals. This issue, however, is currently controversial (Hernandez et al. 2008) and it is possible that the LPS‐induced increase in mEPSC frequency results directly from the presynaptic [Ca2+]i increase exerted by activation of presynaptic group I mGluRs (Fiacco & McCarthy, 2004).
In conclusion, many studies support a role for inflammation in neuronal hyperexcitability (De Simoni et al. 2000; Riazi et al. 2008; Vezzani et al. 2011; Devinsky et al. 2013). Combining our findings with those of previous studies suggests that Kv7/M channels at axon initial segments, and perhaps also at axon terminals, are a critical converging target for inflammation‐induced cascades, mediated by microglia and astrocytes, leading to neuronal hyperexcitability.
Additional information
Competing interests
Authors declare no conflict of interests.
Author contributions
Experiments were performed in the laboratory of Y.Y. and A.M.B. A.T. and Y.B. collected, assembled and analysed the data. A.T., H.L., O.B. S.L. and A.M.B. analysed the data. A.T., Y.Y. and A.M.B. designed the experiments and interpreted the data. A.T., H.L., O.B., Y.B., S.L., Y.Y. and A.M.B. wrote the manuscript. Y.Y. and A.M.B. conceived the study and supervised the project. All authors read and approved the final version of the manuscript. All persons designated as authors qualify for authorship, all persons qualified for authorship are listed as authors.
Funding
Support is gratefully acknowledged from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007‐2013)/ERC grant agreement n 260914 (A.T., H.L. and A.M.B.); Deutsch‐Israelische Projectkooperation program of the Deutsche Forschungsgemeinschaft (DIP) grant agreement BI 1665/1‐1ZI1172/12‐1 (O.B., Y.Y. and A.M.B.) and the Henri J. and Erna D. Leir Chair for Research in Neurodegenerative Diseases (Y.Y.).
Y. Yaari and A. M. Binshtok are co‐senior authors.
Linked articles This article is highlighted by a Perspective by Isbrandt. To read this Perspective, visit http://dx.doi.org/10.1113/JP273252.
Contributor Information
Yoel Yaari, Email: yoely@ekmd.huji.ac.il.
Alexander M. Binshtok, Email: alexanderb@ekmd.huji.ac.il
References
- Abe T, Sugihara H, Nawa H, Shigemoto R, Mizuno N & Nakanishi S (1992). Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J Biol Chem 267, 13361–13368. [PubMed] [Google Scholar]
- Amabile CM & Vasudevan A (2013). Ezogabine: a novel antiepileptic for adjunctive treatment of partial‐onset seizures. Pharmacotherapy 33, 187–194. [DOI] [PubMed] [Google Scholar]
- Amor S & Woodroofe MN (2014). Innate and adaptive immune responses in neurodegeneration and repair. Immunology 141, 287–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, Bergher JP & Swanson RA (2004). ATP‐induced ATP release from astrocytes. J Neurochem 88, 246–256. [DOI] [PubMed] [Google Scholar]
- Angulo MC, Kozlov AS, Charpak S & Audinat E (2004). Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci 24, 6920–6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramori I & Nakanishi S (1992). Signal transduction and pharmacological characteristics of a metabotropic glutamate receptor, mGluR1, in transfected CHO cells. Neuron 8, 757–765. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP & Haydon PG (1998). Glutamate‐dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur J Neurosci 10, 2129–2142. [DOI] [PubMed] [Google Scholar]
- Baude A, Nusser Z, Roberts JD, Mulvihill E, McIlhinney RA & Somogyi P (1993). The metabotropic glutamate receptor (mGluR1α) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction. Neuron 11, 771–787. [DOI] [PubMed] [Google Scholar]
- Boucsein C, Zacharias R, Farber K, Pavlovic S, Hanisch UK & Kettenmann H (2003). Purinergic receptors on microglial cells: functional expression in acute brain slices and modulation of microglial activation in vitro. Eur J Neurosci 17, 2267–2276. [DOI] [PubMed] [Google Scholar]
- Bowman CC, Rasley A, Tranguch SL & Marriott I (2003). Cultured astrocytes express toll‐like receptors for bacterial products. Glia 43, 281–291. [DOI] [PubMed] [Google Scholar]
- Bowser DN & Khakh BS (2004). ATP excites interneurons and astrocytes to increase synaptic inhibition in neuronal networks. J Neurosci 24, 8606–8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowser DN & Khakh BS (2007). Vesicular ATP is the predominant cause of intercellular calcium waves in astrocytes. J Gen Physiol 129, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer JL, Mohanram A, Camaioni E, Jacobson KA & Harden TK (1998). Competitive and selective antagonism of P2Y1 receptors by N 6‐methyl 2'‐deoxyadenosine 3',5'‐bisphosphate. Br J Pharmacol 124, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA & Passmore GM (2009). Neural KCNQ (KV7) channels. Br J Pharmacol 156, 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DR & Kretzschmar HA (1998). The glio‐toxic mechanism of α‐aminoadipic acid on cultured astrocytes. J Neurocytol 27, 109–118. [DOI] [PubMed] [Google Scholar]
- Butt AM (2011). ATP: a ubiquitous gliotransmitter integrating neuron‐glial networks. Semin Cell Dev Biol 22, 205–213. [DOI] [PubMed] [Google Scholar]
- Caspi A, Benninger F & Yaari Y (2009). KV7/M channels mediate osmotic modulation of intrinsic neuronal excitability. J Neurosci 29, 11098–11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Benninger F & Yaari Y (2014). Role of small conductance Ca2+‐activated K+ channels in controlling CA1 pyramidal cell excitability. J Neurosci 34, 8219–8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S & Yaari Y (2008). Spike Ca2+ influx upmodulates the spike afterdepolarization and bursting via intracellular inhibition of KV7/M channels. J Physiol 586, 1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow JC, Young DW, Golenbock DT, Christ WJ & Gusovsky F (1999). Toll‐like receptor‐4 mediates lipopolysaccharide‐induced signal transduction. J Biol Chem 274, 10689–10692. [DOI] [PubMed] [Google Scholar]
- Cruzblanca H, Koh DS & Hille B (1998). Bradykinin inhibits M current via phospholipase C and Ca2+ release from IP3‐sensitive Ca2+ stores in rat sympathetic neurons. Proc Natl Acad Sci USA 95, 7151–7156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML & Gan WB (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8, 752–758. [DOI] [PubMed] [Google Scholar]
- Davies CH, Clarke VR, Jane DE & Collingridge GL (1995). Pharmacology of postsynaptic metabotropic glutamate receptors in rat hippocampal CA1 pyramidal neurones. Br J Pharmacol 116, 1859–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simoni MG, Perego C, Ravizza T, Moneta D, Conti M, Marchesi F, De Luigi A, Garattini S & Vezzani A (2000). Inflammatory cytokines and related genes are induced in the rat hippocampus by limbic status epilepticus. Eur J Neurosci 12, 2623–2633. [DOI] [PubMed] [Google Scholar]
- Devaux JJ, Kleopa KA, Cooper EC & Scherer SS (2004). KCNQ2 is a nodal K+ channel. J Neurosci 24, 1236–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O, Vezzani A, Najjar S, De Lanerolle NC & Rogawski MA (2013). Glia and epilepsy: excitability and inflammation. Trends Neurosci 36, 174–184. [DOI] [PubMed] [Google Scholar]
- Domercq M, Brambilla L, Pilati E, Marchaland J, Volterra A & Bezzi P (2006). P2Y1 receptor‐evoked glutamate exocytosis from astrocytes: control by tumor necrosis factor‐α and prostaglandins. J Biol Chem 281, 30684–30696. [DOI] [PubMed] [Google Scholar]
- Dunn PM & Blakeley AG (1988). Suramin: a reversible P2‐purinoceptor antagonist in the mouse vas deferens. Br J Pharmacol 93, 243–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiacco TA & McCarthy KD (2004). Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J Neurosci 24, 722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov AK, Choi RC, Simon J, Barnard EA & Brown DA (2006). Activation of P2Y1 nucleotide receptors induces inhibition of the M‐type K+ current in rat hippocampal pyramidal neurons. J Neurosci 26, 9340–9348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzjohn SM, Bortolotto ZA, Palmer MJ, Doherty AJ, Ornstein PL, Schoepp DD, Kingston AE, Lodge D & Collingridge GL (1998). The potent mGlu receptor antagonist LY341495 identifies roles for both cloned and novel mGlu receptors in hippocampal synaptic plasticity. Neuropharmacology 37, 1445–1458. [DOI] [PubMed] [Google Scholar]
- Fotuhi M, Standaert DG, Testa CM, Penney JB Jr & Young AB (1994). Differential expression of metabotropic glutamate receptors in the hippocampus and entorhinal cortex of the rat. Brain Res Mol Brain Res 21, 283–292. [DOI] [PubMed] [Google Scholar]
- Freudenberg MA & Galanos C (1990). Bacterial lipopolysaccharides: structure, metabolism and mechanisms of action. Int Rev Immunol 6, 207–221. [DOI] [PubMed] [Google Scholar]
- Gamper N & Shapiro MS (2003). Calmodulin mediates Ca2+‐dependent modulation of M‐type K+ channels. J Gen Physiol 122, 17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Liu Z, Ren W & Jiang W (2014). Acute lipopolysaccharide exposure facilitates epileptiform activity via enhanced excitatory synaptic transmission and neuronal excitability in vitro. Neuropsychiatr Dis Treat 10, 1489–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaschuk O, Yaari Y & Konnerth A (1997). Release and sequestration of calcium by ryanodine‐sensitive stores in rat hippocampal neurones. J Physiol 502, 13–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gereau RW & Conn PJ (1995). Roles of specific metabotropic glutamate‐receptor subtypes in regulation of hippocampal CA1 pyramidal cell excitability. J Neurophysiol 74, 122–129. [DOI] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H & Storm JF (2005). KV7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after‐hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol 566, 689–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen V, Storm‐Mathisen J & Bergersen LH (2015). Neuroglial transmission. Physiol Rev 95, 695–726. [DOI] [PubMed] [Google Scholar]
- Halliwell JV & Adams PR (1982). Voltage‐clamp analysis of muscarinic excitation in hippocampal neurons. Brain Res 250, 71–92. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Sekiyama N, Nakanishi S, Jane DE, Sunter DC, Birse EF, Udvarhelyi PM & Watkins JC (1994). Analysis of agonist and antagonist activities of phenylglycine derivatives for different cloned metabotropic glutamate receptor subtypes. J Neurosci 14, 3370–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez CC, Zaika O, Tolstykh GP & Shapiro MS (2008). Regulation of neural KCNQ channels: signalling pathways, structural motifs and functional implications. J Physiol 586, 1811–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huck S, Grass F & Hatten ME (1984). Gliotoxic effects of α‐aminoadipic acid on monolayer cultures of dissociated postnatal mouse cerebellum. Neuroscience 12, 783–791. [DOI] [PubMed] [Google Scholar]
- Imura Y, Morizawa Y, Komatsu R, Shibata K, Shinozaki Y, Kasai H, Moriishi K, Moriyama Y & Koizumi S (2013). Microglia release ATP by exocytosis. Glia 61, 1320–1330. [DOI] [PubMed] [Google Scholar]
- Jones RT, Faas GC & Mody I (2014). Intracellular bicarbonate regulates action potential generation via KCNQ channel modulation. J Neurosci 34, 4409–4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS & Suh YH (2009). Minocycline and neurodegenerative diseases. Behav Brain Res 196, 168–179. [DOI] [PubMed] [Google Scholar]
- Kingston AE, Ornstein PL, Wright RA, Johnson BG, Mayne NG, Burnett JP, Belagaje R, Wu S & Schoepp DD (1998). LY341495 is a nanomolar potent and selective antagonist of group II metabotropic glutamate receptors. Neuropharmacology 37, 1–12. [DOI] [PubMed] [Google Scholar]
- Koles L, Leichsenring A, Rubini P & Illes P (2011). P2 receptor signaling in neurons and glial cells of the central nervous system. Adv Pharmacol 61, 441–493. [DOI] [PubMed] [Google Scholar]
- Lambrecht G, Braun K, Damer M, Ganso M, Hildebrandt C, Ullmann H, Kassack MU & Nickel P (2002). Structure‐activity relationships of suramin and pyridoxal‐5'‐phosphate derivatives as P2 receptor antagonists. Curr Pharm Des 8, 2371–2399. [DOI] [PubMed] [Google Scholar]
- Leow‐Dyke S, Allen C, Denes A, Nilsson O, Maysami S, Bowie AG, Rothwell NJ & Pinteaux E (2012). Neuronal Toll‐like receptor 4 signaling induces brain endothelial activation and neutrophil transmigration in vitro. J Neuroinflammation 9, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao CK, Wang SM, Chen YL, Wang HS & Wu JC (2010). Lipopolysaccharide‐induced inhibition of connexin43 gap junction communication in astrocytes is mediated by downregulation of caveolin‐3. Int J Biochem Cell B 42, 762–770. [DOI] [PubMed] [Google Scholar]
- Linley JE, Pettinger L, Huang D & Gamper N (2012). M channel enhancers and physiological M channel block. J Physiol 590, 793–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YC, Yeh WC & Ohashi PS (2008). LPS/TLR4 signal transduction pathway. Cytokine 42, 145–151. [DOI] [PubMed] [Google Scholar]
- Lund S, Christensen KV, Hedtjarn M, Mortensen AL, Hagberg H, Falsig J, Hasseldam H, Schrattenholz A, Porzgen P & Leist M (2006). The dynamics of the LPS triggered inflammatory response of murine microglia under different culture and in vivo conditions. J Neuroimmunol 180, 71–87. [DOI] [PubMed] [Google Scholar]
- Luscher C & Huber KM (2010). Group 1 mGluR‐dependent synaptic long‐term depression: mechanisms and implications for circuitry and disease. Neuron 65, 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Main MJ, Cryan JE, Dupere JR, Cox B, Clare JJ & Burbidge SA (2000). Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol Pharmacol 58, 253–262. [DOI] [PubMed] [Google Scholar]
- McBean GJ (1994). Inhibition of the glutamate transporter and glial enzymes in rat striatum by the gliotoxin, α aminoadipate. Br J Pharmacol 113, 536–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miceli F, Soldovieri MV, Martire M & Taglialatela M (2008). Molecular pharmacology and therapeutic potential of neuronal Kv7‐modulating drugs. Curr Opin Pharmacol 8, 65–74. [DOI] [PubMed] [Google Scholar]
- Moran‐Jimenez MJ & Matute C (2000). Immunohistochemical localization of the P2Y1 purinergic receptor in neurons and glial cells of the central nervous system. Brain Res Mol Brain Res 78, 50–58. [DOI] [PubMed] [Google Scholar]
- Olson JK & Miller SD (2004). Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol 173, 3916‐3924. [DOI] [PubMed] [Google Scholar]
- Palmer E, Monaghan DT & Cotman CW (1989). Trans‐ACPD, a selective agonist of the phosphoinositide‐coupled excitatory amino acid receptor. Eur J Pharmacol 166, 585–587. [DOI] [PubMed] [Google Scholar]
- Pan Z, Kao T, Horvath Z, Lemos J, Sul JY, Cranstoun SD, Bennett V, Scherer SS & Cooper EC (2006). A common ankyrin‐G‐based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci 26, 2599–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S & Haydon PG (1994). Glutamate‐mediated astrocyte‐neuron signalling. Nature 369, 744–747. [DOI] [PubMed] [Google Scholar]
- Parpura V & Zorec R (2010). Gliotransmission: Exocytotic release from astrocytes. Brain Res Rev 63, 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual O, Ben Achour S, Rostaing P, Triller A & Bessis A (2012). Microglia activation triggers astrocyte‐mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci USA 109, E197–E205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasti L, Zonta M, Pozzan T, Vicini S & Carmignoto G (2001). Cytosolic calcium oscillations in astrocytes may regulate exocytotic release of glutamate. J Neurosci 21, 477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G & Araque A (2007). Astrocytes potentiate transmitter release at single hippocampal synapses. Science 317, 1083–1086. [DOI] [PubMed] [Google Scholar]
- Peretz A, Sheinin A, Yue C, Degani‐Katzav N, Gibor G, Nachman R, Gopin A, Tam E, Shabat D, Yaari Y & Attali B (2007). Pre‐ and postsynaptic activation of M‐channels by a novel opener dampens neuronal firing and transmitter release. J Neurophysiol 97, 283–295. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi‐Castagnoli P, Layton B & Beutler B (1998). Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088. [DOI] [PubMed] [Google Scholar]
- Raetz CR & Whitfield C (2002). Lipopolysaccharide endotoxins. Annu Rev Biochem 71, 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rannikko EH, Weber SS & Kahle PJ (2015). Exogenous alpha‐synuclein induces toll‐like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci 16, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazi K, Galic MA, Kuzmiski JB, Ho W, Sharkey KA & Pittman QJ (2008). Microglial activation and TNFα production mediate altered CNS excitability following peripheral inflammation. Proc Natl Acad Sci USA 105, 17151–17156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers KM, Hutchinson MR, Northcutt A, Maier SF, Watkins LR & Barth DS (2009). The cortical innate immune response increases local neuronal excitability leading to seizures. Brain 132, 2478–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano C, Sesma MA, McDonald CT, O'Malley K, Van den Pol AN & Olney JW (1995). Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. J Comp Neurol 355, 455–469. [DOI] [PubMed] [Google Scholar]
- Rubio ME & Soto F (2001). Distinct localization of P2X receptors at excitatory postsynaptic specializations. J Neurosci 21, 641–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt TE, Binns KE, Turner JP, Gasparini F & Kuhn R (1999). Antagonism of the mGlu5 agonist 2‐chloro‐5‐hydroxyphenylglycine by the novel selective mGlu5 antagonist 6‐methyl‐2‐(phenylethynyl)‐pyridine (MPEP) in the thalamus. Br J Pharmacol 127, 1057–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzkroin PA (1975). Characteristics of CA1 neurons recorded intracellularly in the hippocampal in vitro slice preparation. Brain Res 85, 423–436. [DOI] [PubMed] [Google Scholar]
- Seifert G & Steinhauser C (2013). Neuron‐astrocyte signaling and epilepsy. Exp Neurol 244, 4–10. [DOI] [PubMed] [Google Scholar]
- Selyanko AA & Brown DA (1996). Intracellular calcium directly inhibits potassium M channels in excised membrane patches from rat sympathetic neurons. Neuron 16, 151–162. [DOI] [PubMed] [Google Scholar]
- Shah M, Mistry M, Marsh SJ, Brown DA & Delmas P (2002). Molecular correlates of the M‐current in cultured rat hippocampal neurons. J Physiol 544, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton MK & McCarthy KD (1999). Mature hippocampal astrocytes exhibit functional metabotropic and ionotropic glutamate receptors in situ. Glia 26, 1–11. [DOI] [PubMed] [Google Scholar]
- Sim JA, Chaumont S, Jo J, Ulmann L, Young MT, Cho K, Buell G, North RA & Rassendren F (2006). Altered hippocampal synaptic potentiation in P2X4 knock‐out mice. J Neurosci 26, 9006–9009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruston N, Schiller Y, Stuart G & Sakmann B (1995). Activity‐dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science 268, 297–300. [DOI] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR & Dawson AP (1990). Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+‐ATPase. Proc Natl Acad Sci USA 87, 2466–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Ma L, Kaarela T & Li Z (2012). Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J Neuroinflammation 9, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, French J, Bartfai T & Baram TZ (2011). The role of inflammation in epilepsy. Nat Rev Neurol 7, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A & Viviani B (2015). Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 96, 70–82. [DOI] [PubMed] [Google Scholar]
- Volterra A, Liaudet N & Savtchouk I (2014). Astrocyte Ca2+ signalling: an unexpected complexity. Nat Rev Neurosci 15, 327–335. [DOI] [PubMed] [Google Scholar]
- von Kugelgen I (2006). Pharmacological profiles of cloned mammalian P2Y‐receptor subtypes. Pharmacol Ther 110, 415–432. [DOI] [PubMed] [Google Scholar]
- Wang F, Smith NA, Xu Q, Goldman S, Peng W, Huang JH, Takano T & Nedergaard M (2013). Photolysis of caged Ca2+ but not receptor‐mediated Ca2+ signaling triggers astrocytic glutamate release. J Neurosci 33, 17404–17412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE & McKinnon D (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M‐channel. Science 282, 1890–1893. [DOI] [PubMed] [Google Scholar]
- Weisman GA, Woods LT, Erb L & Seye CI (2012). P2Y receptors in the mammalian nervous system: pharmacology, ligands and therapeutic potential. CNS Neurol Disord Drug Targets 11, 722–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H & Levitan IB (2002). Calmodulin is an auxiliary subunit of KCNQ2/3 potassium channels. J Neurosci 22, 7991–8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickenden AD, Yu W, Zou A, Jegla T & Wagoner PK (2000). Retigabine, a novel anti‐convulsant, enhances activation of KCNQ2/Q3 potassium channels. Mol Pharmacol 58, 591–600. [DOI] [PubMed] [Google Scholar]
- Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH & Koistinaho J (1999). A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci USA 96, 13496–13500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue C & Yaari Y (2004). KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J Neurosci 24, 4614–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue C & Yaari Y (2006). Axo‐somatic and apical dendritic Kv7/M channels differentially regulate the intrinsic excitability of adult rat CA1 pyramidal cells. J Neurophysiol 95, 3480–3495. [DOI] [PubMed] [Google Scholar]
- Zeng JW, Liu XH, Zhang JH, Wu XG & Ruan HZ (2008). P2Y1 receptor‐mediated glutamate release from cultured dorsal spinal cord astrocytes. J Neurochem 106, 2106–2118. [DOI] [PubMed] [Google Scholar]
- Zhao M, Zhou A, Xu L & Zhang X (2014). The role of TLR4‐mediated PTEN/PI3K/AKT/NF‐κB signaling pathway in neuroinflammation in hippocampal neurons. Neuroscience 269, 93–101. [DOI] [PubMed] [Google Scholar]
- Zhu Y & Kimelberg HK (2001). Developmental expression of metabotropic P2Y1 and P2Y2 receptors in freshly isolated astrocytes from rat hippocampus. J Neurochem 77, 530–541. [DOI] [PubMed] [Google Scholar]