

Abstract
Mitochondrial cytochrome c oxidase (CcO) transfers electrons from cytochrome c (Cyt.c) to O2 to generate H2O, a process coupled to proton pumping. To elucidate the mechanism of electron transfer, we determined the structure of the mammalian Cyt.c–CcO complex at 2.0‐Å resolution and identified an electron transfer pathway from Cyt.c to CcO. The specific interaction between Cyt.c and CcO is stabilized by a few electrostatic interactions between side chains within a small contact surface area. Between the two proteins are three water layers with a long inter‐molecular span, one of which lies between the other two layers without significant direct interaction with either protein. Cyt.c undergoes large structural fluctuations, using the interacting regions with CcO as a fulcrum. These features of the protein–protein interaction at the docking interface represent the first known example of a new class of protein–protein interaction, which we term “soft and specific”. This interaction is likely to contribute to the rapid association/dissociation of the Cyt.c–CcO complex, which facilitates the sequential supply of four electrons for the O2 reduction reaction.
Keywords: cytochrome c, cytochrome c oxidase, electron transfer complex, protein–protein interaction, X‐ray crystallography
Subject Categories: Membrane & Intracellular Transport, Metabolism, Structural Biology
Introduction
Cytochrome c oxidase (CcO) is a typical aa 3‐type CcO, in which electrons are transferred to an active site consisting of heme a 3 and CuB from CuA via heme a. CcO initially accepts electrons from cytochrome c (Cyt.c) to reduce a dioxygen molecule (Ferguson‐Miller & Babcock, 1996; Yoshikawa & Shimada, 2015). Electron transfer (ET) from CuA to the O2 reduction center is coupled to proton pumping across the membrane.
Extensive steady‐state kinetic analyses of oxidation of ferro‐Cyt.c by CcO have revealed two Cyt.c‐binding sites, both of which are actively involved in catalytic turnover (Ferguson‐Miller et al, 1976). Speck et al (1984) proposed a single‐catalytic site model in which one binding site is the catalytic site through which electrons are transferred, whereas the other controls ET in the catalytic site. The amino‐acid residues on the Cyt.c surface that interact with CcO were examined by chemically modifying basic residues of Cyt.c and observing the effect on CcO activity; these experiments revealed the critical involvement of basic residues on the Cyt.c surface (Ferguson‐Miller et al, 1978; Osheroff et al, 1980). The residues that interact with CcO have been investigated more extensively by NMR studies (Sakamoto et al, 2011), which revealed that hydrophobic residues on the surface of Cyt.c make major contributions to complex formation, whereas the charged residues near the hydrophobic core refine the orientation of Cyt.c to precisely control ET. However, as noted above, analyses of the mechanism of ET between Cyt.c and CcO have been largely restricted to the Cyt.c side. With the exception of docking simulation analyses (Roberts & Pique, 1999; Sato et al, 2016), essentially no experimental information is available regarding the Cyt.c‐binding surface of CcO.
Although a significant amounts of data have accumulated regarding ET from Cyt.c to CcO (Speck et al, 1984; Sakamoto et al, 2011), and the X‐ray structures of mammalian CcO (PDB 5B1A) and Cyt.c (Bushnell et al, 1990; De March et al, 2014) have been determined at high resolution, the underlying mechanism of ET remains incompletely understood. A crystal structure of the complex of CcO and Cyt.c would be invaluable for mechanistic studies, but to date no structure of a Cyt.c–CcO complex has been determined other than that of caa 3‐type CcO from Thermus thermophilus (Lyons et al, 2012), which has a covalently tethered cytochrome c domain. Thus, it remains unclear whether this fused Cyt.c has functions analogous to those of the Cyt.c molecules that participate in catalytic turnover in the eukaryotic CcO system.
Two‐dimensional (2D) crystals of the mammalian Cyt.c–CcO complex were prepared at higher pH (7.4–9.0) with both proteins in the oxidized state (Osuda et al, 2016), but these 2D crystals could not provide a structure of sufficient resolution to allow a detailed analysis of the interactions between these proteins. Therefore, in this study, we optimized the three‐dimensional (3D) crystallization conditions for ferri‐Cyt.c and oxidized CcO at high pH and solved the X‐ray structure of the complex at 2.0‐Å resolution. The results revealed a novel mode of protein–protein interaction mediated by three water layers.
Results and Discussion
Crystallization of the Cyt.c–CcO complex
Previously, bovine CcO stabilized with n‐decyl‐β‐D‐maltoside (DM) was crystallized at a pH ≤ 6.8 and analyzed at the atomic level (Tsukihara et al, 1995, 1996). However, no 3D crystallization trial of the Cyt.c–CcO complex has been successful under crystallization conditions similar to those used for CcO at low pH. Therefore, we performed co‐crystallization of Cyt.c and CcO at pH 8.0 under the same conditions used for 2D crystallization of the Cyt.c–CcO complex (Osuda et al, 2016). CcO purified from bovine heart was solubilized with DM and fluorinated octyl‐maltoside (FOM), followed by addition of horse Cyt.c at a Cyt.c/CcO molar ratio of 1.2. The Cyt.c–CcO complex was then co‐crystallized by the batch‐wise method at 277 K (Appendix Fig S1A). Absorption spectral analysis indicated that the resultant crystals contained both Cyt.c and CcO (Appendix Fig S1B). The crystals were soaked in a crystallization solution both containing 50 μM Cyt.c and gradually increasing concentrations of the cryo‐protectant ethylene glycol (EG; 40% at the final step), and then frozen in a cryo‐nitrogen stream at 100 K. The addition of 50 μM Cyt.c prevented the crystal from deterioration due to release of Cyt.c molecules from the complex during soaking. These crystals diffracted X‐rays to a resolution of 1.8 Å (Appendix Fig S1C and D). Statistics of the intensity data and structure refinement at 2.0‐Å resolution are provided in Table 1.
Table 1.
Data collection and refinement statistics of Cyt.c–CcO complex crystals
| Data collection | |
|---|---|
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 113.3, 183.9, 148.9 |
| β (°) | 102.1 |
| Resolution (Å) | 50–2.0 (2.02–2.00) |
| Observed reflections | 1,960,373 |
| Independent reflections | 397,399 (9,885) |
| Averaged redundancy | 4.9 (3.7) |
| I/σ(I) | 17.0 (1.2) |
| Completeness (%) | 99.5 (99.5) |
| R merge | 0.097 (> 1.0) |
| R p.i.m. | 0.044 (0.599) |
| CC 1/2 | 0.903 (0.623) |
| Refinement | |
|---|---|
| Resolution (Å) | 40–2.0 |
| No. reflections (all/free) | 377,338/19,990 |
| R ‖/R free | 0.167/0.207 |
| No. atoms | |
| CcO | 28,969 |
| Cyt.c | 1,738 |
| Others | 4,348 |
| Averaged B‐factors (Å2) | |
| CcO A mol. | 39.7 |
| CcO B mol. | 47.3 |
| Cyt.c A mol. | 88.4 |
| Cyt.c B mol. | 111.6 |
| Others | 67.1 |
| R.m.s.d. bond lengths (Å) | 0.023 |
| R.m.s.d. bond angles (°) | 2.0 |
R.m.s.d., Root‐mean‐square deviation.
Values in parentheses are for highest‐resolution shell.
Structure determination and overall structure of the Cyt.c–CcO complex
Initial phases were determined by the molecular replacement (MR) method (Rossmann & Blow, 1962) using CcO, and Cyt.c molecules were located using the F o–F c difference map and anomalous difference map (Appendix Fig S2A and B). Structure refinement at 2.0‐Å resolution converged well: R/R free = 0.167/0.207; r.m.s.d of bond lengths = 0.023 Å; r.m.s.d. of bond angles = 2.0° (Table 1). The (2F o–F c) electron‐density map for the interface of Cyt.c and CcO clearly shows electron densities of side chains interacting with their counterpart proteins (Appendix Fig S2C).
The asymmetric unit of the monoclinic lattice contains a dimer consisting of two complexes of CcO and Cyt.c. The dimeric structure of CcO of the Cyt.c–CcO complex is almost identical to that of CcO crystallized in an orthorhombic lattice (Tsukihara et al, 1996). As in the CcO orthorhombic crystal (Tomizaki et al, 1999), one of the two CcO molecules in the asymmetric unit had a lower B‐factor than the other, by about 7 Å2, and no significant structural difference was detected between the two complexes. Furthermore, structure refinement was performed under non‐crystallographic symmetry restraint between two CcO molecules; therefore, we focused our structural descriptions on this complex.
Cyt.c is localized on the positive side of a concave surface of CcO (Figs 1 and EV1). Although B‐factors of Cyt.c were significantly higher than those of CcO, all the side chains except for Lys25 were located in the positive density of (2F o–F c) map. Consistent with the results of studies in which Cyt.c was chemically modified at lysyl residues, CcO interacts with the front surface of Cyt.c in a region that includes the exposed heme edge of Cyt.c (Ferguson‐Miller et al, 1978; Osheroff et al, 1980). By contrast, in caa 3‐type CcO, the propionate side of heme group of Cyt.c faces CuA (Lyons et al, 2012). CcO interacts with Cyt.c mainly via subunit II, with 94% of the contact surface of CcO with Cyt.c belonging to subunit II, and 5 and 1% of it belonging to subunits VIb and I, respectively.
Figure 1. Overall structure of the Cyt.c–CcO complex at 2.0‐Å resolution.
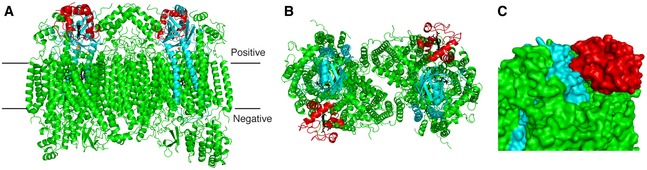
-
A, BRibbon drawing of the Cyt.c–CcO complex, viewed from the trans‐membrane surface (A) and the positive side (B).
-
CClose‐up view of the interface between Cyt.c and the subunit II of CcO, shown as surface representation.
Figure EV1. A cross‐section of surface structures of CcO and Cyt.c–CcO complex.
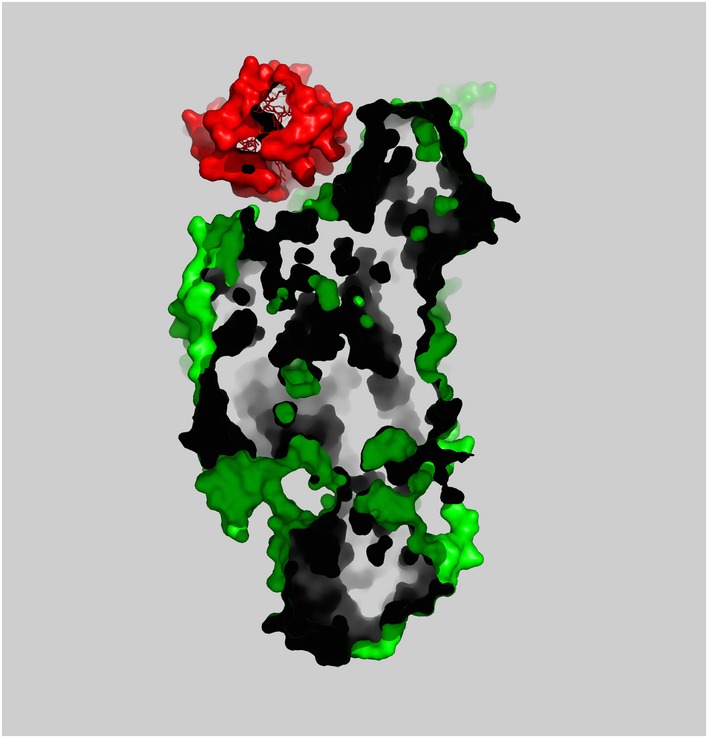
Cyt.c and CcO are shown as surface representation each colored in red and green, respectively. The cross‐section of the surface representation indicates that Cyt.c fits closely with the concave surface.
The closest inter‐atomic distance between Cyt.c and any Cyt.c–CcO complex related by crystallographic symmetry is 6.9 Å. Because Cyt.c does not interact directly with any symmetry‐related Cyt.c–CcO complexes (Appendix Fig S3), molecular packing in the crystal does not perturb the structure of Cyt.c in the Cyt.c–CcO complex. The CcO and Cyt.c structures in the complex superpose well with the previously determined structures of the individual proteins, as shown in Appendix Fig S4. At the current resolution, docking of CcO and Cyt.c results in no significant structural changes in the main chains. In Appendix Fig S5, phospholipids are depicted (sticks) along with the Cα traces of the complex (ribbons). All phospholipids detected in the crystal structure of CcO (PDB 5B1A), but no additional lipids, are present in the Cyt.c–CcO complex. None of these phospholipids are localized near the Cyt.c‐binding site; therefore, Cyt.c does not interact with phospholipids in the crystal of the complex.
A possible electron transfer pathway from heme c to CuA
The concave surface consists of subunit II, which contains CuA, the first loading site for electrons transferred from Cyt.c. The distance between the iron atom of heme c and the copper atom of CuA is 23.0 Å. The dominant ET pathway from the heme c iron to CuA of CcO was explored using the Pathways plugin for VMD (Humphrey et al, 1996; Balabin et al, 2012). The calculations suggest that the most probable ET pathway, as shown in Figs 2 and EV2, proceeds through the iron atom of heme c (Cyt.c)‐Cys14 (Cyt.c)‐Lys13 (Cyt.c)‐Tyr105 (subunit II of CcO)‐Met207 (subunit II of CcO)‐CuA. This ET pathway contains two short through‐space jumps: one from the Nζ atom of Lys13 (Cyt.c) to the Cζ atom of Tyr105 (subunit II of CcO) (a distance of 3.6 Å), and the other from the N atom of the main chain of Tyr105 (subunit II of CcO) to the Sδ atom of Met207 (subunit II of CcO) (a distance of 4.0 Å). The running distance along the pathway is 41.9 Å. Chemical modification of Lys13 of Cyt.c induces drastic inhibition on ET activity (Ferguson‐Miller et al, 1978; Osheroff et al, 1980). All vertebrate Cyt.c proteins contain Lys at the 13th residue and Cys at the 14th residue (Appendix Fig S6A); in addition, Tyr105 and Met207 of CcO subunit II are conserved among vertebrates (Appendix Fig S6B). These observations strongly suggest that the Cyt.c‐binding site in the Cyt.c–CcO complex structure is the catalytic binding site of Cyt.c through which electrons are transferred. Compared with the pathway in caa 3‐type CcO, only the ET from Met207 (subunit II of CcO) to CuA is conserved (Fig EV2) (Lyons et al, 2012).
Figure 2. Electron transfer pathway in the Cyt.c–CcO complex.
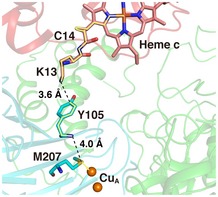
The possible electron transfer (ET) pathway from the heme c iron to CuA was explored using the Pathways plugin for VMD (Humphrey et al, 1996; Balabin et al, 2012). Through‐bond processes and through‐space jumps are represented by yellow solid lines and black dashed lines, respectively. The heme c group and amino‐acid residues of Cyt.c molecule are shown as pink sticks. The amino‐acid residues of subunit II of CcO are shown as cyan sticks. Oxygen, nitrogen, and sulfur atoms are shown in red, blue, and yellow, respectively. Orange spheres indicate CuA. Protein structures are drawn by transparent ribbons in the same colors as those of Fig 1.
Figure EV2. Comparison of the Cyt.c–CcO complex with caa 3‐type CcO (Lyons et al, 2012).
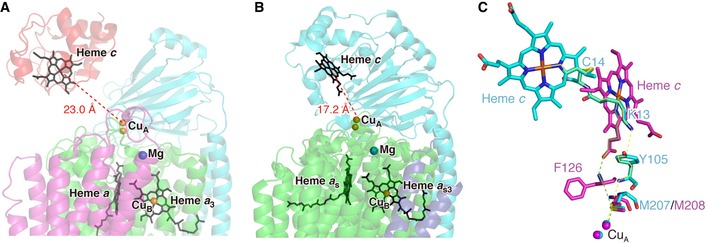
-
A, BArrangement of redox cofactors in the Cyt.c–CcO complex (A) and caa 3‐type CcO (B). Heme groups (c, a, a 3, a s, and a s3) are shown as black sticks. Copper and magnesium ions are represented by orange and blue spheres, respectively. Distances from iron of heme c to CuA are indicated.
-
CComparison of ET pathways from iron of heme c to CuA between the Cyt.c–CcO complex (cyan sticks and yellow lines) and caa 3‐type CcO (magenta sticks and green lines). Both structures are superposed with their CuA‐containing domains. Solid and dashed lines represent through‐bond and through‐space processes, respectively.
It has been proposed that Trp121 in Paracoccus denitrificans CcO (Trp104 in subunit II of bovine CcO) is the electron entry site from Cyt.c, based on W121Q mutation (Witt et al, 1998). The proposal was supported by some docking simulations (Roberts & Pique, 1999; Drosou et al, 2002). However, this mutation is likely to greatly influence the redox potential of CuA, because the side chain of Gln121 in the W121Q mutant is predicted to make hydrogen bonds with both the Sδ atom of Met227 and the Sγ atom of Cys220, which coordinate to copper ions of CuA (Appendix Fig S7B). Furthermore, the present X‐ray structure of the complex shows that Trp104 does not interact tightly with Cyt.c. There is a large gap between the protein surface around Trp104 and Cyt.c, in which Trp104 is separated from the closest atom of heme c by 9.0 Å (Appendix Fig S7A). Thus, the possible electron transfer pathway identified in the present X‐ray structural analyses suggests a significantly more facile electron transfer than the one through the structure including Trp104 (Appendix Fig S7A).
Catalytic binding sites
Speck et al (Speck et al, 1984) proposed a single‐catalytic site model including a catalytic site and a non‐catalytic regulatory site on CcO for Cyt.c to interpret the steady‐state kinetic results indicating two different Michelis–Menten kinetics, without giving any experimental confirmation. In other words, no experimental result has disproven the two‐catalytic site model (Ferguson‐Miller et al, 1976). Following the Speck's definition, the above structure strongly suggests the catalytic binding site, since the Cyt.c–CcO complex shows a facile electron transfer pathway from heme c to CuA. However, following the two‐catalytic site model, this retains both possibilities of the first and the second catalytic sites.
The positive side of the concave surface of CcO is negatively charged, whereas the surface area around the exposed heme edge of Cyt.c is positively charged (Fig 3A and B). Prominent inter‐molecular interactions in this region include six hydrogen bonds or salt bridges between CcO and Cyt.c (Fig 3C and Table 2). Lys8 (Cyt.c) interacts with Asp139 of subunit II (CcO) via a salt bridge, Gln12 (Cyt.c) forms hydrogen bonds with Asp139 (CcO subunit II), Lys13 (Cyt.c) forms hydrogen bonds with Tyr105 and Tyr121 (CcO subunit II) and a salt bridge with Asp119 (CcO subunit II), and Lys87 (Cyt.c) forms hydrogen bonds with Ser117 (CcO subunit II). These four interacting residues of Cyt.c are restricted to the molecular surface near the exposed heme edge (Fig 3B). On the basis of chemical modification and kinetic studies (Ferguson‐Miller et al, 1978), three lysine residues, Lys8, Lys13, and Lys87, were predicted to interact with CcO. Recent site‐directed mutagenesis and kinetics studies of Cyt.c indicated that the ET activities of K13L, K86L/K87L, and K7L/K8L mutants are significantly lower than that of the wild‐type protein (Sato et al, 2016). The side chains of Lys8, Gln12, Lys13, and Lys87 of Cyt.c, as well as the side chains of Tyr105, Asp119, Ser117, Tyr121, and Asp139 of CcO subunit II, provide the physiological electron transfer complex, not an encounter complex under non‐physiological conditions.
Figure 3. Cyt.c–CcO interaction.
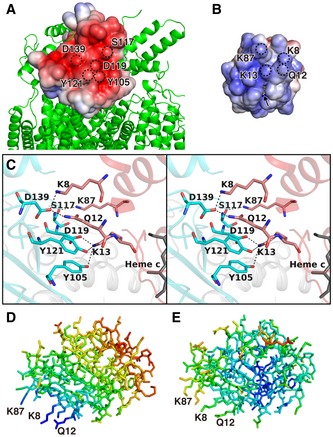
-
A, BOpen‐book view of electrostatic potentials of the interaction surfaces of subunit II (residues 91–227) of CcO (A) and Cyt.c (B) in the Cyt.c–CcO complex. Electrostatic potentials were calculated separately using the program APBS (Baker et al, 2001). The displayed potentials range from −5 (red) to 5 (blue) kTe−1. Heme c (yellow sticks), slightly exposed to the surface, is indicated by an arrow. Dotted circles indicate sites of amino‐acid residues, indicated by single‐letter notation with residue number.
-
CClose‐up view of the interaction site, shown as a stereoscopic pair. Amino‐acid residues involved in the interaction between Cyt.c and CcO are represented by pink (Cyt.c) and cyan sticks (subunit II of CcO). Oxygen and nitrogen atoms are shown in red and blue, respectively. Hydrogen bonds and salt bridges are shown as dashed lines. Amino‐acid residues are indicated by single‐letter notation with residue number.
-
D, EComparison of variation in B‐factors in the Cyt.c molecule. Stick representation of Cyt.c molecule in the Cyt.c–CcO complex (D) and free Cyt.c (PDB 1HRC) (E); colors represent B‐factor ranging from 37.7 Å2 (blue) to 136.0 Å2 (red) in (D) and 5.8 Å2 (blue) to 77.4 Å2 (red) in (E).
Table 2.
Protein–protein distances between Cyt.c and CcO
| Cyt.c | CcO (subunit II) | Distances (Å) |
|---|---|---|
| Heme c Fe | CuA (CU1) | 23.0 |
| Lys8 Nζ | Asp139 Oδ2 | 2.7 |
| Gln12 Nε2 | Asp139 Oδ2 | 2.9 |
| Lys13 Nζ | Tyr105 Oη | 3.3 |
| Lys13 Nζ | Asp119 Oδ2 | 2.6 |
| Lys13 Nζ | Tyr121 Oη | 3.1 |
| Lys87 Nζ | Ser117 O | 2.9 |
A previous NMR study (Sakamoto et al, 2011) detected structural changes in several hydrophobic amino‐acid residues of Cyt.c upon the docking of two proteins, and the authors of that study concluded that Cyt.c interacted with CcO via its non‐polar surface surrounding the heme cleft, as in the cytochrome bc 1 complex (Cyt.bc 1)–Cyt.c (Lange & Hunte, 2002) and Cyt.c–cytochrome c peroxidase (CcP) complexes (Jasion et al, 2012). By contrast, our crystal structure of the Cyt.c–CcO complex has no inter‐molecular interactions between hydrophobic amino acids with an inter‐atomic distance < 5 Å. This is likely because NMR spectroscopy sensitively detected a small structural change undetectable by X‐ray, mediated by an interaction between the residues of Cyt.c and CcO via water molecules present between the two proteins.
The ionic interaction between Lys13 (Cyt.c) and Asp119 (CcO) was predicted by a docking simulation (Roberts & Pique, 1999), and another docking simulation assigned Lys8, Lys13, and Lys87 of Cyt.c as residues interacting with CcO (Sato et al, 2016), as observed in this study. However, inconsistencies remain between the X‐ray structure and the simulated structures of Cyt.c–CcO complex. The former simulation predicted that Lys72 (Cyt.c), which is distant from CcO in the complex structure (Appendix Fig S8), interacts with Gln103 and Asp158 of CcO subunit II. The most probable structure from the latter simulation indicated that the subunit I of CcO had a larger contact surface area with Cyt.c than subunit II of CcO, whereas in our structure most of the contact surface of CcO with Cyt.c belongs to subunit II. These inconsistencies likely arose because water molecules are present between Cyt.c and CcO, but bulk waters were removed from the surfaces of both proteins in the docking simulations.
Amino‐acid residues included in the catalytic binding were assigned based on the Cyt.c–CcO complex structure equilibrated in a solution in which the enzyme exerts its normal catalytic activity (Yonetani & Ray, 1965). The interactions between Cyt.c and CcO elucidated by this crystallographic study are consistent with those revealed for the enzyme–substrate complex under turnover conditions by previous experimental studies involving chemical modifications and kinetics (Ferguson‐Miller et al, 1978) or solution NMR and kinetics for complexes containing wild‐type and mutant Cyt.c proteins (Sakamoto et al, 2011; Sato et al, 2016).
Novel protein–protein interaction scheme
Cyt.c donates electrons to CcO and (CcP) and accepts electrons from (Cyt.bc 1). We compared the interaction scheme of the Cyt.c–CcO complex with those of the Cyt.c–CcP (Pelletier & Kraut, 1992; Jasion et al, 2012) and the Cyt.bc 1–Cyt.c complex (Lange & Hunte, 2002; Solmaz & Hunte, 2008). The shortest distance between two Cα atoms of Cyt.c and CcO is 8.2 Å. By contrast, the shortest distances in the Cyt.c–CcP (PDB 4GED) and Cyt.bc 1–Cyt.c (PDB 3CX5) complexes are much shorter, 5.3 and 5.6 Å, respectively. Thus, Cyt.c in the Cyt.c–CcO complex is farther from CcO than it is from CcP and Cyt.bc 1 in the corresponding complexes. Ahmed et al (2011) compiled 179 X‐ray structures of protein–protein complexes from the RSCB Protein Data Bank (Berman et al, 2000). The inter‐molecular Cα distances of these 179 structures were calculated, and the distribution of the shortest distance in each complex is illustrated in Appendix Fig S9. Notably, the shortest distance in the Cyt.c–CcO complex, 8.2 Å, falls well outside the distribution. Furthermore, the contact surface areas for three ET complexes were calculated by removing surface water molecules. The area of the Cyt.c–CcO complex (222.8 Å2) is approximately one‐third that of the Cyt.c–CcP complex (615.2 Å2), and less than one‐fourth that of Cyt.bc 1–Cyt.c (1008.7 Å2). Thus, fewer direct protein–protein interactions are involved in formation of the Cyt.c–CcO complex than either of the other two complexes. No direct interaction (< 5.0 Å) between hydrophobic residues was detected in the Cyt.c–CcO complex, whereas the other two complexes have several non‐polar groups involved in their inter‐molecular interactions.
The water molecules within 7 Å of both proteins of Cyt.c and CcO fall into three categories, as noted by Ahmed et al (2011): bridging waters that interact with both proteins; non‐bridging waters that interact with one but not both proteins; and non‐interacting waters that are more than 3.5 Å from both proteins. In this study, interactions between waters and proteins atoms were assigned based on a distance of < 3.5 Å between the water oxygen atom and the nearest atom of the protein. As shown in Fig 4 and Appendix Table S1, more water molecules are present between Cyt.c and CcO than between Cyt.c and CcP or Cyt.bc 1 and Cyt.c, and there are a total of 14 non‐interacting water molecules in the Cyt.c–CcO complex. By contrast, the Cyt.c–CcP and Cyt.bc 1–Cyt.c complexes each have only four and two non‐interacting water molecules, respectively. Almost the same numbers of bridging waters are in three ET complexes. Extremely fewer non‐bridging waters are at Cyt.c in the both complexes of Cyt.bc 1–Cyt.c and Cyt.c–CcP than Cyt.c–CcO complex (Fig 4; Appendix Table S1). Hydrophobic residues of Cyt.c in the Cyt.bc 1–Cyt.c complex and the Cyt.c–CcP complex likely contact directly with their counterpart and remove waters in part from the surface of their Cyt.c.
Figure 4. Comparison of the distribution of water molecules between Cyt.c and each redox partner.
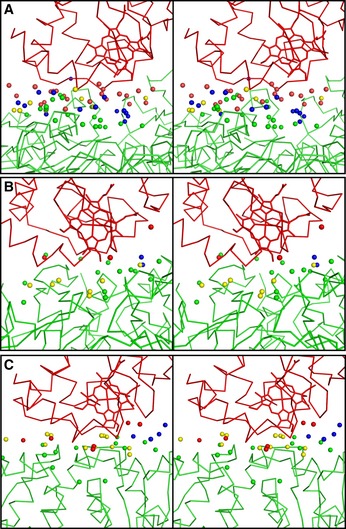
-
A–CStereo views of water molecules between proteins in Cyt.c–CcO (A), Cyt.bc 1–Cyt.c (PDB 3CX5) (B), and Cyt.c–CcP complexes (PDB ID 4GED) (C). Bridging and non‐interacting waters are shown in yellow and blue, respectively. Non‐bridging waters interacting with Cyt.c are red spheres, and that interacting with redox partners are shown in green. Cyt.c and the redox partners are shown by wire models in red and green, respectively.
Any water in the Cyt.c–CcO complex has at least one hydrogen bond with a protein atom or a water molecule. Each of the bridging and non‐bridging waters in the Cyt.c–CcO complex interacts, on average, with three polar atoms or waters and one non‐polar atom (Appendix Table S2). The waters at CcO interact prominently with Asp, whereas those at Cyt.c interact mainly with Lys and Gln (Appendix Table S3). At least two water molecules closely contact a non‐interacting water molecule. These water molecules construct a hydrogen bond network between Cyt.c and CcO (Fig EV3). The averaged B‐factor of 64 waters is 62.7 Å2, which is between that of CcO (37.9 Å2) and that of Cyt.c (88.2 Å2).
Figure EV3. Structure of waters around the direct interaction region between Cyt.c and CcO.
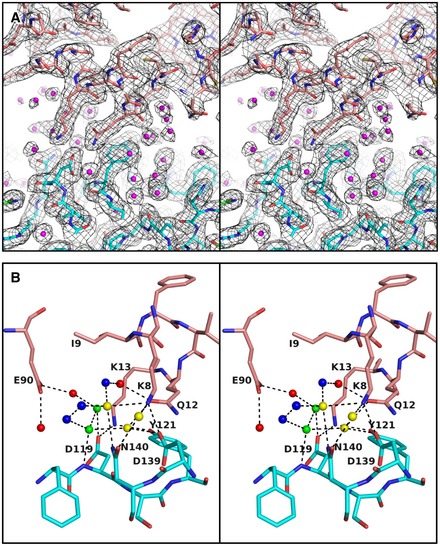
- Stereoscopic view of the 2(F o–F c) map of the Cyt.c–CcO complex, drawn at the 1.0 σ level. Each water molecule is clearly assigned in the map.
- Stereoscopic view of water structure. Each water molecule is drawn in the same color as in Fig 4A. Hydrogen bond networks consisting of water molecules including non‐interacting waters link Cyt.c and CcO.
Out of 19 non‐bridging waters at CcO in the complex, 14 are located at almost the same sites in the CcO crystal (PDB 5B1A), four are in slightly shifted positions, and one water is not assigned in the CcO crystal. Out of 23 non‐bridging waters at Cyt.c in the complex, only five waters are present in the Cyt.c crystal structure (PDB 1HRC). This is probably because the interacting sites of Cyt.c in the Cyt.c–CcO complex are involved in the tight contacts of crystal packing in the Cyt.c crystal, which removes waters from the molecular surface upon crystallization. Because the protein volumes of Cyt.c–CcO and CcO crystals are ~30% of their unit cell volume, significantly lower than that of Cyt.c crystal, more than 40%, non‐bridging water sites are common to the Cyt.c–CcO and CcO crystals.
When Cyt.c docks with CcO, both proteins preserve their main chain folds, and retain water molecules on their surfaces, and they interact with each other via the long arms of side chains (Fig 5A). On the other hand, the docking of Cyt.c and CcP or Cyt.bc 1 leads to the exclusion of water molecules from the surface of each protein (Fig 5B). The chemical shift‐perturbed residues of ferri‐Cyt.c associated with the binding of CcO (Sakamoto et al, 2011) are not affected by direct protein–protein interactions, but are influenced by indirect interactions via the water layers in the crystal structure.
Figure 5. Schematic representation of the distribution of water molecules at the protein–protein interaction site.
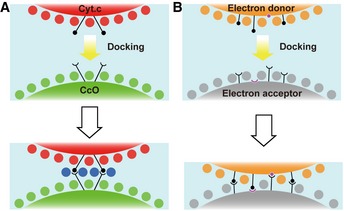
- In the Cyt.c–CcO complex system, water molecules on the surfaces of each protein are preserved to form three layers upon docking, but each protein specifically interacts via the long arms of side chains. Red and green circles represent water molecules on the surfaces of proteins of the corresponding color; blue circles represent water molecules belonging to the non‐interacting water. Black circles and semicircles represent side chains of residues involved in protein–protein interactions.
- In other ET complex systems, electron donor and acceptor proteins form an ET complex by excluding water molecules from the surface of each protein. Orange and gray circles represent water molecules on the surfaces of proteins of the corresponding color. Magenta circles and semicircles represent the main chains of residues involved in protein–protein interactions. Black circles and semicircles represent side chains of residues involved in protein–protein interactions.
The main chain folds of CcO and Cyt.c in Cyt.c–CcO complex are almost identical to those of the corresponding crystals, with Cα r.m.s.d. values of 0.47 and 0.41 Å, respectively. All the CcO side chain structures in the region interacting with Cyt.c are similar to those of the CcO crystal except for Asn203(subunit II of CcO), where the two structures are different from each other by a −90° rotation angle around the Cβ–Cγ bond. By contrast, several side chains of Cyt.c in the interacting region have different orientations between the crystals of Cyt.c–CcO and Cyt.c, probably because of packing effects in the Cyt.c crystal, as noted above for non‐bridging waters at Cyt.c.
The B‐factors of the side chain atoms of Nζ (Lys8), Nε (Gln12), Nζ (Lys13), and Nζ (Lys87) of Cyt.c, which interact with residues of CcO, are 37.7, 39.3, 41.7, and 48.7 Å2, respectively, significantly lower than the average B‐factor of Cyt.c (88.2 Å2) and as low as that of the extracellular domain (residues 91–227) of CcO subunit II (35.2 Å2). By contrast, the B‐factors of Cyt.c atoms increase with distance from these CcO‐interacting residues (Fig 3D). Cyt.c in the Cyt.c–CcO complex undergoes a large fluctuation, using the regions interacting with CcO as a fulcrum. The variation in the B‐factors in Cyt.c molecule of the Cyt.c–CcO complex is another unique feature of the ET complex. However, horse ferri‐ and ferro‐Cyt.c and the Cyt.c molecules in the Cyt.c–CcP and Cyt.bc 1–Cyt.c complexes exhibit low B‐factors at the heme c group (Figs 3E and EV4). The high flexibility of Cyt.c in the Cyt.c–CcO complex is likely to compensate for the entropy loss caused by introduction of more waters upon docking.
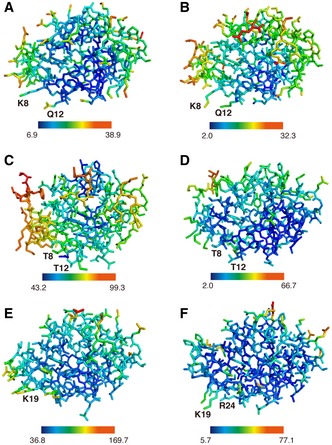
Figure EV4. Distribution of B‐factors of Cyt.c molecules.
-
A–FStick representations of ferri‐Cyt.c (A; PDB 3O1Y), ferro‐Cyt.c (B; PDB 3O20), Cyt.c portion of yeast Cyt.bc 1–Cyt.c complex (C; PDB 3CX5), yeast iso‐1 Cyt.c (D; PDB 1YCC), Cyt.c portion of Leishmania major Cyt.c–CcP complex (E; PDB 4GED), and Leishmania major Cyt.c (F; PDB 4DY9). All structures are viewed from the same direction. The B‐factor color scale is provided below in Å2 unit. Heme c atoms of each Cyt.c have lower B‐factors than those of peripheral residues.
The inter‐molecular interaction between Cyt.c and CcO is characterized by mutual recognition mediated by a few long arms of hydrophilic amino acids, small contact surface, a long span between the two proteins, the presence of three water layers between the two proteins, and a large fluctuation of Cyt.c that uses the regions that interact with CcO as a fulcrum. The non‐interacting water molecules in the Cyt.c–CcO complex exist in vacant spaces around the interacting amino‐acid residues of both proteins (Fig 6) and closely contact with water molecules, thus providing hydrogen bond network between Cyt.c and CcO (Fig EV3). This novel mode of protein–protein interaction, which we term “soft and specific contact”, is not observed in other ET complexes (Table 3) (Shen et al, 1994; Morales et al, 2000; Müller et al, 2001; Axelrod et al, 2002; Darnault et al, 2003; Ashikawa et al, 2006; Sukummar et al, 2006; Dai et al, 2007; Hagelueken et al, 2007; Senda et al, 2007; Nojiri et al, 2009; Fritz‐Wolf et al, 2011; Hiruma et al, 2013; Yukl et al, 2013; Acheson et al, 2014; McGrath et al, 2015).
Figure 6. Locations of four interacting residues in relation to Cyt.c and water molecules that engage in no direct interactions with either protein in the Cyt.c molecule.
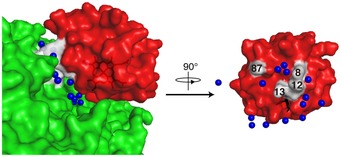
Cyt.c (red) and CcO (green) are shown as surface representations. The heme c group shown as black sticks is slightly exposed to the surface. Blue spheres represent water molecules belonging to the non‐interacting water. Amino‐acid residues involved in interactions between Cyt.c and CcO are shown in white, and figures indicate their residue numbers.
Table 3.
Shortest distance between Cα atoms and contact surface areas of ET complexes
| PDB | Distance (Å) | CSAa (Å2) | Resolution (Å) | References |
|---|---|---|---|---|
| 5IY5b | 8.24 | 222.8 | 2.00 | This study |
| 4GEDc | 5.28 | 615.2 | 1.84 | Jasion et al (2012) |
| 3CX5d | 5.57 | 1008.7 | 1.90 | Solmaz and Hunte (2008) |
| 1E6E | 4.71 | 1157.8 | 2.30 | Müller et al (2001) |
| 1EWY | 4.10 | 1361.1 | 2.38 | Morales et al (2000) |
| 1L9B | 5.30 | 722.8 | 2.40 | Axelrod et al (2002) |
| 1OAO | 4.40 | 2248.3 | 1.90 | Darnault et al (2003) |
| 2DE5 | 4.48 | 932.2 | 1.90 | Ashikawa et al (2006) |
| 2GC4 | 4.36 | 425.2 | 1.90 | Shen et al (1994) |
| 2IAA | 4.91 | 557.1 | 1.95 | Sukummar et al (2006) |
| 2PU9 | 4.67 | 952.0 | 1.65 | Dai et al (2007) |
| 2PVG | 5.53 | 636.6 | 2.40 | Dai et al (2007) |
| 2V3B | 4.76 | 657.1 | 2.45 | Hagelueken et al (2007) |
| 2YVJ | 5.11 | 690.9 | 1.90 | Senda et al (2007) |
| 2ZON | 4.06 | 533.6 | 1.70 | Nojiri et al (2009) |
| 3QFA | 4.81 | 631.7 | 2.20 | Fritz‐Wolf et al (2011) |
| 3W9C | 5.36 | 595.9 | 2.50 | Hiruma et al (2013) |
| 4FA9 | 4.05 | 1433.8 | 2.09 | Yukl et al (2013) |
| 4PIB | 4.29 | 798.7 | 2.05 | Acheson et al (2014) |
| 4PW9 | 4.41 | 739.1 | 2.49 | McGrath et al (2015) |
Contact surface area was calculated with the program AREAIMOL (Lee & Richards, 1971) in CCP4.
Cyt.c–CcO complex.
Cyt.bc 1–Cyt.c complex.
Cyt.c–CcP complex.
The same region of Cyt.c interacts with Cyt.bc 1 in the Cyt.bc 1–Cyt.c complex crystal (Lange & Hunte, 2002; Solmaz & Hunte, 2008) and with CcO in the Cyt.c–CcO complex crystal, as proposed based on the results of a chemical modification study (Rieder & Bosshard, 1980). Cyt.c receives and donates electrons through the same site via a repeated association/dissociation process. The novel mode of protein–protein interaction discovered in this study is likely to decrease the potential barrier caused by structural changes upon association/dissociation of the Cyt.c–CcO complex because conformational change of both proteins and rearrangement of surface waters are not required upon docking. Therefore, soft and specific contact between the two proteins is important for efficient donation of four electrons from Cyt.c to CcO for the O2 reduction reaction. It is remarkable that the X‐ray structure of the interface of Cyt.c–CcO complex facilitating the electron transfer from heme c to CuA is greatly different from that from heme c 1 to heme c, indicating the electron tranfering structures in these two complexes are speciallized for the different electron transfer processes [e.g., two electron transfer from heme c 1 to heme c versus four electron transfer from heme c to CuA; and different molar contents of 3:7:9 for Cyt.bc 1, CcO, and Cyt.c (Hatefi & Galante, 1978)]. Further structural and functional comparisons of these complexes would develop insights in the mechanism of the energy trunsduction by the mitochondrial electron transfer system.
We hypothesize that there are many cases of soft and specific protein–protein interactions involved in various cellular processes. One reason why these interactions were not discovered previously may be related to the need to perform extensive searches for optimal crystallization conditions for these intrinsically unstable protein complexes, as described above. Because single‐particle analysis by cryo‐electron microscopy does not require a crystal, a high‐resolution single‐particle analysis would increase the chance of detecting soft and specific protein–protein interaction.
Materials and Methods
Preparation of horse heart Cyt.c sample
For each crystallization trial, horse heart Cyt.c (Nacalai Tesque) was freshly dissolved in 15 mM sodium phosphate buffer at pH 8.0, and then dialyzed for 1 h against the same buffer to remove remaining salts. The concentration of Cyt.c was calculated from the absorption spectrum of the fully dithionite‐reduced form, using Δε550–535 nm = 19.2 mM−1 cm−1.
Crystallization of the Cyt.c–CcO complex
CcO in the fully oxidized state was purified from bovine heart mitochondria (Tsukihara et al, 1995) and dissolved in 40 mM sodium phosphate buffer (pH 6.8) containing 0.2% (w/v) n‐decyl‐β‐D‐maltoside (DM) (Dojin). CcO was diluted 10‐fold in 15 mM sodium phosphate buffer (pH 8.0) containing 0.7% (w/v) fluorinated octyl‐maltoside (FOM) (Anatrace). CcO at pH 8.0 preserved Cyt.c oxidation activity at ~50% of the level at pH 7.0, as reflected by V max (Yonetani & Ray, 1965). FOM‐treated CcO was concentrated using a membrane filter (Amicon Ultra Centrifugal Filters (100 kDa), Millipore). The concentration of CcO was calculated from the absorption spectrum of the fully dithionite‐reduced form, using Δε604–630 nm = 46.6 mM−1 cm−1. CcO solubilized with DM and FOM was mixed with Cyt.c at a Cyt.c/CcO molar ratio of 1.2. Co‐crystallization of Cyt.c and CcO was performed by the batch‐wise method at 277 K; Cyt.c–CcO (100 mg/ml CcO, ~0.5 mM CcO) was mixed with ~5% polyethylene glycol (PEG) 4000, a precipitant. Rectangular plates of Cyt.c–CcO complex crystals were obtained within 1 day. The crystals were gradually soaked in a crystallization solution containing both 50 μM Cyt.c and ethylene glycol (EG) as cryo‐protectant, reaching final concentrations of 40% EG and 6% PEG 4000. After cryo‐protection, crystals were quickly frozen in a cryo‐nitrogen stream at 100 K.
Structure determination
Crystal screening and X‐ray experiments were carried out at beamline BL26B2 and BL44XU of SPring‐8. The dataset for the structural analysis was obtained at BL44XU, equipped with an MX300HE CCD detector. The X‐ray beam cross‐section for X‐ray diffraction experiments was 50 × 50 μm at the crystal, and the wavelength was 0.9 Å. Photon number at the sample position was 3.0 × 1011 photons/s. For data acquisition at 100 K, the crystals were frozen in a cryo‐nitrogen stream. The dataset was collected with an exposure time of 1 s and a 0.5° oscillation angle over 180°. Diffraction images were processed and scaled with DENZO and SCALEPACK (Otwinowski & Minor, 1997), respectively, and the datasets from the two crystals were merged. A total of 720 images were successfully processed and scaled. The structure factor amplitude (|Fo|) was calculated using the CCP4 program TRUNCATE (French & Wilson, 1978; Weiss, 2001; Winn et al, 2011). The crystal belongs to space group P21, with unit cell dimensions of a = 113.3 Å, b = 183.9 Å, c = 148.9 Å, and β = 102.1°. The asymmetric unit of the crystal lattice contains two complexes of CcO and Cyt.c. The solvent content and V M were 65.6% and 3.58 Å3 Da−1, respectively (Matthews, 1968).
CcO was initially located in the unit cell at 3.0‐Å resolution by the molecular replacement (MR) method (Rossmann & Blow, 1962) using the program MOLREP in CCP4 (Collaborative Computational Project 4, 1994) with the fully oxidized CcO structure, previously determined at 1.8‐Å resolution (PDB 2DYR) (Shinzawa‐Itoh et al, 2007), as a model. Cyt.c was located following the MR search at 3.0‐Å resolution using horse Cyt.c (PDB 1HRC) (Bushnell et al, 1990) as a model, as in the previous case. Initial phases up to 5.0‐Å resolution were calculated with atomic parameters determined by MR and extended to 2.0‐Å resolution by density modification (Wang, 1985) coupled with non‐crystallographic symmetry averaging (Bricogne, 1974, 1976) using the CCP4 program DM (Cowtan, 1994). The resultant phase angles (αMR/DM) were used to calculate the electron‐density map (MR/DM map) with Fourier coefficients |F o| exp(iαMR/DM) and the anomalous difference electron‐density map with Fourier coefficients (|F o +| – |F o −|) exp[i(αMR/DM – π/2), where |F o| is the observed structure amplitude and |F o +| – |F o −| is the Bijvoet difference in |F o|. The anomalous difference electron‐density map indicated the Fe, Cu, and Zn positions, including the heme irons of Cyt.c. The structural model of Cyt.c–CcO was built in the MR/DM map. The structure was refined using the program REFMAC (Winn et al, 2001; Murshudov et al, 2011) at 2.0‐Å resolution. Bulk solvent correction and anisotropic scaling of the observed and calculated structure amplitudes and TLS parameters were incorporated into the refinement calculation. The anisotropic temperature factors for iron, copper, and zinc atoms were imposed on the calculated structure factors. Because the two crystallographically independent monomers packed differently in the crystal, each monomer of CcO was assigned to a single TLS group in the REFMAC refinement. The quality of the structural refinement was characterized by the R and R free values. F o–F c maps were calculated with Fourier coefficients (|F o| – |F c|) exp(iαc), where |F c| and αc are the calculated structure amplitude and phase, respectively, obtained in the structural refinement. Out of 3822 amino‐acid residues, 56 residues of CcO could not be located in the electron‐density maps of the Cyt.c–CcO complex. A total of 33 residues of CcO have multiple conformations. The structure refinement was well converged: R = 0.167 and R free = 0.207. The root‐mean‐square deviations (r.m.s.d.) of bond lengths and angles from their ideal values were 0.023 Å and 2.0°, respectively.
Electrostatic potential was calculated separately for Cyt.c and subunit II of CcO using APBS (Baker et al, 2001). Accessible surface area was calculated with the program AREAIMOL (Lee & Richards, 1971) in CCP4, using a probe with radius 1.4 Å.
Electron transfer pathway calculation
The possible ET pathways from the heme c iron to CuA of CcO were explored by an empirical method, the Pathways plugin for VMD (Humphrey et al, 1996; Balabin et al, 2012), which evaluates the donor‐to‐acceptor tunneling coupling (T DA) value for each pathway. We attached hydrogen atoms based on the charmm22 force field (MacKerell et al, 1998), and performed energy minimization with respect to the hydrogen atoms, which enabled us to positively identify the hydrogen bonds. To evaluate the diversity of the possible ET pathways, we generated 200 candidates, and found that most of the identified ET pathways and their T DA values were similar. In fact, the top 100 solutions contained the route between Lys13 (Cyt.c) and Tyr105 (CcO). Accordingly, we took the ET pathway with the most efficient T DA value as the final solution.
Accession numbers
Atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession code 5IY5.
Author contributions
KS‐I, SY, and TT designed the research; SS, KS‐I, JB, SA, AS, EY, JK, MT, SY, and TT performed the research; SS, KS‐I, and SA performed protein purification and crystallization experiments; SS, KS‐I, JB, SA, AS, EY, and TT performed X‐ray diffraction experiments and analyzed X‐ray data; JK and MT carried out molecular dynamics simulations; SS, KS‐I, AS, MT, SY, and TT wrote the manuscript; and all authors discussed and commented on the results and the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank the staff members of BL26B2/SPring‐8 for their extensive support. This work was supported by JST, CREST (to TT), JSPS KAKENHI Grant 26291033 (to SY), and 25287099 (to MS). SY is a senior visiting scientist at the RIKEN Harima Institute. Diffraction data were collected at BL44XU of the SPring‐8 facility under proposals 2014A6500, 2014B6500, and 2015A6500, and at BL26B2 under proposals 2014A1846 and 2014A1860. Computations for electron transfer pathway were performed using the Computer Center for Agriculture, Forestry, and Fisheries Research, MAFF, Japan.
The EMBO Journal (2017) 36: 291–300
See also: JA Lyons & P Nissen (February 2017)
Contributor Information
Kyoko Shinzawa‐Itoh, Email: shinzawa@sci.u-hyogo.ac.jp.
Tomitake Tsukihara, Email: tsuki@protein.osaka-u.ac.jp.
References
- Acheson JF, Bailey LJ, Elsen NL, Fox BG (2014) Structural basis for biomolecular recognition in overlapping binding sites in a diiron enzyme system. Nat Commun 5: 5009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed MH, Spyrakis F, Cozzini P, Tripathi PK, Mozzarelli A, Scarsdale JN, Safo MA, Kellogg GE (2011) Bound water at protein‐protein interfaces: partner, roles and hydrophobic bubbles as a conserved motif. PLoS ONE 6: e24712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashikawa Y, Fujimoto Z, Noguchi H, Habe H, Omori T, Yamane H, Nojiri H (2006) Electron transfer complex formation between oxygenase and ferredoxin components in Riske nonheme iron oxygenase system. Structure 14: 1779–1789 [DOI] [PubMed] [Google Scholar]
- Axelrod HL, Abresch EC, Okamura MY, Yeh AP, Rees DC, Feher G (2002) X‐ray structure determination of the cytochrome c 2: reaction center electron transfer complex from Rhodobacter sphaeroides . J Mol Biol 319: 501–515 [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and ribosome. Proc Natl Acad Sci USA 98: 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balabin IA, Hu X, Beratan DN (2012) Exploring biological electron transfer pathway dynamics with the pathways plugin for VMD. J Comput Chem 33: 906–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliand G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucl Acids Res 28: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G (1974) Geometric sources of redundancy in intensity data and their use for phase determination. Acta Crystallogr A 30: 395–405 [Google Scholar]
- Bricogne G (1976) Methods and programs for direct‐space exploitation of geometric redundancies. Acta Crystallogr A 32: 832–847 [Google Scholar]
- Bushnell GW, Louie GV, Brayer GD (1990) High‐resolution three‐dimensional structure of the horse heart cytochrome c. J Mol Biol 214: 585–595 [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Cowtan K (1994) “DM”: an automated procedure for phase improvement by density modification. Joint CCP4 ESF‐EACBM Newslett Protein Crystallogr 31: 34–38 [Google Scholar]
- Dai S, Friemann R, Glauser DA, Bourquin F, Manieri W, Schürmann P, Eklund H (2007) Structural snapshots along the reaction pathway of ferredoxin‐thioredoxin reductase. Nature 448: 92–96 [DOI] [PubMed] [Google Scholar]
- Darnault C, Volbeda A, Kim EJ, Legrand P, Vernède X, Lindahl PA, Fontecilla‐Camps JC (2003) Ni‐Zn‐[Fe4‐S4] and Ni‐Ni‐[Fe4‐S4] clusters in closed and open α subunits of acetyl‐CoA synthase/carbon monooxide dehydrogenase. Nat Struct Mol Biol 10: 271–279 [DOI] [PubMed] [Google Scholar]
- De March M, Demitri N, De Zorzi R, Casini A, Gabbiani C, Guerri A, Messori L, Geremia S (2014) Nitrate as a probe of cytochrome c surface: crystallographic identification of crucial “hot spots” for protein‐protein recognition. J Inorg Biochem 135: 58–67 [DOI] [PubMed] [Google Scholar]
- Drosou V, Malatesta F, Ludwig B (2002) Mutations in the docking site for cytochrome c on the Paracoccus heme aa 3 oxidase: electron entry and kinetic phases of the reaction. Eur J Biochem 269: 2980–2988 [DOI] [PubMed] [Google Scholar]
- Ferguson‐Miller S, Brautigan DL, Margoliash E (1976) Correlation of the kinetics of electron transfer activity of various eukaryotic cytochromes c with binding to mitochondrial cytochrome c oxidase. J Biol Chem 251: 1104–1115 [PubMed] [Google Scholar]
- Ferguson‐Miller S, Brautigan DL, Margoliash E (1978) Definition of cytochrome c binding domains by chemical modification III. Kinetics of reaction of carboxydinitrophenyl cytochrome c with cytochrome c oxidase. J Biol Chem 253: 149–159 [PubMed] [Google Scholar]
- Ferguson‐Miller S, Babcock GT (1996) Heme/copper terminal oxidases. Chem Rev 96: 2889–2908 [DOI] [PubMed] [Google Scholar]
- French S, Wilson K (1978) On the treatment of negative intensity observations. Acta Crystallogr A 34: 517–525 [Google Scholar]
- Fritz‐Wolf K, Kehr S, Stumpf M, Rahlfs S, Becker K (2011) Crystal structure of the human thioredoxin reductase‐thioredoxin complex. Nat Commun 2: 383 [DOI] [PubMed] [Google Scholar]
- Hagelueken G, Wiehlmann L, Adams TM, Kolmar H, Heinz DW, Tummler B, Schubert WD (2007) Crystal structure of the electron transfer complex rubredoxin‐rubredoxin reductase of Pseudomonas aeruginosa . Proc Natl Acad Sci USA 104: 12276–12281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatefi Y, Galante YM (1978) Organization of the mitochondrial respiratory chain In Energy conservation in biological membrane, Schaefer G, Klingenberg M. (eds), pp. 19–30. Berlin: Springer‐Verlag; [Google Scholar]
- Hiruma Y, Hass MAS, Kikui Y, Liu WM, Ölmez B, Skinner SP, Blok A, Kloosterman A, Koteishi H, Löhr F, Schwalbe H, Nojiri M, Ubbink M (2013) The structure of the cytochrome P450cam‐putidaredoxin complex determined by paramagnetic NMR spectroscopy and crystallography. J Mol Biol 425: 4353–4365 [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14: 33 38 27–38 [DOI] [PubMed] [Google Scholar]
- Jasion VS, Doukov T, Pineda SH, Li H, Poulos TL (2012) Crystal structure of the Leishmania major peroxidase‐cytochorme c complex. Proc Natl Acad Sci USA 109: 18390–18394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange C, Hunte C (2002) Crystal structure of the yeast cytochrome bc 1 complex with its bound substrate cytochrome c . Proc Natl Acad Sci USA 99: 2800–2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Richards FM (1971) The interpretation of protein structures: estimation of static accessibility. J Mol Biol 55: 379–400 [DOI] [PubMed] [Google Scholar]
- Lyons JA, Aragao D, Slattery O, Pisliakov AV, Soulimane T, Caffrey M (2012) Structural insights into electron transfer in caa 3‐type cytochrome oxidase. Nature 487: 514–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph‐McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE et al (1998) All‐atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102: 3586–3616 [DOI] [PubMed] [Google Scholar]
- Matthews BW (1968) Solvent content of protein crystals. J Mol Biol 33: 491–497 [DOI] [PubMed] [Google Scholar]
- McGrath AP, Laming EL, Garcia GPC, Kvansakul M, Guss JM, Trewhella J, Calmes B, Bernhardt PV, Hanson GR, Kappler U, Maher MJ (2015) Structural basis of interprotein electron transfer in bacterial sulfite oxidation. eLife 4: e09066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales R, Kachalova G, Vellieux F, Charon MH, Frey M (2000) Crystallographic studies of the interaction between the ferredoxin‐NADP+ reductase and ferredoxin from the cyanobacterium Anabaena: looking for the elusive ferredoxin molecule. Acta Crystallogr D 56: 1408–1412 [DOI] [PubMed] [Google Scholar]
- Müller JJ, Lapko A, Bourenkov G, Ruckpaul K, Heinemann U (2001) Adrenodoxin reductase‐adrenodoxin complex structure suggests electron transfer path in steroid biosynthesis. J Biol Chem 276: 2786–2789 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D 67: 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojiri M, Koteishi H, Nakagami T, Kobayashi K, Inoue T, Yamaguchi K, Suzuki S (2009) Structural basis of inter‐protein electron transfer for nitrite reduction in denitrification. Nature 462: 117–120 [DOI] [PubMed] [Google Scholar]
- Osheroff N, Brautigan DL, Margoliash E (1980) Definition of enzymic interaction domains on cytochrome c . J Biol Chem 255: 8245–8251 [PubMed] [Google Scholar]
- Osuda Y, Shinzawa‐Itoh K, Tani K, Maeda S, Yoshikawa S, Tsukihara T, Gerle C (2016) Two‐dimensional crystallization of monomeric bovine cytochrome c oxidase with bound cytochrome c in reconstituted lipid membranes. Microscopy 65: 263–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Pelletier H, Kraut J (1992) Crystal structure of a complex between electron transfer partners, cytochrome c peroxidase and cytochrome c . Science 252: 1285–1288 [DOI] [PubMed] [Google Scholar]
- Rieder R, Bosshard R (1980) Comparison of the binding sites on cytochrome c for cytochrome c oxidase, cytochrome bc 1, and cytochrome c 1 . J Biol Chem 255: 4732–4739 [PubMed] [Google Scholar]
- Roberts VA, Pique ME (1999) Definition of the interaction domain for cytochrome c on cytochrome c oxidase: III. Prediction of the docked complex by a complete, systematic search. J Biol Chem 274: 38051–38060 [DOI] [PubMed] [Google Scholar]
- Rossmann MG, Blow DM (1962) The detection of sub‐units within the crystallographic asymmetric unit. Acta Crystallogr 15: 24–31 [Google Scholar]
- Sakamoto K, Kamiya M, Imai M, Shinzawa‐Itoh K, Uchida T, Kawano K, Yoshikawa S, Ishimori K (2011) NMR basis for interprotein electron transfer gating between cytochrome c and cytochrome c oxidase. Proc Natl Acad Sci USA 108: 12271–12276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato W, Hitaoka S, Inoue K, Imai M, Saio T, Uchida T, Shinzawa‐Itoh K, Yoshikawa S, Yoshizawa K, Ishimori K (2016) Energetic Mechanism of Cytochrome c‐Cytochrome c Oxidase Electron Transfer Complex Formation under Turnover Conditions Revealed by Mutational Effects and Docking Simulation. J Biol Chem 291: 15320–15331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senda M, Kishigami S, Kimura S, Fukuda M, Ishida T, Senda T (2007) Molecular mechanism of the redox‐dependent interaction between NADH‐dependent ferredoxin reductase and Rieske type [2Fe‐2S] ferredoxin. J Mol Biol 373: 382–400 [DOI] [PubMed] [Google Scholar]
- Shen L, Durley RC, Mathews FS, Davidson VL (1994) Structure of an electron transfer complex: methylamine dehydrogenase, amicyanin, and cytochrome c551i. Science 264: 86–90 [DOI] [PubMed] [Google Scholar]
- Shinzawa‐Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, Yamasaki A, Sugimura T, Kurono S, Tsujimoto K, Mizushima T, Yamashita E, Tsukihara T, Yoshikawa S (2007) Structures and physiological roles of all the integral lipids of bovine heart cytochrome c oxidase. EMBO J 26: 1713–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solmaz SR, Hunte C (2008) Structure of complex III with bound cytochrome c in reduced state and definition of a minimal core interface for electron transfer. J Biol Chem 283: 17542–17549 [DOI] [PubMed] [Google Scholar]
- Speck SH, Dye D, Margoliash E (1984) Single catalytic site model for the oxidation of ferrocytochrome c by mitochondrial cytochrome c oxidase. Proc Natl Acad Sci USA 81: 347–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukummar N, Chen ZW, Ferrari D, Merli A, Rossi GL, Bellamy HD, Chistoserdov A, Davidson VL, Mathews FS (2006) Crystal structure of an electron transfer complex between aromatic amine dehydrogenase and azurin from Alcaligenes faecalis . Biochemistry 45: 13500–13510 [DOI] [PubMed] [Google Scholar]
- Tomizaki T, Yamashita E, Yamaguchi H, Aoyama H, Tsukihara T, Shinzawa‐Itoh K, Nakashima R, Yaono R, Yoshikawa S (1999) Structure analysis of bovine heart cytochrome c oxidase at 2.8 Å resolution. Acta Crystallogr D 55: 31–45 [DOI] [PubMed] [Google Scholar]
- Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa‐Itoh K, Nakashima R, Yaono R, Yoshikawa S (1995) Structure of metal sites of oxidized cytochrome c oxidase at 2.8 Å. Science 269: 1069–1074 [DOI] [PubMed] [Google Scholar]
- Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa‐Itoh K, Nakashima R, Yaono R, Yoshikawa S (1996) The whole structure of the 13‐subunit oxidized cytochrome c oxidase at 2.8 Å. Science 272: 1136–1144 [DOI] [PubMed] [Google Scholar]
- Wang BC (1985) Resolution of phase ambiguity in macromolecular crystallography. Methods Enzymol 115: 90–113 [DOI] [PubMed] [Google Scholar]
- Weiss MS (2001) Global indicators of X‐ray data quality. J Appl Cryst 34: 130–135 [Google Scholar]
- Winn M, Isupov M, Murshudov GN (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D 57: 122–133 [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D 67: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt H, Malatesta F, Nicoletti F, Brunori M, Ludwig B (1998) Tryptophan 121 of subunit II is the electron entry site to cytochrome‐c oxidase in Paracoccus denitrificans . J Biol Chem 273: 5132–5136 [DOI] [PubMed] [Google Scholar]
- Yonetani T, Ray GS (1965) Studies on cytochrome oxidase IV. Kinetics of aerobic oxidation of ferrocytochrome c by cytochrome oxidase. J Biol Chem 240: 3392–3398 [PubMed] [Google Scholar]
- Yoshikawa S, Shimada A (2015) Reaction mechanism of cytochrome c oxidase. Chem Rev 115: 1936–1989 [DOI] [PubMed] [Google Scholar]
- Yukl ET, Liu F, Krzystek J, Shin S, Jensen LMR, Davidson VL, Wilmot CM, Liu A (2013) Diradical intermediate within the context of tryptophan tryptophan tryptophylquinone biosynthesis. Proc Natl Acad Sci USA 110: 4569–4573 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File