

Abstract
In the mitotic spindle, kinetochore microtubules form k‐fibers, whereas overlap or interpolar microtubules form antiparallel arrays containing the cross‐linker protein regulator of cytokinesis 1 (PRC1). We have recently shown that an overlap bundle, termed bridging fiber, links outermost sister k‐fibers. However, the relationship between overlap bundles and k‐fibers throughout the spindle remained unknown. Here, we show that in a metaphase spindle more than 90% of overlap bundles act as a bridge between sister k‐fibers. We found that the number of PRC1‐GFP‐labeled bundles per spindle is nearly the same as the number of kinetochore pairs. Live‐cell imaging revealed that kinetochore movement in the equatorial plane of the spindle is highly correlated with the movement of the coupled PRC1‐GFP‐labeled fiber, whereas the correlation with other fibers decreases with increasing distance. Analysis of endogenous PRC1 localization confirmed the results obtained with PRC1‐GFP. PRC1 knockdown reduced the bridging fiber thickness and interkinetochore distance throughout the spindle, suggesting a function of PRC1 in bridging microtubule organization and force balance in the metaphase spindle.
Keywords: bridging fiber, interpolar microtubules, k‐fiber, mitosis, overlap microtubules
Subject Categories: Cell Cycle
Introduction
The mitotic spindle is a highly dynamic and complex machinery that orchestrates progression through mitosis and cytokinesis 1. Kinetochores are protein complexes assembled on two sides of the chromosome's centromeric region, which are necessary for interaction of spindle microtubules with chromosomes and proper chromosome segregation 2. Microtubules that bind to kinetochores with their plus ends become k‐fibers that can exert forces on chromosomes 3. Meanwhile, non‐kinetochore microtubules interact in an antiparallel fashion in the central part of the spindle, thus forming overlap regions. In addition to k‐fibers, it is thought that non‐kinetochore microtubules comprise the majority of microtubules in mammalian spindles 4. During metaphase, non‐kinetochore microtubules bundle together 30–50 nm apart in groups of 2–6, with antiparallel interactions apparently preferred 4. Their antiparallel region contains motor and non‐motor cross‐linking proteins. Motors contribute to antiparallel microtubule sliding, whereas passive cross‐linkers take part in maintenance of the overlap integrity 5, 6.
PRC1 is a conserved non‐motor cross‐linking protein localized in the antiparallel overlaps of microtubules in vitro 7, 8, 9 and of the spindle midzone 10, 11, 12, 13, 14 where it plays an essential role in regulating its formation and cytokinesis 15. Orthologs of PRC1 with conserved function include Ase1 (anaphase spindle elongation 1) in yeasts 14, 16, SPD‐1 (spindle defective 1) in Caenorhabditis elegans 17, Feo (Fascetto) in Drosophila melanogaster 18, and MAP65 (microtubule‐associated protein 65) in plants 19, all of which fall in a conserved family of non‐motor microtubule‐associated proteins (MAPs).
Even though it is widely accepted that during metaphase‐to‐anaphase transition a conspicuous network of antiparallel non‐kinetochore interdigitating microtubules assembles between separating chromosomes 4, 11, 15, very little is known of PRC1‐containing overlap fibers in metaphase. The affinity of PRC1 to bind to microtubules is regulated by phosphorylation and dephosphorylation events precisely timed throughout mitosis 11, 20. Due to dephosphorylation, PRC1 forms dimers and binds to microtubules to form cross‐linkages between neighboring interdigitating fibers 11, 21. It was shown that only in anaphase an organized central spindle midzone forms between separating chromosomes and consists of a dense network of overlapping antiparallel microtubules cross‐linked by PRC1 4, 11, 12, 20. By combining structural flexibility and rigidity, PRC1 stabilizes antiparallel overlaps while not impeding sliding between them 22.
We have recently shown that a bundle of overlap microtubules which contains PRC1 links outermost sister k‐fibers 23, 24, 25. This fiber, termed “bridging fiber”, balances the tension between sister kinetochores and helps the spindle to obtain a rounded shape. However, the fraction of overlap bundles that function as bridging fibers, as well as the fraction of kinetochore pairs that have a microtubule bridge between them, remained unexplored.
Here, we show that the number of PRC1‐labeled overlap fibers is nearly the same as the number of kinetochore pairs in a spindle during metaphase. Dynamic properties of PRC1‐labeled bundles and sister kinetochores revealed that the majority of overlap bundles are associated with a pair of sister kinetochores. The endogenous PRC1 visualized by immunofluorescence was predominantly localized in the central part of metaphase spindles and co‐localized with bridging fibers throughout the spindle. PRC1 knockdown by siRNA generated thinner bridging fibers, as well as a reduced interkinetochore distance. Taken together, our results indicate that in metaphase nearly all overlap fibers exist as bridging fibers between pairs of sister kinetochores and that PRC1 plays a key role in linking antiparallel microtubules in the bridging fibers.
Results and Discussion
The number of PRC1‐labeled bundles in a metaphase spindle is correlated with the number of chromosomes per cell
To study the relationship between overlap bundles and kinetochores during metaphase, we used HeLa cells stably expressing PRC1‐GFP from a bacterial artificial chromosome (BAC) 26, which were transiently transfected with mRFP‐CENP‐B to visualize kinetochores. We acquired z‐stacks of images that cover a whole metaphase spindle in fixed cells. Metaphase was identified by the alignment of sister kinetochores on the metaphase plate, and kinetochores were defined as sisters if the bi‐orientation was estimated within individual planes. We measured the number of PRC1‐labeled overlap bundles and kinetochore pairs by using two approaches: spindles with their long axis oriented roughly parallel to the imaging plane (horizontal spindles) and spindles with their long axis oriented roughly perpendicular to the imaging plane (vertical spindles).
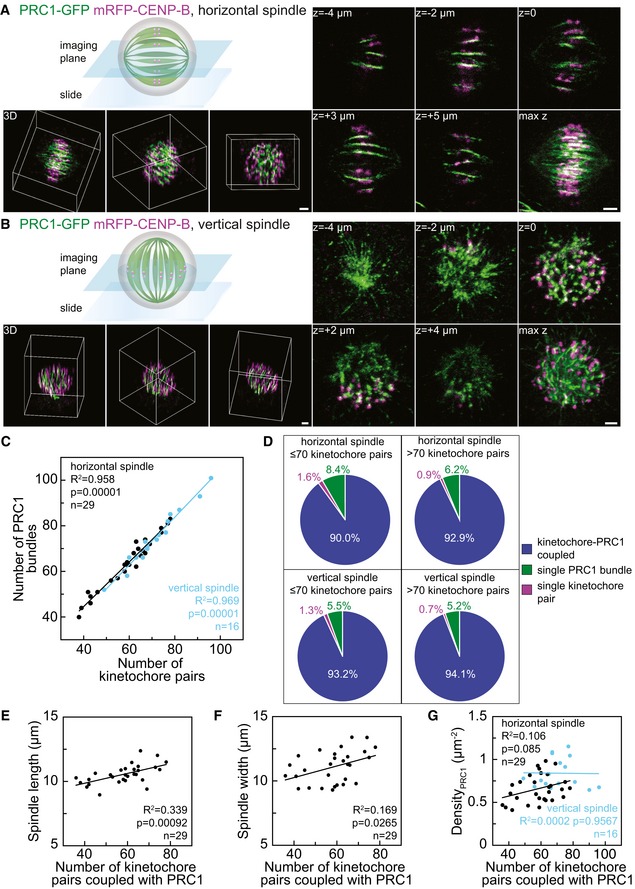
In the first approach, we analyzed horizontal spindles, which was the most frequent spindle orientation (Figs 1A and EV1A, Video EV1). Individual PRC1‐labeled overlaps appeared as slightly curved lines with a broader central part, gradually narrowing in both directions toward the spindle poles (Figs 1A and EV1A). The number of PRC1‐labeled bundles per spindle was 63 ± 2 and the number of kinetochore pairs 59 ± 2 (all results are mean ± s.e.m. unless otherwise indicated, n = 29 spindles). These numbers may be somewhat underestimated due to occasional overlaying of neighboring sister kinetochores or neighboring PRC1‐labeled bundles in the images. We conclude that the mean number of PRC1‐labeled bundles is roughly the same as the mean number of chromosomes.
Figure 1. Most PRC1‐labeled overlap bundles are coupled with sister kinetochores in metaphase.
-
ASpindle in a fixed HeLa cell expressing PRC1‐GFP (green) and mRFP‐CENP‐B (magenta) oriented horizontally with respect to the imaging plane, as shown in the scheme. Images of different z‐slices (central plane of the spindle z = 0, two images below, z = −4 μm and z = −2 μm, and above, z = +3 μm and z = +5 μm), maximum projection of a z‐stack (max z), and 3D projections (3D) with the 3D coordinate system represented as a cuboidal box that indicates different spindle orientations are shown. Additional z‐slices of this spindle are shown in Fig EV1A.
-
BSpindle in a fixed HeLa cell expressing PRC1‐GFP (green) and mRFP‐CENP‐B (magenta) oriented vertically with respect to the imaging plane, as shown in the scheme. Legend as in (A). Additional z‐slices of this spindle are shown in Fig EV1B.
-
CCorrelation between the number of PRC1‐labeled bundles and the number of pairs of sister kinetochores counted throughout horizontal (black) and vertical (blue) spindles of fixed HeLa cells in metaphase. Data points represent individual spindles, lines show linear fits.
-
DPie charts showing the fraction of PRC1‐labeled bundles associated with kinetochore pairs (blue), PRC1‐labeled bundles not associated with kinetochores (green) and kinetochores not associated with PRC1 bundles (magenta) in horizontal (top row) and vertical spindles (bottom row) from cells with up to 70 chromosomes (left column) and more than 70 chromosomes (right column). Horizontal spindles contained a total of 1,786 kinetochore‐PRC1 pairs (n = 29 cells) and vertical spindles 1,133 kinetochore‐PRC1 pairs (n = 16 cells).
-
E, FSpindle length and spindle width as a function of the number of kinetochore pairs coupled with PRC1‐labeled bundles.
-
GNumber of kinetochore pairs coupled with PRC1‐labeled bundles per unit area of the cross section of the central part of horizontal (black) and vertical (blue) spindles as a function of the number of kinetochore pairs coupled with PRC1‐labeled bundles in the spindle.
Figure EV1. Additional characterization of kinetochore‐PRC1 pairs.
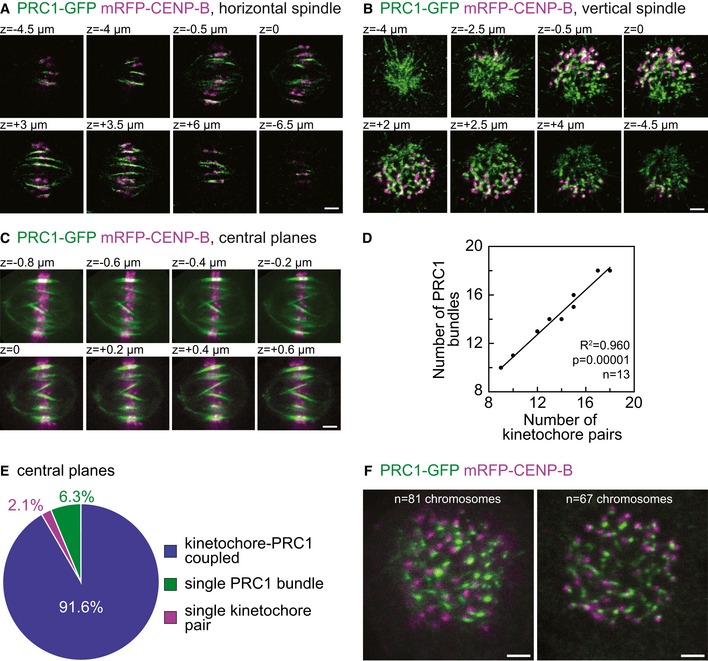
-
A–CIndividual z‐stack images acquired in fixed HeLa cells stably expressing PRC1‐GFP (green) and transiently mRFP‐CENP‐B (magenta). Z‐stack images of (A) horizontal spindle (same spindle shown in Fig 1A), (B) vertical spindle (same spindle shown in Fig 1B), and (C) spindle imaged with different settings (line averaged 16 times with 200 nm spacing between z‐slices). Regardless of depicted approaches, images reveal majority of sister kinetochores positioned in close proximity of PRC1‐labeled fibers.
-
DGraph shows correlation between the number of sister kinetochores and PRC1 signals counted in the central z‐plane of fixed HeLa cells in metaphase.
-
EPie chart shows what fraction of PRC1‐labeled fibers and sister kinetochores are linked (blue) in the central z‐plane of fixed HeLa cells in metaphase. Percentage of single kinetochores and single PRC1 signals are shown in magenta and green, respectively.
-
FA cross section of vertical spindles with 81 (left) and 67 (right) chromosomes.
In the second approach, we analyzed vertical spindles, which were found occasionally in the field of view (Figs 1B and EV1B, Video EV2). The average number of PRC1‐labeled bundles per spindle was 75 ± 3 and the number of kinetochore pairs 72 ± 3 (n = 16 spindles). Images of vertically oriented spindles confirmed our observation in horizontally oriented spindles that the mean number of PRC1‐labeled bundles is nearly the same as the number of kinetochore pairs. In this approach, the number of sister kinetochores and PRC1‐labeled fibers was larger due to less frequent overlaying of neighboring bundles. Notably, the majority of kinetochores were positioned right next to PRC1‐labeled bundles.
In HeLa cells, both the number of chromosomes and their structure show abnormalities 27, 28, 29. The number of chromosomes has been shown to vary between 56 and 179 30, 31, 32, 33. We found the number of kinetochore pairs per cell to be in the range of 38–96 (Fig 1C), in agreement with previous studies. We used this inherently unstable HeLa karyotype to determine whether the number of PRC1‐labeled bundles is correlated with the number of chromosomes (kinetochore pairs). Indeed, we found the number of PRC1‐labeled bundles to be roughly equal to the number of kinetochore pairs in individual spindles, both horizontal and vertical ones (Fig 1C), which further prompted us to investigate their association.
Nearly all overlap bundles are associated with sister kinetochores, acting as a bridge between them
Next, we explored how many PRC1‐labeled overlap bundles are associated with kinetochore pairs and vice versa. A PRC1‐labeled bundle and a kinetochore pair were defined as associated if the distance between them was smaller than 0.3 μm (see Materials and Methods), based on a previous measurement of this distance for outermost kinetochores 23. We found that > 90% of PRC1‐labeled fibers were associated with a kinetochore pair, both in horizontal and vertical spindles (n > 1,000 PRC1‐labeled bundles in each approach, Fig 1D). Conversely, our analysis revealed only a small fraction of PRC1‐labeled bundles and kinetochore pairs that were not mutually linked (Fig 1D). To test whether the association between PRC1‐labeled bundles and kinetochore pairs depends on the number of chromosomes in a cell, we grouped the cells into two groups: those with ≤ 70 and those with > 70 kinetochore pairs. In each group, we found that > 90% of PRC1‐labeled fibers were associated with a kinetochore pair (Fig 1D), indicating that the association between PRC1‐labeled bundles and kinetochores does not depend on the number of chromosomes in the spindle. Moreover, by using different imaging settings (higher signal‐to‐noise ratio, 200 nm spacing between z‐slices, see Fig EV1C) to image only the central planes of horizontal spindles, we obtained similar results as above (n = 13 spindles, Fig EV1C–E). We conclude that nearly all PRC1‐labeled overlap bundles are associated with pairs of sister kinetochores, acting as bridges that link sister k‐fibers.
Spindles with more chromosomes have a larger length and width
Our finding that spindles with more chromosomes contain more overlap bundles prompted us to ask how spindle length and width vary to accommodate these differences. To answer this question, we used only horizontal spindles because spindle length and width could not have been precisely measured in vertically oriented spindles due to the variable tilt of the spindle long axis. We measured the average spindle length to be 10.65 ± 0.16 μm, and the width 11.21 ± 0.25 μm (n = 29), consistent with previous measurements 23, 34. Both spindle length (Fig 1E) and width (Fig 1F) increased with the number of coupled kinetochore pairs and PRC1‐labeled fibers. The increase in width was similar to the increase in length, which indicates that spindles accommodate a larger number of chromosomes by increasing their length and width to a similar extent.
To examine the spatial distribution of bridging fibers in spindles with different numbers of chromosomes, we measured the density of PRC1‐labeled fibers coupled with kinetochores, that is, their number per unit area in the equatorial plane of horizontal and vertical spindles. We found that the density does not depend significantly on the number of coupled pairs (Figs 1G and EV1F). Thus, by accommodating its width the spindle maintains the neighboring bridging fibers at similar distances regardless of the total number of chromosomes in the spindle.
Dynamic properties of PRC1‐labeled fibers and kinetochores in vertical spindles confirm their association
In order to understand the dynamic interplay between neighboring PRC1‐labeled fibers and kinetochores, we acquired time series of vertical spindles and tracked individual bundles and kinetochores in the spindle (Fig 2A and Video EV3). We analyzed the dynamics in the transversal cross section and found that in the majority of the associated pairs, the PRC1‐labeled bundle and the kinetochores moved along identical trajectories or moved in the same direction and passed similar distances, whereas some pairs showed movements in mutually independent directions (Fig 2B).
Figure 2. Dynamic properties of PRC1‐labeled bundles and kinetochores in vertical spindles confirm their association.
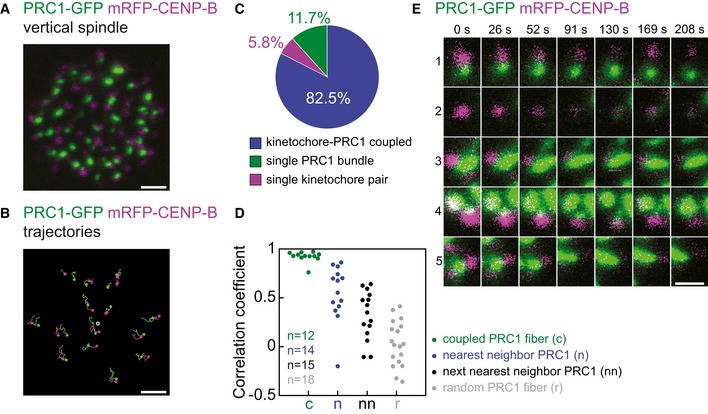
- Central plane of the spindle in a live HeLa cell expressing PRC1‐GFP (green) and mRFP‐CENP‐B (magenta) oriented vertically with respect to the imaging plane.
- Examples of trajectories, with respect to the spindle's center of the mass, of individual PRC1‐GFP (green) and corresponding mRFP‐CENP‐B (magenta) signals from the spindle in (A) that moved together for at least 200 s. Dots represent starting points of trajectories, t = 0 s. Trajectories finish at t = 200 s. Gray circle represents the center of mass of the spindle.
- Pie chart showing the fraction of PRC1‐labeled fibers and sister kinetochores that moved together for at least 60 s (blue, n = 226 pairs in five cells). A small fraction of PRC1‐labeled fibers (green, n = 32 in five cells) and kinetochores (magenta, n = 16 in five cells) did not move together.
- Correlation coefficients between trajectories (with respect to the spindle's center of the mass) of a kinetochore pair and the trajectories of the PRC1‐labeled fiber coupled with the kinetochore pair (green), the nearest neighbor PRC1‐labeled fiber (blue), the next nearest neighbor (black) and a randomly chosen PRC1‐labeled fiber (gray); n, the number of trajectories from three cells.
- Examples of scenarios observed in dynamic interplay of PRC1‐labeled bundles (green) and kinetochores (magenta) from the spindle in (A). Row 1: PRC1‐labeled bundle and kinetochore move together until they separate at 130 s. Row 2: kinetochore is free of any PRC1‐labeled bundle until it appears at 130 s. Row 3: kinetochore and PRC1‐labeled bundle move together. Row 4: kinetochore moves with PRC1‐labeled bundle which merges with neighboring bundle at 169 s. Row 5: two kinetochores move together with a single PRC1‐labeled bundle.
In order to distinguish the fraction of overlap bundles and kinetochores that moved together, we tracked all PRC1‐labeled fibers and kinetochores within the spindle. We observed a dynamic interaction between PRC1‐labeled fibers and kinetochores in their vicinity. A PRC1‐labeled fiber and a kinetochore were termed associated if they moved together for at least five time frames (~1 min) (n = 274 associated and individual PRC1‐labeled fibers and kinetochores from five cells). We found 82.5 ± 2.7% (n = 226) of mutually associated fibers and kinetochores, whereas 11.7 ± 2.3% (n = 32) of PRC1‐labeled fibers did not have a coupled kinetochore with which they moved along the same or similar trajectories, and 5.8 ± 0.8% (n = 16) of kinetochores were observed as free of any PRC1‐labeled fiber (Figs 2C and EV2A and B).
Figure EV2. Dynamics of PRC1‐labeled bundles and kinetochores in the spindle cross section.
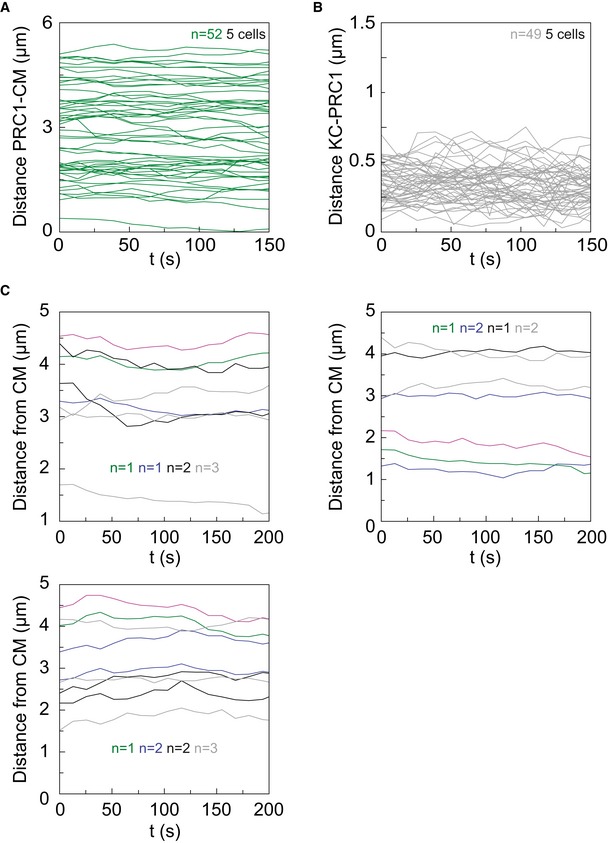
- Dynamics of PRC1‐labeled bundles in live HeLa cells expressing PRC1‐GFP and mRFP‐CENP‐B. The distance between PRC1‐labeled bundles and center of the mass of the spindle (CM) is plotted over t = 150 s. n, number of pairs.
- Dynamics between PRC1‐labeled bundles and corresponding kinetochores in live HeLa cells expressing PRC1‐GFP and mRFP‐CENP‐B. The distance between them is plotted over t = 150 s. n, number of pairs.
- Examples of trajectories of a kinetochore pair (magenta), its coupled PRC1‐labeled bundle (green), nearest PRC1 neighbors (blue), next nearest PRC1 neighbors (black), and randomly chosen PRC1 bundles (gray) with respect to the spindle's center of the mass (CM) over t = 200 s in three cells. n, number of PRC1‐labeled bundles.
To quantify to which extent the kinetochores move in a correlated manner with different PRC1‐labeled fibers in the spindle, we performed cross‐correlation analysis 35 on the acquired trajectories. Our analysis revealed high correlation of movement between a kinetochore pair and the coupled PRC1‐labeled fiber, with a median correlation coefficient of 0.93 (n = 12). The correlation coefficient decreased with an increasing distance between the PRC1‐labeled fiber and the kinetochore pair: The median correlation coefficient was 0.61 for the nearest neighbor fiber that was not coupled with the kinetochore pair, 0.35 for the next nearest neighbor, and 0.02 for a randomly chosen fiber (n = 14–18 fibers in each group, Fig 2D; examples of trajectories are shown in Fig EV1C). These results indicate that kinetochores typically move together with their coupled PRC1‐labeled fiber, whereas the correlation of movement with neighboring fibers decreases in a distance‐dependent manner and vanishes for remote fibers.
To determine the dynamic events between PRC1‐labeled bundles and kinetochores in more detail, we analyzed only the outermost PRC1‐labeled fibers and kinetochores, because those were most easily distinguished from their neighbors. Within this group, we observed several types of behavior of neighboring PRC1‐labeled fibers and kinetochores. Due to the better clarity of events in this region, we used a more strict criterion for determining the interaction of PRC1‐labeled fibers and kinetochores: PRC1‐labeled fiber and kinetochore were termed associated if they were moving along the same or similar trajectories during the entire acquired video (~5 min). We found that 65.7 ± 4.1% of PRC1‐labeled fibers and kinetochores were mutually associated. Within this group, we included the following occasional events as well: one PRC1‐labeled fiber moved together with two kinetochores which do not seem to be sisters; PRC1‐labeled fiber moved together with sister kinetochores until they both disappeared from the imaged planes in the z‐direction at the same time. Other scenarios in which we term a kinetochore and PRC1‐labeled fiber uncoupled (34.3 ± 4.1%) were as follows: first, the kinetochore seemed to be free of any PRC1‐labeled fiber, and after a certain time, a PRC1 signal appeared and they started moving together; a kinetochore and PRC1‐labeled fiber moved together and eventually separated to a distance greater than 0.3 μm; kinetochore first moved alone and at a certain time point moved to the vicinity of a neighboring PRC1 and they started moving together; a PRC1‐labeled fiber merged with the neighboring bundle and they appeared as a single PRC1‐labeled fiber; kinetochore moved with the PRC1 until PRC1 disappeared; kinetochore moved with the PRC1, at one point separated from it, and subsequently associated with the same bundle to continue moving together. For events described above, see Fig 2E and Video EV3. The observed events reveal the dynamic nature of the interactions between PRC1‐labeled fibers and kinetochores. However, most of the time PRC1‐labeled bridging fibers and kinetochores are in close proximity and move along the same or similar trajectories.
Endogenous PRC1 localizes to the central part of the bridging fibers in metaphase
The results described above were obtained on a cell line that expresses PRC1‐GFP in addition to the endogenous PRC1. Western blot analysis showed 1.64 ± 0.10 times higher expression of PRC1 in this cell line compared with unlabeled HeLa cells (n = 6 independent experiments, P = 0.0004) while tubulin‐GFP cell line showed the same expression level of PRC1 as determined in unlabeled cells (Fig EV3A and B, and Table 1). Previous studies have shown that PRC1 exhibits a slightly different localization when overexpressed, for example, a substantial fraction of the protein is cytosolic and localizes to brightly stained ring‐shaped arrays around the interphase nucleus 11. Thus, we set out to define the localization and distribution of endogenous PRC1 in metaphase spindles and compare it with the localization of PRC1‐GFP.
Figure EV3. Additional characterization of PRC1‐labeled bundles.
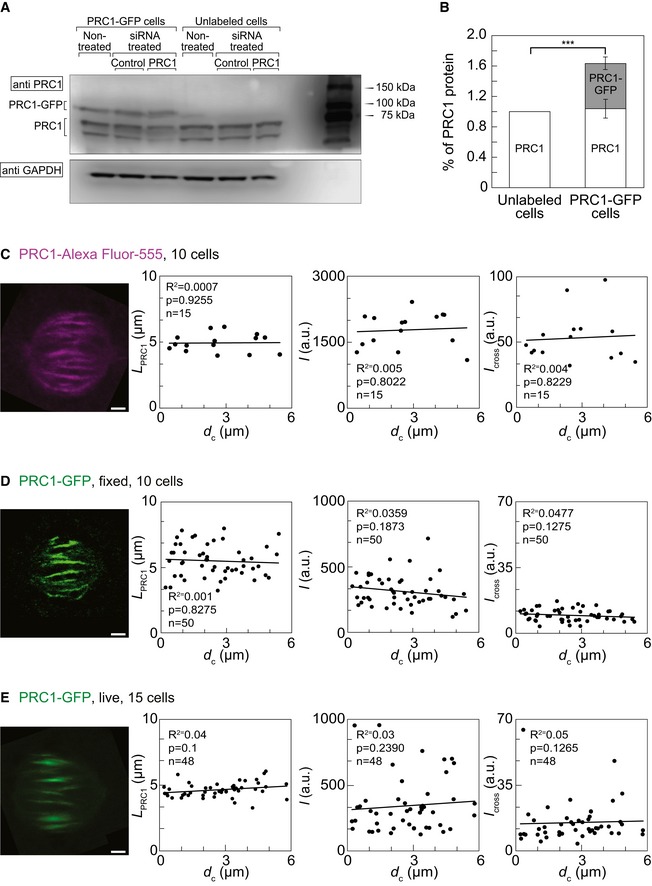
-
A, BWestern blot for PRC1 in various HeLa cell lines and conditions, as denoted in the figure. PRC1 expression in PRC1‐GFP HeLa cells was 60.97 ± 10.01% higher than in unlabeled cells. Synchronized cells were lysed in lysis buffer 29 h after transfection. Cell lysates were subjected to SDS‐PAGE (12% polyacrylamide), transferred on to a nitrocellulose membrane and immunoblotted with anti‐PRC1 and anti‐GAPDH antibodies used as a loading control. Intensities of the Western blot bands of endogenous PRC1 isoforms (61–71 kDa) and PRC1‐GFP (98 kDa) were quantified as described in Materials and Methods. P‐value was 0.0004.
-
CFrom left to right: image of the immunostained PRC1 in the spindle of a HeLa cell expressing tubulin‐GFP and immunostained for PRC1; dependence of the length of immunostained PRC1 signal in HeLa cells expressing tubulin‐GFP and immunostained for PRC1 on the distance from the spindle long axis; dependence of the signal intensity I of immunostained PRC1 signal in HeLa cells expressing tubulin‐GFP and immunostained for PRC1 on the distance from the spindle long axis; dependence of I cross of immunostained PRC1 signal in HeLa cells expressing tubulin‐GFP and immunostained for PRC1 on the distance from the spindle long axis.
-
DFrom left to right: image of PRC1‐GFP in the spindle of a fixed HeLa cell expressing PRC1‐GFP and mRFP‐CENP‐B; dependence of the length of PRC1‐GFP signal in fixed HeLa cells expressing PRC1‐GFP on the distance from the spindle long axis; dependence of the signal intensity I of PRC1‐GFP signal in fixed HeLa cells expressing PRC1‐GFP on the distance from the spindle long axis; dependence of I cross of PRC1‐GFP signal in fixed HeLa cells expressing PRC1‐GFP and on the distance from the spindle long axis.
-
EFrom left to right: image of PRC1‐GFP in the spindle of a live HeLa cell expressing PRC1‐GFP and mRFP‐CENP‐B; dependence of the length of PRC1‐GFP signal in live HeLa cells expressing PRC1‐GFP on the distance from the spindle long axis; dependence of the signal intensity I of PRC1‐GFP signal in live HeLa cells expressing PRC1‐GFP on the distance from the spindle long axis; dependence of I cross of PRC1‐GFP signal in live HeLa cells expressing PRC1‐GFP and on the distance from the spindle long axis.
Table 1.
Properties of the PRC1‐labeled overlap measured by different approaches
| Tubulin‐GFP cells immunostained for PRC1 | PRC1‐GFP cells fixed | PRC1‐GFP cells live | |
|---|---|---|---|
| I/au | 1778.78 ± 101.37 | 311.34 ± 16.49 | 343.69 ± 30.23 |
| I cross/au | 53.03 ± 4.85 | 9.56 ± 0.44 | 15.01 ± 1.51 |
| L PRC1/μm | 4.95 ± 0.18 | 5.49 ± 0.17 | 4.64 ± 0.08 |
| Spindle length/μm | ND | 10.65 ± 0.16 | ND |
| Spindle width/μm | ND | 11.21 ± 0.25 | ND |
Au, arbitrary units; ND, not determined.
All values are given as mean ± s.e.m. and measured as in Materials and Methods.
We used HeLa cells stably expressing tubulin‐GFP, which allowed us to identify the k‐fibers and bridging fibers, and immunostained them for PRC1 (see Materials and Methods). We were interested in the distribution of PRC1 in metaphase, so we ensured that the cells are in metaphase by arresting them using the proteasome inhibitor MG132 (see Materials and Methods). We found that endogenous PRC1 localizes to the metaphase spindle with enrichment in its central part (Fig 3A), as previously shown 11, 23. We determined the individual bridging fibers throughout metaphase spindles by following the tubulin‐GFP signal that spans the region between sister k‐fibers. Localization of endogenous PRC1 in the same cells showed that 97.8 ± 0.1% of the bridging fibers were immunostained for endogenous PRC1 (n = 46 bridges in 10 cells; Fig 3B). Thus, endogenous PRC1 localizes to the bridging fibers throughout metaphase spindles. Furthermore, we observed that the endogenous PRC1 signal extends to the region of k‐fibers (determined by tubulin‐GFP signal), proximal to the estimated position of the kinetochore, which independently suggests that PRC1‐labeled fibers are bound to sister k‐fibers.
Figure 3. Endogenous PRC1 localizes to the metaphase spindle midzone.
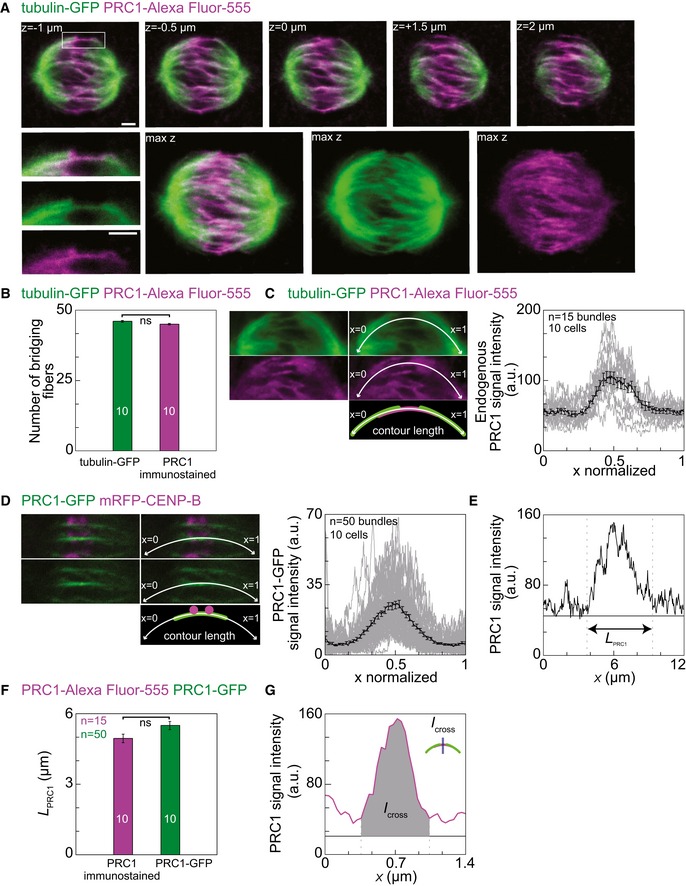
- Top row: MG132 arrested HeLa cells stably expressing tubulin‐GFP (green) and immunostained for endogenous PRC1 (Alexa Fluor‐555 shown in magenta) in five individual z‐images. Enlargements of the boxed region (top: merge, middle: GFP, bottom: Alexa Fluor‐555) below the first image show the bridging fiber determined as the line connecting two ends of the sister k‐fibers in green channel and localization of endogenous PRC1 (magenta). Corresponding maximum projections are shown to the right (max z, left: merge, middle: GFP, right: Alexa Fluor‐555).
- Quantification of tubulin‐GFP bridging fibers (green bar) immunostained for endogenous PRC1 (magenta bar).
- Images show pole‐to‐pole tracking (white curve) of tubulin signal (top, green), which was used to measure the endogenous PRC1 signal along the same contour (middle, magenta), and the corresponding scheme (bottom) from a HeLa cell expressing tubulin‐GFP and immunostained for PRC1. Images of the same spindle without (left) and with the tracked contour (right) are shown. The graph shows normalized pole‐to‐pole intensity profiles (each intensity profile was scaled so that one pole is at x = 0 and the other at x = 1) of endogenous PRC1 (gray lines) acquired in tubulin‐GFP HeLa cells immunostained for PRC1. Black line shows the mean value.
- Images show pole‐to‐pole tracking (white curve) of PRC1‐GFP signal (top, merge; middle, GFP; bottom, scheme) from a HeLa cell expressing PRC1‐GFP and mRFP‐CENP‐B. Images of the same spindle without (left) and with the tracked contour (right) are shown. The graph shows normalized pole‐to‐pole intensity profiles (legend as in C) of PRC1‐GFP acquired in HeLa cells expressing PRC1‐GFP and mRFP‐CENP‐B.
- Example of length measurement of the immunostained PRC1 signal (L PRC1) in HeLa cell expressing tubulin‐GFP and immunostained for PRC1.
- Comparison of the length L PRC1 between PRC1‐GFP signal (green corresponds to the PRC1 fused to GFP from a PRC1‐GFP cell line) and PRC1‐Alexa Fluor‐555 (magenta corresponds to the endogenous immunostained PRC1) (P = 0.10).
- Cross section signal intensity of the immunostained endogenous PRC1 (magenta line) from a HeLa cell expressing tubulin‐GFP and immunostained for PRC1. Area under the peak is defined as I cross, measured at the position of the blue line as in scheme. Horizontal lines mark the background signal (black line), and vertical lines delimit the area (gray) where the signal was measured.
Next, we quantified the signal of endogenous immunostained PRC1 and compared it with the signal of PRC1‐GFP. We tracked pole‐to‐pole tubulin‐GFP signals of the sister k‐fibers and the corresponding bridging fiber in the green channel, and measured intensity profiles of endogenous PRC1 in the red channel (see Fig 3C), which confirmed localization of endogenous PRC1 in the central part of the spindle (n = 15 bridges from 10 cells; Fig 3C). Similarly, pole‐to‐pole intensity profiles of PRC1‐labeled bundles in the cell line expressing PRC1‐GFP showed localization of PRC1‐GFP in the central part of the spindle (n = 50 bridges from 10 cells; Fig 3D). We defined the length of the PRC1 signal, L PRC1, as the width of the peak in the intensity profile (Fig 3E). The length of the endogenous PRC1 signal was 4.95 ± 0.18 μm (n = 15 bridges from 10 cells), whereas the length of the PRC1‐GFP signal was 5.49 ± 0.17 μm (n = 50 bridges from 10 cells, P = 0.10; Fig 3F and Table 1). These results confirm that PRC1‐GFP localizes in the same regions as endogenous PRC1. Thus, antiparallel microtubule overlaps that bind PRC1 are present in the central part of bridging fibers in metaphase spindles and extend over a well‐defined region.
K‐fibers at the periphery of the spindle are longer and more curved than those near the spindle long axis. Thus, we asked whether PRC1 signal parameters in the bridging fiber depend on the distance of the fiber from the spindle long axis. We defined two additional measures of the signal intensity: I was defined as the total intensity in the pole‐to‐pole intensity profile divided by the contour length of this intensity profile (Fig 3C and Table 1, Materials and Methods), and I cross as the total intensity under the peak in the intensity profile acquired transversely to the PRC1 signal (Fig 3G, Materials and Methods). We measured L PRC1, I and I cross of the endogenous immunostained PRC1, as well as of PRC1‐GFP in fixed and live cells throughout the spindle (Table 1). In all these conditions, the parameters of the PRC1 signal did not depend on the distance from the spindle long axis (Fig EV3C–E). These results suggest that all bridging fibers in the spindle have a similar length of the PRC1‐bound antiparallel overlap zone and a similar amount of PRC1, regardless of the length and curvature of the associated k‐fibers.
PRC1 knockdown results in thinner bridging fibers and smaller interkinetochore distance
Previous experiments showed that knockdown of PRC1 by small interfering RNA (siRNA) reduces the thickness of the bridging fiber on the outermost part of the spindle 23. To characterize the effect of PRC1 silencing on the whole metaphase spindle, we used mild PRC1 knockdown in order to maintain spindle integrity in HeLa cells expressing tubulin‐GFP and mRFP‐CENP‐B (Materials and Methods). Images of cells treated with PRC1 siRNA and control siRNA were acquired 24 h after transfection (Fig 4A). Western blot analysis showed that in unlabeled HeLa cells treated with PRC1 siRNA the amount of PRC1 was reduced by 51.10 ± 5.49% compared to control cells (n = 3 independent experiments, P = 0.0092), whereas in cells expressing tubulin‐GFP the amount of PRC1 was reduced by 53.40 ± 11.76% (n = 6 independent experiments, P = 0.0478, Figs 4B and EV4A, and Table 2).
Figure 4. PRC1 silencing reduces bridging fiber thickness and interkinetochore distance.
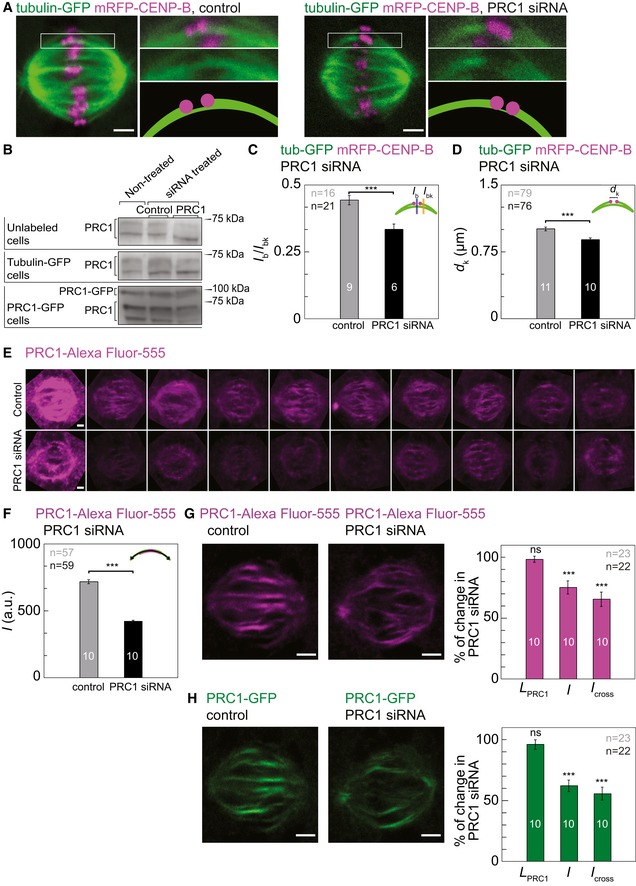
- Images of HeLa cells stably expressing tubulin‐GFP (green) and transiently mRFP‐CENP‐B (magenta) treated with control siRNA (left images) and siRNA targeting PRC1 (right images). Enlargements of the boxed region, shown to the right of the image of the whole spindle, are focused on kinetochores and bridging fiber (top: merge; middle: GFP, bottom: scheme).
- Western blot showing PRC1 protein from unlabeled cells, tubulin‐GFP HeLa cell line, and PRC1‐GFP HeLa cell line. Detailed measurements are shown in Fig EV4.
- Mean ratio of signal intensities of the bridging fiber (I b, measured at the position of a blue line as in scheme) and sum of the bridging and k‐fiber (I bk, measured at the position of the orange line as in scheme), measured in control cells (gray bar) and PRC1 siRNA‐treated cells (black bar) (P = 0.0003).
- Mean interkinetochore distance (d k, see scheme) measured in control cells (gray bar) and PRC1 siRNA‐treated cells (black bar) (P = 0.0001).
- Images of individual spindles of HeLa cells expressing tubulin‐GFP and immunostained for PRC1 obtained from control cells (top) and PRC1 siRNA‐treated cells (bottom). All cells were imaged with the same imaging parameters. Only Alexa Fluor‐555 channel is shown.
- Mean signal intensity I of the immunostained PRC1 in control cells (gray bar) and PRC1 siRNA‐treated cells (black bar) in HeLa cells expressing tubulin‐GFP and immunostained for PRC1 (P = 0.0001).
- Images of HeLa cells stably expressing PRC1‐GFP treated with control (left image) and siRNA targeting PRC1 (right image) immunostained for PRC1 (magenta). Graph showing the length of the immunostained PRC1 signal, L PRC1 (P = 0.5167), signal intensity, I (P = 0.0003), and I cross (P = 0.0001) in siRNA targeting PRC1 in comparison with control HeLa cells expressing PRC1‐GFP and immunostained for PRC1.
- Images of HeLa cells stably expressing PRC1‐GFP (green) treated with control (left image) and siRNA targeting PRC1 (right image) immunostained for PRC1. Graph showing the length of the PRC1‐GFP signal, L PRC1 (P = 0.3407), signal intensity, I (P = 0.0001), and I cross (P = 0.0001) in siRNA targeting PRC1 in comparison with control HeLa cells expressing PRC1‐GFP and immunostained for PRC1.
Figure EV4. Additional characterization of PRC1 silencing effect.
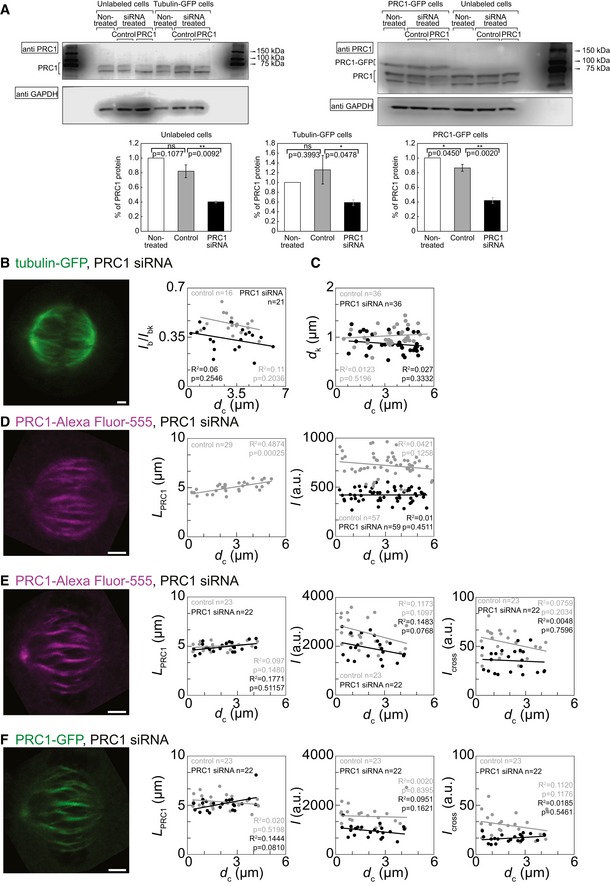
- Western blot for PRC1 in various HeLa cell lines and conditions, as denoted in the figure. Graphs show quantification of Western blots. Please note a decrease of the amount of PRC1 after treatment with PRC1 siRNA. HeLa cells grown on 6‐well plates were transfected with 200 nM control or PRC1 siRNA. Synchronized cells were lysed in lysis buffer 29 h after transfection. Cell lysates were subjected to SDS‐PAGE (12% polyacrylamide), transferred on to a nitrocellulose membrane and immunoblotted with anti‐PRC1 and anti‐GAPDH antibodies. Percent of PRC1 protein was calculated from Western blot band intensities measured in Image Studio Lite program after normalizing to the corresponding GAPDH band intensity. The data were acquired from three to six independent experiments. Please note that the tubulin‐GFP HeLa was the least viable of the cell lines and thus the data varied. P‐values are given in the graphs.
- Left: Image of tubulin‐GFP in the spindle of a HeLa cell expressing tubulin‐GFP and mRFP‐CENP‐B, treated with PRC1 siRNA. Right: Dependence of the ratio of signal intensities of the bridging fiber and the sum of the bridging and k‐fiber, I b/I bk measured in control cells (gray) and PRC1 siRNA‐treated (black) HeLa cells expressing tubulin‐GFP and mRFP‐CENP‐B on the distance from the spindle long axis.
- Dependence of the distance between sister kinetochores in control cells (gray) and PRC1 siRNA‐treated (black) cells expressing tubulin‐GFP and mRFP‐CENP‐B on the distance from the spindle long axis.
- From left to right: image of PRC1‐Alexa Fluor‐555 in the spindle of a HeLa cell expressing tubulin‐GFP and immunostained for PRC1, treated with PRC1 siRNA; dependence of the length of immunostained PRC1 signal in HeLa cells expressing tubulin‐GFP and immunostained for PRC1 on the distance from the spindle long axis; dependence of the signal intensity I of immunostained PRC1 signal in HeLa cells expressing tubulin‐GFP and immunostained for PRC1 on the distance from the spindle long axis. Control cells (gray) and PRC1 siRNA‐treated cells (black) are shown.
- From left to right: image of PRC1‐Alexa Fluor‐555 in the spindle of a HeLa cell expressing PRC1‐GFP and immunostained for PRC1, treated with PRC1 siRNA; dependence of the length, signal intensity I, and I cross of immunostained PRC1 signal in HeLa cells expressing PRC1‐GFP and immunostained for PRC1, measured in control cells (gray) and PRC1 siRNA‐treated cells (black) on the distance from the spindle long axis.
- From left to right: image of PRC1‐GFP in the spindle of a HeLa cell expressing PRC1‐GFP and immunostained for PRC1, treated with PRC1 siRNA; dependence of the length, signal intensity I, and I cross of PRC1‐GFP signal in HeLa cells expressing PRC1‐GFP and immunostained for PRC1, measured in control cells (gray) and PRC1 siRNA‐treated cells (black) on the distance from the spindle long axis.
Table 2.
Properties of different cell lines treated with siRNA
| Control | PRC1 siRNA | % of change | P‐value | |
|---|---|---|---|---|
| Unlabeled cells | ||||
| Western blot | 0.81 ± 0.08 | 0.40 ± 0.01 | 51.10 ± 5.49 | 0.0092 |
| Tubulin‐GFP‐labeled cells | ||||
| Western blot | 1.25 ± 0.29 | 0.58 ± 0.05 | 53.40 ± 11.76 | 0.0476 |
| I b | 13.16 ± 1.40 | 9.45 ± 1.40 | 28.19 ± 13.09 | 0.0744 |
| I bk | 29.12 ± 2.77 | 26.94 ± 3.58 | 7.48 ± 15.11 | 0.6503 |
| I k = I bk − I b | 15.95 ± 1.49 | 17.49 ± 2.30 | 8.80 ± 14.71 | 0.6042 |
| I b /I bk | 0.44 ± 0.01 | 0.33 ± 0.01 | 25.00 ± 2.84 | 0.0003 |
| d k/μm | 1.00 ± 0.02 | 0.88 ± 0.02 | 12.00 ± 2.64 | 0.0001 |
| Spindle length/μm | 10.79 ± 0.10 | 11.06 ± 0.09 | 2.45 ± 1.26 | 0.0722 |
| Spindle width/μm | 11.42 ± 0.29 | 11.63 ± 0.43 | 1.81 ± 4.55 | 0.6835 |
| Tubulin‐GFP‐labeled cells immunostained for PRC1 | ||||
| L PRC1/μm | 4.96 ± 0.08 | |||
| I/au | 717.77 ± 15.27 | 421.18 ± 8.58 | 41.33 ± 1.70 | 0.0001 |
| PRC1‐GFP‐labeled cells immunostained for PRC1 | ||||
| Western blot | 0.86 ± 0.04 | 0.41 ± 0.04 | 51.78 ± 5.40 | 0.0020 |
| L PRC1 immunostained/μm | 4.99 ± 0.06 | 4.91 ± 0.10 | 1.60 ± 2.54 | 0.51667 |
| L PRC1‐GFP/μm | 5.36 ± 0.14 | 5.15 ± 0.16 | 3.91 ± 3.89 | 0.3407 |
| I immunostained PRC1/au | 2550.80 ± 125.34 | 1918.98 ± 99.89 | 25.77 ± 5.38 | 0.0003 |
| I PRC1‐GFP/au | 1551.59 ± 83.97 | 965.30 ± 51.57 | 37.79 ± 4.73 | 0.0001 |
| I cross immunostained/au | 54.06 ± 2.99 | 35.43 ± 2.48 | 34.46 ± 5.84 | 0.0001 |
| I cross PRC1‐GFP/au | 29.92 ± 2.42 | 16.66 ± 0.81 | 44.31 ± 5.25 | 0.0001 |
| Spindle length/μm | 10.81 ± 0.23 | 10.59 ± 0.32 | 2.03 ± 3.57 | 0.5897 |
| Spindle width/μm | 9.51 ± 0.33 | 9.20 ± 0.24 | 3.25 ± 4.16 | 0.4592 |
All values are given as mean ± s.e.m. and measured as described in the Materials and Methods section (au = arbitrary units).
Next, we used signal intensities of tubulin‐GFP in the bridging fiber and the k‐fiber to determine the reduction of the number of microtubules in the bridging fiber induced by PRC1 siRNA. Following the method described in 23, we measured the signal intensity of tubulin‐GFP between sister kinetochores, I b, and across the k‐fiber, laterally of kinetochore, I bk. We interpret I b as the signal of the bridging fiber, and I bk as the signal of the bundle consisting of the bridging fiber and the k‐fiber together. In cells treated with control siRNA (control cells), the ratio I b/I bk was 0.44 ± 0.01 (n = 16 bridges in nine cells, see Table 2 for absolute values of I b and I bk), consistent with previous results 23. PRC1 knockdown reduced the ratio I b/I bk to 0.33 ± 0.01 (n = 21 bridges in six cells, P = 0.0003, Fig 4C and Table 2). The level of reduction was constant regardless of the distance of the bridging fiber from the spindle long axis (Fig EV4B). The signal intensity I b was reduced by roughly 28% after PRC1 knockdown, which we interpret as the reduction in the number of microtubules in the bridging fiber (Table 2). On the contrary, the signal intensity I k = I bk − I b, which corresponds to the number of microtubules in the k‐fiber, was not affected significantly by PRC1 siRNA treatment (P = 0.6, Table 2). Thus, our results suggest that PRC1 knockdown reduces the number of microtubules in the bridging fibers throughout the spindle.
Next, we tested whether PRC1 siRNA affected the interkinetochore distance and spindle length and width. We found that the distance between centers of sister kinetochores, d k, was reduced from 1.00 ± 0.02 μm in control cells (n = 79 pairs of sister kinetochores in 11 cells) to 0.88 ± 0.02 μm in cells treated with PRC1 siRNA (n = 76 pairs of sister kinetochores in 10 cells, P = 0.0001, Fig 4D and Table 2). The distance between sister kinetochores did not depend on their distance from the spindle long axis, in both control cells and those treated with PRC1 siRNA (Fig EV4C). Spindle length and width did not change significantly after PRC1 knockdown (Table 2). We conclude that PRC1 knockdown results in a decreased distance between sister kinetochores, which we interpret as a decrease in interkinetochore tension.
To investigate the changes of the microtubule overlap region in the bridging fiber induced by PRC1 knockdown, we used HeLa cells expressing tubulin‐GFP, and immunostained them for PRC1 (Materials and Methods). An overall reduction in the PRC1 signal in the metaphase spindle was found in cells treated with PRC1 siRNA compared with control cells (Fig 4E). In control cells, the length of immunostained PRC1 signal, L PRC1, was 4.96 ± 0.08 μm (n = 29 bridges in 10 cells), which was similar to L PRC1 in non‐treated synchronized cells described above (P = 0.95), and it did not depend on the distance from the spindle long axis (Fig EV4D and Table 2). L PRC1 of immunostained PRC1 could not be determined in cells treated with PRC1 siRNA due to its low signal. PRC1 signal intensity, I, was found to be reduced by 41.33 ± 1.70% (P = 0.0001, Fig 4F and Table 2), which was also independent of the distance from the spindle long axis (Fig EV4D). These data confirm the reduction in PRC1 intensity in the overlap regions due to PRC1 knockdown.
To further determine the resulting difference between overlap fibers in control and PRC1 siRNA‐treated cells, we examined HeLa cells expressing PRC1‐GFP from a BAC, and immunostained them for PRC1. Western blots showed that in PRC1‐GFP cell line the amount of PRC1 was reduced by 51.78 ± 5.40% in cells treated with PRC1 siRNA compared to control cells (n = 3 individual experiments, P = 0.0020, Figs 4B and EV4A, and Table 2). Next, we quantified signals of both immunostained PRC1 and PRC1‐GFP in control and PRC1 siRNA cells as described above. Interestingly, neither the length, L PRC1, of immunostained PRC1 nor the length of PRC1‐GFP was altered by PRC1 knockdown (Figs 4G and EV4E and F, and Table 2). As expected, both PRC1 signal intensity, I, and the signal intensity of the cross section, I cross, were reduced by PRC1 knockdown (Fig 4G and H, and Table 2). Spindle length and width were not affected by PRC1 knockdown (Table 2). Taken together, our data show that a mild knockdown of PRC1 (~50%) leads to a reduction in PRC1 signal intensity in the metaphase spindle, whereas the length of the PRC1‐labeled regions remains unchanged. Thus, our results suggest that the number of microtubules in the antiparallel overlap zones is reduced, whereas the length of their overlap region remains the same after PRC1 knockdown.
Previous studies have suggested that the formation of PRC1 homodimers, which is required for the interaction with kinesin‐4 and for microtubule binding, is triggered at the onset of anaphase 10, 13, 15. Yet, antiparallel overlaps containing PRC1 have also been reported in metaphase spindles 11, 23. Our observation of PRC1‐decorated fibers is not limited to overexpression of PRC1‐GFP as it was confirmed by immunofluorescence. The quantification of endogenous PRC1 signal in non‐synchronized cells expressing tubulin‐GFP and treated with control siRNA indicates that the measured endogenous PRC1 is not a result of the applied synchronization treatment.
The dynamics of live spindles revealed that PRC1‐labeled fibers spend most of the time moving together with their associated kinetochores, with occasional uncoupling and recoupling events. Thus, the small fraction of PRC1‐labeled fibers that were found not coupled with kinetochores in fixed cells likely represents bridging fibers that were in a dissociated state from their kinetochores at the time of cell fixation. Uncoupled fibers and kinetochores may be found more frequently in prophase, during the formation of the structure comprising sister kinetochores, k‐fibers, and their bridging fiber 36.
While the kinetochores moved in the equatorial plane of the spindle in a highly correlated manner with their coupled PRC1‐labeled fiber, there was a moderate correlation with neighboring fibers. Similarly, a previous study has shown that neighboring kinetochore pairs oscillate in a correlated manner, which was explained by elastic linkages between k‐fibers 37. Our results support the existence of lateral connections between adjacent bundles consisting of bridging and k‐fibers.
PRC1 knockdown resulted in thinner bridging fibers and reduced interkinetochore distance, but the spindle shape did not change significantly. According to our model 23, the compression in the bridging fiber counteracts the tension at the end of the k‐fiber. Thus, when both forces are reduced, the spindle shape can remain unchanged, which is in agreement with our measurements.
In summary, we have shown that in a metaphase spindle, nearly all overlap microtubule bundles are associated with kinetochores and act as a bridge between sister k‐fibers (Fig 5). Our results indicate that PRC1 plays a key role in linking antiparallel microtubules in the bridging fibers. It will be interesting to identify the motor proteins bound in the antiparallel overlap zone of the bridging fiber and their role in the force balance of the metaphase spindle.
Figure 5. Overlap bundles act as bridges between sister k‐fibers in a metaphase spindle.
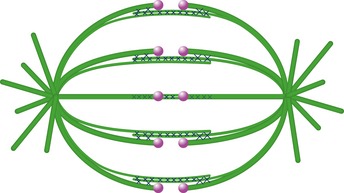
Our work shows that virtually all overlap microtubule bundles containing PRC1 are linked to sister k‐fibers during metaphase. Likewise, each pair of sister kinetochores is associated with a single overlap bundle, which connects their k‐fibers as a bridge. Microtubules are represented in green, kinetochores in magenta, and PRC1 cross‐linkers as crosses.
Materials and Methods
Cell culture and sample preparation
HeLa‐TDS cells were permanently transfected and stabilized (courtesy of Mariola Chacon) using pEGFP‐α‐tubulin plasmid, which was acquired from Frank Bradke (Max Planck Institute of Neurobiology, Martinsried). HeLa‐Kyoto BAC lines stably expressing PRC1‐GFP 26 were courtesy of Ina Poser and Tony Hyman (MPI‐CBG, Dresden). Cells were grown in DMEM (1 g/l d‐glucose, l‐glutamine, pyruvate obtained from Sigma, St. Louis, MO, USA) with 50 μg/ml geneticin (Santa Cruz Biotechnology, Inc., Dallas, USA) and appropriate supplements. The cells were kept at 37°C and 5% CO2 in a Galaxy 170 R CO2 humidified incubator (Eppendorf, Hamburg, Germany).
HeLa cells were transfected by electroporation using Nucleofector Kit R (Lonza, Basel, Switzerland) with the Nucleofector 2b Device (Lonza, Basel, Switzerland), using the high‐viability O‐005 program. Transfection protocol provided by the manufacturer was followed. Cells were transfected with mRFP‐CENP‐B plasmid (pMX234) provided by Linda Wordeman (University of Washington). 1 × 106 cells and 2 μg of plasmid DNA were used. Transfection of PRC1‐GFP BAC line cells with mRFP‐CENP‐B (2.5 μg DNA) was performed 25–35 h before imaging.
For PRC1 siRNA, 1 × 106 cells at 50–60% confluency were transfected with 200 nM targeting or control non‐targeting siRNA raw constructs diluted in a Nucleofector solution R together with 2.5 μg mRFP‐CENP‐B plasmid. The constructs used were as follows: siGENOME SMART pool for human PRC1 (M‐019491‐00‐0005) and siGENOME control pool (D‐001206‐13‐05), both from Dharmacon (Lafayette, CO, USA). To prepare samples for microscopy, following the transfection, HeLa cells were seeded and cultured in 1.5 ml DMEM medium with supplements at 37°C and 5% CO2 on uncoated 35‐mm glass coverslip dishes, No 1.5 coverglass (MatTek Corporation, Ashland, MA, USA). Before live‐cell imaging, the medium was replaced with Leibovitz's L‐15 CO2‐independent medium supplemented with fetal bovine serum (FBS, Life Technologies, Carlsbad, CA, USA). For experiments with the fixed samples, cells were fixed in ice‐cold methanol for 3 min, washed three times with phosphate‐buffered saline (PBS, Merck, Darmstadt, Germany) and immunostained.
Immunostaining
Cells were fixed in ice‐cold methanol (100%) for 3 min and washed. To permeabilize cell membranes, cells were incubated in triton (0.5% in PBS) for 25 min at room temperature. Unspecific binding of antibodies was blocked in blocking solution (1% normal goat serum (NGS) in PBS) for 1 h at 10°C. Cells were incubated in 250 μl of primary antibody solution (4 mg/ml in 1% NGS in PBS) for 48 h at 10°C. Rabbit polyclonal anti‐PRC1 antibody (H‐70; sc‐8356, Santa Cruz Biotechnology, USA) was used. After washing of primary antibody solution, cells were incubated in 250 ml of the secondary antibody solution (4 μg/ml in 2% NGS in PBS; Alexa Fluor‐555 F‐conjugated goat anti‐rabbit IgG, A21430; Thermo Fisher Scientific, Waltham, MA, USA) for 1 h at room temperature, protected from light. After each incubation step, washing was performed three times for 5 min in PBS softly shaken at room temperature.
Synchronization
Cells were seeded at 40% confluency in uncoated 35‐mm glass coverslip dishes, No 1.5 coverglass (MatTek Corporation, Ashland, MA, USA) with 2 ml DMEM medium with supplements. At 4 pm the day before imaging, thymidine (Sigma‐Aldrich, St. Louis, MO, USA) was added at a final concentration of 2 mM. Cells were left in thymidine for 17 h, and at 9 am each dish was washed three times with warm PBS and 2 ml of fresh DMEM medium with supplements was added. At 12:30 pm, RO‐3306 (Calbiochem, Merck Millipore, Billerica, MA, USA) was added at a final concentration of 9 mM. At 7 pm, the dishes were washed three times with warm PBS. Then, the cells were left in the incubator with 2 ml DMEM medium with supplements for 30 min to recover.
At 7:30 pm, the medium was replaced with L‐15 with appropriate supplements and 20 mM of the proteasome inhibitor MG‐132 (Sigma‐Aldrich, St. Louis, MO, USA) to arrest the cells in metaphase. Cells were fixed in ice‐cold methanol 30 min after adding MG‐132.
Image acquisition
HeLa cells were imaged by using a Leica TCS SP8 X laser scanning confocal microscope with a HC PL APO 63×/1.4 oil immersion objective (Leica, Wetzlar, Germany) heated with an objective integrated heater system (Okolab, Burlingame, CA, USA). Excitation and emission lights were separated with Acousto‐Optical Beam Splitter (AOBS, Leica, Wetzlar, Germany). For live‐cell imaging, cells were maintained at 37°C in Okolab stage top heating chamber (Okolab, Burlingame, CA, USA). For excitation, a 488‐nm line of a visible gas Argon laser and a gated STED supercontinuum visible white light laser at 575 nm were used for GFP and mRFP/Alexa Fluor‐555, respectively. GFP and mRFP/Alexa Fluor‐555 emissions were detected with HyD (hybrid) detectors in ranges of 498–558 and 585–665 nm, respectively. Pinhole diameter was set to 0.8 μm. In experiments with PRC1‐GFP BAC line cells (counting and coupling experiments), images were acquired at 25–35 focal planes with 0.5 μm spacing and 400 Hz unidirectional xyz scan mode. For experiments with dynamic properties in vertical spindles, live PRC1‐GFP cells transiently transfected with mRFP‐CENP‐B were imaged at five focal planes with 0.5 μm spacing and 600 Hz unidirectional xyzt scan mode with time interval set to 13 s. In the cases when the transiently expressed mRFP‐CENP‐B significantly bleached during the experiment, the power of the white light laser (575 nm) was increased during the acquisition, which did not affect the measurements because the mRFP‐CENP‐B signal intensity was not quantified. In experiments with tubulin‐GFP cells (MG132 arrested cells and siRNA experiments), images were acquired at 4–10 focal planes with 0.5 μm spacing and 400 Hz unidirectional xyz scan mode. The system was controlled with the Leica Application Suite X software (LASX, 1.8.1.13759, Leica, Wetzlar, Germany).
Lysate preparation and Western blot analyses
HeLa cells grown on six‐well plates were transfected with 200 nM control siRNA (non‐targeting) or PRC1 siRNA. Non‐treated samples were not transfected. Following transfection and synchronization, the cells were washed with sterile PBS, and harvested by addition of RIPA buffer (Sigma, St. Louis, MO, USA) containing protease inhibitors (Complete TM; Roche, Basel, Switzerland). SDS–PAGE was performed using 12% gels and blotted onto nitrocellulose membranes (BIO‐RAD, Hercules, CAL, USA). Membranes were blocked in 5% bovine serum albumin and probed using rabbit anti‐PRC1 (sc‐8356; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or rabbit anti‐GAPDH antibody (G9545; Sigma, St. Louis, MO, USA), which was used as a loading control. Bound primary antibodies were detected using peroxidase‐conjugated anti‐rabbit immunoglobulin G (A0545. Sigma, St. Louis, MO, USA) and Clarity ECL Western Blotting substrate (Bio‐Rad, Hercules, CAL, USA). Images were acquired using the C‐DiGit blot scanner (LI‐COR, Bad Homburg, Germany). Images were analyzed using Image Studio software (LI‐COR, Bad Homburg, Germany). Percent of PRC1 protein was calculated from Western blot band intensities of all the PRC1 isoform bands in one gel line after normalizing to the corresponding GAPDH band intensity. The data were acquired from 3 to 6 independent experiments.
Image analysis
Image processing and measurements were performed in ImageJ (National Institutes of Health, Bethesda, MD, USA). Quantification, data analysis, and scientific graphing were performed in SciDAVis (Free Software Foundation Inc., Boston, MA, USA). 3D projections were deconvolved and rendered in Huygens Software (Scientific Volume Imaging B.V., Laapersveld, VB, the Netherlands) with the coordinate system represented as a cuboidal box indicating spindle orientation. Cross‐correlation analysis was performed in MATLAB (MathWorks, Natick, MA, USA). Figures and schemes were assembled in Adobe Illustrator CC (Adobe Systems, Mountain View, CA, USA). Statistical analysis was performed using Student's t‐test. Data are given as mean ± s.e.m., unless otherwise indicated.
We used acquired z‐stack images of whole spindles to quantify the number of kinetochore pairs and PRC1‐labeled fibers. In spindles that were oriented with their long axis roughly parallel to the imaging plane both kinetochore pairs and PRC1‐labeled fibers were observed in each z‐slice of individual spindle. In spindles with their long axis oriented roughly perpendicular to the imaging plane, an individual PRC1‐labeled bundle appeared as a bright green dot that spans about 10 z‐slices (5 μm), whereas kinetochores were observed only in central planes that correspond to the metaphase plate. We counted each kinetochore pair and PRC1‐labeled fiber throughout the spindle minding the presence of its signal in the upper and lower z‐plane with respect to the plane in which it had highest signal intensity.
Kinetochore pairs and PRC1‐labeled fibers were defined associated if the distance between the central part of the fiber and the midpoint between centers of sister kinetochores was smaller than 0.3 μm. Spindle length was calculated as the distance between the spindle poles, whereas spindle width was calculated as the distance between the midpoints of the outermost sister kinetochores on the opposite spindle sides. The density of kinetochore pairs coupled with PRC1‐labeled bundles was calculated as the number of kinetochore‐PRC1 pairs in a cross section of the central part of the spindle divided by the cross‐sectional area.
Trajectories of kinetochores and PRC1‐labeled bundles in spindles with their long axis oriented roughly perpendicular to the imaging plane were acquired by using Low Light Tracking Tool, an ImageJ plugin 38. Tracking of kinetochores and PRC1‐labeled bundles in the xy plane was performed on maximum‐intensity projections of up to four planes. To avoid the possible effect of trajectories being the result of the entire spindle moving as a cohesive unit, we calculated the trajectories of kinetochore pairs and of PRC1‐labeled bundles with respect to the spindle's center of mass in each image. Cross‐correlation was calculated with the MATLAB inbuilt function normxcorr2, which includes normalization by dividing with the product of the local standard deviation 35. We acquired only correlation coefficients at unshifted positions, that is, at lag = 0.
In HeLa cells stably expressing tubulin‐GFP that were immunostained for PRC1, we tracked a 5‐pixel‐thick pole‐to‐pole contour of tubulin‐GFP signal of the sister k‐fibers and the corresponding bridging fiber that spans between them. The positions of the spindle poles were estimated as the merging points of k‐fibers. The bundles were tracked manually, point‐by‐point along the curved line, following the tubulin‐GFP signal path (note that the bundles that disappeared in the z‐direction were not tracked). We used this contour to measure intensity profiles of endogenous PRC1 in the red channel (immunostaining).
In cells expressing PRC1‐GFP we tracked the pole‐to‐pole contour of PRC1‐GFP by using approximately 30 points, and measured the intensity profile in the green channel. The positions of the spindle poles were estimated as the merging points of k‐fibers in the maximum‐intensity projection of all z‐slices covering the entire spindle. The mean value of the background signal present in the cytoplasm was subtracted from the intensity profiles. The length of the PRC1‐labeled overlap region, L PRC1, was manually determined as the width of the peak of the PRC1‐GFP signal intensity in the central part of the contour. The width of the peak was defined as the distance between the positions at the base of the PRC1‐GFP peak where the PRC1‐GFP signal intensity is roughly equal to the mean value of the PRC1‐GFP signal intensity along the contour on either side of the peak. We defined signal intensity I as the total sum of intensities in the intensity profile of a 5‐pixel‐thick pole‐to‐pole contour of PRC1‐GFP signal, divided by the contour's total length, and I cross as the total intensity under the peak in the intensity profile along a 5‐pixel‐thick line drawn transversely to the PRC1‐GFP signal. The same method was used for quantification of endogenous PRC1‐immunostained bundles.
The signal intensity of a cross section of a bridging fiber in cells expressing tubulin‐GFP and mRFP‐CENP‐B was measured by drawing a 3‐pixel‐thick line between sister kinetochores and perpendicular to the line joining centers of the two kinetochores, whereas a cross section of the bundle consisting of a bridging fiber and a k‐fiber was measured about 1 μm laterally from either kinetochore. The mean value of the background signal present in the cytoplasm was subtracted from the intensity profile.
The distance from the center was measured between the central part of the overlap fiber and the spindle long axis, perpendicular to the spindle long axis. Distance between sister kinetochores was measured as the distance between their centers, acquired by optimizing and tracking with Low Light Tracking Tool 38.
Author contributions
IMT conceived and supervised the project. BP carried out all experiments except Western blots, PR analyzed the data, SL performed Western blot experiments and analysis. BP and PR wrote the paper, with input from IMT and SL.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Video EV1
Video EV2
Video EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank Ina Poser, Tony Hyman, Frank Bradke, Linda Wordeman, and Mariola Chacon for cell lines and plasmids; Igor Weber, Marko Šoštar, Vedrana Filić Mileta, Maja Marinović, and the entire Weber group (Ruđer Bošković Institute, Zagreb) for help with using the microscope at its full potential; Marijeta Kralj, Marko Marjanović, Iva Guberović, Lidija Uzelac, Katja Ester, and the entire Kralj group (Ruđer Bošković Institute, Zagreb) for help with cell culture; Nenad Pavin, Matko Glunčić, Marcel Prelogović, Maja Novak, and the entire Pavin group (Department of Physics, University of Zagreb) for discussions; Juraj Simunić, Kruno Vukušić, Renata Buđa, and Ana Milas for helpful discussions, Lejla Ferhatović Hamzić, Jelena Martinčić, and the entire Tolić group (Ruđer Bošković Institute, Zagreb) for extensive help, advice, and constructive comments on the manuscript; and Ivana Šarić for the drawings. We thank the European Research Council (ERC, GA Number 647077) for funding.
EMBO Reports (2017) 18: 217–230
References
- 1. Pavin N, Tolic IM (2016) Self‐organization and forces in the mitotic spindle. Annu Rev Biophys 45: 279–298 [DOI] [PubMed] [Google Scholar]
- 2. Cheeseman IM, Desai A (2008) Molecular architecture of the kinetochore‐microtubule interface. Nat Rev Mol Cell Biol 9: 33–46 [DOI] [PubMed] [Google Scholar]
- 3. Rieder CL, Salmon ED (1998) The vertebrate cell kinetochore and its roles during mitosis. Trends Cell Biol 8: 310–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mastronarde DN, McDonald KL, Ding R, McIntosh JR (1993) Interpolar spindle microtubules in PTK cells. J Cell Biol 123(6 Pt 1): 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Manning AL, Compton DA (2008) Structural and regulatory roles of nonmotor spindle proteins. Curr Opin Cell Biol 20: 101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Glotzer M (2009) The 3Ms of central spindle assembly: microtubules, motors and MAPs. Nat Rev Mol Cell Biol 10: 9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bieling P, Telley IA, Surrey T (2010) A minimal midzone protein module controls formation and length of antiparallel microtubule overlaps. Cell 142: 420–432 [DOI] [PubMed] [Google Scholar]
- 8. Subramanian R, Ti SC, Tan L, Darst SA, Kapoor TM (2013) Marking and measuring single microtubules by PRC1 and kinesin‐4. Cell 154: 377–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kapitein LC, Janson ME, van den Wildenberg SM, Hoogenraad CC, Schmidt CF, Peterman EJ (2008) Microtubule‐driven multimerization recruits ase1p onto overlapping microtubules. Curr Biol 18: 1713–1717 [DOI] [PubMed] [Google Scholar]
- 10. Jiang W, Jimenez G, Wells NJ, Hope TJ, Wahl GM, Hunter T, Fukunaga R (1998) PRC1: a human mitotic spindle‐associated CDK substrate protein required for cytokinesis. Mol Cell 2: 877–885 [DOI] [PubMed] [Google Scholar]
- 11. Mollinari C, Kleman JP, Jiang W, Schoehn G, Hunter T, Margolis RL (2002) PRC1 is a microtubule binding and bundling protein essential to maintain the mitotic spindle midzone. J Cell Biol 157: 1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kurasawa Y, Earnshaw WC, Mochizuki Y, Dohmae N, Todokoro K (2004) Essential roles of KIF4 and its binding partner PRC1 in organized central spindle midzone formation. EMBO J 23: 3237–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhu C, Jiang W (2005) Cell cycle‐dependent translocation of PRC1 on the spindle by Kif4 is essential for midzone formation and cytokinesis. Proc Natl Acad Sci USA 102: 343–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pellman D, Bagget M, Tu YH, Fink GR, Tu H (1995) Two microtubule‐associated proteins required for anaphase spindle movement in Saccharomyces cerevisiae. J Cell Biol 130: 1373–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu C, Lau E, Schwarzenbacher R, Bossy‐Wetzel E, Jiang W (2006) Spatiotemporal control of spindle midzone formation by PRC1 in human cells. Proc Natl Acad Sci USA 103: 6196–6201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Janson ME, Loughlin R, Loiodice I, Fu C, Brunner D, Nedelec FJ, Tran PT (2007) Crosslinkers and motors organize dynamic microtubules to form stable bipolar arrays in fission yeast. Cell 128: 357–368 [DOI] [PubMed] [Google Scholar]
- 17. Verbrugghe KJ, White JG (2004) SPD‐1 is required for the formation of the spindle midzone but is not essential for the completion of cytokinesis in C. elegans embryos. Curr Biol 14: 1755–1760 [DOI] [PubMed] [Google Scholar]
- 18. Verni F, Somma MP, Gunsalus KC, Bonaccorsi S, Belloni G, Goldberg ML, Gatti M (2004) Feo, the Drosophila homolog of PRC1, is required for central‐spindle formation and cytokinesis. Curr Biol 14: 1569–1575 [DOI] [PubMed] [Google Scholar]
- 19. Smertenko A, Saleh N, Igarashi H, Mori H, Hauser‐Hahn I, Jiang CJ, Sonobe S, Lloyd CW, Hussey PJ (2000) A new class of microtubule‐associated proteins in plants. Nat Cell Biol 2: 750–753 [DOI] [PubMed] [Google Scholar]
- 20. Hu CK, Ozlu N, Coughlin M, Steen JJ, Mitchison TJ (2012) Plk1 negatively regulates PRC1 to prevent premature midzone formation before cytokinesis. Mol Biol Cell 23: 2702–2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Neef R, Gruneberg U, Kopajtich R, Li X, Nigg EA, Sillje H, Barr FA (2007) Choice of Plk1 docking partners during mitosis and cytokinesis is controlled by the activation state of Cdk1. Nat Cell Biol 9: 436–444 [DOI] [PubMed] [Google Scholar]
- 22. Subramanian R, Wilson‐Kubalek EM, Arthur CP, Bick MJ, Campbell EA, Darst SA, Milligan RA, Kapoor TM (2010) Insights into antiparallel microtubule crosslinking by PRC1, a conserved nonmotor microtubule binding protein. Cell 142: 433–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kajtez J, Solomatina A, Novak M, Polak B, Vukusic K, Rudiger J, Cojoc G, Milas A, Sumanovac IS, Risteski P et al (2016) Overlap microtubules link sister k‐fibres and balance the forces on bi‐oriented kinetochores. Nat Commun 7: 10298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Milas A, Tolic IM (2016) Relaxation of interkinetochore tension after severing of a k‐fiber depends on the length of the k‐fiber stub. Matters doi: 10.19185/matters.201603000025
- 25. Tolic IM, Pavin N (2016) Bridging the gap between sister kinetochores. Cell Cycle 15: 1169–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Poser I, Sarov M, Hutchins JR, Heriche JK, Toyoda Y, Pozniakovsky A, Weigl D, Nitzsche A, Hegemann B, Bird AW et al (2008) BAC TransgeneOmics: a high‐throughput method for exploration of protein function in mammals. Nat Methods 5: 409–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Landry JJ, Pyl PT, Rausch T, Zichner T, Tekkedil MM, Stutz AM, Jauch A, Aiyar RS, Pau G, Delhomme N et al (2013) The genomic and transcriptomic landscape of a HeLa cell line. G3 (Bethesda) 3: 1213–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adey A, Burton JN, Kitzman JO, Hiatt JB, Lewis AP, Martin BK, Qiu R, Lee C, Shendure J (2013) The haplotype‐resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature 500: 207–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ruess D, Ye LZ, Grond‐Ginsbach C (1993) HeLa D98/aH‐2 studied by chromosome painting and conventional cytogenetical techniques. Chromosoma 102: 473–477 [DOI] [PubMed] [Google Scholar]
- 30. Bottomley RH, Trainer AL, Griffin MJ (1969) Enzymatic and chromosomal characterization of HeLa variants. J Cell Biol 41: 806–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heneen WK (1976) HeLa cells and their possible contamination of other cell lines: karyotype studies. Hereditas 82: 217–248 [DOI] [PubMed] [Google Scholar]
- 32. Macville M, Schröck E, Padilla‐Nash H, Keck C, Ghadimi BM, Zimonjic D, Popescu N, Ried T (1999) Comprehensive and definitive molecular cytogenetic characterization of HeLa cells by Spectral Karyotyping. Cancer Res 59: 141–150 [PubMed] [Google Scholar]
- 33. Lavappa KS, Macy ML, Shannon JE (1976) Examination of ATCC stocks for HeLa marker chromosomes in human cell lines. Nature 259: 211–213 [DOI] [PubMed] [Google Scholar]
- 34. Hori A, Morand A, Ikebe C, Frith D, Snijders AP, Toda T (2015) The conserved Wdr8‐hMsd1/SSX2IP complex localises to the centrosome and ensures proper spindle length and orientation. Biochem Biophys Res Commun 468: 39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lewis JP (1995) Fast normalized cross‐correlation. Vision Interface 10: 120–123 [Google Scholar]
- 36. Simunic J, Tolic IM (2016) Mitotic spindle assembly: building the bridge between sister k‐fibers. Trends Biochem Sci 41: 824–833 [DOI] [PubMed] [Google Scholar]
- 37. Vladimirou E, Mchedlishvili N, Gasic I, Armond JW, Samora CP, Meraldi P, McAinsh AD (2013) Nonautonomous movement of chromosomes in mitosis. Dev Cell 27: 60–71 [DOI] [PubMed] [Google Scholar]
- 38. Krull A, Steinborn A, Ananthanarayanan V, Ramunno‐Johnson D, Petersohn U, Tolic‐Norrelykke IM (2014) A divide and conquer strategy for the maximum likelihood localization of low intensity objects. Opt Express 22: 210–228 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Video EV1
Video EV2
Video EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4