

Abstract
An important distinction is frequently made between constitutively expressed housekeeping genes versus regulated genes. Although generally characterized by different DNA elements, chromatin architecture and cofactors, it is not known to what degree promoter classes strictly follow regulatability rules and which molecular mechanisms dictate such differences. We show that SAGA‐dominated/TATA‐box promoters are more responsive to changes in the amount of activator, even compared to TFIID/TATA‐like promoters that depend on the same activator Hsf1. Regulatability is therefore an inherent property of promoter class. Further analyses show that SAGA/TATA‐box promoters are more dynamic because TATA‐binding protein recruitment through SAGA is susceptible to removal by Mot1. In addition, the nucleosome configuration upon activator depletion shifts on SAGA/TATA‐box promoters and seems less amenable to preinitiation complex formation. The results explain the fundamental difference between housekeeping and regulatable genes, revealing an additional facet of combinatorial control: an activator can elicit a different response dependent on core promoter class.
Keywords: chromatin, gene regulation, SAGA, TFIID, transcription
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Transcription
Introduction
First coined almost 40 years ago, the term “housekeeping gene” is widely used to distinguish genes with constitutive expression from genes whereby expression is regulated upon environmental, cell type or developmental cues. Genome‐wide analyses have facilitated the identification of housekeeping genes, resulting in the characterization of general properties such as gene structure, evolutionary conservation and promoter features (Eisenberg & Levanon, 2013). Despite the long‐standing distinction that has been made between housekeeping and regulatable genes, the degree to which such a functional division strictly applies is not clear. In addition, the molecular mechanisms behind such functional differences are not understood.
As well as being grouped based on function, genes can also be classified according to the DNA elements present in their promoters. Besides cis‐regulatory elements for binding transcriptional activators, promoters also harbour core promoter elements (Juven‐Gershon & Kadonaga, 2010; Müller & Tora, 2014). One such core promoter motif is the TATA‐box. The presence or absence of such core promoter elements has been linked to function. For example, mammalian genes without a well‐defined TATA‐box but with a CpG island promoter are generally associated with housekeeping function (Deaton & Bird, 2011). Functional distinction based on core promoter type is not clear‐cut however. For example, many tissue‐specific genes are also driven by CpG island promoters (Deaton & Bird, 2011).
A less complex regulatory system is offered by the yeast Saccharomyces cerevisiae where a similar dichotomy is present. Here promoters with a TATA‐like element rather than a consensus TATA‐box are also generally associated with housekeeping function (Basehoar et al, 2004; Huisinga & Pugh, 2004). A related dichotomy is based on the co‐activator used to nucleate assembly of the RNA polymerase II preinitiation complex (PIC) at the different core promoter types (Lee et al, 2000; Huisinga & Pugh, 2004). An essential and early step in PIC formation is TATA‐binding protein (TBP) recruitment (Davison et al, 1983; Buratowski et al, 1989; Roeder, 1996). Whereas TATA‐box‐containing promoters generally depend on the co‐activator SAGA to recruit TBP, TATA‐like promoters are enriched for the co‐activator TFIID for TBP recruitment (Basehoar et al, 2004; Huisinga & Pugh, 2004; Rhee & Pugh, 2012).
In yeast, there is therefore a general distinction between housekeeping genes with a TATA‐like element and preferential usage of TFIID on the one hand, versus regulatable/responsive genes with a consensus TATA‐box and preferential usage of SAGA on the other. A clear explanation for the regulatory difference between these two broad classes of promoters is missing however. Several concepts are possible. For example, on average, individual SAGA‐dominated/TATA‐box promoters seem to be targeted by more regulators than TFIID‐dominated/TATA‐like promoters (Huisinga & Pugh, 2004; Tirosh et al, 2006; Venters et al, 2011) offering a possible straightforward explanation for their higher regulatability. There are also general differences in nucleosome configuration between the two promoter classes (Albert et al, 2007; Tirosh & Barkai, 2008; Cairns, 2009; Jiang & Pugh, 2009; Rhee & Pugh, 2012). Nucleosome occupancy and positioning differences have previously been associated with diverse regulatory properties including responsiveness (Lam et al, 2008; Tirosh & Barkai, 2008; Raveh‐Sadka et al, 2009). Whether chromatin architecture explains regulatability differences between SAGA‐ and TFIID‐dominated promoters is still an open question however (Nocetti & Whitehouse, 2016). Furthermore, neither the involvement of chromatin architecture nor a difference in the number of regulators explains the apparent involvement of the two co‐activators TFIID and SAGA in regulatability differences. This is particularly poignant given that an association between TATA‐box type and constitutive versus regulatable expression was first reported 30 years ago (Struhl, 1986).
Here we characterize differences in regulatability between SAGA‐ and TFIID‐dominated genes and study the molecular mechanisms that dictate this property. Most of what is known about the relationship between core promoter type and responsiveness is derived from correlations between genome‐scale datasets. We therefore first set up a tractable experimental system that allows for a clear measure of promoter responsiveness, is amenable to mechanistic analyses and is capable of addressing the role of core promoter type with minimal potential interference of different regulator use. By focusing on a single transcriptional activator that serves genes from both classes of promoters, a clear difference in regulatability is demonstrated. Whereas SAGA/TATA‐box promoters are highly responsive to the presence of the activator Hsf1, TFIID/TATA‐like promoters are much less dynamically responsive to changes of the same activator. On its own, this clearly establishes that there is indeed an inherent difference in regulatability between the two promoter classes, not dictated by use of different numbers or types of transcriptional activators. The system is then further analysed to uncover the molecular mechanisms that dictate the difference in responsiveness. Evidence for two contributing mechanisms is presented. The first explains the differential involvement of TFIID and SAGA on constitutive and responsive promoters, respectively, and involves a general negative regulator of TBP binding, Mot1 (Pereira et al, 2003). The second mechanism involves differences in the nucleosome architecture changes observed upon activator absence or presence on the two promoter classes. The consequence of both mechanisms is that promoter output on SAGA/TATA‐box promoters is more dynamically dependent on the presence of an activator because of dynamic negative regulation particular to this promoter class. The results reveal an additional facet of combinatorial control and provide a molecular mechanistic explanation for the fundamental difference between housekeeping and regulatable genes.
Results
Previously reported differences in responsiveness between SAGA‐ and TFIID‐dominated genes, or between TATA‐box and TATA‐like promoters, were based on general observations of higher variation in expression levels for SAGA‐dominated/TATA‐like genes under a variety of experimental and genetic perturbations (Huisinga & Pugh, 2004; Tirosh et al, 2006; Venters et al, 2011). Such differences may simply reflect a difference between the type or number of transcriptional regulators used by the two classes of promoters, as has indeed been suggested (Huisinga & Pugh, 2004; Tirosh et al, 2006; Venters et al, 2011). We therefore first determined whether there is a transcriptional activator that regulates genes from both promoter classes. Based on genome‐wide binding of most yeast transcription factors (MacIsaac et al, 2006), a possible candidate is heat‐shock factor 1 (Hsf1). Cofactor binding data indicate that approximately half of the promoters bound by Hsf1 are dominated by SAGA and the other half by TFIID (Rhee & Pugh, 2012). This suggests that Hsf1 and its targets would likely form a good model to investigate inherent differences in regulatability between SAGA‐ and TFIID‐dominated promoters. Since this assessment is based on genome‐wide binding data, it was important to first verify which targets are indeed dependent on Hsf1 and whether these direct targets represent both classes of core promoters.
Rapid nuclear depletion of Hsf1
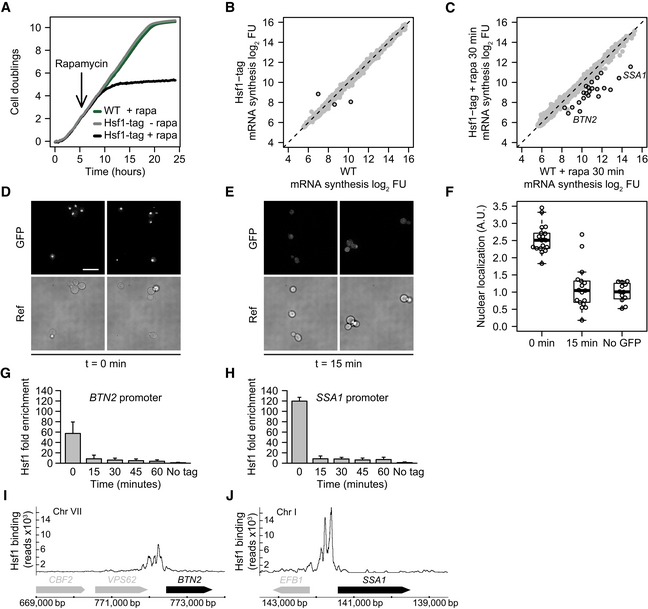
Hsf1 is required for the induction of heat‐shock proteins but also binds some promoters under standard growth conditions (Hahn et al, 2004). This is likely the reason why HSF1 is essential for cell viability even in the absence of a heat shock (Wiederrecht et al, 1988), a proposal that has recently been confirmed (Solís et al, 2016). Previous studies have mostly used temperature‐sensitive HSF1 mutant alleles to study Hsf1 function (Smith & Yaffe, 1991; Zarzov et al, 1997; Imazu & Sakurai, 2005). To circumvent the secondary effects associated with such approaches, an inducible Hsf1 nuclear depletion strain was constructed based on the “anchor‐away” technique developed in the Laemmli laboratory (Haruki et al, 2008), but using the S288C/BY4742 genetic background. Besides the FRB (FK506 binding protein–rapamycin binding domain) tag required to induce nuclear depletion, GFP was also added to monitor cellular location. To ensure that FRB‐GFP tagging of Hsf1 does not interfere with its function, growth of the tagged strain was compared to an untagged background strain (wild type: WT). Growth was found to be identical (Fig 1A). As a more stringent test, genome‐wide mRNA synthesis was compared between WT and the tagged strain (Fig 1B). Only three genes show a significant difference in mRNA synthesis (P < 0.01 and fold change (FC) > 1.7). The two genes with apparently decreased levels are HSF1 itself and a dubious open reading frame (ORF) overlapping the altered 3' end of HSF1. The HSF1 gene is considerably longer because of the FRB‐GFP tag, making cDNA synthesis and cRNA amplification of the region encompassing the microarray probe less efficient and thereby causing an apparent drop in transcription rate compared to WT. The gene with increased transcription is the dubious ORF YDL196W, known to have highly variable expression in WT strains (Kemmeren et al, 2014). The small number of expression changes (Fig 1B) and identical growth compared to WT (Fig 1A) indicates that tagging of Hsf1 in this way does not compromise its function under standard growth conditions.
Figure 1. Dynamics of Hsf1 nuclear depletion.
-
AGrowth curves of WT and the HSF1 FRB‐GFP‐tagged strain with and without rapamycin, the inducing agent for nuclear depletion (Haruki et al, 2008). WT is the parental S288C/BY4742 strain for anchor‐away that has been genetically desensitized to rapamycin.
-
BScatterplot of 4tU‐derived mRNA synthesis in the Hsf1‐tagged strain compared to WT. The x‐ and y‐axes plot the log2 fluorescent dye intensities of the microarray probes representing each gene (dots), averaged over four replicates. Black circles indicate genes with significantly altered synthesis (FC > 1.7 and P < 0.01, calculated using limma).
-
CmRNA synthesis, 30 min after induction of Hsf1 nuclear depletion versus same treatment in WT. Black circles indicate genes with significantly decreased synthesis (FC > 1.7 and P < 0.01, calculated using limma).
-
DFluorescence microscopy images of Hsf1‐FRB‐GFP at t = 0 for induced depletion. Scale bar: 10 μm.
-
EFluorescence microscopy images of Hsf1‐FRB‐GFP 15 min after induction of depletion.
-
FBoxplot showing the quantification of Hsf1 depletion from the nucleus. Dots represent individual cell measurements. Solid horizontal lines show the median, the box represents the interquartile range and the whiskers are at the most extreme data point no further away from the closest quartile than 1.5 times the interquartile range.
-
G, HDynamics of Hsf1 depletion from the SSA1 promoter (SAGA‐dominated) and BTN2 promoter (TFIID‐dominated), as measured by ChIP‐qPCR. Error bars show the standard deviation of four independent replicates.
-
I, JHsf1 binding to the SSA1 and BTN2 promoters as measured by ChIP‐seq.
Yeast Hsf1 is a nuclear protein (Morano et al, 2012). Anchor‐away with the FRB‐GFP‐tagged strain facilitates induced nuclear depletion. HSF1 FRB‐GFP‐tagged cells cease growth approximately five hours after starting depletion (Fig 1A). This agrees with the previously established essential role of Hsf1 under standard growth conditions (Wiederrecht et al, 1988). Given that it is the lack of expression of target genes that causes HSF1 gene deletion lethality (Solís et al, 2016), growth cessation is an indirect measure of Hsf1 loss‐of‐function dynamics. Fluorescence microscopy was therefore applied to more precisely determine the dynamics of Hsf1 nuclear depletion. In contrast to cessation of growth (5 h: Fig 1A), nuclear depletion of Hsf1 is much faster, with nuclear GFP signal dropping to background levels within 15 min (Fig 1D–F), in agreement with Hsf1 nuclear depletion in a different genetic background (Solís et al, 2016). To establish whether any residual Hsf1 remains bound to target promoters after 15 min, at levels undetectable by fluorescence microscopy, chromatin immunoprecipitation (ChIP) was performed on two Hsf1 bound promoters (MacIsaac et al, 2006): BTN2, which is TFIID‐dominated, and SSA1, which is SAGA‐dominated (Rhee & Pugh, 2012). The dynamics of Hsf1 loss from both promoters (Fig 1G and H) corresponds very well with the dynamics of nuclear depletion determined by fluorescence microscopy (Fig 1E and F), also reaching background levels after 15 min of depletion (Fig 1G and H). Taken together, these results demonstrate that Hsf1 can be rapidly depleted from target promoters and the nucleus within 15 min.
Direct targets of Hsf1 during standard growth conditions
To determine the direct transcriptional targets of Hsf1, mRNA synthesis rates were monitored upon Hsf1 depletion and compared to Hsf1 genome‐wide localization derived from ChIP‐seq. mRNA synthesis rates were determined using 4‐thiouracil (4tU) which is incorporated into newly synthesized mRNAs (Sun et al, 2012). This eliminates the confounding effect of differential mRNA decay rates interfering with target gene identification and is a more direct measure of promoter output, better suited to the goals of this study. A specific set of genes show reduced synthesis rates 30 min after induction of Hsf1 depletion (P < 0.01 and FC > 1.7, Fig 1C). To determine whether all these effects are direct, Hsf1 binding was monitored by ChIP‐seq under the same growth condition. All genes with decreased mRNA synthesis (Fig 1C) also show strong Hsf1 promoter binding. Examples of TFIID‐ and SAGA‐dominated targets are shown in Fig 1I and J. Integration of the mRNA synthesis and Hsf1 binding data leads to identification of 21 direct targets (Materials and Methods). These are enriched for the Gene Ontology (GO) term protein folding (P < 2 × 10−24), in agreement with the established role of Hsf1 in regulating the expression of protein chaperones, also under non‐heat‐shock conditions (Gross et al, 1990; Solís et al, 2016). The 21 targets are a subset of the promoters previously reported to be bound by Hsf1 under standard and heat‐shock conditions (Hahn et al, 2004) and correspond almost exactly with Hsf1 direct targets also recently defined using anchor‐away (Solís et al, 2016). Most importantly, half of these targets are dominated by SAGA (11/21) and the other half by TFIID (10/21) (Rhee & Pugh, 2012). This is in itself an important finding. The fact that a single activator can equally service genes from both promoter classes already indicates that different regulatability properties associated with the different classes are indeed inherent to promoter type rather than dependent on the number or type of activator. Such a conclusion can only be drawn if the Hsf1 targets actually exhibit differences in regulatability between the two promoter classes. We therefore next set out to investigate regulatability using the Hsf1 target genes as a model.
SAGA‐ and TFIID‐dominated genes are dependent on Hsf1 in different ways
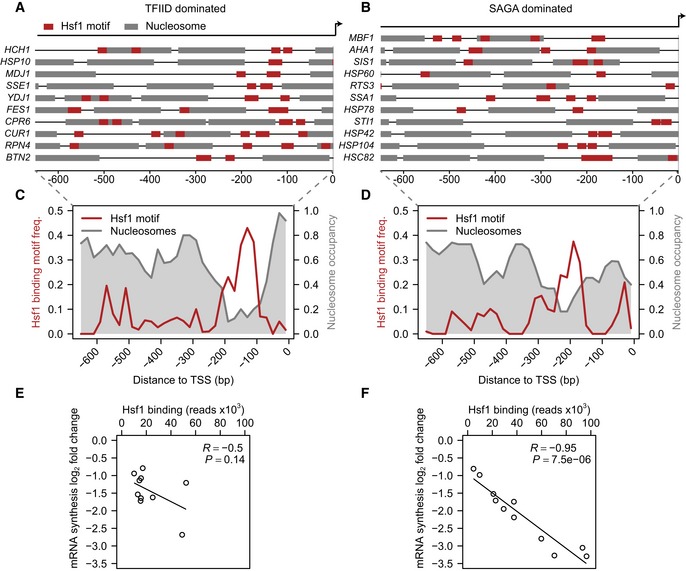
To serve as a model, it is also essential that the Hsf1 direct targets exhibit the properties that are characteristic for each promoter class (Albert et al, 2007; Tirosh & Barkai, 2008; Cairns, 2009; Rhee & Pugh, 2012). TFIID‐dominated promoters are characterized by a nucleosome‐depleted region (NDR) followed by a well‐positioned +1 nucleosome. SAGA‐dominated genes generally show a less uniformly organized promoter, with fuzzier nucleosome positioning and a smaller, less pronounced NDR. Based on publicly available nucleosome occupancy data (Jiang & Pugh, 2009), the Hsf1 targets defined here reflect these established differences (Fig 2A–D). The average location of Hsf1 binding motifs also shows a clear difference, with a location approximately 60 bp more downstream in TFIID‐ compared to SAGA‐dominated promoters (Fig 2C and D). A similar difference in motif location of other activators has been described before, for promoters with and without a TATA‐box (Erb & van Nimwegen, 2011). Together, the attributes displayed by the two sets of Hsf1 targets defined here fit very well with the attributes previously defined on TFIID‐ and SAGA‐dominated promoters genome‐wide, further indicating their utility to study functional differences between the two classes, irrespective of activator usage.
Figure 2. SAGA‐ and TFIID‐dominated genes are differentially sensitive to loss of the same activator.
-
A–DMap of nucleosome positions (Jiang & Pugh, 2009) and trimeric Hsf1 binding motif locations for TFIID‐ (A) and SAGA‐dominated (B) promoters, with the average shown using 10 bp bins in (C) and (D), respectively.
-
E, FScatterplots for the amount of bound Hsf1 on each of the TFIID‐ (E) and SAGA‐dominated (F) Hsf1 targets under standard growth conditions versus the drop in mRNA synthesis rates 30 min after induction of Hsf1 nuclear depletion. To quantify the correlation between the binding and expression changes, R‐ and P‐values were calculated using the function “cor.test” in the statistical language R. Table EV1 contains the underlying values.
By definition, all the 21 direct target genes of Hsf1 are dependent for mRNA synthesis on Hsf1 presence, regardless of promoter class. To investigate promoter responsiveness, we first asked whether there is a relationship between Hsf1 binding levels under standard growth conditions, and the drop in mRNA synthesis observed upon Hsf1 depletion. Strikingly, SAGA‐dominated genes exhibit a strong correlation between the amount of Hsf1 initially bound to the promoter and the change in transcriptional output upon Hsf1 depletion (Fig 2F, the correlation of the SAGA‐dominated genes is robust to outliers; Table EV1). This is not the case for TFIID‐dominated genes. Here there is no statistically significant correlation and the value depicted is dependent on a single data point (Fig 2E and Table EV1). On average, the Hsf1 binding levels are twofold higher on SAGA‐dominated promoters. This difference is not statistically significant however (P = 0.07, two‐sided t‐test), and sequence motif analyses did not reveal strong differences either. Furthermore, although a few of the SAGA‐dominated promoters exhibit high initial Hsf1 binding levels, the stronger response to Hsf1 loss is also exhibited by those SAGA‐dominated promoters with lower initial Hsf1 binding levels (Fig 2F). These comparisons indicate that for SAGA‐dominated promoters, there is a tight relationship between the amount of Hsf1 that is bound and the transcriptional output, whereas for TFIID‐dominated genes such a clear relationship is lacking. This difference fits with the idea that SAGA‐dominated promoters are more regulatable than TFIID‐dominated promoters.
SAGA‐dominated promoters are more responsive to activator presence
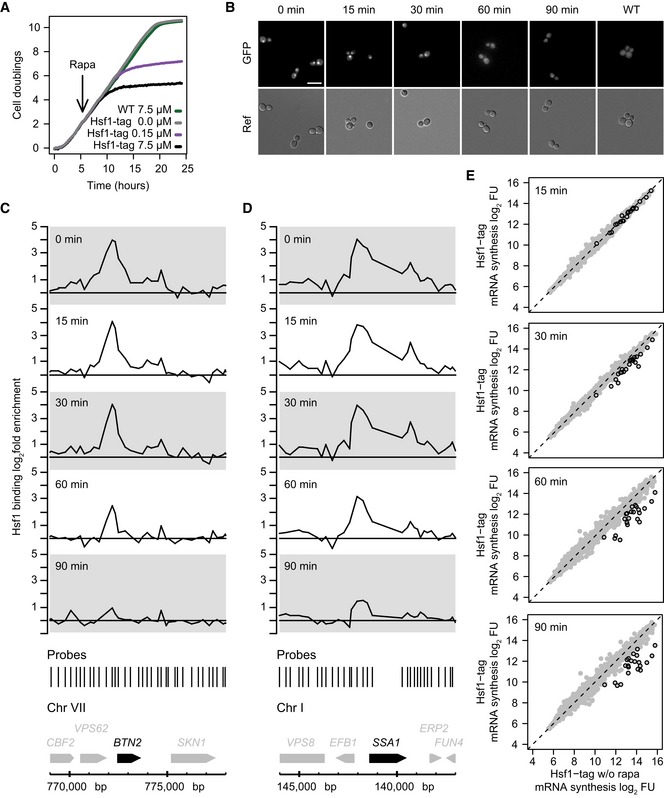
A limitation of the previous analysis is that it does not determine how each individual promoter responds to different amounts of Hsf1. To investigate the difference between TFIID‐ and SAGA‐dominated Hsf1 targets in more detail, individual promoter responses were determined by measuring Hsf1 binding and transcriptional output over a range of nuclear Hsf1 concentrations. Because depletion of Hsf1 from the nucleus is rapid (Fig 1D–F), especially in comparison with the six‐minute window of 4tU label incorporation, a slow nuclear depletion experiment was performed. This enables measurement of mRNA synthesis rates at multiple Hsf1 levels. Slow depletion was achieved by adding a fifty‐fold lower concentration of the inducing agent, resulting in a delayed impact on growth compared to the high concentration (Fig 3A). Monitoring the slow depletion using fluorescence microscopy shows that it takes approximately 90 min to deplete Hsf1 from the nucleus (Fig 3B). Promoter‐bound Hsf1 was monitored during the time course (examples in Fig 3C and D) and the corresponding promoter output was measured by 4tU labelling at multiple time points (Fig 3E). Hsf1 binding and the promoter output both show a gradual decrease, corresponding to a slower depletion of Hsf1 from the nucleus. When the Hsf1 binding and mRNA synthesis data are combined, a clear contrast between the two promoter classes is revealed (Fig 4). Figure 4A depicts for each Hsf1 target, the fold change in mRNA synthesis rates (log2 FC, y‐axis) versus the fold change in Hsf1 binding (log2 FC, x‐axis). The slope of the line fitted through these data points is a measure for the response in promoter output to a change in the levels of bound Hsf1: promoter responsiveness. Determining responsiveness in this manner yields a value that is independent of the starting amounts of either promoter‐bound Hsf1 or initial mRNA synthesis rates. Comparing the responsiveness between the two classes (Fig 4B) shows that the SAGA‐dominated genes have a higher responsiveness than the TFIID‐dominated promoters. The difference between TFIID‐ and SAGA‐dominated promoters is also reflected upon ranking the responsiveness (Fig 4A): almost all TFIID‐dominated promoters (blue) have a lower responsiveness than the SAGA‐dominated promoters (orange). This agrees with the differences observed in the fast depletion experiment (Fig 2E and F), extending the observation to promoters individually. Given the use of an identical activator by the two promoter classes, these experiments demonstrate that responsiveness is inherent to promoter type.
Figure 3. Slow nuclear depletion of Hsf1 using a lower concentration of rapamycin.
-
AGrowth curve of Hsf1‐tagged strain after exposure to a 50‐fold reduced rapamycin concentration, compared to untreated, and to WT or the tagged strain with the high dose.
-
BFluorescence microscopy images of Hsf1 depletion at low rapamycin concentration. Scale bar: 10 μm.
-
C, DSlow depletion of Hsf1 from BTN2 (C) and SSA1 (D) promoters as measured by high‐density tiling arrays, using four independent replicates.
-
E4tU mRNA synthesis changes upon slow depletion of Hsf1 from the nucleus. The x‐ and y‐axes plot the log2 fluorescent dye intensities of the probes representing each gene (dots), averaged over four replicates, during the time course (y‐axis) versus the tagged strain before depletion (x‐axis). Circled dots indicate direct Hsf1 targets.
Figure 4. SAGA‐dominated Hsf1 targets are more responsive than TFIID‐dominated Hsf1 targets.
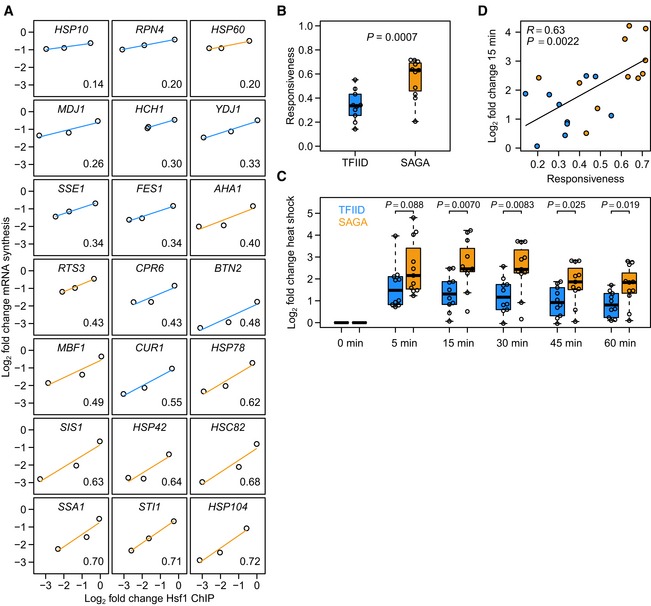
- Responsiveness (measured as the slope of the line fit to Hsf1 binding change versus mRNA synthesis change) of Hsf1 targets ordered from low to high. Blue lines are TFIID‐regulated genes and orange lines are SAGA‐regulated genes. The responsiveness of each target is shown in the bottom right corner.
- Boxplot of responsiveness values shown in (A). The P‐value was calculated using a linear model with the log2 binding ratio as a continuous covariate. Solid horizontal lines show the median, the box represents the interquartile range and the whiskers are at the most extreme data point no further away from the closest quartile than 1.5 times the interquartile range.
- Log2 fold expression changes of the TFIID‐dominated (blue) and SAGA‐dominated (orange) Hsf1 targets in response to heat shock. To determine the significance of difference in upregulation between the two promoter classes, P‐values were calculated using a two‐sided t‐test. Each time point was grown as biological replicate and measured in technical replicate, yielding four measurements per gene.
- Expression changes upon 15 min of heat shock versus the responsiveness of the direct Hsf1 targets. Colours are the same as in (A). Heat‐shock expression data were taken from O'Duibhir et al (2014). R‐ and P‐values were calculated using the function “cor.test” in the statistical language R. Fig EV1 encompasses additional analyses of responsiveness.
SAGA‐dominated Hsf1 targets are also more responsive during heat shock
Before setting out to determine the molecular mechanism that underlies differential responsiveness, it was first determined whether the differential responsiveness observed between SAGA and TFIID Hsf1 targets in the depletion experiments (Figs 2E and F, and 4A and B) is also reflected during physiological changes. Hsf1 is an activator of the heat‐shock response (Morano et al, 2012), activating its targets when cells are exposed to heat stress. Based on published heat‐shock gene expression data (O'Duibhir et al, 2014), the TFIID‐ and SAGA‐dominated Hsf1 targets indeed behave differently upon heat stress, with the SAGA‐dominated Hsf1 targets showing a larger expression increase compared to the TFIID‐dominated Hsf1 targets (Fig 4C). This agrees well with the higher responsiveness of the SAGA‐dominated targets observed upon Hsf1 depletion under non‐heat‐shock conditions (Fig 4A and B). There is also good correspondence between the responsiveness determined by nuclear depletion of Hsf1 and the heat‐shock response when analysed individually (Fig 4D). This is especially striking when bearing in mind that a physiological heat shock involves many more cellular changes besides upregulation of heat‐shock genes (Morano et al, 2012) and that the heat‐shock response was monitored without 4tU (O'Duibhir et al, 2014). The concordance (Fig 4D) shows that the higher responsiveness measured for SAGA‐dominated Hsf1 targets by nuclear depletion is physiologically relevant and reflected by the differential behaviour of these targets upon heat shock.
Molecular mechanisms of activator responsiveness
Having established that SAGA and TFIID promoters are differentially responsive to the presence of an identical activator (Fig 4), we next investigated the molecular mechanisms underlying the difference in responsiveness. First, several trivial explanations were explored. Importantly, the rate of Hsf1 promoter depletion from the two classes is not measurably different (Fig EV1A), ruling out differential promoter depletion as a possible explanation. Class differences in initial or final mRNA synthesis rates were also ruled out (Fig EV1B and C). There is also no significant correlation between the initial synthesis rates and the responsiveness for either class (Fig EV1D and E), as expected given the way in which responsiveness is calculated. Also, no significant Hsf1 binding motif differences or differential presence of poly[dA:dT] motifs were found between the two classes. Neither was there a differential enrichment for any of the general regulatory factors (GRFs) Abf1, Rap1 or Reb1 (Rhee & Pugh, 2011; Kasinathan et al, 2014). This indicates that motif differences or binding of GRFs do not account for the difference in responsiveness observed.
Figure EV1. Depletion rate and mRNA synthesis levels do not explain differences in responsiveness.
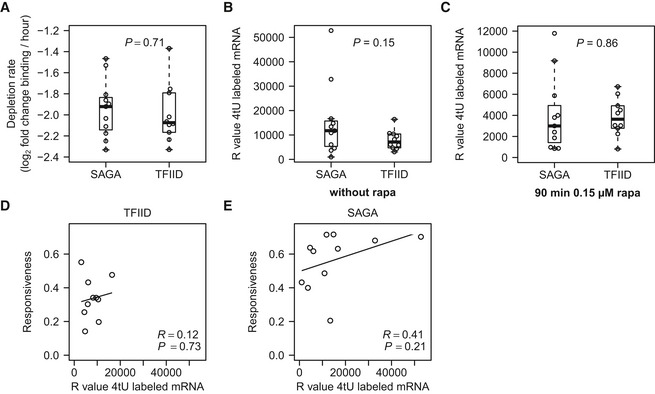
-
ABoxplot showing the comparison of the depletion rate of Hsf1 from the promoter of the SAGA‐ and TFIID‐dominated genes. Depletion rate was calculated by plotting Hsf1 binding versus time and taking the slope of the line fitted through these points.
-
BBoxplot of initial mRNA synthesis before addition of rapamycin, as measured by the raw microarray intensity values (R values), compared between the SAGA‐ and TFIID‐dominated genes.
-
CBoxplot with the mRNA synthesis rates (R values) after 90 min of slow depletion.
-
D, ECorrelation of the mRNA synthesis before depletion (R values) with the responsiveness of (D) the TFIID‐dominated and (E) the SAGA‐dominated targets.
Many activators exert a positive influence on transcription through direct recruitment of PIC components (Struhl, 1995; Ptashne & Gann, 1997). One of the essential components of the PIC is TFIIB, which directly contacts DNA, TBP and RNA polymerase II (Grünberg & Hahn, 2013; Sainsbury et al, 2015). For both the TFIID‐ and SAGA‐dominated Hsf1 targets, there is a good correlation between TFIIB binding and Pol II presence (Fig 5A and B). This suggests that any PIC formation steps following TFIIB binding and that lead to Pol II recruitment proceed similarly for the two promoter classes. Interestingly, this concordance between the two promoter classes is not observed when comparing TFIIB binding and Hsf1 binding. Whereas on SAGA‐dominated promoters the amount of TFIIB correlates well with the amount of Hsf1, for TFIID promoters this relationship is almost absent (Fig 5C and D). This suggests that Hsf1 is important for PIC formation or stability on SAGA promoters, but less so for TFIID‐dominated genes and that the distinguishing step takes place at or before TFIIB binding.
Figure 5. Hsf1 SAGA‐dominated promoters are sensitive to negative regulation by Mot1.
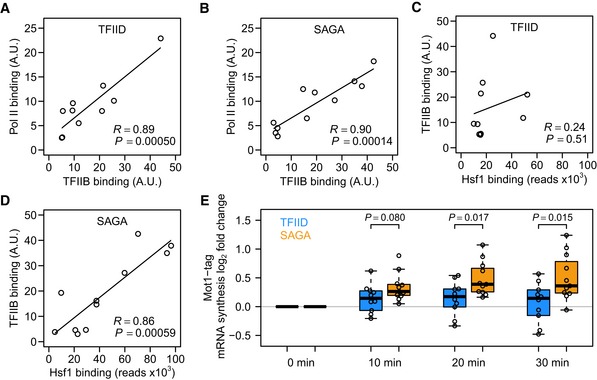
-
A, BCorrelation between the amount of TFIIB and Pol II bound to the TFIID‐dominated (A) and SAGA‐dominated (B) Hsf1 targets.
-
CLack of correlation between the amount of TFIIB binding and the amount of Hsf1 binding for TFIID‐dominated Hsf1 targets.
-
DCorrelation between the amount of TFIIB binding and the amount of Hsf1 binding for SAGA‐dominated targets. TFIIB and Pol II binding data are from Rhee and Pugh (2012).
-
ELog2 fold change in 4tU mRNA synthesis rates for the TFIID (blue) and SAGA (orange) Hsf1 targets immediately following Mot1 nuclear depletion, all compared to the same strain before depletion. The P‐values were by two‐sided t‐test. Each time point was grown as biological replicate and measured in technical replicate, yielding four measurements per gene. Solid horizontal lines show the median, the box represents the interquartile range and the whiskers are at the most extreme data point no further away from the closest quartile than 1.5 times the interquartile range.
Negative regulation by Mot1 is associated with higher responsiveness
The disconnect between Hsf1 binding and TFIIB presence at TFIID‐dominated promoters suggests that there is a difference in PIC formation or stability at the two promoter classes. A key step in PIC formation is TBP recruitment (Davison et al, 1983; Buratowski et al, 1989; Roeder, 1996), which proceeds differently for the two promoter classes due to different preferential co‐activator use (Basehoar et al, 2004; Huisinga & Pugh, 2004; Rhee & Pugh, 2012). A known regulator of TBP promoter dynamics is Mot1 (Tora & Timmers, 2010; Viswanathan & Auble, 2011), which removes TBP preferentially from SAGA‐dominated/TATA‐box‐containing promoters (Zentner & Henikoff, 2013). Differential TBP turnover can potentially explain a difference in responsiveness. When TBP is more rapidly removed from one of the classes of promoters, transcription would be more efficiently shut down in this class, leading to a larger response upon depletion of an activator. Indeed, TBP turnover has been shown to be high in SAGA‐dominated promoters and low in TFIID‐dominated promoters (van Werven et al, 2009). Furthermore, genes that become derepressed upon Mot1 inactivation are enriched for SAGA‐dominated genes (Spedale et al, 2012). Since Mot1 evicts TBP from SAGA‐dependent promoters and given the indications that there is a difference in PIC stability between the two promoter classes (Fig 5C and D), it was next tested whether negative regulation by Mot1 underlies the difference in responsiveness.
Mot1 was tagged with FRB‐GFP in the S288C background and mRNA synthesis was monitored with 4tU immediately after induction of Mot1 nuclear depletion (Fig 5E). In this experiment too, SAGA‐dominated Hsf1 targets behave differently, showing more sensitivity in the form of increased mRNA synthesis rates upon loss of Mot1 function (Fig 5E). This fits very well with the idea that continuous Mot1‐mediated TBP removal on SAGA‐dominated promoters makes such genes more dependent on activator presence and therefore more responsive. As is discussed later, the differential sensitivity of the two promoter classes to Mot1 also explains why TFIID is linked to housekeeping genes and SAGA to regulatable genes.
Nucleosome repositioning also underlies higher responsiveness
Sensitivity to Mot1 loss of function is significantly different for the two Hsf1‐dependent promoter classes (Fig 5E), but the difference is modest. This is in part due to an inability to monitor changes after longer periods of Mot1 nuclear depletion because of the onset of indirect effects (O'Duibhir et al, 2014). Nevertheless, mechanisms other than TBP eviction potentially also play a role, including promoter chromatin architecture. Nucleosome configurations generally differ between the two promoter classes (Albert et al, 2007; Tirosh & Barkai, 2008; Cairns, 2009; Rhee & Pugh, 2012), see also Fig 2). This is of interest since chromatin can have diverse roles in shaping regulatory properties including responsiveness (Lam et al, 2008; Tirosh & Barkai, 2008; Raveh‐Sadka et al, 2009).
To investigate chromatin in the presence and absence of Hsf1, nucleosome positions were determined genome‐wide using micrococcal nuclease followed by sequencing (MNase‐seq), both before and 30 min after induction of Hsf1 nuclear depletion. Because the sensitivity of nucleosomes to MNase varies (Weiner et al, 2010; Henikoff et al, 2011; Kent et al, 2011; Xi et al, 2011; Kubik et al, 2015; Mieczkowski et al, 2016), chromatin was digested with four different concentrations of MNase. Figure 6 shows nucleosomal DNA midpoints for the average TFIID‐dominated Hsf1 target (left), in either the presence (grey) or absence of Hsf1 (line), and for the four different MNase concentrations (0.05–3 U, top to bottom). The corresponding average SAGA‐dominated Hsf1 target is depicted on the right of Fig 6, with three examples of each promoter class depicted in Fig 7. Figure EV2 contains plots for all 21 individual Hsf1‐dependent promoters.
Figure 6. Hsf1 removal results in nucleosomal repositioning on SAGA‐dominated promoters.
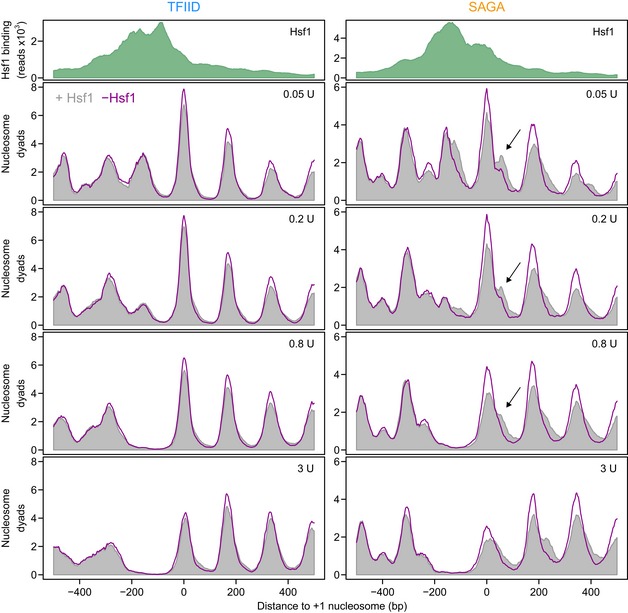
Average profiles of MNase‐seq nucleosomal DNA midpoint positions for the TFIID‐dominated (left) and SAGA‐dominated (right) Hsf1 targets. The upper panels display the average Hsf1 binding by ChIP‐seq (green). The lower panels show nucleosomal DNA midpoints using 31 bp smoothing in the presence (grey) and absence (purple line) of Hsf1 (30 min of nuclear depletion). Each profile is the average of three biological replicates each scaled to 10 million mapped reads. The values in the top right corner indicate the amount of MNase used to digest the chromatin. The data are aligned to the +1 nucleosome. The arrows indicate the downstream +1 nucleosome in the SAGA‐dominated promoters, that shifts upstream upon depletion of Hsf1.
Figure 7. Individual examples of nucleosome repositioning on SAGA‐dominated promoters.
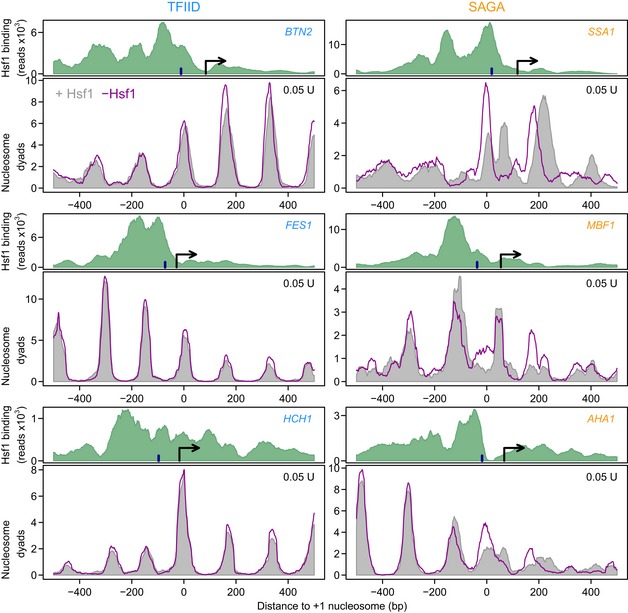
As in Fig 6, but for three individual promoters from each class as indicated by the gene name in each panel. The three genes on the left are TFIID‐dominated and the three genes on the right are SAGA‐dominated. The arrow denotes the transcription start site (Nagalakshmi et al, 2008). The dark blue bar indicates the location of the TATA (‐like) element (Rhee & Pugh, 2012). The examples shown were from the experiments digested using 0.05 units of MNase. All 21 Hsf1 target promoters are depicted in Fig EV2.
Figure EV2. Hsf1 removal results in nucleosomal repositioning on SAGA‐dominated promoters.
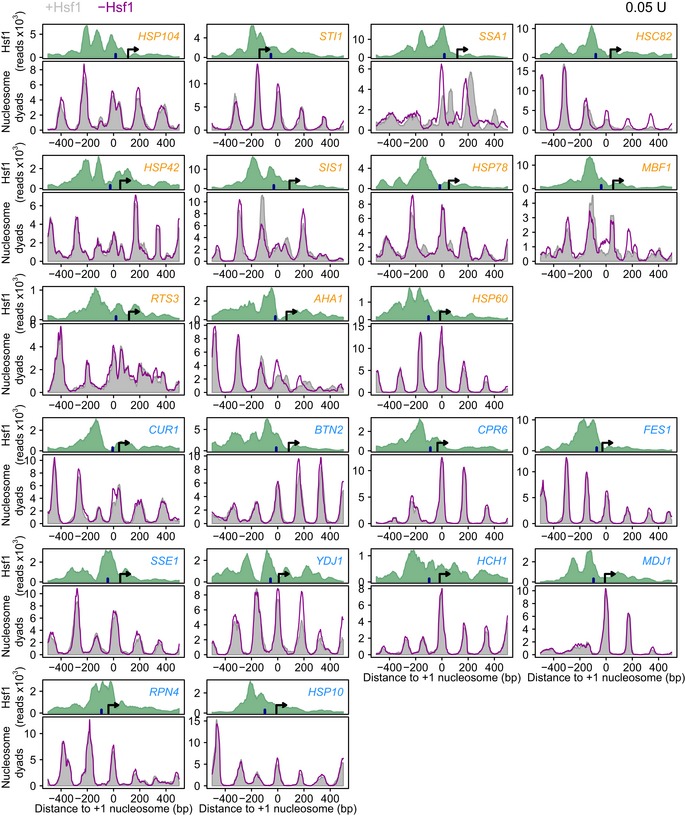
MNase‐seq nucleosomal DNA midpoint mapping with and without Hsf1 for all 11 SAGA‐dominated targets (orange) and all 10 TFIID‐dominated targets (blue). The arrow denotes the transcriptional start site (Nagalakshmi et al, 2008). The dark blue bar indicates the location of the TATA(‐like) element (Rhee & Pugh, 2012). For STI1 either the TATA‐box or the start site is likely incorrectly called. All the tracks are aligned to the +1 nucleosome. These tracks were used to create the average plot shown in Fig 6.
Nucleosome occupancy and positioning on the TFIID‐dominated targets are only mildly affected by Hsf1 depletion, with only a slight increase in occupancy visible (Fig 6 left), consistent with reduced transcription rates. The high degree of activator independence on TFIID‐dominated promoters is clear from both the average plot (Fig 6 left) and each individual promoter (Fig 7 left and Fig EV2). In contrast, nucleosomes on the SAGA‐dominated Hsf1 targets are much more strongly affected by Hsf1 (Figs 6 and 7 right). On the SAGA‐dominated targets, the +1 nucleosome forms two peaks in the presence of Hsf1, whereby the minor, most downstream peak (Fig 6 right, arrow), shifts back upstream upon Hsf1 removal. Due to averaging (Fig 6), some extremely large repositioning differences on individual SAGA‐dominated promoters are lost (Fig 7 right). On the SSA1 promoter for example, two different cell populations of +1 nucleosome DNA fragment midpoints are observed in the presence of Hsf1, with the most downstream population completely disappearing upon Hsf1 removal (Fig 7 right). This indicates that the +1 nucleosome is in the downstream position when the gene is active, and in the upstream position when the gene is inactive. We infer that the two nucleosome positions represent different cell populations because the spacing (60 bp) is too small for simultaneous binding. Upon removal of Hsf1, nucleosomes shift from the downstream (active) position to the upstream (inactive) position. Most of the SAGA‐dominated Hsf1 targets show similar effects, although to a lesser extent (Figs 7 and EV2). This is in stark contrast to the TFIID‐dominated promoters, only one of which shows slight nucleosome repositioning: CUR1, the most responsive TFIID‐dominated promoter (Fig EV2).
The results show that Hsf1 removal has different consequences at the two promoter classes. For the TFIID‐dominated targets, Hsf1 removal has little immediate effect on nucleosome positioning. On SAGA‐dominated genes, Hsf1 removal results in repositioning of the +1 nucleosome upstream into what is apparently a transcriptionally less favourable position. These results agree with the idea that transcriptional plasticity is linked to nucleosome configuration (Lam et al, 2008; Tirosh & Barkai, 2008; Raveh‐Sadka et al, 2009), as well as with the idea that an appropriately positioned +1 nucleosome is important for transcription (Rhee & Pugh, 2012). Notably, Fig 6 shows that removal of an activator results in nucleosomal repositioning on SAGA‐dominated targets, but not on TFIID‐dominated targets, even though genes from both classes depend on the same activator for promoter output. Besides differential TBP turnover, differences in nucleosome configuration therefore likely also contribute to the different responsiveness found between TFIID housekeeping and SAGA regulatable genes.
Discussion
The central question addressed here is regulatability of transcription and what dictates this property. The two different core promoter types, TATA‐box/SAGA‐dominated and TATA‐like/TFIID‐dominated, have in the past been associated with increased regulatability and housekeeping transcription, respectively (Basehoar et al, 2004; Huisinga & Pugh, 2004; Tirosh et al, 2006; Tirosh & Barkai, 2008). This association was based on an enrichment for TATA‐box/SAGA‐dominated promoters in genes with higher expression variation. An important finding of this study is that the property of responsiveness is directly inherent to core promoter type. Genes dependent on a single transcriptional activator show different degrees of responsiveness and this difference in regulatory dynamics is due to core promoter class. This is important to emphasize since previous explanations put forward for the higher variation in gene expression observed for TATA‐box/SAGA‐dominated genes include increased competition between activators and nucleosomes, combined with promoter targeting by a greater number of regulatory factors. Although it cannot be formally excluded that the differences are specific to Hsf1 targets, the analyses presented here indicate that responsiveness is directly associated with core promoter type rather than the number or type of regulators. Evidence for two molecular mechanisms is presented. Both involve dynamic negative regulation that makes SAGA/TATA‐box promoters more dynamically dependent on an activator.
Increased turnover of TBP would serve to make a promoter more responsive to activator presence because on such a promoter transcription would otherwise be much more quickly shut down. Both this and the converse SAGA/TATA‐box promoter upregulation upon Mot1 inactivation are observed here. The detailed analyses of a single transcription factor regulon fits well with previous genome‐wide and biochemical studies showing that Mot1 increases TBP turnover preferentially at SAGA‐dominated/TATA‐box promoters (van Werven et al, 2009; Tora & Timmers, 2010; Viswanathan & Auble, 2011; Spedale et al, 2012; Zentner & Henikoff, 2013). This mechanism also explains the long‐standing observation that TATA‐box type is associated with regulatability properties (Struhl, 1986). Firstly, a consensus TATA‐box puts more strain on DNA which may contribute to Mot1‐mediated TBP release (Tora & Timmers, 2010). Flanking DNA sequences likely also play a role (Viswanathan et al, 2016). In addition, structural analyses indicate that TBP is protected from Mot1 interaction in the context of TFIID, but not in the context of SAGA (Bagby et al, 2000; Wollmann et al, 2011; Anandapadamanaban et al, 2013; Ravarani et al, 2016). This therefore also contributes to our understanding of why TFIID is associated with housekeeping genes, as TBP in the TFIID complex, bound to a consensus TATA‐box, is less susceptible to removal.
TATA‐binding protein turnover is unlikely to be the only mechanism contributing to promoter responsiveness. The differential effects on nucleosome repositioning between TFIID/TATA‐like and SAGA/TATA‐box promoters upon activator loss also implicate differences in nucleosome configuration. The most straightforward interpretation is that the activator Hsf1 is important for keeping SAGA/TATA‐box promoters in a nucleosomal configuration that is amenable to transcription. Upon Hsf1 loss, nucleosome positioning reverts to a configuration that is less favourable for transcription. In this sense, the mechanism is similar to increased TBP turnover, that is increased responsiveness due to more dynamic state transformations of SAGA/TATA‐box genes. A correctly positioned +1 nucleosome is thought to be important for transcription (Rhee & Pugh, 2012) and previous studies have indeed shown differences in nucleosome organization between SAGA/TATA‐box and TFIID/TATA‐like promoters that includes the +1 nucleosome (Albert et al, 2007; Tirosh & Barkai, 2008; Cairns, 2009; Jiang & Pugh, 2009; Rhee & Pugh, 2012). Models implicating nucleosomes in regulatability (Lam et al, 2008; Tirosh & Barkai, 2008; Raveh‐Sadka et al, 2009) often apply direct competition as mechanism. The position of Hsf1 binding on the SAGA‐dominated Hsf1 targets is only compatible with a direct +1 nucleosome competition model in a few cases (Fig EV2) and recruitment of nucleosome remodelling activity by Hsf1 may therefore be involved (Shivaswamy & Iyer, 2008; Erkina et al, 2010).
The findings presented here fit with recent studies showing the importance of an appropriate interplay between transcriptional regulators and chromatin architecture during gene expression (Kubik et al, 2015; Reja et al, 2015; Nocetti & Whitehouse, 2016). As in other studies, here too SAGA‐dominated promoters show more changes in nucleosome positioning compared to TFIID‐dominated promoters. It has been reported that generally, TATA‐less genes have regions of lower bendability in their promoters (Tirosh et al, 2007). This disfavours nucleosome binding and stimulates a more inherently open nucleosome configuration. Lack of such regions may cause SAGA‐dominated genes to be more dependent on nucleosome repositioning through activator‐dependent recruitment of remodelers.
Responsiveness is correlated to gene expression noise and may be mechanistically linked (Lehner, 2010). This agrees with the higher responsiveness of SAGA/TATA‐box promoters since the presence of a TATA‐box increases gene expression noise (Raser & O'Shea, 2004; Murphy et al, 2010). Related to this, the TATA‐box has a major role in determining transcriptional burst size which may in fact reflect responsiveness (Hornung et al, 2012). The latter study also highlights that individual promoter features are not always unambiguously associated with promoter properties, an aspect also illustrated here. For example, HSP60 is distinct from other SAGA/TATA‐box promoters since it is less responsive than most of the TFIID/TATA‐less promoters analysed (Fig 4A), indicating that there is still much to be learnt regarding gene regulation rules. In line with this, it is important to point out that TFIID does not function exclusively at TATA‐like promoters (Basehoar et al, 2004; Huisinga & Pugh, 2004; Rhee & Pugh, 2012). Within the 21 Hsf1 targets defined here, there are two TFIID‐dominated genes with a TATA‐box and there are two SAGA‐dominated genes that lack a TATA‐box. This exemplifies that the difference between SAGA‐ and TFIID‐dominated genes is not black‐and‐white and also explains why some TFIID‐dominated genes show increased expression upon depletion of Mot1 (Fig 5E). Combinatorial control is a well‐established concept in regulation of gene expression that refers to multiple transcription factors acting in synergistic combinations to specify unique gene expression patterns. This study adds an additional facet to combinatorial control mechanisms: the action of an identical activator can have a different outcome depending on promoter context.
Materials and Methods
Strain creation
The anchor‐away system (Haruki et al, 2008) was recreated in the Saccharomyces cerevisiae S288C/BY4742 strain. To achieve this, FPR1 was deleted, a tor1‐1 mutation was introduced to desensitize the strain to rapamycin and RPL13A was tagged with 2xFKBP12 as in the original anchor‐away system (Haruki et al, 2008). This strain was subsequently used here as the parental strain to create each specific anchor‐away strain. This strain is referred to as wild type (WT) throughout the manuscript. In order to create the specific anchor‐away strains, Hsf1 was tagged with an FRB‐yEGFP tag and Mot1 with an FRB‐yEGFP‐3V5 tag. Proper tagging was checked using PCR, Western blotting and microscopy.
Growth curves
Strains were streaked from −80°C stocks onto appropriate selection plates and grown for 3 days. Liquid cultures were inoculated with independent colonies and grown overnight (ON) in synthetic complete (SC) medium: 2 g/l dropout mix complete and 6.71 g/l yeast nitrogen base without AA, carbohydrate & w/AS (YNB) from US Biologicals (Swampscott, USA) with 2% D‐glucose. The following day, ON cultures were used to inoculate 1.5 ml of SC media and cells were grown in an Infinite F200 plate reader (Tecan) alongside a WT parental strain at 30°C. Starting OD595 of cultures was 0.15 ± 0.05 and the OD595 was measured every 10 min. Cells were diluted 1:3 after two doublings (OD = 0.60) by removing 1 ml of medium and adding 1 ml of fresh SC medium containing 2% glucose and various concentrations of rapamycin. This dilution procedure was repeated several times. These measurements were used to calculate doubling times and to determine the rapamycin concentration to use for slow depletion experiments.
Anchor‐away depletion
Hsf1 and Mot1 were depleted from the nucleus by adding rapamycin, dissolved in DMSO, to a final concentration of 7.5 μM. To achieve a slow depletion, a 50× lower concentration of rapamycin was used (final concentration 0.15 μM). For the “t = 0” samples, no rapamycin, but the same volume of DMSO was added.
Microscopy
Nuclear Hsf1 depletion rates were determined in exponentially growing cells by fluorescence microscopy. A non‐tagged parental strain was used as a negative control. All the images were set to have identical min and max brightness values. Circles were manually drawn around cells to measure pixel intensities, and the skewness of these intensities was calculated using the ImageJ tool. Nuclear GFP intensity was quantified for every cell at each time point as the skewness of the GFP signal (pixel intensities) + 1.5 in each cell.
RNA labelling and extraction
4‐Thiouracil (4tU) was added to cell cultures at a final concentration of 5 mM, 6 min prior to mRNA extraction. Cells were then harvested by centrifugation (3,952 × g for 3 min), and pellets were immediately frozen in liquid nitrogen after removal of supernatant. Frozen cells (−80°C) were resuspended in 500 μl acid phenol–chloroform (Sigma, 5:1, pH 4.7). Immediately, an equal volume of TES buffer (TES: 10 mM Tris pH 7.5, 10 mM EDTA, 0.5% SDS) was added. Samples were vortexed hard for 20 s and incubated in a water bath for 10 min at 65°C and vortexed again. Samples were then put in a thermomixer (65°C, 1,400 rpm) for 50 min. Samples were spun for 20 min in an Eppendorf bench top centrifuge at 18,407 × g at 4°C. Phenol extraction was repeated once, followed by a chloroform–isoamyl‐alcohol (25:1) extraction. RNA was precipitated with sodium acetate (NaAc 3M, pH 5.2) and ethanol (96%, −20°C). The pellet was washed with ethanol and dissolved in sterile water (MQ) to 1 μg/μl final concentration. RNA samples were heated for 10 min at 60°C and then immediately put on ice for 2 min. 100 μg RNA was then biotinylated with 200 μl of 1 mg/ml biotin‐HDPD and unbound biotin was removed using chloroform extraction. Labelled RNA was separated from total RNA on a μMACs column containing streptavidin‐conjugated magnetic beads. The bound RNA was washed 6× with 65°C washing buffer (100 mM Tris pH 7.5, 10 mM EDTA, 1 M NaCl, 0.1% Tween‐20) and eluted using 200 μl of 100 mM DTT. The RNA was then purified with an RNeasy MinElute Cleanup Kit from Qiagen.
Microarray profiling
Dual‐channel 70‐mer oligonucleotide arrays were employed with a 4tU common reference WT RNA. For most microarray experiments, each measurement point is the average of two biological replicates, each profiled as a technical replicate in dye‐swap, yielding four replicates that were averaged and statistically analysed by limma (Smyth et al, 2005) versus either wild‐type or the same strain at t = 0. For the fast nuclear depletion of Hsf1, one biological replicate was used. Apart from 4tU labelling and mRNA extraction, all procedures were identical and are described in detail in Kemmeren et al (2014). Calculations were done using the statistical language R version 3.0.1 on a Linux machine running CentOS 5.5. Expression changes are the log2 ratios relative to the median of the four wild types at the same time point, or of the same strain at t = 0.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was carried out as previously described (van Bakel et al, 2008) with some modifications. In short, 250 ml of mid‐log growing yeast cells (OD595 = 1.0) was cross‐linked with 2% formaldehyde for 30 min at 30°C, the reaction was quenched with glycine (final concentration = 125 mM), and cells were collected by centrifugation. Rapamycin addition was staggered during the time course experiment to allow harvesting of all samples at the same time and OD. Subsequently, cells were spheroplasted according to the protocol of the Rando laboratory (Rando, 2010) and then directly sonicated (Bioruptor, Diagenode: 10 cycles, 30 sec on/off, medium setting). About 200 μl chromatin extract was incubated with 10 μl of anti‐GFP antiserum (3 h, RT) which had been coupled to protein G agarose beads (Roche 11 243 233 01) overnight at 4°C. After incubation with the antibody, the beads were washed twice in FA lysis buffer (50 mM HEPES‐KOH pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X‐100, 0.1% Na‐deoxycholate, 0.1% SDS), twice with FA lysis buffer containing 0.5 M NaCl, and twice with 10 mM Tris at pH 8.0, 0.25 mM LiCl, 1 mM EDTA, 0.5% Nonidet P‐40 and 0.5% Na‐deoxycholate. Cross‐links of the ChIP samples were reversed overnight at 65°C in 150 μl 10 mM Tris–HCl (pH 8.0), 1 mM EDTA, 1% SDS. Samples were treated with RNAse and proteinase K, and DNA was recovered for further analysis using phenol extraction.
ChIP‐seq and peak finding
Prior to library preparation, the samples were sub‐sheared to obtain fragments in the optimal size range (Mokry et al, 2010). Both ChIP and input samples were sequenced single end (50 bp) using a SOLiD Wildfire platform. The reads were aligned to the sacCer3 genome assembly (February 2011) using bwa (Li & Durbin, 2009) with the settings “‐c –l 25 –k 2 –n 10”, yielding 32.5 million mapped reads (48%) for the ChIP sample, and 48.1 million mapped reads (60%) for the input sample. Hsf1‐binding peaks were detected using the program CisGenome (Ji et al, 2008). The maximal log2(fold change) between IP and WT DNA fragment counts (max log2(fold change)), peak width and the normalized number of reads in the peak were used for further analysis.
ChIP‐qPCR
DNA samples were recovered after ChIP as described above. The fold change of two Hsf1 targets (BTN2 and SSA1) over two control regions (HMR and POL1) was measured by qPCR, which were performed in 384‐well plates using 6 μl IQ SYBR Green super mix (Bio‐Rad), 2 μl of each primer set and 2 μl of DNA. The total volume per reaction was 10 μl. To create a standard curve, 10 μl of each of the input samples was combined and used undiluted and diluted at concentrations of 1:10, 1:100 and 1:1,000. The qPCRs were run on a 7900HT fast real‐time PCR machine (Applied Biosystems). IP and mock samples were first normalized to their corresponding input samples. The fold change of BTN2 and SSA1 was then calculated by dividing the signal from these genes by the average signal of the two negative control genes (HMR and POL1). All samples were measured as biological triplicates and technical quadruplicates. All qPCR primers are shown in Table EV2.
ChIP‐chip time course
For the slow depletion time course, cells were grown for almost two doublings (from OD = 0.15 to OD = 0.5). Subsequently, rapamycin was added to a final concentration of 0.15 μM. Additions were staggered so that all the time points (WT, t0, t15, t30, t60, t90) were ready at the same OD and time. For the t0, no rapamycin but a similar volume of DMSO was added for 30 min. ChIP‐chip hybridizations and normalizations were done as described (van Bakel et al, 2008; van Werven et al, 2009). Four independently grown cultures were used for each time point, two of which were done in dye‐swap. To control for non‐specific antibody binding, the WT track was subtracted from all other tracks. Peaks were found by searching for genomic locations where two adjacent probes had at least a fold change of 2, with a P‐value lower than 0.05. For each peak, the probe with the highest fold change was taken and subsequently followed during the time course. If two adjacent probes switched between being the biggest fold changer within the peak during the time course, the average of the fold change and P‐value of these two probes was taken for subsequent analysis. The depletion rate was calculated by plotting the log2 fold change in Hsf1 binding against time and fitting a line through these points. The slope of this line was used as the depletion rate from the promoters.
Motifs
To find de novo motifs in the 1,000 bp upstream promoter regions of the Hsf1 targets genes, the MEME tool was used (Bailey et al, 2009). The default settings were used except for minimum motif size, which was set to 5, minimum width to 5 and the distributions of motifs was set to “any number of repetitions”. Sites were identified based on 57 high confidence Hsf1‐bound targets. These targets were bound in both the ChIP‐chip and ChIP‐seq experiments.
Mapping of binding sites
The RSAT tool (Turatsinze et al, 2008) was used to map significant (P‐value < 0.001) presence of a found motif to the promoter regions of the Hsf1 targets. As a motif‐input fasta sequences, files were given that were found by MEME and used by MEME to create the motif. The standard settings were used except for the background, which was set to Saccharomyces cerevisiae upstream regions without ORFs. For the scanning options, sequence origin was set to “start”, return to “site + pval” and P‐value was set to 0.001. The identified motifs were mapped to 650 bp promoters of the function Hsf1 targets. In parallel, published nucleosome positions (Jiang & Pugh, 2009) were also mapped. The average presence of the motif and published nucleosomes was calculated for the SAGA‐ and TFIID‐dominated genes and shown in 10 bp bins.
Direct Hsf1 targets
The direct Hsf1 targets are a subset of the genes bound in both the ChIP‐seq and the ChIP‐chip experiments (57 targets) that also robustly change in expression (FC > 1.7, P < 0.01) in both the fast and the slow Hsf1 depletion experiments (22 targets). HSP82 is omitted from all analysis (except the motif discovery) due to transcript cross‐hybridization with its orthologue HSC82. As HSC82 is far more highly expressed, HSP82 is considered to have a negligible effect on the measured mRNA levels of HSC82 (Borkovich et al, 1989). Therefore, HSC82 is included in the analysis, resulting in 21 direct targets.
Responsiveness
To calculate the responsiveness, the ChIP‐chip and 4tU slow depletion time course data of the direct Hsf1 targets were used. The log2(fold change) values of the ChIP‐chip time course were plotted against the log2(fold change) values of the 4tU labelling time course, in each case compared to the corresponding t = 0 sample. A line was fitted through the last three time points t30, t60 and t90. The slope of this line was used as a measure for responsiveness. The significance of the slope difference in the mRNA expression versus binding plots was calculated using the linear model:
with E the log2(fold change) in mRNA expression; G the effect of the gene; C the effect of the gene class, a dichotomous variable that is either SAGA or TFIID; B the log2(binding ratio), which is a continuous covariate; CB the interaction between terms C and B; and lastly the residual error term . The overall fit is 0.88 (adjusted R 2; P = 1.15e‐15). The only term of interest in the model is the interaction term CB. Its effect is equal to the average difference in slope between the two classes of genes; its P‐value is 0.00066. Residuals are normally distributed (P = 0.28, Shapiro test) and show no dependency on the independent or fitted values. A simple t‐test of the difference, per gene class, in the slopes of their regression lines yields a P‐value of 0.002. However, this approach is incorrect as it ignores the variance structure of underlying data.
TFIIB and Pol II occupancy
For the comparison of TFIIB with polymerase II and Hsf1 binding, genome‐wide ChIP‐exo data of PIC components were used (Rhee & Pugh, 2012). TFIIB and Pol II occupancy as provided in “Rhee_SuppData1” was taken and scaled down for visualization purposes (all occupancy values were divided by 100 and 10 for TFIIB and Pol II, respectively).
Heat shock
Log2 expression values of previously published expression data of heat‐shocked BY4742 cells were used (O'Duibhir et al, 2014). The difference in expression changes between SAGA‐ and TFIID‐dominated Hsf1 targets was calculated using a two‐tailed t‐test.
Correlations
All correlations and corresponding P‐values were calculated using the function “cor.test” of the statistical language R, which uses the fact that √((n−2)× r 2/(1−r 2)) is t‐distributed with (n−2) degrees of freedom.
Micrococcal nuclease chromatin digestion
Isolation of mono‐nucleosomal DNA was done essentially as described (Kubik et al, 2015) with some modifications. In short, 70 ml of yeast cells was grown in SC at 30°C to an OD595 of 1.0 in three independent biological replicates. The cells were cross‐linked at RT for 5 min using formaldehyde at a final concentration of 1% and quenched for 5 min with glycine (final concentration = 125 mM). Subsequently, the cells were spun down and washed once with sorbitol (1 M) and spheroblasted for 8 min using 1 ml of spheroblasting buffer (1 M sorbitol, 1 mM β‐mercaptoethanol, 10 mg/ml Zymolyase 100T (USB)). The spheroblasts were spun down and washed twice before resuspending them in 1 ml of MNase digestion buffer (1 M sorbitol, 50 mM NaCl, 10 mM Tris pH 7.5, 5 mM MgCl2, 1 mM CaCl2, 0.075% Nonidet P‐40, 1 mM β‐mercaptoethanol, 0.5 mM spermidine). The samples were divided over 4× four‐fold different MNase amounts (Sigma‐Aldrich): 0.046875 U, 0.1875 U, 0.75 U and 3.0 U and incubated at 37°C for 45 min. The digestion was stopped by putting the samples on ice and immediately adding 15 μl of 0.5M EDTA and subsequently 15 μl of 10% SDS. The proteins were degraded by adding 15 μl of (10 mg/ml) proteinase K and incubating the samples for 1 h at 37°C. The cross‐links were reversed by putting the samples at 65°C overnight. The next day, nucleosomal DNA was isolated using phenol extraction and RNA was digested using RNase A/T1 (Thermo Scientific; final concentration 0.2 mg/ml and 250 U/ml). The extent of digestion was evaluated on a Bioanalyzer 2100 high sensitivity chip.
Sequencing and mapping of nucleosomal DNA
Sequencing libraries were created from the purified nucleosomal DNA using the NextflexTM rapid DNA‐Seq kit (Bioo Scientific) using a modified protocol. The libraries were sequenced paired end (2 × 75 bp) on a NextSeq500 platform. The paired‐end reads were mapped to the sacCer3 (February 2011) genome assembly using bowtie2 (Langmead & Salzberg, 2012) (with the settings “ –no‐discordant –no‐contain –maxins 1980 –trim5 5 –trim3 15 –end‐to‐end –sensitive”. Only read pairs with an insert size between 95 and 225 bp were used for subsequent analyses. To compare the occupancy of all samples, they were scaled to 10 million mapped paired‐end reads per sample using genomecov from the bedtools2 suite version 2.250 (Quinlan & Hall, 2010). The middle of each mate pair was used as the position of the nucleosome dyad. The three replicates for each digestion and time point showed only minimal difference in genome‐wide occupancy; hence, for the rest of the analysis, these replicates were merged. The nucleosome dyads were smoothed using a 31 bp running average. The 31 bp window was chosen because it provides clearly separated peaks, but keeps positions with a double peak (in other words: a position with two populations of nucleosomes) that are merged into a single peak to a minimum. The Hsf1 targets were manually aligned to the +1 nucleosome, which was defined as the first nucleosome downstream of the nucleosome‐depleted region (as is visible in the 0.75 U and 3.0 U digestion samples). The summit of the most upstream +1 peak in the Hsf1‐depleted, 0.1875 U MNase‐digested samples was used as the position of the +1 nucleosome dyad. The script that was used for the centring and smoothing is available on https://github.com/plijnzaad/phtools/blob/master/ngs/center+smooth.pl. The TSS location in Fig 6 was taken from (Nagalakshmi et al, 2008); however, the location of the TSS in the promoter of STI1 was likely wrongly annotated. The TATA locations were taken from (Rhee & Pugh, 2012) except for AHA1. Here the TATA is annotated as a TATA‐like element although there is a TATA‐box without mismatches closer to the canonical position; hence, this TATA‐box is shown.
Data availability
All sequencing and microarray data are available on GEO through the accession number GSE81481.
Author contributions
EOD, WJJ and FCPH contributed to conceptualization; EOD, WJJ, DL and MJAGK performed investigation; EOD, WJJ, and PL performed formal analysis; EOD, WJJ and FCPH contributed to writing; PK and FCPH involved in supervision; and PK and FCPH contributed to funding acquisition.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Acknowledgements
We would like to thank Slawomir Kubik and David Shore for help with the MNase‐seq protocol; Michael Tolstorukov for advice regarding the MNase titration; Michal Levo and Eran Segal for modelling; the Utrecht sequencing facility for sequencing; and the Holstege and Kemmeren group members for assistance and discussions. This work was supported by the Netherlands Organisation for Scientific Research (NWO) grants 016108607 (FH), 91106009 (FH) and 86411010 (PK) and by the European Research Council (ERC) grant 671174 DynaMech.
The EMBO Journal (2017) 36: 274–290
See also: S Kubik et al (February 2017)
References
- Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF (2007) Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 446: 572–576 [DOI] [PubMed] [Google Scholar]
- Anandapadamanaban M, Andresen C, Helander S, Ohyama Y, Siponen MI, Lundström P, Kokubo T, Ikura M, Moche M, Sunnerhagen M (2013) High‐resolution structure of TBP with TAF1 reveals anchoring patterns in transcriptional regulation. Nat Struct Mol Biol 20: 1008–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagby S, Mal TK, Liu D, Raddatz E, Nakatani Y, Ikura M (2000) TFIIA‐TAF regulatory interplay: NMR evidence for overlapping binding sites on TBP. FEBS Lett 468: 149–154 [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bakel H, van Werven FJ, Radonjic M, Brok MO, van Leenen D, Holstege FCP, Timmers HTM (2008) Improved genome‐wide localization by ChIP‐chip using double‐round T7 RNA polymerase‐based amplification. Nucleic Acids Res 36: e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basehoar AD, Zanton SJ, Pugh BF (2004) Identification and distinct regulation of yeast TATA box‐containing genes. Cell 116: 699–709 [DOI] [PubMed] [Google Scholar]
- Borkovich KA, Farrelly FW, Finkelstein DB, Taulien J, Lindquist S (1989) hsp82 is an essential protein that is required in higher concentrations for growth of cells at higher temperatures. Mol Cell Biol 9: 3919–3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratowski S, Hahn S, Guarente L, Sharp PA (1989) Five intermediate complexes in transcription initiation by RNA polymerase II. Cell 56: 549–561 [DOI] [PubMed] [Google Scholar]
- Cairns BR (2009) The logic of chromatin architecture and remodelling at promoters. Nature 461: 193–198 [DOI] [PubMed] [Google Scholar]
- Davison BL, Egly JM, Mulvihill ER, Chambon P (1983) Formation of stable preinitiation complexes between eukaryotic class B transcription factors and promoter sequences. Nature 301: 680–686 [DOI] [PubMed] [Google Scholar]
- Deaton AM, Bird A (2011) CpG islands and the regulation of transcription. Genes Dev 25: 1010–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg E, Levanon EY (2013) Human housekeeping genes, revisited. Trends Genet 29: 569–574 [DOI] [PubMed] [Google Scholar]
- Erb I, van Nimwegen E (2011) Transcription factor binding site positioning in yeast: proximal promoter motifs characterize TATA‐less promoters. PLoS ONE 6: e24279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkina TY, Zou Y, Freeling S, Vorobyev VI, Erkine AM (2010) Functional interplay between chromatin remodeling complexes RSC, SWI/SNF and ISWI in regulation of yeast heat shock genes. Nucleic Acids Res 38: 1441–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross DS, English KE, Collins KW, Lee SW (1990) Genomic footprinting of the yeast HSP82 promoter reveals marked distortion of the DNA helix and constitutive occupancy of heat shock and TATA elements. J Mol Biol 216: 611–631 [DOI] [PubMed] [Google Scholar]
- Grünberg S, Hahn S (2013) Structural insights into transcription initiation by RNA polymerase II. Trends Biochem Sci 38: 603–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn J‐S, Hu Z, Thiele DJ, Iyer VR (2004) Genome‐wide analysis of the biology of stress responses through heat shock transcription factor. Mol Cell Biol 24: 5249–5256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, Laemmli UK (2008) The anchor‐away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell 31: 925–932 [DOI] [PubMed] [Google Scholar]
- Henikoff JG, Belsky JA, Krassovsky K, MacAlpine DM, Henikoff S (2011) Epigenome characterization at single base‐pair resolution. Proc Natl Acad Sci 108: 18318–18323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung G, Bar‐Ziv R, Rosin D, Tokuriki N, Tawfik DS, Oren M, Barkai N (2012) Noise‐mean relationship in mutated promoters. Genome Res 22: 2409–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisinga KL, Pugh BF (2004) A genome‐wide housekeeping role for TFIID and a highly regulated stress‐related role for SAGA in Saccharomyces cerevisiae . Mol Cell 13: 573–585 [DOI] [PubMed] [Google Scholar]
- Imazu H, Sakurai H (2005) Saccharomyces cerevisiae heat shock transcription factor regulates cell wall remodeling in response to heat shock. Eukaryot Cell 4: 1050–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH (2008) An integrated software system for analyzing ChIP‐chip and ChIP‐seq data. Nat Biotechnol 26: 1293–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Pugh BF (2009) A compiled and systematic reference map of nucleosome positions across the Saccharomyces cerevisiae genome. Genome Biol 10: R109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juven‐Gershon T, Kadonaga JT (2010) Regulation of gene expression via the core promoter and the basal transcriptional machinery. Dev Biol 339: 225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasinathan S, Orsi GA, Zentner GE, Ahmad K, Henikoff S (2014) High‐resolution mapping of transcription factor binding sites on native chromatin. Nat Methods 11: 203–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemmeren P, Sameith K, van de Pasch LAL, Benschop JJ, Lenstra TL, Margaritis T, O'Duibhir E, Apweiler E, van Wageningen S, Ko CW, van Heesch S, Kashani MM, Ampatziadis‐Michailidis G, Brok MO, Brabers NACH, Miles AJ, Bouwmeester D, van Hooff SR, van Bakel H, Sluiters E et al (2014) Large‐scale genetic perturbations reveal regulatory networks and an abundance of gene‐specific repressors. Cell 157: 740–752 [DOI] [PubMed] [Google Scholar]
- Kent NA, Adams S, Moorhouse A, Paszkiewicz K (2011) Chromatin particle spectrum analysis: a method for comparative chromatin structure analysis using paired‐end mode next‐generation DNA sequencing. Nucleic Acids Res 39: e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubik S, Bruzzone MJ, Jacquet P, Falcone J‐L, Rougemont J, Shore D (2015) Nucleosome stability distinguishes two different promoter types at all protein‐coding genes in yeast. Mol Cell 60: 422–434 [DOI] [PubMed] [Google Scholar]
- Lam FH, Steger DJ, O'Shea EK (2008) Chromatin decouples promoter threshold from dynamic range. Nature 453: 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Causton HC, Holstege FC, Shen WC, Hannett N, Jennings EG, Winston F, Green MR, Young RA (2000) Redundant roles for the TFIID and SAGA complexes in global transcription. Nature 405: 701–704 [DOI] [PubMed] [Google Scholar]
- Lehner B (2010) Conflict between noise and plasticity in yeast. PLoS Genet 6: e1001185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E (2006) An improved map of conserved regulatory sites for Saccharomyces cerevisiae . BMC Bioinformatics 7: 113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieczkowski J, Cook A, Bowman SK, Mueller B, Alver BH, Kundu S, Deaton AM, Urban JA, Larschan E, Park PJ, Kingston RE, Tolstorukov MY (2016) MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat Commun 7: 11485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokry M, Hatzis P, de Bruijn E, Koster J, Versteeg R, Schuijers J, van de Wetering M, Guryev V, Clevers H, Cuppen E (2010) Efficient double fragmentation ChIP‐seq provides nucleotide resolution protein‐DNA binding profiles. PLoS ONE 5: e15092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morano KA, Grant CM, Moye‐Rowley WS (2012) The response to heat shock and oxidative stress in Saccharomyces cerevisiae . Genetics 190: 1157–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller F, Tora L (2014) Chromatin and DNA sequences in defining promoters for transcription initiation. Biochim Biophys Acta 1839: 118–128 [DOI] [PubMed] [Google Scholar]
- Murphy KF, Adams RM, Wang X, Balázsi G, Collins JJ (2010) Tuning and controlling gene expression noise in synthetic gene networks. Nucleic Acids Res 38: 2712–2726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M (2008) The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocetti N, Whitehouse I (2016) Nucleosome repositioning underlies dynamic gene expression. Genes Dev 30: 660–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Duibhir E, Lijnzaad P, Benschop JJ, Lenstra TL, van Leenen D, Groot Koerkamp MJ, Margaritis T, Brok MO, Kemmeren P, Holstege FC (2014) Cell cycle population effects in perturbation studies. Mol Syst Biol 10: 732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira LA, Klejman MP, Timmers HTM (2003) Roles for BTAF1 and Mot1p in dynamics of TATA‐binding protein and regulation of RNA polymerase II transcription. Gene 315: 1–13 [DOI] [PubMed] [Google Scholar]
- Ptashne M, Gann A (1997) Transcriptional activation by recruitment. Nature 386: 569–577 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando OJ (2010) Genome‐wide mapping of nucleosomes in yeast. Methods Enzymol 470: 105–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raser JM, O'Shea EK (2004) Control of stochasticity in eukaryotic gene expression. Science 304: 1811–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravarani CNJ, Chalancon G, Breker M, de Groot NS, Babu MM (2016) Affinity and competition for TBP are molecular determinants of gene expression noise. Nat Commun 7: 10417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh‐Sadka T, Levo M, Segal E (2009) Incorporating nucleosomes into thermodynamic models of transcription regulation. Genome Res 19: 1480–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reja R, Vinayachandran V, Ghosh S, Pugh BF (2015) Molecular mechanisms of ribosomal protein gene coregulation. Genes Dev 29: 1942–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF (2011) Comprehensive genome‐wide protein‐DNA interactions detected at single‐nucleotide resolution. Cell 147: 1408–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF (2012) Genome‐wide structure and organization of eukaryotic pre‐initiation complexes. Nature 483: 295–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder RG (1996) The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem Sci 21: 327–335 [PubMed] [Google Scholar]
- Sainsbury S, Bernecky C, Cramer P (2015) Structural basis of transcription initiation by RNA polymerase II. Nat Rev Mol Cell Biol 16: 129–143 [DOI] [PubMed] [Google Scholar]
- Shivaswamy S, Iyer VR (2008) Stress‐dependent dynamics of global chromatin remodeling in yeast: dual role for SWI/SNF in the heat shock stress response. Mol Cell Biol 28: 2221–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BJ, Yaffe MP (1991) A mutation in the yeast heat‐shock factor gene causes temperature‐sensitive defects in both mitochondrial protein import and the cell cycle. Mol Cell Biol 11: 2647–2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK, Michaud J, Scott HS (2005) Use of within‐array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 21: 2067–2075 [DOI] [PubMed] [Google Scholar]
- Solís EJ, Pandey JP, Zheng X, Jin DX, Gupta PB, Airoldi EM, Pincus D, Denic V (2016) Defining the essential function of yeast Hsf1 reveals a compact transcriptional program for maintaining eukaryotic proteostasis. Mol Cell 63: 60–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spedale G, Meddens CA, Koster MJE, Ko CW, van Hooff SR, Holstege FCP, Timmers HTM, Pijnappel WWMP (2012) Tight cooperation between Mot1p and NC2β in regulating genome‐wide transcription, repression of transcription following heat shock induction and genetic interaction with SAGA. Nucleic Acids Res 40: 996–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K (1986) Constitutive and inducible Saccharomyces cerevisiae promoters: evidence for two distinct molecular mechanisms. Mol Cell Biol 6: 3847–3853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K (1995) Yeast transcriptional regulatory mechanisms. Annu Rev Genet 29: 651–674 [DOI] [PubMed] [Google Scholar]
- Sun M, Schwalb B, Schulz D, Pirkl N, Etzold S, Larivière L, Maier KC, Seizl M, Tresch A, Cramer P (2012) Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res 22: 1350–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Weinberger A, Carmi M, Barkai N (2006) A genetic signature of interspecies variations in gene expression. Nat Genet 38: 830–834 [DOI] [PubMed] [Google Scholar]
- Tirosh I, Berman J, Barkai N (2007) The pattern and evolution of yeast promoter bendability. Trends Genet 23: 318–321 [DOI] [PubMed] [Google Scholar]
- Tirosh I, Barkai N (2008) Two strategies for gene regulation by promoter nucleosomes. Genome Res 18: 1084–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tora L, Timmers HTM (2010) The TATA box regulates TATA‐binding protein (TBP) dynamics in vivo. Trends Biochem Sci 35: 309–314 [DOI] [PubMed] [Google Scholar]
- Turatsinze J‐V, Thomas‐Chollier M, Defrance M, van Helden J (2008) Using RSAT to scan genome sequences for transcription factor binding sites and cis‐regulatory modules. Nat Protoc 3: 1578–1588 [DOI] [PubMed] [Google Scholar]
- Venters BJ, Wachi S, Mavrich TN, Andersen BE, Jena P, Sinnamon AJ, Jain P, Rolleri NS, Jiang C, Hemeryck‐Walsh C, Pugh BF (2011) A comprehensive genomic binding map of gene and chromatin regulatory proteins in Saccharomyces. Mol Cell 41: 480–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan R, Auble DT (2011) One small step for Mot1; one giant leap for other Swi2/Snf2 enzymes? Biochim Biophys Acta 1809: 488–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan R, True JD, Auble DT (2016) Molecular mechanism of Mot1, a TATA‐binding protein (TBP)‐DNA dissociating enzyme. J Biol Chem 291: 15714–15726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner A, Hughes A, Yassour M, Rando OJ, Friedman N (2010) High‐resolution nucleosome mapping reveals transcription‐dependent promoter packaging. Genome Res 20: 90–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Werven FJ, van Teeffelen HAAM, Holstege FCP, Timmers HTM (2009) Distinct promoter dynamics of the basal transcription factor TBP across the yeast genome. Nat Struct Mol Biol 16: 1043–1048 [DOI] [PubMed] [Google Scholar]
- Wiederrecht G, Seto D, Parker CS (1988) Isolation of the gene encoding the S. cerevisiae heat shock transcription factor. Cell 54: 841–853 [DOI] [PubMed] [Google Scholar]
- Wollmann P, Cui S, Viswanathan R, Berninghausen O, Wells MN, Moldt M, Witte G, Butryn A, Wendler P, Beckmann R, Auble DT, Hopfner K‐P (2011) Structure and mechanism of the Swi2/Snf2 remodeller Mot1 in complex with its substrate TBP. Nature 475: 403–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y, Yao J, Chen R, Li W, He X (2011) Nucleosome fragility reveals novel functional states of chromatin and poises genes for activation. Genome Res 21: 718–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarzov P, Boucherie H, Mann C (1997) A yeast heat shock transcription factor (Hsf1) mutant is defective in both Hsc82/Hsp82 synthesis and spindle pole body duplication. J Cell Sci 110(Pt 16): 1879–1891 [DOI] [PubMed] [Google Scholar]
- Zentner GE, Henikoff S (2013) Mot1 redistributes TBP from TATA‐containing to TATA‐less promoters. Mol Cell Biol 33: 4996–5004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Data Availability Statement
All sequencing and microarray data are available on GEO through the accession number GSE81481.