

Abstract
Ribonuclease activity of topoisomerase I (Top1) causes DNA nicks bearing 2′,3′‐cyclic phosphates at ribonucleotide sites. Here, we provide genetic and biochemical evidence that DNA double‐strand breaks (DSBs) can be directly generated by Top1 at sites of genomic ribonucleotides. We show that RNase H2‐deficient yeast cells displayed elevated frequency of Rad52 foci, inactivation of RNase H2 and RAD52 led to synthetic lethality, and combined loss of RNase H2 and RAD51 induced slow growth and replication stress. Importantly, these phenotypes were rescued upon additional deletion of TOP1, implicating homologous recombination for the repair of Top1‐induced damage at ribonuclelotide sites. We demonstrate biochemically that irreversible DSBs are generated by subsequent Top1 cleavage on the opposite strand from the Top1‐induced DNA nicks at ribonucleotide sites. Analysis of Top1‐linked DNA from pull‐down experiments revealed that Top1 is covalently linked to the end of DNA in RNase H2‐deficient yeast cells, supporting this model. Taken together, these results define Top1 as a source of DSBs and genome instability when ribonucleotides incorporated by the replicative polymerases are not removed by RNase H2.
Keywords: double‐strand breaks, homologous recombination, ribonucleotide excision repair, RNase H2, topoisomerase I
Subject Categories: DNA Replication, Repair & Recombination
Introduction
Recent biochemical and genetic studies show that DNA polymerases (Pols), such as Pols α, δ, and ε, readily incorporate ribonucleotides into DNA (Nick McElhinny et al, 2010a,b; Williams et al, 2013). While DNA polymerases can discriminate against insertion of ribonucleotides, studies in both yeast and human cells show that the ribonucleotide concentration is much higher than the deoxyribonucleotide concentration (Traut, 1994; Nick McElhinny et al, 2010b), increasing the probability of ribonucleotide insertion. Consequently, ribonucleotides are estimated to be the most common non‐canonical nucleotide incorporated into DNA during replication (Nick McElhinny et al, 2010b; Reijns et al, 2012). This has important implications for genome stability, because ribonucleotides are more susceptible to spontaneous hydrolysis (Li & Breaker, 1999), generating DNA ends that cannot be directly ligated. They also have structural effects on DNA, including altered helical parameters and elastic properties (Ban et al, 1994; DeRose et al, 2012; Chiu et al, 2014). While newly incorporated ribonucleotides might serve as signals for DNA repair processes, such as mismatch repair (Ghodgaonkar et al, 2013; Lujan et al, 2013), there are clearly negative consequences of unrepaired ribonucleotides [discussed in detail in several recent reviews (Potenski & Klein, 2014; Williams & Kunkel, 2014; Cerritelli & Crouch, 2016; Williams et al, 2016)].
The major pathway for ribonucleotide removal is ribonucleotide excision repair (RER). RER is initiated when RNase H2 incises on the 5′ side of a ribonucleotide, and additional proteins work in concert to remove the ribonucleotide‐containing DNA segment and synthesize ribonucleotide‐free DNA (Rydberg & Game, 2002; Sparks et al, 2012). Although RNase H2 is dispensable for viability in yeast, RNase H2‐null strains harbor high levels of genomic ribonucleotides and 2–5 base pair (bp) deletions at DNA sequences consisting of short repeats (Nick McElhinny et al, 2010a; Kim et al, 2011; Potenski et al, 2014). RNase H2‐null strains also exhibit hyper‐recombination and replication stress (Nick McElhinny et al, 2010a; Lazzaro et al, 2012; Potenski et al, 2014). In mice, loss of RNase H2 leads to embryonic lethality (Hiller et al, 2012; Reijns et al, 2012), while in humans, mutations in RNase H2 lead to Aicardi–Goutières syndrome (AGS), a neuroinflammatory disease (Rabe, 2013; Reijns & Jackson, 2014).
Genetic analyses using budding yeast as a model organism have identified Top1 as a key contributor to genome instability phenotypes that arise upon RER inactivation. These phenotypes include genome integrity checkpoint activation, replication stress, elevated mutagenesis, hyper‐recombination, and increased chromosomal rearrangements (Kim et al, 2011; Lazzaro et al, 2012; Williams et al, 2013; Potenski et al, 2014), all of which are reduced or eliminated upon deletion of TOP1. As Top1 possesses a ribonuclease activity (Sekiguchi & Shuman, 1997; Kim et al, 2011; Huang et al, 2015; Sparks & Burgers, 2015), the Top1‐induced DNA nicks at genomic ribonucleotides can initiate the observed genome instabilities. Recent reports have described a mechanism by which the 2–5 bp deletions are generated by Top1 (Huang et al, 2015; Sparks & Burgers, 2015; Cho et al, 2016). However, how Top1 causes replication stress, hyper‐recombination, and checkpoint activation has remained unclear.
Double‐strand breaks (DSBs) are among the most genotoxic lesions [reviewed in (Mehta & Haber, 2014)] resulting from endogenous sources, such as replication fork collapse, or from exogenous sources, including ionizing radiation and chemotherapeutic agents. DSBs lead to mutations, genetic rearrangements, and cancer. In response to DSBs, cells activate checkpoints and invoke repair pathways, including both non‐homologous end joining (NHEJ) and homologous recombination (HR). In yeast, the predominant DSB repair pathway is HR, a highly regulated, multi‐step process that is template‐dependent and involves precise coordination between multiple protein factors, including Rad51 and Rad52. Several lines of evidence suggest that DSBs contribute to genome instabilities in the absence of RER. RNase H2‐null mouse embryonic fibroblasts exhibit increased γH2AX foci, chromosomal rearrangements, and micronuclei, coupled with a p53‐dependent DNA damage response (Hiller et al, 2012; Reijns et al, 2012), consistent with the formation of DSBs. Moreover, RNase H2‐null yeast strains exhibit elevated rates of recombination and chromosomal rearrangements (Allen‐Soltero et al, 2014; O'Connell et al, 2015) that are Top1‐dependent (Potenski et al, 2014; Conover et al, 2015; Epshtein et al, 2016). Also, transcriptional profiling of RNase H2‐null yeast cells reveals up‐regulation of genes involved in DSB repair as well as recombination (Arana et al, 2012).
Here, using both in vitro and in vivo approaches, we present evidence establishing that Top1 induces DSBs at ribonucleotide sites and that HR is critical for repair of these genomic lesions. Our data provide a mechanism whereby sequential Top1 cleavage events on opposite DNA strands induce DSBs at unrepaired ribonucleotides, resulting in checkpoint activation and replication stress.
Results
Unrepaired single genomic ribonucleotides cause Top1‐dependent Rad52‐YFP foci
To investigate the role of Top1 in the formation of spontaneous DSBs induced by unrepaired ribonucleotides in S. cerevisiae cells, we constructed strains expressing Rad52‐YFP in RNase H2‐proficient or RNase H2‐deficient strains with or without additional inactivation of TOP1. Rad52 is an indicator of DNA damage, as it forms foci at sites of DSBs during HR repair (Lisby et al, 2001). We measured the percentage of cells containing one or more Rad52‐YFP foci. Consistent with a previous report (Potenski & Klein, 2014), deletion of RNH201, the gene encoding the catalytic subunit of RNase H2, resulted in a higher percentage of cells containing one or more spontaneous Rad52‐YFP foci (Fig 1A). Importantly, additional inactivation of TOP1 (rnh201Δ top1Δ) significantly decreased the percentage of cells with a Rad52‐YFP focus compared to the rnh201Δ single knockout strain, while inactivation of TOP1 alone (top1Δ) shows little impact (Fig 1A). These results are consistent with a model wherein spontaneous DSBs are generated by the action of Top1 at unrepaired ribonucleotides in the absence of RER.
Figure 1. Spontaneous Rad52‐YFP foci are elevated in a RNase H2‐deficient yeast strain.
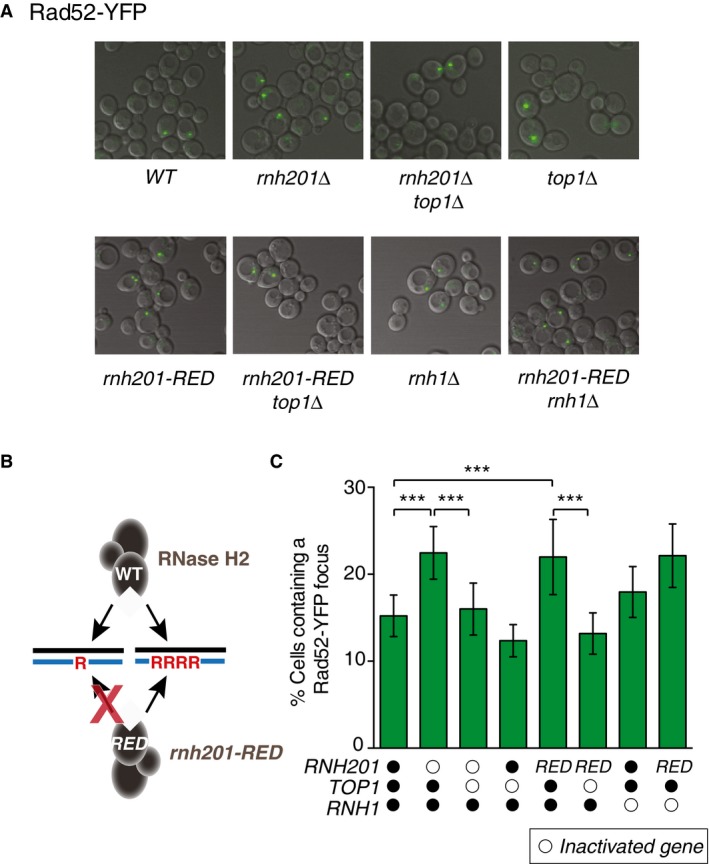
- A representative merged image (DIC and YFP) by live cell microscopy from each indicated strain shows the percentage of cells containing a spontaneous Rad52‐yellow fluorescent protein (YFP) focus.
- Diagram illustrating the functional difference in wild‐type RNase H2 and RNase H2‐Ribonucleotide Excision Defective (rnh201‐RED) mutant.
- The percentage of cells containing a spontaneous Rad52‐YFP focus was determined from at least 3,200 cells for each strain. The bar graph represents the mean values ± SD. ***P ≤ 0.001 (Student's t‐test in GraphPad Prism). Open circles indicate inactivated genes for each strain, and RED indicates rnh201‐RED strain.
RNase H2 functions to remove both single genomic ribonucleotides and stretches of consecutive ribonucleotides, including R‐loops generated during transcription. Because unresolved RNA–DNA hybrids such as R‐loops can cause DNA DSBs, we constructed a RNase H2‐Ribonucleotide Excision Defective (rnh201‐RED) strain that is defective in resolving single ribonucleotides yet proficient in resolving stretches of ribonucleotides (illustrated in Fig 1B; Chon et al, 2013). We observed in the rnh201‐RED strain a similar increase in cells with a Rad52‐YFP focus as in the rnh201Δ strain, and this increase was Top1 dependent (Fig 1A, quantified in Fig 1C). This analysis was extended to include a RNase H1‐deficient strain (rnh1Δ), as RNase H1 is active on RNA–DNA hybrids but not on substrates containing < 4 consecutive ribonucleotides (Cerritelli & Crouch, 2009). Deletion of RNH1 caused a small increase in the percentage of cells containing a spontaneous Rad52‐YFP focus compared to wild‐type cells (WT) (Fig 1A). This result is consistent with an increase in gross chromosomal rearrangements observed in a rnh1Δ mutant (Wahba et al, 2011), suggesting that processing of RNA–DNA hybrids by RNase H1 may be important for preventing spontaneous DSBs. Importantly, the rnh201‐RED rnh1Δ double mutant gave rise to a similar level of spontaneous Rad52‐YFP foci as the rnh201‐RED single mutant, suggesting that failure to repair single genomic ribonucleotides is the predominant source of Rad52‐YFP foci.
Rad52 is essential for tolerance of Top1‐induced DNA damage at unrepaired single genomic ribonucleotides incorporated by the replicative polymerase on the leading strand
To further assess the importance of HR in the repair of Top1‐induced DNA damage at unrepaired ribonucleotides, we compared the effect of inactivating RAD52, a crucial factor broadly involved in HR (Krogh & Symington, 2004), in different genetic backgrounds. Previous studies have shown that by mutating the residues of the DNA polymerases near the active site “steric gate,” the levels of ribonucleotide incorporation by a polymerase can be modulated. In addition to inactivating RNH201, we mutated different DNA polymerases (pol2‐M644G for Pol ε, pol1‐L868M for Pol α and pol3‐L612M for Pol δ) so that all mutator allele strains incorporated significantly higher levels of ribonucleotides specifically on either the leading or lagging strand according to the division of labor by the polymerases at the replication fork.
Tetrad dissection of heterozygous RAD52/rad52Δ diploid strains showed that inactivating RAD52 had differential effects depending on which polymerase mutator allele was also present (Fig 2A and Appendix Fig S1). Based on the size of the spore colonies, inactivating RAD52 in the rnh201Δ background gave rise to less‐healthy strains (Appendix Fig S1). Similarly, the pol1‐L868M rnh201Δ rad52Δ and pol3‐L612M rnh201Δ rad52Δ haploid cells gave rise to smaller spore colonies but remained viable (Fig 2A, dissections 7–12, green circles). By contrast, Rad52 is essential in the pol2‐M644G rnh201Δ cells as the pol2‐M644G rnh201Δ rad52Δ haploid spore progeny was inviable (Fig 2A, dissections 1–3, green circles). The microscopic images of individual spore colonies confirmed that the pol2‐M644G rnh201Δ rad52Δ spores germinated but were unable to divide more than a few times and arrested as large G2/M cells (Fig 2B). Since the pol2‐M644G mutator allele predominantly incorporates a high level of ribonucleotides into the nascent leading strand, while the pol1‐L868M and pol3‐L612M mutator alleles primarily do so into the nascent lagging strand (Lujan et al, 2013; Williams et al, 2015), our results suggest that Rad52 is essential for the repair of DNA damage initiated by unrepaired ribonucleotides incorporated into the nascent leading strand.
Figure 2. RAD52 is essential in the presence of high levels of genomic ribonucleotides present in nascent leading strand DNA in a Top1‐dependent manner.
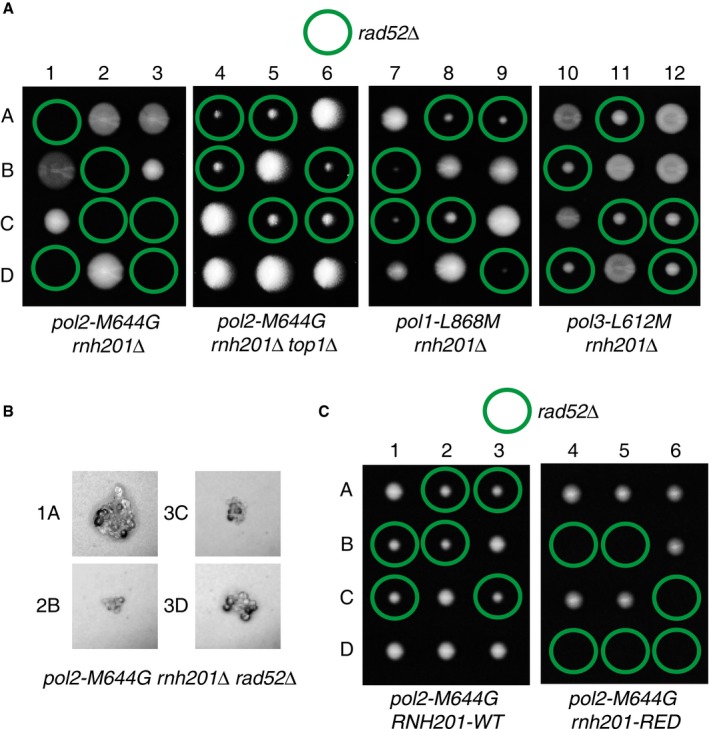
- Tetrad analysis of RAD52/rad52Δ diploids. 1 to 12 are tetrad dissections, and A–D are haploid spore colonies. Plates were imaged after 5 days of growth on YPDA at 30°C.
- Microscopy images reveal that the pol2‐M644G rnh201Δ rad52Δ haploid derivatives sporulated but failed to divide more than a few times and arrested as large G2/M cells.
- Tetrad analysis of RAD52/rad52Δ diploids in a rnh201‐RED mutant reveals that the presence of unrepaired single ribonucleotides is the cause of synthetic lethality in a pol2‐M644G rnh201Δ or pol2‐M644G rnh201‐RED strain background. Plates were imaged after 4 days of growth. pol2‐M644G rnh201‐RED rad52Δ haploid derivatives arrested as large G2/M cells.
More importantly, when we carried out the same tetrad dissection (RAD52/rad52Δ diploid) with the additional deletion of TOP1 (pol2‐M644G rnh201Δ top1Δ), the pol2‐M644G rnh201Δ top1Δ rad52Δ haploid cells were viable (Fig 2A, dissections 4–6, green circles), indicating that Rad52 was no longer essential. Our results suggest that Rad52 is essential for the repair of Top1‐induced DNA damage in the presence of a high level of nascent leading strand ribonucleotides incorporated by Pol ε. This is consistent with our recent finding that Top1 incision at ribonucleotides appears to be specific to ribonucleotides incorporated into the nascent leading strand by Pol ε (Williams et al, 2015).
To determine whether single genomic ribonucleotides or stretches of consecutive ribonucleotides are involved in Top1‐induced DNA damage that requires Rad52, we carried out tetrad dissections with the rnh201‐RED mutant strain as well. The results demonstrated that pol2‐M644G rnh201‐RED rad52Δ haploid cells were inviable (Fig 2C, dissections 4–6, green circles), while pol2‐M644G RNH201‐WT rad52Δ cells were viable (Fig 2C, dissections 1–3, green circles). This is consistent with the elevated level of rnh201‐RED cells bearing a Rad52‐YFP focus (Fig 1C), suggesting that Top1‐induced DNA damage at ribonucleotides mainly arises from single genomic ribonucleotides and not from R‐loops. Furthermore, Rad52 plays an essential role in the repair of Top1‐induced DNA damage at single ribonucleotide incorporated into DNA by Pol ε, suggesting that Top1 induces DSBs in vivo when ribonucleotides are not removed by RNase H2.
Rad51 counteracts the genome instability caused by Top1‐induced DNA damage at unrepaired single genomic ribonucleotides
Next, we evaluated the impact of inactivating RAD51, an enzyme critical for recombination filament formation and strand invasion during DSB repair by HR [reviewed in Krogh and Symington (2004)]. Inactivating RAD51 in a wild‐type DNA polymerase background (± RNH201) showed no obvious effect in unperturbed conditions (Appendix Fig S2A). In contrast, pol2‐M644G rnh201Δ rad51Δ cells were viable, but showed an increased doubling time and abnormal cell morphology (small and heterogeneous colonies) as compared to either pol2‐M644G rnh201Δ or pol2‐M644G rad51Δ cells (Fig 3A). Inactivation of RAD51 generally gives rise to a milder phenotype than inactivation of RAD52, potentially because Rad51‐independent recombination is known to occur and Rad52 is still required in these alternative pathways. This is also consistent with a previous report that rnh1Δ rnh201Δ rad51Δ cells showed higher hydroxyurea sensitivity, but rnh1Δ rnh201Δ rad52Δ cells were inviable (Lazzaro et al, 2012).
Figure 3. Deletion of RAD51 in a yeast strain containing a large number of unrepaired leading strand ribonucleotides (pol2‐M644G rnh201Δ) causes genome instability phenotypes that are Top1 dependent.
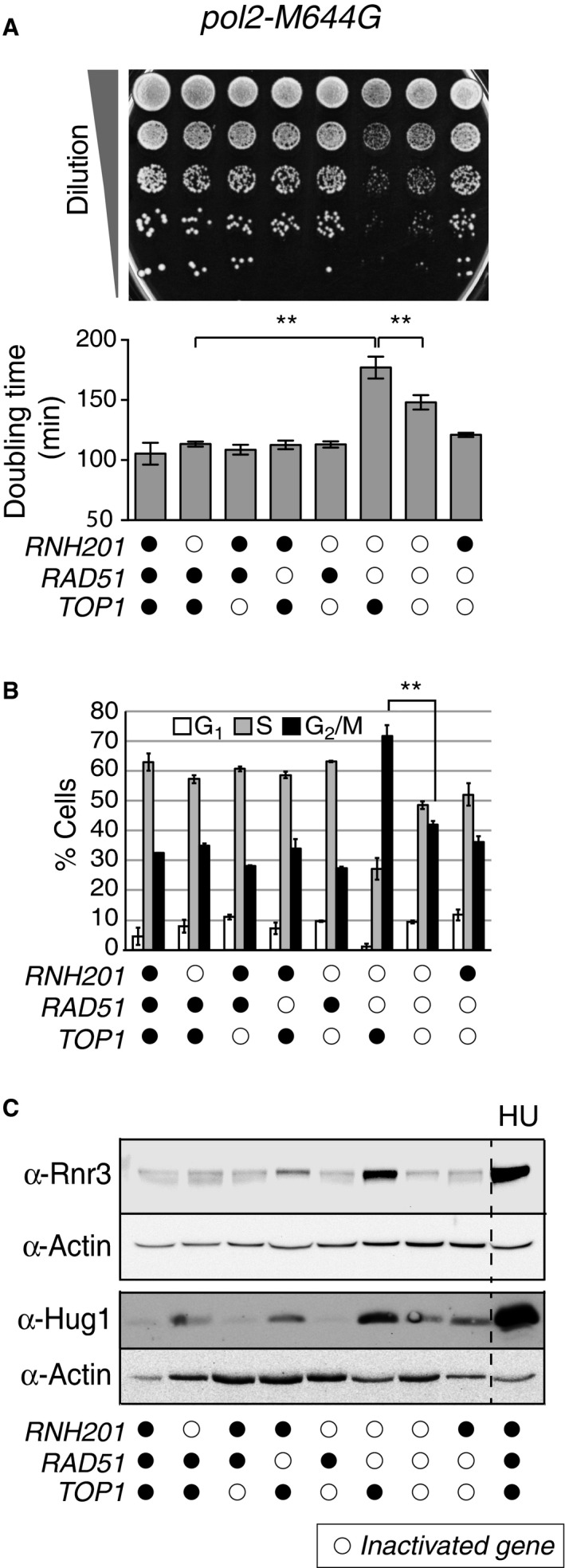
- Slow growth caused by loss of RAD51 in a pol2‐M644G rnh201Δ strain is Top1 dependent. Upper panel: Tenfold serial dilutions of exponentially growing yeast strains with combinations of indicated gene deletions were spotted onto a YPDA agar plate and grown for 2 days at 30°C. Lower panel: Doubling times (D t) were calculated for respective strains in the logarithmic phase of growth in rich medium at 30°C. **P ≤ 0.01 (Student's t‐test in GraphPad Prism). The experiment was performed in triplicate. Data are displayed as the mean D t ± standard deviation (SD).
- Flow cytometry analysis of cell cycle progression reveals that deletion of RAD51 in a pol2‐M644G rnh201Δ strain causes accumulation of cells in G2/M phase in a Top1‐dependent manner. Representative histograms are presented in Appendix Fig S2B. Bar graphs display the percentage of cells in G1, S, or G2/M phase. The experiment was performed twice, and mean values are plotted ± SD. **P ≤ 0.01 (Student's t‐test in GraphPad Prism).
- Activation of the genome integrity checkpoint was measured by immunoblotting for Rnr3 and Hug1 in whole cell extracts from the indicated strains. Increase in Rnr3 and Hug1 expression levels serves as a sensitive indicator of genome integrity checkpoint activation (Basrai et al, 1999; Kumar et al, 2010; Davidson et al, 2012). Hydroxyurea (HU)‐treated (200 mM for 3 h) cells served as a positive control.
The fact that pol2‐M644G rnh201Δ rad51Δ cells were viable allowed further analysis of the role of Top1 in the induction of replication stress in rnh201Δ cells. Notably, cell growth significantly recovered upon additional deletion of TOP1, and the small and heterogeneous colony morphology was alleviated (Fig 3A). These Rnh201‐, Rad51‐, and Top1‐dependent effects on growth are consistent with the formation of Top1‐induced DSBs at ribonucleotide sites and the consequent involvement of DSB repair by HR. This decreased growth rate suggests that Rad51 is important for processing DNA damage that results from a high density of unrepaired ribonucleotides.
Consistent with the observed effect on growth rate, flow cytometry analysis demonstrated that the cell cycle distribution profile for the pol2‐M644G rnh201Δ rad51Δ strain was significantly altered (Fig 3B and Appendix Fig S2B). Over twice as many pol2‐M644G rnh201Δ rad51Δ cells accumulated in the G2/M phase compared to other strains (Fig 3B, 70% versus 30%). Under these conditions, deletion of TOP1 (pol2‐M644G rnh201Δ rad51Δ top1Δ) reduced the percentage of cells in the G2/M phase down to 42% (Fig 3B and Appendix Fig S2B). These results show that Rad51‐mediated repair is important for Top1‐induced damage in the context of newly incorporated ribonucleotides.
The Top1‐dependent slow growth and cell cycle defects of the pol2‐M644G rnh201Δ rad51Δ strain were accompanied by constitutive genome integrity checkpoint activation. Levels of both Rnr3 and Hug1 were substantially increased in the pol2‐M644G rnh201Δ rad51Δ strain when compared to control strains (Fig 3C). Rnr3 is a subunit of ribonucleotide reductase and Hug1 is also known as a MEC1‐mediated checkpoint protein, and both proteins are up‐regulated in response to checkpoint activation and replication stress (Basrai et al, 1999; Kumar et al, 2010; Davidson et al, 2012). Consistent with the effects observed on growth rates and cell cycle distribution, additional deletion of TOP1 (pol2‐M644G rnh201Δ rad51Δ top1Δ) attenuated the checkpoint activation (Fig 3C).
To distinguish between the effect of unrepaired of R‐loops and single ribonucleotides, we carried out the same genetic analyses using the pol2‐M644G rnh201‐RED rad51Δ strain, and we observed similar increases in doubling time, accumulation in the G2/M phase, and replication stress as in pol2‐M644G rnh201Δ rad51Δ cells (Fig EV1). These results indicate that the unrepaired single ribonucleotides are sufficient to cause these observed phenotypic responses.
Figure EV1. Deletion of RAD51 in a rnh201‐RED strain causes genome instability phenotypes that are Top1 dependent.
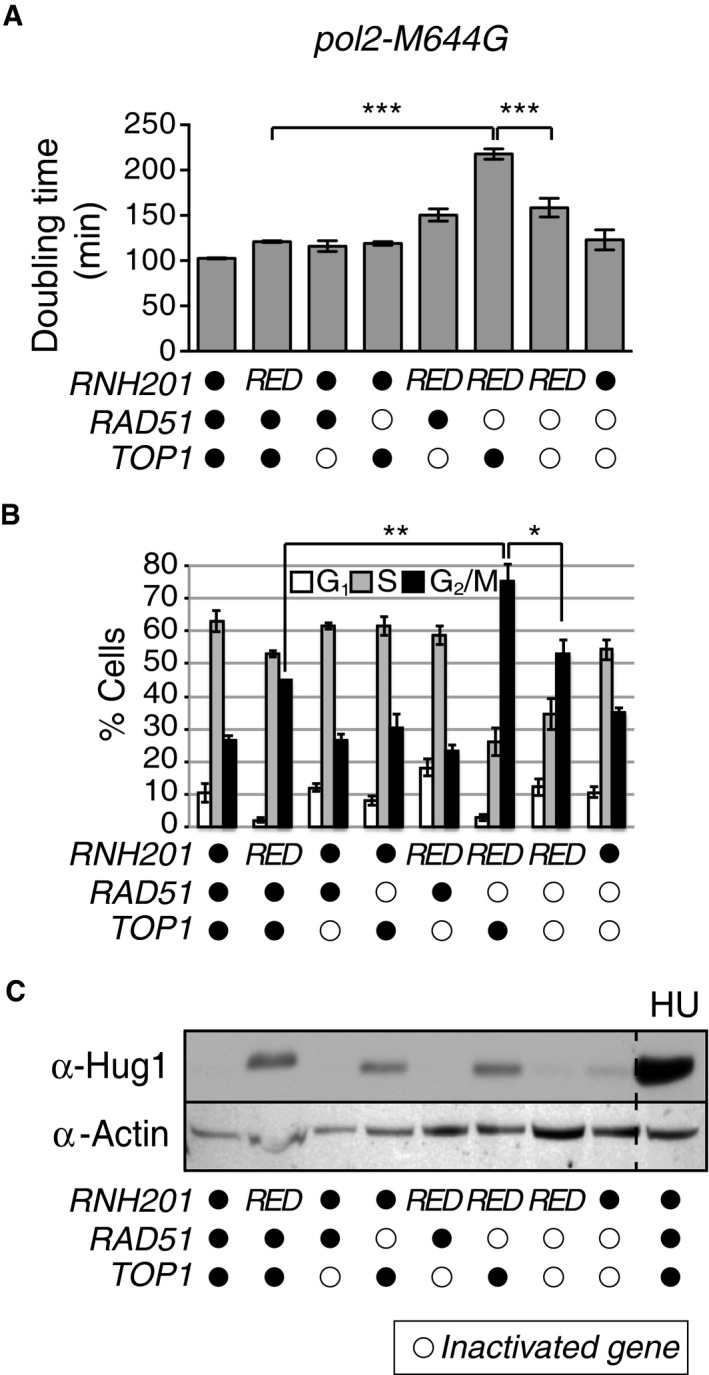
- Slow growth caused by loss of RAD51 in a pol2‐M644G rnh201‐RED strain is Top1 dependent. Upper panel: Doubling times (D t) were calculated for strains in the logarithmic phase of growth in rich medium at 30°C. ***P ≤ 0.001 (unpaired Student's t‐test in GraphPad Prism). The experiment was performed in triplicate. Data are displayed as the mean D t ± standard deviation (SD).
- Flow cytometry analysis of cell cycle progression reveals that deletion of RAD51 in a pol2‐M644G rnh201Δ strain causes accumulation of cells in G2/M phase in a Top1‐dependent manner. Bar graphs display the percentage of cells in G1, S, or G2/M phase. The experiment was performed twice, and mean values ± SD are plotted. **P ≤ 0.01 and *P ≤ 0.05 (unpaired Student's t‐test in GraphPad Prism).
- Activation of the genome integrity checkpoint was measured by immunoblotting for Hug1 in whole cell extracts from the indicated strains. Increase in Hug1 expression levels serves as a sensitive indicator of genome integrity checkpoint activation. Hydroxyurea (HU)‐treated (200 mM for 3 h) cells served as a positive control.
In summary, constitutive checkpoint activation depends on the presence of Top1, the inability of RNase H2 to remove single ribonucleotides (but not R‐loops) and the absence of Rad51. These results suggest that Rad51 is important in repairing Top1‐induced damage at newly incorporated ribonucleotides in order to maintain genome integrity. This is consistent with a model in which Rad51‐mediated HR is important for the repair of Top1‐induced DSBs when high levels of persistent ribonucleotides have been incorporated into the genome by a replicative DNA polymerase.
Sequential Top1 cleavage on the opposite strand from the Top1‐induced nick at a single ribonucleotide site leads to irreversible DSBs
What is the mechanism of Top1‐induced DSBs at unrepaired ribonucleotide sites? One possibility is that replication of the nicked strand generates a DSB. Another possibility is that two sequential Top1 cleavage events on the opposite strands can create DSBs at ribonucleotide sites, leaving one side with a 2′,3′‐cyclic phosphate end and the other side with a covalently linked Top1, which is prone to religate/recombine with DNA bearing 5′‐OH groups. Figure 4 shows the proposed model. In either scenario, Top1‐induced DSBs would be generated via a mechanism that is distinct from the reported mechanism through which Top1 induces 2–5 bp deletions at repetitive sequences via two sequential Top1 cleavage events on the same ribonucleotide‐containing strand (Nick McElhinny et al, 2010a; Kim et al, 2011; Williams et al, 2013, 2015; Huang et al, 2015; Sparks & Burgers, 2015). Consistent with this idea, we found that the loss of HR does not appear to impact the rate of Top1‐dependent 2–5 bp deletions at repetitive sequences in rnh201Δ cells (Appendix Table S1 and Fig EV2), thus suggesting a novel mechanism for how Top1 generates DSBs at ribonucleotide sites.
Figure 4. Molecular mechanisms for the formation of a DSB by sequential Top1 ribonuclease activity and Top1 cleavage events on the opposite DNA strand from the newly incorporated ribonucleotides.
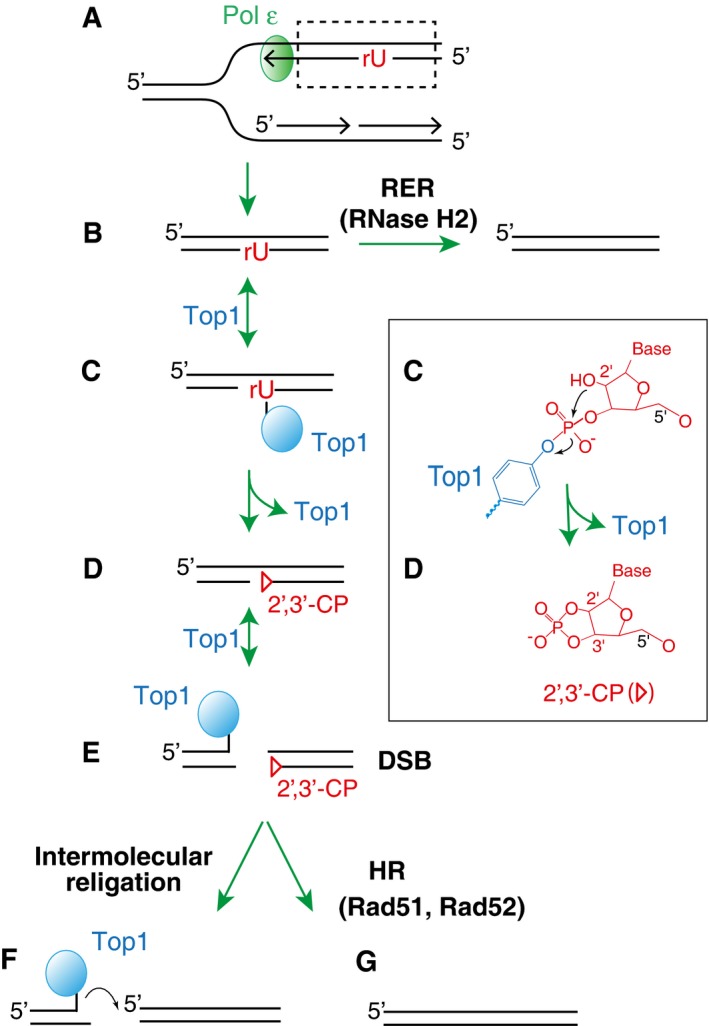
- Ribonucleotides are preferentially incorporated on the leading strand when Pol ε is genetically altered.
- Newly incorporated ribonucleotides are normally removed by RER initiated by RNase H2 incision at the ribonucleotide site.
- In the absence of RNase H2, Top1 forms a cleavage complex at the ribonucleotide site.
- The attack by 2′‐hydroxyl group on the phosphotyrosyl bond releases Top1, converting the ribonucleotides into nicks with 2′,3′‐cyclic phosphate ends (triangle). A detailed biochemical reaction is shown in the box on the right.
- Subsequent cleavage by Top1 on the opposite strand of the existing nick leads to a DSB.
- The resulting DSB with covalently linked Top1 at the end is prone to religate/recombine with other DNA ends.
- Alternatively, homologous recombination (dependent on Rad51 and Rad52) repairs the DSB.
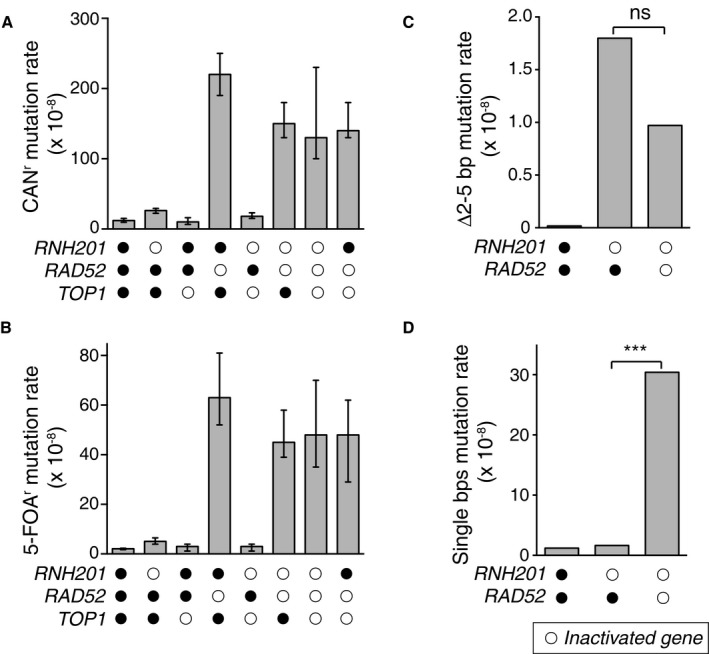
Figure EV2. Top1‐dependent short deletions at unrepaired ribonucleotides occur independent of DSB repair.
-
A, B(A) Overall CAN1 mutation rates for strains with deletions of indicated genes. Spontaneous mutation rates were determined using fluctuation analysis as described previously (Shcherbakova & Kunkel, 1999). (B) Same as in panel (A), but for 5‐FOA‐resistance (5‐FOAr) conferred by mutation of the URA3 reporter gene in orientation 2 adjacent to the ARS306 replication origin (Nick McElhinny et al, 2008). Displayed are mean values ± the 95% confidence interval. The experiment was performed twice and mutation rate measurements were calculated for 24 independent isolates per genotype per experiment. Displayed are median values ± the 95% confidence interval.
-
CDisplayed are 2–5 bp deletion mutation rates comparing WT, rnh201Δ, and rnh201Δ rad52Δ strains calculated from data listed in Appendix Table S1 for the URA3‐OR2 reporter. This rate was calculated using mutation spectra from Nick McElhinny et al (2010a) for the WT strain and from Clark et al (2011) for the rnh201Δ strain. 2–5 bp deletion rates were calculated by multiplying the proportion of this mutation type by the overall mutation rate for each strain. ns: P = 0.089 (Monte Carlo simulation) (Lujan et al, 2013). This demonstrates that the 2–5 bp deletion rate is not statistically significantly different when comparing the rnh201Δ strain to the rnh201Δ rad5Δ strain.
-
DDisplayed are base pair substitution rates comparing WT, rnh201Δ, and rnh201Δ rad52Δ strains calculated from data listed in Appendix Table S1 for the URA3‐OR2 reporter. ***P ≤ 0.001 (the rate is calculated as described in panel C).
To establish biochemically whether Top1 can induce DSBs via the mechanism shown in Fig 4, we examined cleavage products by recombinant Top1 enzyme on a DNA construct containing the (AG)4 hot spot from the CAN1 reporter sequence (Kim et al, 2011), where the non‐transcribed strand (NTS) bored either a single ribonucleotide or a deoxyribonucleotide (position X in Fig 5A). To establish ribonuclease activity of Top1 in this context (see box in Fig 4), we first carried out a cleavage and reversal assay with the DNA substrate labeled on the NTS strand (Appendix Fig S3). When the formation of Top1 cleavage complexes (Top1cc) reaches equilibrium, addition of a high concentration of NaCl prevents the formation of new Top1cc, yet allowing the dissociation of existing Top1cc in the reverse direction to continue (Pourquier et al, 1997). Thus, transient Top1cc would result in bands whose intensities decrease with time in this assay. The results show that in the absence of the specific Top1 poison camptothecin (CPT), Top1 induces an irreversible, permanent break at the site corresponding to position X only when a ribonucleotide was incorporated at position X (site b in Appendix Fig S3). Thus, as expected, Top1 acted as a ribonuclease and generated a permanent DNA nick at the ribonucleotide site in this sequence context.
Figure 5. Top1 induces DSBs in close proximity to ribonucleotide sites.
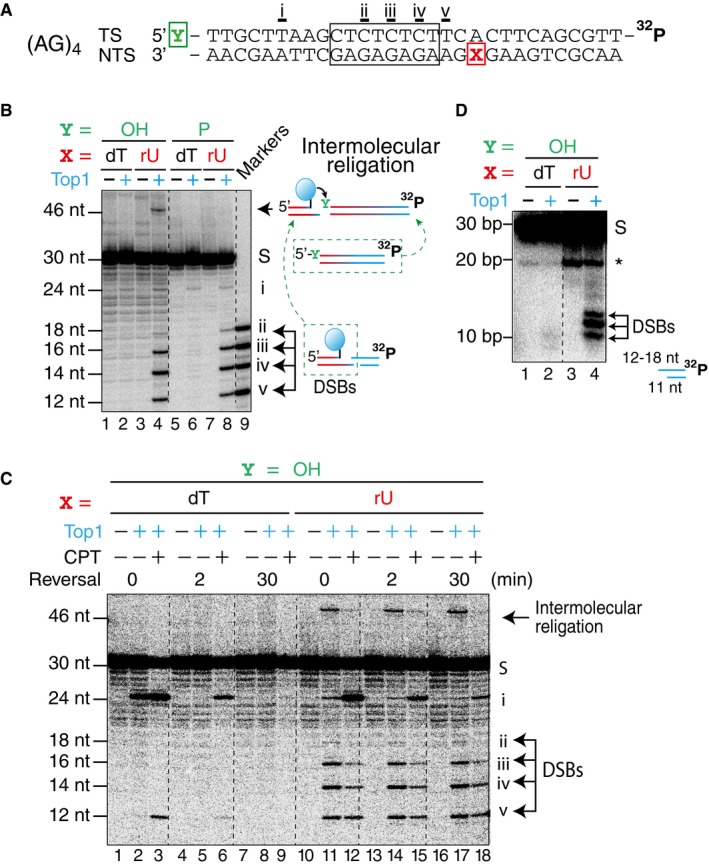
- DNA constructs containing the sequence from the CAN1 (AG)4 site (marked by a box) with the transcribed strand (TS) radiolabeled at the 3′‐end. Position X in the non‐transcribed strand (NTS) contained either a deoxythymidine (dT) or a ribouridine (rU). Position Y of the TS contained either a hydroxyl or a phosphate group.
- A representative gel image of Top1 cleavage assay with the construct shown in panel (A), resolved on a denaturing sequencing gel. Top1‐induced products are labeled (i–v), with the corresponding Top1 cleavage site labeled below the sequence in panel (A). DNA markers of the expected lengths were loaded in lane 9.
- A representative gel image of Top1 cleavage and reversal assay with construct shown in panel (A) with or without the presence of camptothecin (CPT).
- Top1 cleavage assay with the same construct resolved on a native gel. A Top1‐independent band (labeled by *) in lanes 3 and 4 is attributed to a partially melted DNA flap after the substrate has undergone spontaneous hydrolysis at the ribonucleotide site.
Next, we examined Top1 cleavage products with the DNA construct labeled on the transcribed strand (TS) in order to detect Top1‐induced DSBs. One weak Top1‐induced cleavage product was detectable when the NTS contained only deoxyribonucleotides (Fig 5B, lanes 2 and 6, site i), while five Top1‐induced cleavage products were detectable when the position X in the NTS contained a ribonucleotide (Fig 5B, lanes 4 and 8, sites i–v). The nucleotide position that is covalently linked to Top1 in each instance is underlined in the sequence in Fig 5A (sites i–v). Additionally, a Top1‐induced intermolecular religation product was generated when position X contained a ribonucleotide (Fig 5B, lane 4, 46 nt). When Top1 is covalently linked to DNA end, it is prone to religate with DNA containing a free 5′‐hydoxyl group. As the diagram on the right side of Fig 5B shows, one way the 46 nt product can be generated is through religation of Top1‐linked DSB with full‐length TS substrate. We reasoned that if the 5′‐end of the TS was phosphorylated, the Top1‐dependent religation product would be blocked. Accordingly, when the position Y in the TS contained a phosphate instead of a hydroxyl group, the Top1‐dependent recombination product was not observed, while the other Top1‐induced cleavage sites remained unchanged (Fig 5B, lane 8).
Top1 cleavage and reversal assays in the presence or absence of CPT with the (AG)4 construct (Fig 5A) showed that when position X contained a deoxyribonucleotide, both CPT‐dependent and CPT‐independent sites were fully reversible after 30 min (Fig 5C, lanes 1–9). However, when position X contained a ribonucleotide, the intensity of site i decreased as the reversal reaction progressed, but the intensities of sites ii–v did not change over time (Fig 5C, lanes 10–18), consistent with irreversible Top1‐induced DSBs. Because denaturing sequencing gels cannot differentiate between DNA single‐ and double‐strand breaks, we resolved the Top1 cleavage products of the (AG)4 construct on native gels. When position X contains a ribonucleotide, the expected Top1‐induced double‐strand fragment should be a 11 nt fragment of the NTS annealed to a 12 to 18 nt fragment of the TS, depending on the site of Top1 cleavage. As expected, only when position X contained a ribonucleotide, the Top1 cleavage products gave rise to three bands with the expected sizes (Fig 5D, lane 4, between 10 and 20 bp). The lower resolution of the native gel and the partial melting of the DNA duplex likely contributed to why we only observed three instead of four DSB products in the native gel. We also performed the Top1 cleavage assays on a construct containing the (AT)2 hotspot from the CAN1 sequence resolved in both denaturing sequencing and native gels (Appendix Fig S4). Similar results were observed.
As shown in the proposed model in Fig 4, the first step of Top1‐induced DSB formation requires the ribonuclease activity of Top1. Canonical Top1 cleavage activity is characterized by a broad and relaxed sequence dependence (Porter & Champoux, 1989; Jaxel et al, 1991). To probe whether the presence of ribonucleotides affects the frequency of Top1 cleavage, we compared the effect of ribonucleotides to different Top1 trapping poisons, traditionally used to capture transient Top1 cleavage complexes. Using DNA constructs incorporating ribonucleotides at different positions, we found that Top1 cleaves at ribonucleotide sites at high frequencies, revealing sites that were undetectable using any of the three Top1 poisons tested (Fig EV3). Taken together, our biochemical results demonstrate that Top1 displays frequent ribonuclease activity at unrepaired ribonucleotide sites and that abundant DSBs are readily generated by a subsequent Top1 cleavage event on the opposite strand.
Figure EV3. Top1 ribonuclease activity cleaves at ribonucleotide sites at a high frequency, but increasing number of consecutive ribonucleotides diminishes Top1 cleavage sites gradually.
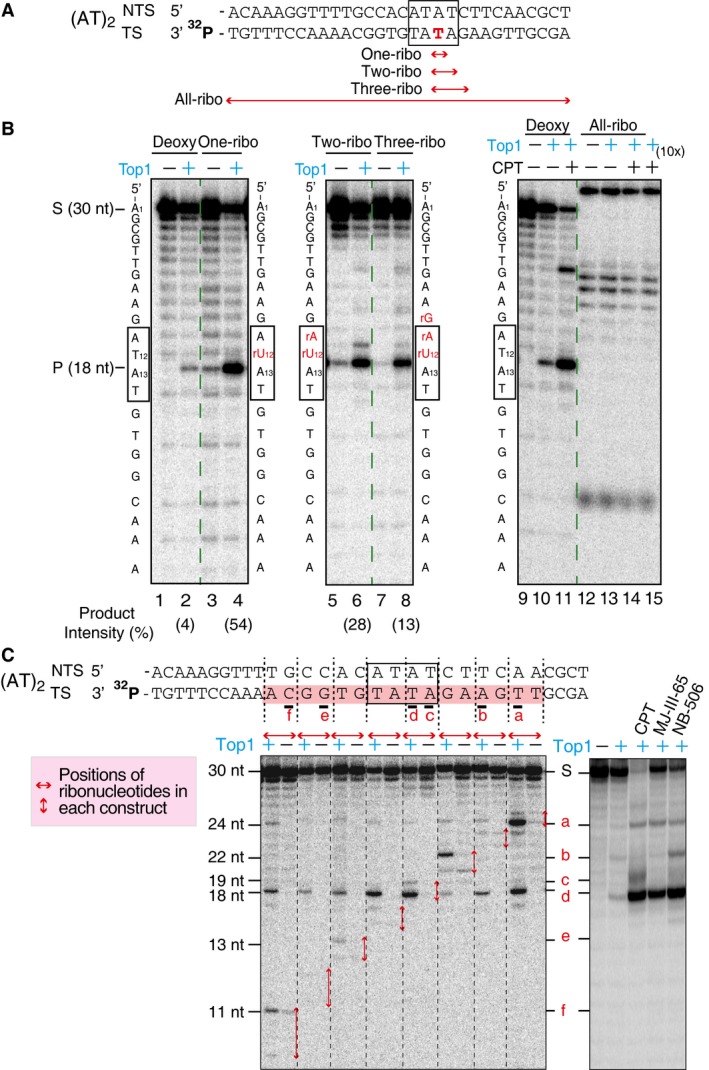
- DNA constructs contained sequences from the (AT)2 site (marked by the black boxes) in the CAN1 reporter gene, and the transcribed strand (TS) of each construct is radiolabeled on the 3′‐end. Red double arrows indicate the positions of ribonucleotides in each construct. The constructs contain either zero, one, two, three, or all ribonucleotides at indicated positions.
- Top1 cleavage assay with DNA constructs with different number of ribonucleotides. The positions of the ribo‐substitutions in each construct are indicated by red letters next to the gel panels. Product intensity is the amount of 18 nt Top1 cleavage products [P/(S + P)]. The results show that the presence of one ribonucleotide increased the Top1 cleavage product from 4% to 54%, while successive addition of ribonucleotides gradually decreases the percentage of Top1 cleavage product. All Top1 cleavage products are blocked with the All‐ribo construct despite the presence of CPT and higher concentration of Top1 enzyme. The reactions contained 70 nM Top1 and 10 μM CPT where indicated, and the last sample with the All‐ribo construct (lane 15) contained 700 nM Top1 recombinant protein (10×).
- To assess the frequency of Top1‐induced DNA nicks at ribonucleotide sites, we carried out Top1 cleavage assays using DNA constructs with ribonucleotides incorporated at different positions. The constructs contained the (AT)2 repeat sequence (marked by a box) from the CAN1 reporter gene and were radiolabeled on the 3′‐end of transcribed strand (TS). Each of the set of eight constructs contains two unique ribonucleotide substitutions (Appendix Table S3), and in total, a stretch of 16 nucleotides (shaded) on the TS sequence was examined. Lower left panel: representative gel image showing Top1 cleavage patterns for different constructs. Horizontal red double arrows mark the positions of ribonucleotide substitutions in each construct. The Top1‐induced cleavage sites within ribonucleotide substitutions are labeled a–f next to the gel panels and below TS. The six Top1 sites identified include sites with Top1 covalent linkage to all four DNA bases, A, T, C, and G, deviating from the reported bias for a thymidine at the −1 position (covalently linked to Top1) (Jaxel et al, 1991; Parker & Champoux, 1993). Interestingly, the presence of ribonucleotides in some constructs, even at a distance, enhances Top1 cleavage (see site d), likely attributable to alteration in DNA local structures induced by ribonucleotides. Lower right panel: Top1 cleavage sites in the presence of three different Top1‐trapping drugs with DNA construct containing only deoxyribonucleotides of the (AT)2 sequence, radiolabeled on the 3′‐end on the TS. The last three lanes contain additions of either CPT, indenoisoquinoline LMP774 (MJ‐III‐65) or indolocarbazole (NB‐506). Only four out of six Top1 cleavage sites identified by ribonucleotide scanning were detected this way, labeled a–d next to the gel panels.
Unrepaired ribonucleotides induce covalently linked Top1 to DNA ends in vivo
To further demonstrate the sequential Top1 cleavage mechanism for the generation of DSBs at unrepaired ribonucleotide sites in vivo, we probed for DNA fragments that are covalently linked to Top1 at their 3′‐end (Fig 4E). For this, we tagged the endogenous Top1 protein with a 5×FLAG‐epitope tag in RNase H2‐proficient or RNase H2‐deficient strains and pulled‐down DNA fragments covalently linked to Top1 from unfixed cell lysates using an anti‐FLAG antibody. The efficiency of pull‐down was verified by immunoblotting followed by probing for the remaining Top1 in the supernatant post‐pull‐down (Fig EV4A). We further verified that the pulled‐down DNA was covalently linked to Top1 by treatment with tyrosyl‐DNA phosphodiesterase 1 (TDP1), which specifically cleave the phosphotyrosyl bond of Top1cc (Fig EV4B). If the covalently linked Top1 is located at an internal position, the DNA fragment would contain a nick at the Top1 cleavage site after proteinase K treatment, making it susceptible to subsequent S1 nuclease digestion (Fig 6A). By contrast, a DNA fragment with the covalently linked Top1 located at the 3′‐end would not contain DNA nick and thus would be resistant to S1 nuclease (Fig 6B).
Figure EV4. Analysis of DNA covalently linked with Top1 by immuno‐pull‐down.
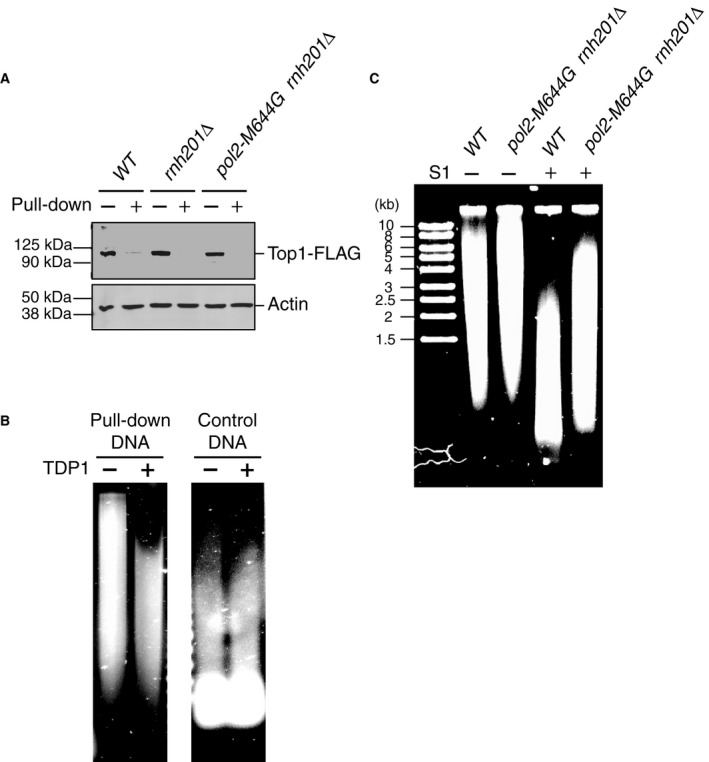
- Immunoblot of yeast cell lysates of different strains probing for the presence of 5×FLAG‐tagged Top1 before (−) and after (+) immuno‐pull‐down with anti‐FLAG antibodies.
- The DNA covalently linked with Top1 purified by immuno‐pull‐down (after proteinase K digestion) was further treated with 3 μM recombinant TDP1 overnight or mock‐treated (Huang et al, 2013). TDP1‐treated DNA showed increased gel mobility. The control DNA was purified from the same sample after the immuno‐pull‐down procedure and TDP1 treatment showed no effect on the control DNA.
- A representative gel image of mock‐treated or S1‐treated pulled‐down DNA from either wild‐type (WT) or pol2‐M644G rnh201Δ strains.
Figure 6. Analysis of DNA covalently linked with Top1 by immuno‐pull‐down.
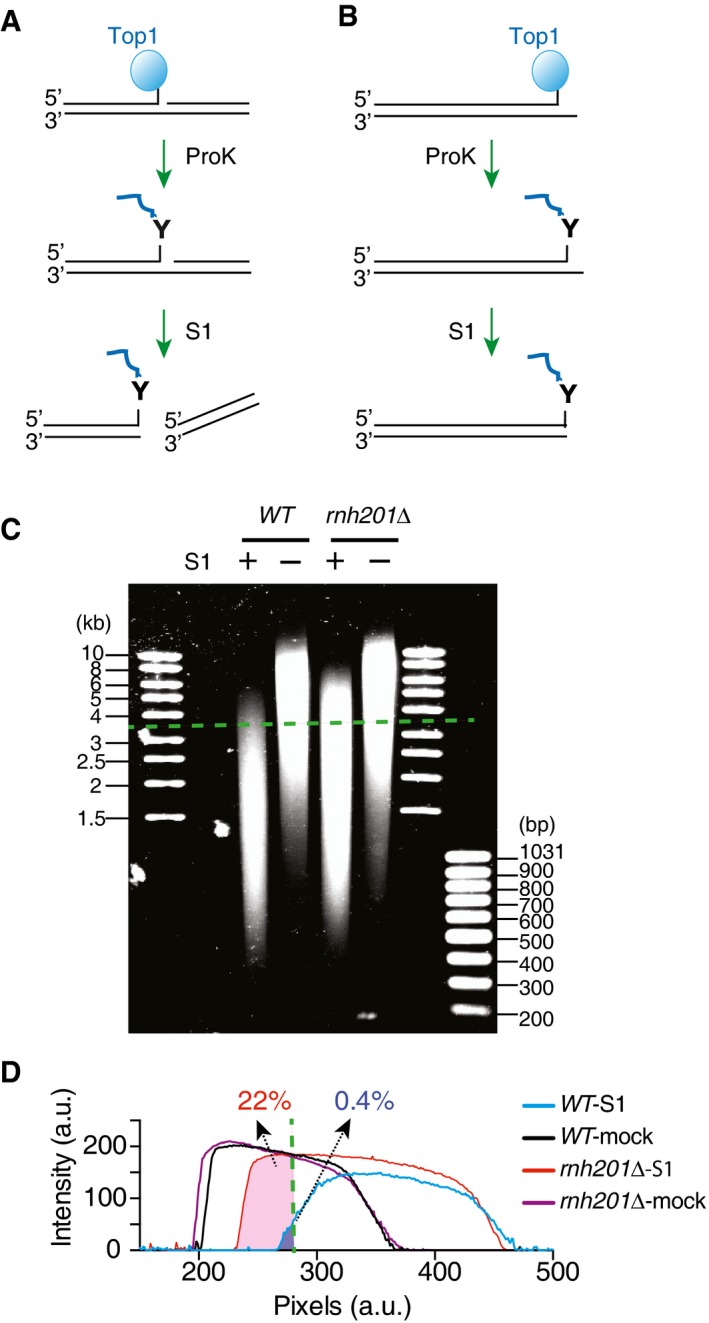
- Diagram depicting the pulled‐down DNA that are covalently linked to Top1 at an internal position. After proteinase K digestion, the resulting DNA nick is cleaved by nuclease S1.
- If the covalently linked Top1 is located at the end and no other nicks are present in the pulled‐down DNA, it is resistant to cleavage by nuclease S1.
- S1‐treated or mock‐treated pulled‐down DNA from either wild‐type (WT) or rnh201Δ strains resolved on a 1% agarose gel and stained with SYBR Gold. A representative gel image is shown, and dashed green line marks the position for DNA of 3.5 kb.
- Traces of DNA intensities for the samples from the gel image in panel (C). Dashed line marks the position for DNA of 3.5 kb, and the listed numbers show the percentage of DNA density longer than 3.5 kb calculated from the total area under the curves.
We treated the pulled‐down DNA fragments from yeast strains with or without RNase H2 with low concentrations of nuclease S1 that effectively cleaved nicked DNA but not double‐strand DNA. S1‐treated DNA from wild‐type cells consistently migrated faster than the S1‐treated DNA from rnh201Δ or pol2‐M644G rnh201Δ cells (Figs 6C and D, and EV4C). The tracing of the S1‐treated DNA showed almost no DNA from wild‐type cells longer than 3.5 kb, as opposed to ~22% of DNA from rnh201Δ cells (Fig 6C and D, green dash line). Under similar conditions, the mock‐treated DNA from different yeast strains showed no difference in migration (Figs 6C and D, and EV4C). The greater resistance of the pulled‐down DNA from rnh201Δ or pol2‐M644G rnh201Δ cells to nuclease S1 digestion provides evidence that Top1 is more frequently covalently linked to the end of DNA in RNase H2‐deficient yeast cells in vivo. These biochemical experiments are consistent with the sequential Top1 cleavage model for DSB formation when newly incorporated ribonucleotides are left unrepaired in the genome.
Discussion
A large body of experimental evidence in recent years demonstrated that ribonucleotides are frequently incorporated into DNA during replication, making them the most common non‐canonical nucleotides in the genome (Nick McElhinny et al, 2010b; Potenski & Klein, 2014; Reijns & Jackson, 2014; Williams & Kunkel, 2014; Cerritelli & Crouch, 2016; Williams et al, 2016). Both persistent and transient ribonucleotides in the genome have potentially far‐reaching consequences. Elucidating their deleterious consequences and the processes by which they are repaired is currently a particularly active area of research. Here, we provide genetic evidence that DSBs are generated by Top1, an essential and abundant enzyme (Pommier et al, 2016), at sites of single genomic ribonucleotide in the absence of RER. We also provide in vitro and in vivo biochemical evidence that sequential Top1 ribonuclease activity (Sekiguchi & Shuman, 1997; Kim et al, 2011) and Top1 cleavage events on opposite DNA strands can generate DSBs.
The predominant pathway for the removal of genomic ribonucleotides is RER (Fig 4B), in which RNase H2 initiates the process by nicking the DNA backbone on the 5′‐end of a newly incorporated ribonucleotide. RNase H2 is conserved through all domains of life, and RNase H2‐null mice are embryonic lethal (Hiller et al, 2012; Reijns et al, 2012). Unicellular organisms do not require RNase H2 for viability, however, making it possible to elucidate the consequences of RER deficiency in model organisms such as yeast. Convergent studies have established that Top1 produces short deletions at ribonucleotide sites (Kim et al, 2011; Potenski et al, 2014), which have been attributed to the ribonuclease activity of Top1 (Sekiguchi & Shuman, 1997; Kim et al, 2011; Sparks & Burgers, 2015; Huang et al, 2015) (Fig EV5). When a Top1 covalent cleavage is formed at a ribonucleotide, the 2′‐hydroxyl group carries out a nucleophilic attack on the phosphotyrosyl bond, nicking the DNA and simultaneously freeing Top1 (Figs 4 and EV5). Our data show that the frequency of Top1 ribonuclease activity at single genomic ribonucleotide sites is potentially very high. We revealed more Top1 sites in our test sequence using substrates that contain ribonucleotides at different positions than using Top1 trapping drugs and substrates containing only deoxyribonucleotides (Fig EV3C). Furthermore, the ribonucleotide sites where Top1 nicks present a broader sequence diversity compared to preferred sites by Top1 trapping drugs that tend to trap Top1 cleavage at thymidines (Jaxel et al, 1991; Parker & Champoux, 1993). As Top1 is coupled with replication (Pommier et al, 2016), the capacity of Top1 to either nick or generate a DSB at sites of transient or persistent ribonucleotides introduced during replication emerges as a previously unanticipated threat to genome stability.
Figure EV5. Scheme of the different outcomes resulting from Top1 activity at incorporated ribonucleotides.
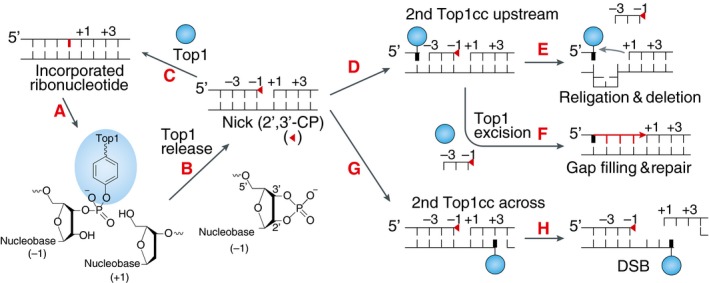
- Following ribonucleotide incorporation into the DNA (shown in red) during replication, a Top1cc forms at a ribonucleotide site.
- Top1cc is reversed by the nucleophilic attack of the 2′‐ribose hydroxyl, which generates a 2′,3′‐cyclic phosphate (2′,3′‐CP) end (red triangle) and releases catalytically active Top1.
- Top1 can then bind to the nick, enabling nucleophilic attack of the 5′‐ribose hydroxyl on 2′,3′‐CP and facilitating religation of DNA backbone independently of its enzymatic activity (Huang et al, 2015).
- Sequential Top1 cleavage on the same strand, upstream of the 2′,3′‐CP by the released Top1 or by another Top1 can lead to the loss of short DNA fragment containing 2′,3′‐CP and persistent Top1cc.
- Alternatively, endonucleolytic cleavage (not shown) or excision of the Top1cc by tyrosyl‐DNA phosphodiesterase 1 (TDP1) (Sparks & Burgers, 2015) followed by gap filling can repair the DNA while eliminating the ribonucleotide.
- When the sequential Top1 cleavage is on the opposite strand to the 2′,3′‐CP, a DSB is formed.
- Such Top1‐linked DSBs can cause replication stress and lead to recombinations. Adapted from Pommier et al (2016).
It is now well established that in the absence of RER, Top1‐initiated ribonucleotide removal results in 2–5 bp deletions in short tandem repeats (Nick McElhinny et al, 2010a; Clark et al, 2011; Kim et al, 2011; Potenski et al, 2014), presumably through a second Top1 cleaving event on the same DNA strand followed by strand slippage and faulty religation across the DNA gap (Huang et al, 2015; Sparks & Burgers, 2015) (Fig EV5D and E). When the ribonuclease activity of Top1 occurs away from tandem repeats, the resulting nicks (Fig 4D) could not effect strand slippage and would need to be repaired, potentially through DNA gap‐filling mechansims (Potenski et al, 2014; Sparks & Burgers, 2015; Fig EV5F). Left unrepaired, these Top1‐induced nicks could also lead to severe forms of DNA damage.
Here, we demonstrate that an alternative mechanism, where a second Top1 cleaving event on the opposite strand from the nick, induces a DSB with a covalently attached Top1 molecule (Figs 4E and EV5G and H). This configuration is prone to religate/recombine with a free 5′‐hydroxyl group (Fig 4F). Detailed analysis of mutation rates and specificity supports that Top1‐induced 2–5 bp deletions and Top1‐induced DSBs arise through distinct mechanisms (Fig EV2). We note that the 2–5 bp deletions occur at low frequency in vivo, and in vitro biochemical work suggests that only a fraction of the second Top1‐cleavage products leads to strand slippage and faulty religations that result in 2–5 bp deletions (Huang et al, 2015). On the other hand, we demonstrate here that multiple DSB species are generated from a single unrepaired ribonucleotide in vitro. Thus, it is plausible that Top1‐induced DSBs occur in vivo.
Our genetic analyses indicate that HR, the major DSB repair pathway in yeast, is critical for repairing Top1‐induced damage at ribonucleotide sites. During HR, 5′–3′ resection generates single‐strand DNA that is then coated by RPA. The Rad51 recombinase subsequently displaces RPA to form filaments, and Rad52 is required in this process. Both Rad51 and Rad52 form discrete microscopic foci that represent specialized DSB repair centers (Lisby et al, 2001, 2003; Miyazaki et al, 2004). We observed a higher percentage of cells with Rad52 foci in a rnh201Δ strain than in the WT strain, and further inactivation of TOP1 reversed this trend (Fig 1), consistent with the formation of DSBs at DNA nicks generated by Top1 ribonuclease activity at unrepaired single ribonucleotide sites. Accordingly, we found that inactivating RAD52 or RAD51 impaired the ability of the cell to cope with Top1‐induced damage at ribonucleotides (Figs 2 and 3).
Our findings are also consistent with the Top1‐induced elevated rates of recombination and chromosomal rearrangements in RNase H2‐null yeast strains (Potenski et al, 2014; Conover et al, 2015; Epshtein et al, 2016). However, multiple pathways of processing ribonucleotides on the leading and lagging DNA strands can lead to genome destablizing recombination‐initiating events, including DSBs. For example, as Top1 generates DNA nicks at ribonucleotide sites, it is possible that replication or transcription machineries encountering the Top1‐generated DNA nicks contribute to DSB formation in vivo. Recent reports also suggested that unresolved R‐loops could trigger Top1‐induced recombination events in RNase H2‐deficient yeasts (O'Connell et al, 2015), although the effect of incorporated ribonucleotides could not be ruled out as rnh201‐RED strains still showed elevated recombination rates (Epshtein et al, 2016). Here, we focused on Top1‐dependent DSBs initiated by ribonucleotide incorporation into nascent leading strand by Pol ε, which was shown to induce high recombination rates in a study using different mutator alleles of polymerase in RNase H2‐deficient yeast (Conover et al, 2015). Based on our experiments with the RNase H2 mutant lacking R‐loop removal activity [the rnh201‐RED strains (Chon et al, 2013)], we conclude that the unrepaired single genomic ribonucleotides are sufficient to induce Top1‐dependent DSBs (Figs 1, 2C, and EV1).
To further support our proposed mechanism (Fig 4), we analyzed the DNA covalently linked to Top1 from the pull‐down experiment, which showed that Top1 is more frequently covalently linked to the end of DNA in vivo in the absence of RER (Fig 6). Although our data from pol2‐M644G strains clearly implicate Top1‐induced DSBs in the leading strands, we cannot rule out that Top1 might induce DNA damage that requires HR in the lagging strands. This may be reflected in the slow growth observed for the haploid spore colonies formed in the pol1‐L868M rnh201Δ rad52Δ and pol3‐L612M rnh201Δ rad52Δ strains (Fig 2A). Additionally, while TOP1 deletion rescued the inviability of the pol2‐M644G rnh201Δ rad52Δ mutant, colonies formed for the pol2‐M644G rnh201Δ rad52Δ top1Δ strain were small and showed reduced growth rate, which could be attributed to unresolved DNA breaks, potentially resulting in genome rearrangements.
In keeping with our model, RNase H2 knockout mouse embryonic fibroblasts and cells from several embryonic tissues show increased levels of γH2AX (Hiller et al, 2012; Reijns et al, 2012). Furthermore, the most significantly up‐regulated genes in RNase H2 knockout murine cells are targets of p53 (Hiller et al, 2012; Reijns et al, 2012), which is known to be critical for multiple aspects of DSB repair. Importantly, the danger posed by Top1‐induced DNA damage at ribonucleotide sites does not appear to be limited to RNase H2‐deficient cells. Previous reports have shown that high levels of transcription also promote the Top1‐induced 2–5 bp deletions (Lippert et al, 2011; Takahashi et al, 2011; Cho et al, 2013). It is possible that DNA repair introduces ribonucleotides at highly transcribed regions and that abundance of Top1 at these regions [reviewed in (Pommier et al, 2016)] leads to Top1 acting with newly incorporated ribonucleotides before their removal by RNase H2‐dependent RER.
Reduction‐of‐function mutations in RNase H2 in humans lead to AGS, an inflammatory disease that affects primarily the brain and the skin, and is associated with by increased interferon α (Rabe, 2013; Reijns & Jackson, 2014). A recent report also linked mutations in RNase H2 to heightened risk of systemic lupus erythematosus (SLE) (Gunther et al, 2015), an autoimmune disease that is also characterized by increased interferon α. Fibroblast cells from SLE patients with RNase H2 mutations display more γH2AX foci and higher levels of p53 (Gunther et al, 2015), consistent with data from the RNase H2‐null mouse embryonic fibroblasts (Hiller et al, 2012; Reijns et al, 2012). Mutations in several other genes also lead to AGS, including TREX1, SAMHD1, ADAR1, and IFIH1/MDA5, all of which are involved in metabolism of nucleic acids or recognition of viral genomic fragments (Rabe, 2013; Reijns & Jackson, 2014; Gunther et al, 2015). It is believed that accumulation of endogenous nucleic acid species triggers innate immune responses. In the case of RNase H2, the molecular mechanism of how interferon α is up‐regulated is currently unknown. Based on our biochemical data and genetic analysis in yeast, investigation into whether Top1‐induced DSBs in the RNase H2‐deficient cells potentially contribute to the stimulation of the characteristic immune responses of AGS is warranted.
Materials and Methods
Yeast strains
Saccharomyces cerevisiae strains are isogenic derivatives of strain Δ|(‐2)|‐7B‐YUNI300 (MATa CAN1 his7‐2 leu2Δ::kanMX ura3Δ trp1‐289 ade2‐1 lys2‐ΔGG2899‐2900) (Pavlov et al, 2001). Strain genotypes are listed in Appendix Table S2. The URA3 reporter gene was introduced in either orientation 1 (OR1) or orientation 2 (OR2) at position AGP1 (Shcherbakova & Kunkel, 1999) by transformation of a PCR product containing URA3 and its endogenous promoter flanked by sequence targeting the reporter to AGP1. rad51Δ and rad52Δ variants were generated by deletion replacement of the endogenous genes via transformation with a product containing the nourseothricin‐resistance cassette (natMX4) amplified from pAG25 and flanked by 60 nucleotides of sequence homologous to the intergenic regions upstream and downstream of the RAD51 or RAD52 open reading frame. To generate rnh201‐RED strains, a plasmid containing Rnh201 C‐terminally tagged with 5×FLAG and flanked by upstream and downstream sequences was mutagenized using the Quikchange II site‐directed mutagenesis kit (Agilent Technologies). Following PCR amplification, this construct was transformed into yeast to replace RNH201. Gene replacement was confirmed by marker selection and PCR analysis. The presence of the P45D and Y219A mutations was confirmed by sequencing, and protein expression was confirmed by Western blotting using an anti‐FLAG M2 (mouse) antibody (Sigma F1804).
Construction of diploid strains heterogeneous for rad52::natMX4 was performed by deletion replacement of 1 copy of RAD52. Transformants that arose from homologous recombination were verified by PCR analysis and appropriate marker selection. Tetrad dissection was performed following a 3‐ to 4‐days incubation of diploid strains on sporulation medium (0.1% potassium acetate, 250 mg adenine, 2% agar) at 25°C. Strains expressing Rad52‐YFP were constructed by transformation with plasmid pWJ1344 (Lisby et al, 2001) and selected on synthetic complete (SC) medium lacking leucine (SC‐Leu).
Fluorescence microscopy
10 ml cultures of each strain were grown overnight at 25°C in selective medium (SC‐Leu). Cell pellets were washed once with 1× PBS and mounted on glass slides. Two independent experiments were performed, and between 9 and 15 images were captured for each strain (for a total of > 3,200 cells in which foci were scored for each). Images were captured on a Zeiss LSM510‐UV meta (Carl Zeiss Inc, Oberkochen, Germany) using a Plan‐Apochromat 63×/1.4 Oil DIC objective. The 514‐nm laser line from an argon laser at 6% power was used for excitation of the Rad52‐YFP, and a 530‐ to 600‐nm band pass emission filter was used to collect the images of the YFP with a pinhole setting of 1 Airy unit. Image processing and foci quantitation were performed using ImageJ v1.49p (http://imagej.nih.gov/ij/). Z‐sections were merged into one maximum intensity projection image where the “Analyze Particles” module of ImageJ was used to count thresholded features. The number of cells containing YFP foci was normalized to total number of cells counted.
Phenotype analyses
Strains were grown in YPDA medium (1% yeast extract, 2% bacto‐peptone, 250 mg/l adenine, 2% dextrose, 2% agar for plates) at 30°C. Doubling time (D t) values were calculated from cultures in the logarithmic phase of growth. The experiment was performed in triplicate; data are displayed as the mean D t ± SD. Spot assays were performed by plating 10‐fold serial dilutions of exponentially growing cells onto YPDA agar. Plates were incubated at 30°C and photographed after 2 days of growth. Two independent biological replicates of this test were performed, with a representative panel shown. Flow cytometry analysis of DNA content was performed as previously described (Williams et al, 2013). The percentage of cells in G1, S and G2/M was determined using data from two experiments using Modfit software (Verity Software House, Topsham, ME).
Immunoblotting
Whole cell extracts were prepared from exponentially growing cells as described (Williams et al, 2013). Actin was probed as a loading control. Gel electrophoresis and Western blotting for Rnr3 and Actin were performed as described (Tumbale et al, 2014). Blotting for Hug1 was performed using a custom‐made polyclonal rabbit antibody at a dilution of 1:500. The positive control is an extract prepared from wild‐type cells treated with 200 mM hydroxyurea (HU) for 3 h.
Generation of biochemical substrates
Custom‐synthesized oligonucleotides (IDT, Coralville, IA) were 3′‐labeled by terminal deoxynucleotidyl transferase (Invitrogen, Carlsbad, CA, USA) and [α‐32P] cordycepin‐5′‐triphosphate (PerkinElmer, Inc. Waltham, MA, USA). Quick Spin Oligo Columns (Roche Applied Science) removed residual [α‐32P] cordycepin‐5′‐triphosphate, and the 3′‐radiolabeled oligos were annealed to its unlabeled complementary strands at 1:1 ratio. The sequences of all the oligonucleotides used in the Top1 cleavage assays are listed in Appendix Table S3.
Top1 cleavage assays
Recombinant human Top1 was overexpressed and purified using a baculovirus expression system from Sf9 insect cells as described (Pourquier et al, 1999). The Top1 cleavage and reversal assays were carried out as described (Dexheimer & Pommier, 2008; Kim et al, 2011).
Analysis of DNA covalently linked with Top1 by immuno‐pull‐down
Half a liter of Saccharomyces cerevisiae cells with Top1 tagged with 5×FLAG‐epitope was cultured to mid‐log phase before harvest. All subsequent steps were carried out at 4°C. The pellets were immediately washed twice in denaturing buffer (50 mM HEPES, pH = 7.3, 150 mM NaCl, 1 mM EDTA, 1% Triton, 0.1% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, supplemented with protease inhibitor cocktail) followed by two washes in lysis buffer (50 mM HEPES, pH = 7.3, 150 mM NaCl, 1 mM EDTA, 1% Triton, 1 mM PMSF, supplemented with protease inhibitor cocktail). The pellets were then resuspended in lysis buffer and lysed on a bead beater in the presence of glass beads. The lysates were then clarified before sonication with a Bioruptor on medium–low setting. The sheared chromatins were clarified again and incubated with lysis buffer‐washed Sepharose slurry for 1 h followed by clarification again. Dynabeads were washed trice with PBST plus 0.5% BSA and then incubated with anti‐FLAG antibodies overnight before combining with the clarified chromatins for incubation overnight. The Dynabeads were then washed twice with lysis buffer, twice with lysis buffer plus 300 mM NaCl, once with wash buffer (10 mM Tris–HCl, pH = 8, 250 mM LiCl, 0.1% Nonidet P‐40, 1 mM EDTA) and finally twice in 1× TE. The Dynabeads were resuspended in 1× TE with 0.25% SDS supplemented with 500 μg/ml proteinase K and incubated at 55°C overnight. The resulting supernatant was then extracted by phenol:chloroform:isoamyl alcohol (25:24:1) solution followed by ethanol precipitation in the presence of glycogen. The DNA pellets were resuspended in 1× TE. Equal amounts of DNA (~100 ng) were treated with nuclease S1 (0.4 unit) or mock‐treated at 25°C for 1 h before inactivation of the enzymes at 70°C for 10 min. The resulting DNA samples were analyzed on a 1% agarose gel stained with SYBR Gold.
Author contributions
S‐yNH and JSW designed and conducted the experiments. MEA constructed yeast strains and determined mutational spectra. TAK and YP conceived and supervised the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Michael Lisby (University of Copenhagen) for the pWJ1344 plasmid expressing Rad52‐YFP. We acknowledge the Fluorescence Microscopy and Imaging Center and the Cytometry Center at NIEHS for microscopy and FACS analysis, respectively. We are grateful to Jeff Tucker for assistance with microscopy analysis and Scott Lujan and Grace Kissling for statistical expertise. We thank Martin Zofall and Laura Baranello of NCI for sharing advice and experience on the pull‐down assays. This work was supported by Project Z01 BC006161 to Y.P. from the Center for Cancer Research at National Cancer Institute and by Project Z01 ES065070 to T.A.K. from the Division of Intramural Research of the NIEHS, NIEHS.
The EMBO Journal (2017) 36: 361–373
References
- Allen‐Soltero S, Martinez SL, Putnam CD, Kolodner RD (2014) A Saccharomyces cerevisiae RNase H2 interaction network functions to suppress genome instability. Mol Cell Biol 34: 1521–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arana ME, Kerns RT, Wharey L, Gerrish KE, Bushel PR, Kunkel TA (2012) Transcriptional responses to loss of RNase H2 in Saccharomyces cerevisiae . DNA Repair (Amst) 11: 933–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban C, Ramakrishnan B, Sundaralingam M (1994) A single 2'‐hydroxyl group converts B‐DNA to A‐DNA. Crystal structure of the DNA‐RNA chimeric decamer duplex d(CCGGC)r(G)d(CCGG) with a novel intermolecular G‐C base‐paired quadruplet. J Mol Biol 236: 275–285 [DOI] [PubMed] [Google Scholar]
- Basrai MA, Velculescu VE, Kinzler KW, Hieter P (1999) NORF5/HUG1 is a component of the MEC1‐mediated checkpoint response to DNA damage and replication arrest in Saccharomyces cerevisiae . Mol Cell Biol 19: 7041–7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerritelli SM, Crouch RJ (2009) Ribonuclease H: the enzymes in eukaryotes. FEBS J 276: 1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerritelli SM, Crouch RJ (2016) The balancing act of ribonucleotides in DNA. Trends Biochem Sci 41: 434–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu HC, Koh KD, Evich M, Lesiak AL, Germann MW, Bongiorno A, Riedo E, Storici F (2014) RNA intrusions change DNA elastic properties and structure. Nanoscale 6: 10009–10017 [DOI] [PubMed] [Google Scholar]
- Cho JE, Kim N, Li YC, Jinks‐Robertson S (2013) Two distinct mechanisms of Topoisomerase 1‐dependent mutagenesis in yeast. DNA Repair (Amst) 12: 205–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JE, Huang SN, Burgers PM, Shuman S, Pommier Y, Jinks‐Robertson S (2016) Parallel analysis of ribonucleotide‐dependent deletions produced by yeast Top1 in vitro and in vivo. Nucleic Acids Res 44: 7714–7721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chon H, Sparks JL, Rychlik M, Nowotny M, Burgers PM, Crouch RJ, Cerritelli SM (2013) RNase H2 roles in genome integrity revealed by unlinking its activities. Nucleic Acids Res 41: 3130–3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AB, Lujan SA, Kissling GE, Kunkel TA (2011) Mismatch repair‐independent tandem repeat sequence instability resulting from ribonucleotide incorporation by DNA polymerase epsilon. DNA Repair (Amst) 10: 476–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover HN, Lujan SA, Chapman MJ, Cornelio DA, Sharif R, Williams JS, Clark AB, Camilo F, Kunkel TA, Argueso JL (2015) Stimulation of chromosomal rearrangements by ribonucleotides. Genetics 201: 951–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson MB, Katou Y, Keszthelyi A, Sing TL, Xia T, Ou J, Vaisica JA, Thevakumaran N, Marjavaara L, Myers CL, Chabes A, Shirahige K, Brown GW (2012) Endogenous DNA replication stress results in expansion of dNTP pools and a mutator phenotype. EMBO J 31: 895–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRose EF, Perera L, Murray MS, Kunkel TA, London RE (2012) Solution structure of the Dickerson DNA dodecamer containing a single ribonucleotide. Biochemistry 51: 2407–2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexheimer TS, Pommier Y (2008) DNA cleavage assay for the identification of topoisomerase I inhibitors. Nat Protoc 3: 1736–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epshtein A, Potenski C, Klein H (2016) Increased spontaneous recombination in RNase H2‐ deficient cells arises from multiple contiguous rNMPs and not from single rNMP residues incorporated by DNA polymerase epsilon. Microbial Cell 3: 248–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodgaonkar MM, Lazzaro F, Olivera‐Pimentel M, Artola‐Boran M, Cejka P, Reijns MA, Jackson AP, Plevani P, Muzi‐Falconi M, Jiricny J (2013) Ribonucleotides misincorporated into DNA act as strand‐discrimination signals in eukaryotic mismatch repair. Mol Cell 50: 323–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther C, Kind B, Reijns MA, Berndt N, Martinez‐Bueno M, Wolf C, Tungler V, Chara O, Lee YA, Hubner N, Bicknell L, Blum S, Krug C, Schmidt F, Kretschmer S, Koss S, Astell KR, Ramantani G, Bauerfeind A, Morris DL et al (2015) Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J Clin Invest 125: 413–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller B, Achleitner M, Glage S, Naumann R, Behrendt R, Roers A (2012) Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J Exp Med 209: 1419–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SY, Murai J, Dalla Rosa I, Dexheimer TS, Naumova A, Gmeiner WH, Pommier Y (2013) TDP1 repairs nuclear and mitochondrial DNA damage induced by chain‐terminating anticancer and antiviral nucleoside analogs. Nucleic Acids Res 41: 7793–7803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SY, Ghosh S, Pommier Y (2015) Topoisomerase I alone is sufficient to produce short DNA deletions and can also reverse nicks at ribonucleotide sites. J Biol Chem 290: 14068–14076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaxel C, Capranico G, Kerrigan D, Kohn KW, Pommier Y (1991) Effect of local DNA sequence on topoisomerase I cleavage in the presence or absence of camptothecin. J Biol Chem 266: 20418–20423 [PubMed] [Google Scholar]
- Kim N, Huang S‐yN, Williams JS, Li YC, Clark AB, Cho J‐E, Kunkel TA, Pommier Y, Jinks‐Robertson S (2011) Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science 332: 1561–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh BO, Symington LS (2004) Recombination proteins in yeast. Annu Rev Genet 38: 233–271 [DOI] [PubMed] [Google Scholar]
- Kumar D, Viberg J, Nilsson AK, Chabes A (2010) Highly mutagenic and severely imbalanced dNTP pools can escape detection by the S‐phase checkpoint. Nucleic Acids Res 38: 3975–3983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaro F, Novarina D, Amara F, Watt DL, Stone JE, Costanzo V, Burgers PM, Kunkel TA, Plevani P, Muzi‐Falconi M (2012) RNase H and postreplication repair protect cells from ribonucleotides incorporated in DNA. Mol Cell 45: 99–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Breaker RR (1999) Kinetics of RNA degradation by specific base catalysis of transesterification involving the 2′‐hydroxyl group. J Am Chem Soc 121: 5326–5372 [Google Scholar]
- Lippert MJ, Kim N, Cho J‐E, Larson RP, Schoenly NE, O'Shea SH, Jinks‐Robertson S (2011) Role for topoisomerase 1 in transcription‐associated mutagenesis in yeast. Proc Natl Acad Sci USA 108: 698–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Rothstein R, Mortensen UH (2001) Rad52 forms DNA repair and recombination centers during S phase. Proc Natl Acad Sci USA 98: 8276–8282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Mortensen UH, Rothstein R (2003) Colocalization of multiple DNA double‐strand breaks at a single Rad52 repair centre. Nat Cell Biol 5: 572–577 [DOI] [PubMed] [Google Scholar]
- Lujan SA, Williams JS, Clausen AR, Clark AB, Kunkel TA (2013) Ribonucleotides are signals for mismatch repair of leading‐strand replication errors. Mol Cell 50: 437–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Haber JE (2014) Sources of DNA double‐strand breaks and models of recombinational DNA repair. Cold Spring Harbor Perspect Biol 6: a016428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T, Bressan DA, Shinohara M, Haber JE, Shinohara A (2004) In vivo assembly and disassembly of Rad51 and Rad52 complexes during double‐strand break repair. EMBO J 23: 939–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Gordenin DA, Stith CM, Burgers PM, Kunkel TA (2008) Division of labor at the eukaryotic replication fork. Mol Cell 30: 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Kumar D, Clark AB, Watt DL, Watts BE, Lundström E‐B, Johansson E, Chabes A, Kunkel TA (2010a) Genome instability due to ribonucleotide incorporation into DNA. Nat Chem Biol 6: 774–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Watts B, Kumar D, Watt DL, Lundström E‐B, Burgers PMJ, Johansson E, Chabes A, Kunkel TA (2010b) Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc Natl Acad Sci USA 107: 4949–4954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell K, Jinks‐Robertson S, Petes TD (2015) Elevated genome‐wide instability in yeast mutants lacking RNase H activity. Genetics 201: 963–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker LH, Champoux JJ (1993) Analysis of the biased distribution of topoisomerase I break sites on replicating simian virus 40 DNA. J Mol Biol 231: 6–18 [DOI] [PubMed] [Google Scholar]
- Pavlov YI, Shcherbakova PV, Kunkel TA (2001) In vivo consequences of putative active site mutations in yeast DNA polymerases alpha, epsilon, delta, and zeta. Genetics 159: 47–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, Sun Y, Huang SN, Nitiss JL (2016) Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol 17: 703–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter SE, Champoux JJ (1989) Mapping in vivo topoisomerase I sites on simian virus 40 DNA: asymmetric distribution of sites on replicating molecules. Mol Cell Biol 9: 541–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenski CJ, Klein HL (2014) How the misincorporation of ribonucleotides into genomic DNA can be both harmful and helpful to cells. Nucleic Acids Res 42: 10226–10234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenski CJ, Niu H, Sung P, Klein HL (2014) Avoidance of ribonucleotide‐induced mutations by RNase H2 and Srs2‐Exo1 mechanisms. Nature 511: 251–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourquier P, Pilon A, Kohlhagen G, Mazumder A, Sharma A, Pommier Y (1997) Trapping of mammalian topoisomerase I and recombinations induced by damaged DNA containing nicks or gaps: importance of DNA end phosphorylation and camptothecin effects. J Biol Chem 272: 26441–26447 [DOI] [PubMed] [Google Scholar]
- Pourquier P, Ueng L‐M, Fertala J, Wang D, Park H‐J, Essigmann JM, Bjornsti M‐A, Pommier Y (1999) Induction of reversible complexes between eukaryotic DNA topoisomerase I and DNA‐containing oxidative base damages. J Biol Chem 274: 8516–8523 [DOI] [PubMed] [Google Scholar]
- Rabe B (2013) Aicardi‐Goutieres syndrome: clues from the RNase H2 knock‐out mouse. J Mol Med 91: 1235–1240 [DOI] [PubMed] [Google Scholar]
- Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, Devenney PS, Sexton D, Grimes G, Holt IJ, Hill RE, Taylor MS, Lawson KA, Dorin JR, Jackson AP (2012) Enzymatic removal of ribonucleotides from DNA is essential for Mammalian genome integrity and development. Cell 149: 1008–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns MA, Jackson AP (2014) Ribonuclease H2 in health and disease. Biochem Soc Trans 42: 717–725 [DOI] [PubMed] [Google Scholar]
- Rydberg B, Game J (2002) Excision of misincorporated ribonucleotides in DNA by RNase H (type 2) and FEN‐1 in cell‐free extracts. Proc Natl Acad Sci USA 99: 16654–16659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi J, Shuman S (1997) Site‐specific ribonuclease activity of eukaryotic DNA topoisomerase I. Mol Cell 1: 89–97 [DOI] [PubMed] [Google Scholar]
- Shcherbakova PV, Kunkel TA (1999) Mutator phenotypes conferred by MLH1 overexpression and by heterozygosity for mlh1 mutations. Mol Cell Biol 19: 3177–3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks JL, Chon H, Cerritelli SM, Kunkel TA, Johansson E, Crouch RJ, Burgers PM (2012) RNase H2‐initiated ribonucleotide excision repair. Mol Cell 47: 980–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks JL, Burgers PM (2015) Error‐free and mutagenic processing of topoisomerase 1‐provoked damage at genomic ribonucleotides. EMBO J 34: 1259–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Burguiere‐Slezak G, Van der Kemp PA, Boiteux S (2011) Topoisomerase 1 provokes the formation of short deletions in repeated sequences upon high transcription in Saccharomyces cerevisiae . Proc Natl Acad Sci USA 108: 692–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traut TW (1994) Physiological concentrations of purines and pyrimidines. Mol Cell Biochem 140: 1–22 [DOI] [PubMed] [Google Scholar]
- Tumbale P, Williams JS, Schellenberg MJ, Kunkel TA, Williams RS (2014) Aprataxin resolves adenylated RNA‐DNA junctions to maintain genome integrity. Nature 506: 111–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Amon JD, Koshland D, Vuica‐Ross M (2011) RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol Cell 44: 978–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Smith DJ, Marjavaara L, Lujan SA, Chabes A, Kunkel TA (2013) Topoisomerase 1‐mediated removal of ribonucleotides from nascent leading‐strand DNA. Mol Cell 49: 1010–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Kunkel TA (2014) Ribonucleotides in DNA: origins, repair and consequences. DNA Repair (Amst) 19: 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Clausen AR, Lujan SA, Marjavaara L, Clark AB, Burgers PM, Chabes A, Kunkel TA (2015) Evidence that processing of ribonucleotides in DNA by topoisomerase 1 is leading‐strand specific. Nat Struct Mol Biol 22: 291–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Lujan SA, Kunkel TA (2016) Processing ribonucleotides incorporated during eukaryotic DNA replication. Nat Rev Mol Cell Biol 17: 350–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File