

Abstract
MicroRNA (miRNA)‐guided mRNA repression, mediated by the miRNA‐induced silencing complex (miRISC), is an important component of post‐transcriptional gene silencing. However, how miRISC identifies the target mRNA in vivo is not well understood. Here, we show that the nucleoporin Nup358 plays an important role in this process. Nup358 localizes to the nuclear pore complex and to the cytoplasmic annulate lamellae (AL), and these structures dynamically associate with two mRNP granules: processing bodies (P bodies) and stress granules (SGs). Nup358 depletion disrupts P bodies and concomitantly impairs the miRNA pathway. Furthermore, Nup358 interacts with AGO and GW182 proteins and promotes the association of target mRNA with miRISC. A well‐characterized SUMO‐interacting motif (SIM) in Nup358 is sufficient for Nup358 to directly bind to AGO proteins. Moreover, AGO and PIWI proteins interact with SIMs derived from other SUMO‐binding proteins. Our study indicates that Nup358–AGO interaction is important for miRNA‐mediated gene silencing and identifies SIM as a new interacting motif for the AGO family of proteins. The findings also support a model wherein the coupling of miRISC with the target mRNA could occur at AL, specialized domains within the ER, and at the nuclear envelope.
Keywords: annulate lamellae, Argonaute, miRNA, nucleoporin, Nup358
Subject Categories: RNA Biology
Introduction
Regulation of gene expression at the translational level is shown to be involved in diverse cellular processes and has emerged as an area of intense investigation. Small non‐coding RNAs, particularly microRNAs (miRNAs), appear to significantly contribute to this layer of regulation. miRNAs, which are of ~22 nucleotides length, suppress translation of mRNAs that possess partial or complete sequence complementarity, mostly at the 3′‐untranslated region (UTR) 1. Predictions based on sequence analysis have indicated that miRNAs could target over 50% of human protein‐coding genes 2. The genes encoding miRNAs are generally transcribed by RNA polymerase II to produce primary miRNAs (pri‐miRNAs), which are processed into precursor miRNAs (pre‐miRNAs) by the microprocessor complex containing Drosha and DGCR8 in the nucleus 3. The pre‐miRNA, in complex with exportin‐5 and RanGTP, is exported through the nuclear pore complex (NPC) into the cytoplasm, where it is further processed by Dicer into double‐stranded miRNA. One of the strands stably associates with Argonaute (AGO) protein to generate a functional miRISC. Humans have four AGO isoforms: AGO1–AGO4 4. A glycine–tryptophan (GW)‐rich protein, GW182 (also called TNRC6), interacts directly with AGO proteins and is essential for miRISC‐mediated translational repression and/or degradation of target mRNAs through recruitment of deadenylation and decapping complexes. The suppression and/or degradation of target mRNAs is believed to occur in the cytoplasmic foci termed as “processing bodies (P bodies)” 5, 6. As downstream effectors, GW182 family of proteins directly bind to AGO proteins through conserved GW/WG sequence. This motif is also present in other AGO‐interacting proteins and is referred to as “AGO hook” 7, 8.
Argonaute proteins have a highly conserved role in RNA silencing 4. AGO family is divided into two clades based on their functions: AGO and PIWI subfamilies. As described earlier, AGO subfamily proteins are present ubiquitously and are involved in small interfering RNA (siRNA)‐mediated cleavage of mRNA or miRNA‐mediated suppression of mRNA translation 4. However, the members of PIWI subclade are mostly present in germ cells and are involved in silencing transposons, maintenance of genome integrity, and gametogenesis 9.
Although the subcellular location where the loading of miRNAs to AGO proteins (miRISC formation) and association of miRISC with the target mRNAs occur is not well understood, recent studies have indicated a role for endoplasmic reticulum (ER) in these processes. It was shown that Arabidopsis AGO1 associates peripherally with ER, and miRISC could inhibit the translation of target mRNAs on the ER 10. Another study indicated that rough ER could be the site for miRNA and siRNA loading to AGO proteins and translational regulation of target mRNAs 11. A central question that is yet unresolved is how miRISC identifies the target mRNAs in vivo. Although a sorting mechanism could be envisaged that couples the RNAs exported from the nucleus with the miRISC, there is no available evidence for the existence of such machinery.
The nuclear envelope (NE) that encircles the nucleus is an extension of ER and is made up of a double‐layered membrane. Nuclear pore complexes (NPCs) form the molecular gates on the NE, through which the transport of macromolecules between the nucleus and the cytoplasm occurs 12. The protein components of NPCs are termed as nucleoporins (Nups), and each mammalian NPC contains around 30 different nucleoporins in multiple copies 13. The spatial distribution of individual nucleoporins within the NPC structure could vary 14. Although the nucleoporins are fundamentally expected to be involved in the regulation of nucleo‐cytoplasmic transport, several of them are shown to have multiple other functions 15, 16.
Apart from the localization to NPCs on the NE, some nucleoporins also accumulate in annulate lamellae (AL), which are stacked ER membrane‐containing pore‐like structures 17, 18, 19. These AL pore complexes show gross structural similarities to that of NPCs at electron microscopy level 17, 19. Although AL structures have been extensively analyzed in male and female gametes, other proliferating non‐germ cells also possess varying quantities of AL 17, 19. The functional role for these structures in any cellular processes is unclear. Previous studies have implicated AL as the storehouse of excess nucleoporins to be supplied as and when the cell requires, for example, to meet the increasing demand for nucleoporins in the assembly of new NPCs during initial zygotic cell divisions. However, there is experimental evidence arguing against such a function 20. Consistent with being a part of the endoplasmic reticulum, electron microscopic studies also have suggested AL to often have RNA and ribosomes in their close vicinity 17, 19. Previous studies have shown that AL associate with MEX‐3‐positive RNP granules in the arrested Caenorhabditis elegans oocytes and that several nucleoporins play a role in the complete assembly of these RNP granules 21. However, whether AL associate with other mRNP granules and play a role in their functions is not known.
Nup358 is a nucleoporin that localizes to the cytoplasmic side of the NPC and has been implicated in several functions 22, 23, 24, 25, 26, 27, 28, 29, 30, 31. Depletion of Nup358 does not appear to grossly affect transport of macromolecules across the NE, although some studies suggest a role for this nucleoporin in specific receptor‐ and cargo‐dependent transport 32, 33, 34, 35, 36. Nup358 has been identified as a small ubiquitin‐like modifier (SUMO) E3 ligase 28 and is shown to mediate in vivo SUMOylation of substrates such as topoisomerase II 37, borealin 38, and Ran 39.
SUMO is a small protein that gets covalently conjugated to target proteins through specific lysine residues and modulates their function 40, 41. SUMO pathway is shown to be involved in multiple cellular processes 42. In humans, there are four SUMO isoforms: SUMO1–4. In addition to the covalent interaction, SUMO associates with other proteins through directly binding to specific SUMO‐interacting motif (SIM), which is characterized by a conserved set of hydrophobic amino acids 40, 41. Multiple SIMs have been identified in many SUMO‐interacting proteins and functionally validated 43. The presence of a stretch of negatively charged amino acids adjacent to the N‐ or C‐terminus of the hydrophobic sequence (SIM) is shown to contribute to the strength, orientation, and paralog specificity of SUMO binding 42.
SUMO conjugation to the substrate lysine requires concerted action of SUMO‐specific E1 (Aos1/Uba2 heterodimer), E2 (Ubc9), and multiple E3 ligases 42. RanGTPase‐activating protein (RanGAP) is the first SUMO substrate identified 44, 45, 46. SUMO gets attached to lysine 524 of human RanGAP, which targets it to the NPC through binding to Nup358. Structural and functional analyses showed that SUMO‐RanGAP interacts with Nup358 through a region having internal repeats (IR) harboring two SIMs 47, 48. Nup358‐IR also possesses the SUMO E3 ligase activity 28. Each of the two repeats, IR1 and IR2, has a SIM‐binding and a Ubc9‐binding domain 49, 50. However, studies have shown that IR1 (SIM1) is involved in SUMO~RanGAP1 interaction, which is stabilized by Ubc9 as it directly binds to IR1, RanGAP1, and SUMO 47, 51. In vitro studies have illustrated that SUMO‐RanGAP and Ubc9 form a stable complex with IR1, and not with IR2 51, 52, 53. Although no conclusive evidence exists, it is believed that SUMO‐dependent binding of RanGAP1 to Nup358 would enhance RanGAP's ability to activate the hydrolysis of GTP on Ran in the export complex 54, 55. Endogenously, bulk of RanGAP is SUMO‐modified and has been shown to associate with Nup358 throughout the cell cycle 25, 56.
Here, we show that Nup358‐positive AL structures dynamically associate with cytoplasmic mRNPs such as P bodies and stress granules (SGs). Furthermore, our study reveals interaction between Nup358 and components of miRISC, AGO, and GW182. The results suggest an unanticipated function for this nucleoporin in miRNA‐mediated gene silencing by aiding in the coupling of miRISC with target mRNAs. The results also indicate a possible role for AL in the miRISC–mRNA coupling process. Interestingly, characterization of Nup358–AGO interaction led to identification of SIM as a new conserved interaction motif for AGO family of proteins. Our data also suggest that Nup358–AGO interaction is essential for miRNA‐mediated suppression of mRNA translation.
Results
Nup358‐positive AL structures and NE associate with SGs and P bodies
Localization of endogenous Nup358 in HeLa cells using a specific antibody showed that, in addition to NE, this nucleoporin is present in cytoplasmic punctate structures along with RanGAP1, a known interacting partner of Nup358 (Fig 1A) 45, 46. To validate whether the cytoplasmic Nup358‐positive structures represented AL, we immunostained for other nucleoporins and found these entities to contain Nup214 (Fig 1A) and Nup62 (Fig EV1A), but not Nup153 (Fig EV1A). Moreover, these Nup358‐positive structures were associated with ER, particularly marking distinct domains within the ER (Fig EV1B). Co‐localization with a set of nucleoporins and association with ER indicated that Nup358‐positive cytoplasmic structures represented the previously characterized AL 57, 58. Exogenously expressed GFP‐tagged Nup358 (GFP‐Nup358) also accumulated in AL, as confirmed by its co‐localization with AL‐specific nucleoporins such as Nup214 and Nup62 (Fig EV1C).
Figure 1. Nup358‐positive AL structures associate with P bodies and SGs, and Nup358 depletion leads to impairment of P body assembly.
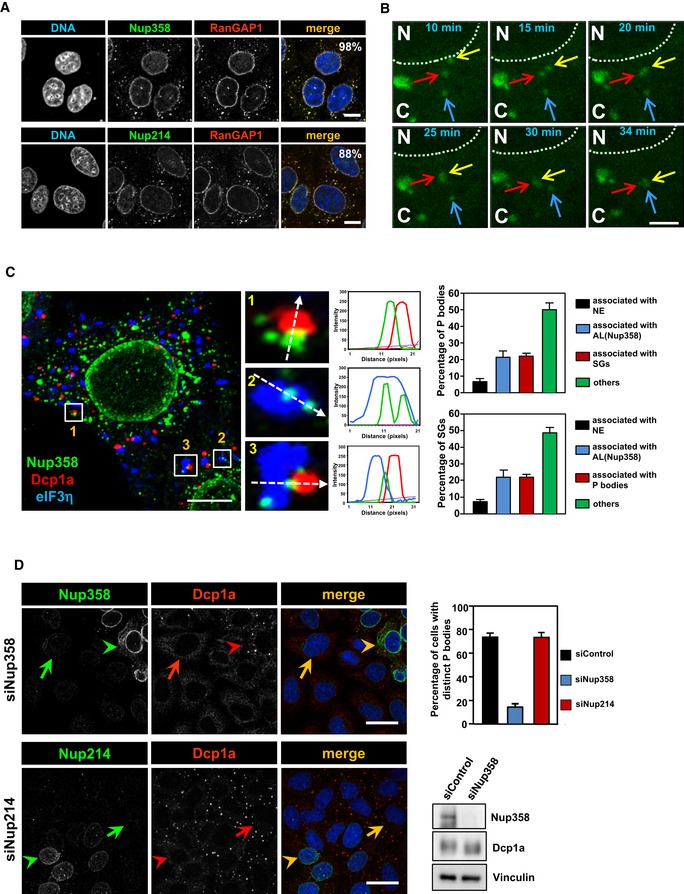
- Confocal microscopic image showing HeLa cells immunostained for Nup358 (green, upper panel) or Nup214 (green, lower panel) and RanGAP1 (red) using specific antibodies. DNA was stained with Hoechst 33342 (blue). Scale bars, 10 μm.
- COS‐7 cells were transfected with GFP‐Nup358 and one of the cells was subjected to live imaging using confocal microscopy, and the frames at the indicated time points have been provided. Arrows denoted by different colors indicate individual AL structures. Note that one of the AL structures (marked by yellow arrow) originated from the NE (dotted line) and fused with another AL structure (marked by red arrow), which later fused with a different AL structure (marked by blue arrow). N, nucleus; C, cytoplasm. Scale bar, 5 μm.
- Maximum‐intensity projection confocal image of a sodium arsenite‐treated HeLa cell immunostained for endogenous Nup358 (green), P bodies (red, Dcp1a as a marker), and SGs (blue, eIF3η as a marker) using specific antibodies. Scale bar, 10 μm. The histograms represent fluorescence intensity profile along the dotted arrows. Adjacent graph represents quantitative data showing percentage of P bodies (top) or SGs (bottom) associated with nuclear envelope (NE), Nup358‐positive AL, with each other or unassociated with any of the other mentioned structures (others). Data are presented as mean ± SD (n = 3).
- HeLa cells were transfected with Nup358 siRNA (siNup358) or Nup214 siRNA (siNup214). Cells were fixed and stained for endogenous Nup358 (green, upper panel) or Nup214 (green, lower panel) and endogenous P body marker (Dcp1a, red). Graph represents the quantitative data as mean ± SD. The data were obtained from three independent experiments, and in each experiment, 100 cells were counted from different fields for the presence of intact microscopically distinct P bodies and expressed as percentage. Western blots (WB) indicate the extent of Nup358 depletion and the level of Dcp1a. Arrow indicates Nup358/Nup214 depleted cell and arrow head shows non‐depleted cell. Scale bar, 10 μm.
Figure EV1. Endogenous and overexpressed Nup358‐positive cytoplasmic puncta represent AL .
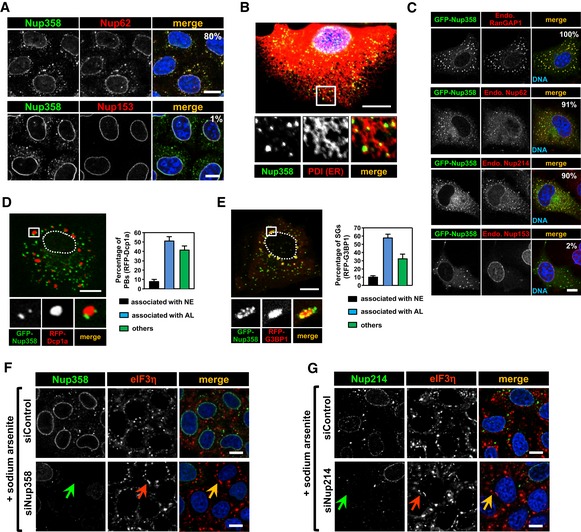
- HeLa cells were immunostained for Nup62 (red, upper panel) or Nup153 (red, lower panel) along with Nup358 (green) using specific antibodies. DNA was stained with Hoechst 33342 (blue, merge). Percentages of Nup358‐positive cytoplasmic puncta co‐localizing with Nup62 (upper panel) and Nup358 with Nup153 (lower panel) are indicated. Scale bars, 10 μm.
- HeLa cells were fixed and stained for endogenous Nup358 (green) and PDI (red, as an ER marker). Scale bar, 10 μm.
- COS‐7 cells were transfected with GFP‐Nup358 (green) and stained for endogenous RanGAP1, Nup214, Nup62, or Nup153 (red) using specific antibodies. DNA is shown in blue (merge). Percentages of GFP‐Nup358‐positive cytoplasmic puncta co‐localizing with respective proteins are indicated. Scale bar, 10 μm.
- COS‐7 cells expressing GFP‐Nup358 (green) and RFP‐Dcp1 (red) were fixed and analyzed for their relative distribution within the cytoplasm. Often GFP‐Nup358‐positive AL structures are in close proximity/associated with P bodies (RFP‐Dcp1a). The region marked by square is magnified (inset). Dotted line indicates nuclear periphery. Graph represents quantitative data showing percent of P bodies (RFP‐Dcp1a) associated with nuclear envelope (NE), GFP‐Nup358‐positive AL, or unassociated with either of the structures (free). Scale bar, 10 μm. Data are presented as mean ± SD (n = 3).
- COS‐7 cells expressing GFP‐Nup358 (green) and RFP‐G3BP1 (red) were fixed and analyzed by fluorescence microscopy. Often GFP‐Nup358‐positive AL structures are in close proximity/associated with SGs induced by RFP‐G3BP1 expression. The region marked by square is magnified (inset). Dotted line indicates nuclear periphery. Graph represents quantitative data showing percent of SGs (RFP‐G3BP1) associated with nuclear envelope (NE), GFP‐Nup358‐positive AL, or unassociated with either of the structures (free). Scale bar, 10 μm. Data are presented as mean ± SD (n = 3).
- HeLa cells were transfected with control siRNA (siControl) or siRNA against Nup358 (siNup358). Cells were treated with 0.5 mM sodium arsenite for 30 min prior to fixation and stained for endogenous Nup358 (green) and eIF3η (red) as a SG marker using specific antibodies. DNA was stained with Hoechst 33342 (merge, blue). Arrows indicate a Nup358‐depleted cell. Scale bars, 10 μm.
- HeLa cells were transfected with control siRNA (siControl) or Nup214 siRNA (siNup214). Cells were treated as indicated in (A) and were fixed and stained for endogenous Nup358 (green) and a SG marker (eIF3η, red). DNA was stained with Hoechst 33342 (merge, blue). Arrows indicate a Nup214‐depleted cell. Scale bars, 10 μm.
The nature and origin of AL have been unclear, and to monitor these, we analyzed the dynamics of GFP‐Nup358‐labeled AL using live cell imaging (Movie EV1). We observed that AL were highly dynamic and often underwent homotypic fusion with neighboring AL structures. Interestingly, we noticed that some AL structures were budding off from the NE and fusing with the pre‐existing cytoplasmic AL (Fig 1B and Movie EV2). These results suggested that cytoplasmic AL could originate from NE and are extensively dynamic entities.
Further, we sought to investigate the distribution of AL in relation to other cytoplasmic structures. Interestingly, we found that two cytoplasmic messenger ribonucleoprotein (mRNP) granules, namely SGs and P bodies, were often associated with or present juxtaposed to AL (Fig 1C). We subjected HeLa cells to oxidative stress through sodium arsenite treatment to induce SGs 59 and immunostained for endogenous Nup358, eIF3η (SG marker), and Dcp1a (P body marker). P bodies and SGs were found often juxtaposed to each other in the cytoplasm as previously reported 59. Interestingly, we observed that many individual Nup358‐positive AL structures were present adjacent to SGs or P bodies, and in some cases, all three structures appeared to physically associate with each other (Fig 1C). Quantitative analysis suggested that ~20% of P bodies were associated with either Nup358‐positive AL or SGs, whereas ~50% of them were found to be associated with neither AL nor SGs, and ~6% were associated with the NE (Fig 1C). Similarly, ~20% of SGs were associated with either Nup358‐positive AL or P bodies, whereas ~50% of them were found to be associated with neither AL nor P bodies, and ~10% were associated with the NE (Fig 1C). In unstressed cells, ~16% of endogenous P bodies associated with Nup358‐positive AL structures. The physical association was much more striking when GFP‐Nup358 was exogenously expressed along with RFP‐Dcp1a (P body marker) or RFP‐G3BP1 (SG marker) (Fig EV1D and E). Under this condition, ~47% of P bodies associated with Nup358‐positive AL, whereas < 10% associated with the NE (Fig EV1D). Similarly, ~58% of SGs were present juxtaposed to Nup358‐positive AL and ~10% were associated with NE (Fig EV1E). Live cell imaging, interestingly, indicated a dynamic interplay between the two mRNP granules and Nup358‐positive AL/NE (Movies EV3 and EV4).
Depletion of Nup358 disrupts P body formation
The dynamic association of Nup358‐positive AL/NE with SGs and P bodies prompted us to investigate whether any functional link exists between these entities. Toward this, we tested whether siRNA‐mediated Nup358 depletion caused any effect on the assembly of mRNP granules. Removal of Nup358 did not have any gross effect on SG assembly (assessed by SG‐specific marker, eIF3η) as compared to control cells (Fig EV1F). Neither did depletion of Nup214, another nucleoporin present on the cytoplasmic face of NPC and AL, show any effect on SG assembly (Fig EV1G). Interestingly, knockdown of Nup358, but not Nup214, led to dramatic impairment of P body assembly as assessed by Dcp1a staining (Fig 1D). However, the levels of Dcp1a were comparable between control and Nup358 knockdown cells (Fig 1D). These results demonstrated a specific requirement for Nup358 in P body formation, and possibly in its function.
Nup358 is required for miRNA‐mediated translation suppression
Previous studies have shown that mRNAs suppressed by miRISC localize to P bodies 5, 6, and disturbances in miRNA pathway lead to disruption of microscopically visible distinct P body structures 60. We sought to find out whether Nup358 depletion affected the miRNA pathway. As let‐7a is one of the abundant miRNAs expressed in HeLa cells, a Renilla luciferase (RL) reporter construct that expresses RL mRNAs containing three imperfect let‐7a binding sites in its 3′‐UTR (RL‐3xBulge) was used to monitor let‐7a‐mediated translation suppression in HeLa cells 61. Compared to control Renilla luciferase (RL‐control) mRNAs that did not have any let‐7a binding site, RL‐3xBulge generally showed ~60% suppression (Fig 2B). We measured the RL activity in cells depleted of Dicer, Nup214, or Nup358 (Fig 2A) and found that similar to Dicer knockdown, Nup358 depletion caused significant reversal of miRNA‐mediated suppression (Fig 2B). Cells with Nup214 knockdown, however, showed no significant change in the reporter activity as compared to control siRNA‐treated cells (Fig 2B). These results indicated a specific role for Nup358 in miRNA function.
Figure 2. Nup358 is required for miRNA function.
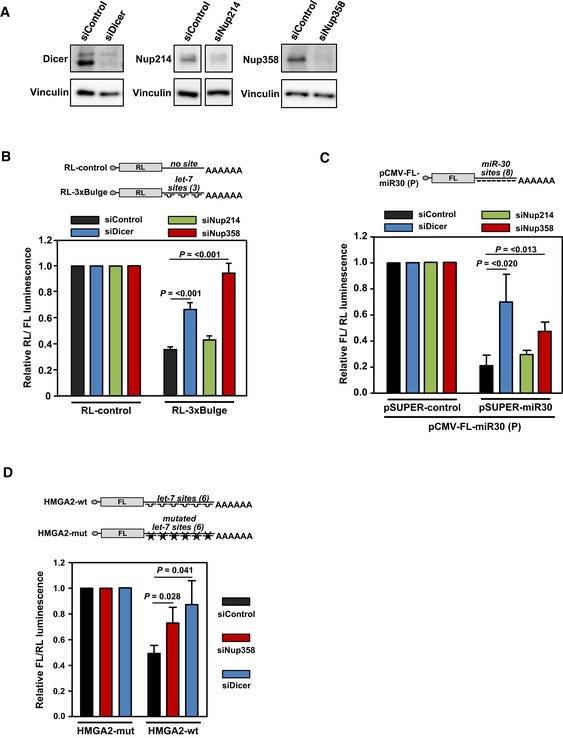
- Western analysis of HeLa cells, treated with control (siControl), Dicer (siDicer), Nup214 (siNup214), or Nup358 (siNup358) siRNA, for assessing the extent of protein depletion using indicated antibodies. Vinculin was used as loading control.
- HeLa cells were initially transfected with indicated siRNAs, followed by Renilla luciferase (RL) reporter constructs: RL‐control (no let‐7a binding site in the 3′‐UTR) or RL‐3xBulge (3 imperfect let‐7a binding sites in the 3′‐UTR). Firefly luciferase (FL) was co‐transfected to serve as internal control. RL/FL luminescence ratio was calculated. Data are presented as mean ± SD (n = 3), P‐values were calculated using Student's t‐test.
- HEK293T cells were transfected with indicated siRNAs, followed by pCMV‐FL‐miR30 (P) reporter with either pSUPER‐control or pSUPER‐miR30 constructs. RL was co‐transfected as internal control. FL/RL luminescence ratio was calculated. Data are presented as mean ± SD (n = 3), P‐values were calculated using Student's t‐test.
- HeLa cells were initially transfected with control (siControl), Dicer (siDicer), or Nup358 (siNup358)‐specific siRNAs, followed by FL constructs containing wild‐type (HMGA2‐wt) or mutant (HMGA2‐mut) HMGA2 3′‐UTR, along with RL as internal control. Graph was plotted using data from three independent experiments. FL/RL luminescence ratio was calculated. Data are presented as mean ± SD (n = 3), P‐values were calculated using Student's t‐test.
To test the generality of Nup358 function in miRNA pathway, we utilized another miRNA reporter system in HEK293T cells, involving firefly luciferase (FL) that contains eight miR‐30a perfect binding sites (sequence with complete complementarity to miR‐30a) in the 3′‐UTR 62. Nup358 depletion also significantly impaired miR‐30a activity (Fig 2C). Additionally, Nup358 was required for let‐7‐mediated suppression of FL mRNAs engineered to contain the 3′‐UTR of HMGA2 that harbors multiple functional let‐7 binding sites (Fig 2D) 63. Moreover, knockdown of Nup358 using three independent siRNAs targeted to different regions of Nup358 gene led to significant de‐repression of RL‐3xBulge reporter mRNA (Fig EV2A). When different concentrations of siRNAs were used to deplete Nup358 to varying levels, the de‐repression occurred in a dose‐dependent manner (Fig EV2B). Ectopic expression of GFP‐Nup358 in HeLa cells enhanced miR‐30‐mediated suppression of FL‐reporter mRNA as compared to GFP‐control (Fig EV2C). Moreover, exogenous expression of Nup358 also rescued the de‐repression caused by Nup358 knockdown (Fig EV2D). Taken together, these results suggested that Nup358 plays an important role in miRNA pathway.
Figure EV2. Nup358 depletion impairs and overexpression enhances miRNA‐mediated mRNA suppression.
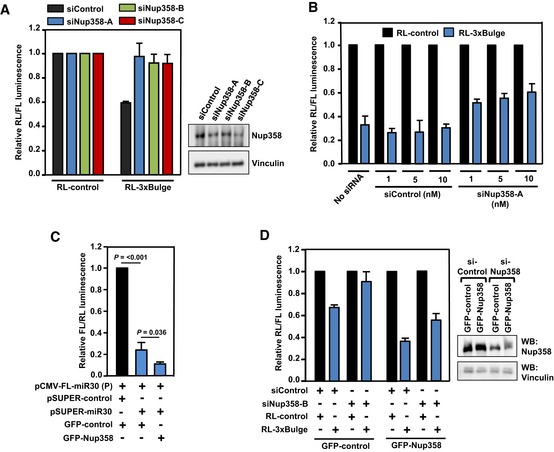
- HeLa cells were transfected with siRNAs targeted against different regions of Nup358 transcript (siNup358‐A, siNup358‐B, or siNup358‐C), and subsequently with indicated RL reporter constructs along with FL as internal control. Left panel: RL/FL luminescence ratio was calculated. Data are presented as mean ±SD (n = 3). Right panel: Western analysis of lysates prepared from cells transfected with three different siRNAs as indicated, for monitoring the extent of Nup358 depletion. Vinculin was used as loading control. Nup358‐A was used for depleting Nup358 (as siNup358) in all the experiments in the study, except where mentioned otherwise.
- HeLa cells were initially transfected with no (no siRNA), or varying concentrations of control (siControl) or Nup358‐specific (siNup358) siRNA in different concentrations (in nM) as indicated. Subsequently, the cells were transfected with RL‐3xBulge reporter along with FL as internal control. RL/FL luminescence ratio was calculated. Data are presented as mean ± SD (n = 3).
- HeLa cells were co‐transfected with indicated constructs and dual‐luciferase assay was performed. RL/FL luminescence ratio was calculated. Data are presented as mean ± SD (n = 3), P‐values were calculated using Student's t‐test.
- HeLa cells were sequentially transfected with control (siControl) or siRNA targeting the 3′‐UTR of Nup358 (siNup358‐B), followed by RL‐3xBulge reporter construct, along with FL as internal control. These were co‐transfected with either GFP‐control or GFP‐Nup358 constructs. Left panel: RL/FL luminescence ratio was calculated. Data are presented as mean ± SD (n = 3). Right panel: Western blot analysis of the lysates prepared from cells transfected with indicated siRNAs and expression constructs, using anti‐Nup358 antibodies. Vinculin was used as loading control.
Nup358 depletion does not affect mature miRNA levels
To test whether Nup358 knockdown affected pre‐miRNA export and/or maturation, we measured the levels of mature miRNAs using different methods. Northern blot analysis with specific 32P radiolabeled miRNA probe indicated that Nup358 depletion did not grossly affect the levels of mature let‐7a miRNA in HeLa (Fig 3A, upper panel). As expected, we observed a reduction in the level of mature let‐7a in Dicer‐depleted cells in comparison with control siRNA‐treated cells. These Northern blot results were further confirmed by a TaqMan‐based real‐time quantitative PCR (qPCR) assay (Fig 3A, lower panel). We also performed deep sequencing of small RNAs and assessed the relative levels of miRNAs between control and Nup358‐depleted HEK293T cells. The results indicated that out of 629 miRNAs commonly detected in control and Nup358 knockdown cells, 494 miRNAs (~78%) showed no significant change in the level of their expression. Among the remaining miRNAs analyzed, ~10% were downregulated and ~12% were upregulated in Nup358‐depleted cells as compared to control cells. The relative levels of 50 most abundant miRNAs in HEK293T cells under control and Nup358 knockdown conditions are shown in Fig EV3A. Collectively, these results suggested that Nup358 does not play any significant role in miRNA biogenesis.
Figure 3. Nup358 interacts with AGO and GW182 and is required for coupling miRISC with target mRNAs.
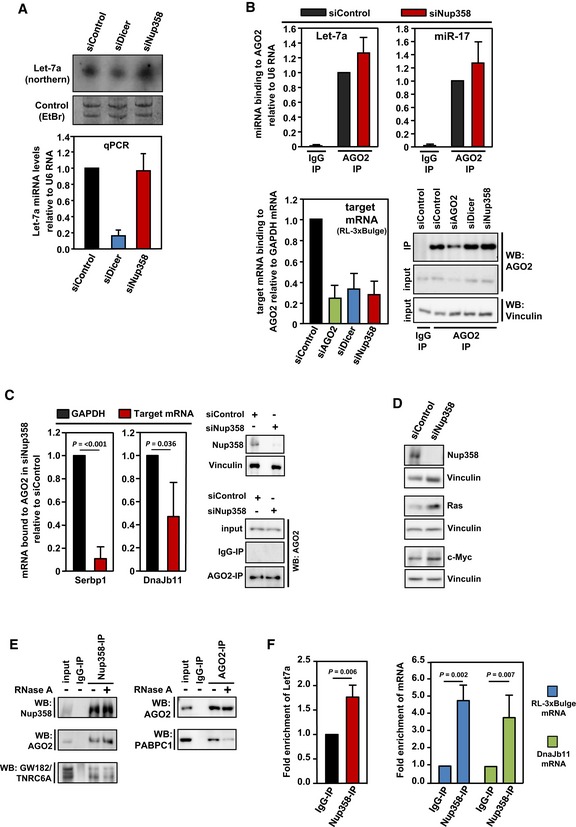
- HeLa cells were transfected with control, Dicer, or Nup358‐specific siRNAs, as indicated. Upper panel: Total RNA was isolated and analyzed by Northern blotting for let‐7a using radiolabeled probe. Ethidium bromide (EtBr)‐stained gel indicates equal loading of RNA samples. Bottom panel: Total RNA was isolated from control, Dicer‐, or Nup358 siRNA‐treated cells and was reverse‐transcribed using TaqMan microRNA reverse transcription kit. The levels of let‐7a were quantified by qPCR following the manufacturer's instructions. Graph represents the relative levels of miRNAs as compared to U6 RNA control. The values were further normalized to let‐7a levels in control siRNA knockdown condition. Data are presented as mean ± SD (n = 3).
- HeLa cells were transfected with indicated siRNAs, followed by RL‐3xBulge construct. Immunoprecipitation (IP) was performed with control mouse IgG (IgG‐IP) or anti‐AGO2 antibody (AGO2‐IP) using lysates prepared from these cells. Total RNA was isolated from control and AGO2 immunoprecipitates and the levels of AGO2‐associated let‐7a (top left panel) and miR‐17 (top right panel) were quantified by qPCR. Graph represents the extent of miRNAs associated with AGO2 as compared to U6 RNA (as a negative control) with AGO2. Bottom left panel: RNA isolated from IgG control or AGO2 immunoprecipitate samples was reverse‐transcribed using oligo(dT) primer. RL‐3xBulge mRNA association with AGO2 was quantified by qPCR and normalized to the level of GAPDH mRNA associated with AGO2. Bottom right panel: AGO2 immunoprecipitates were analyzed by Western blotting using AGO2‐specific antibody. Vinculin was used as loading control. Data are presented as mean ± SD (n = 3).
- HeLa cells were transfected with control (siControl) or Nup358‐specific siRNA (siNup358) for 96 h. Cells were lysed and subjected to immunoprecipitation with control IgG (IgG‐IP) or anti‐AGO2 (AGO2‐IP). Total RNA was extracted from initial lysate and IP samples and analyzed by qPCR for validated miRNA targets, Serbp1 and Dnajb11. GAPDH was considered as negative control. Data expressed as the relative amount of target mRNA associated with AGO2 as compared to GAPDH mRNA. Western blots indicate the extent of Nup358 depletion and AGO2 immunoprecipitation in siControl and siNup358 samples. Vinculin was used as loading control. Data are presented as mean ± SD (n = 3), P‐values were calculated using Student's t‐test.
- HeLa cells were treated with indicated siRNAs for 96 h. Cells were lysed and analyzed for extent of Nup358 depletion and for the levels of validated miRNA targets, Ras and c‐Myc, by Western analysis. Vinculin was used as loading control.
- HeLa cells were lysed and immunoprecipitation was performed with control (IgG‐IP) or Nup358 (Nup358‐IP, left panel) or AGO2 (AGO2‐IP, right panel) using specific antibodies. The immunoprecipitates were washed with a buffer containing (+) or not containing (−) RNase A and probed for the presence of indicated proteins by Western blotting.
- HeLa cells were transfected with RL‐3xBulge construct and immunoprecipitation was performed using control (IgG‐IP) or Nup358 (Nup38‐IP) antibodies. Total RNA was isolated from the immunoprecipitates and analyzed for let‐7a miRNA (left panel), RL‐3xBulge mRNA, and the endogenous miRNA target Dnajb11 using qPCR (right panel). Fold enrichment in Nup358‐IP as compared to IgG control was calculated, and the data are presented as mean ± SD (n = 3), P‐values were obtained using Student's t‐test.
Figure EV3. Nup358 depletion does not grossly alter mature miRNA levels or nucleo‐cytoplasmic distribution of mRNAs and key proteins involved in miRNA pathway.
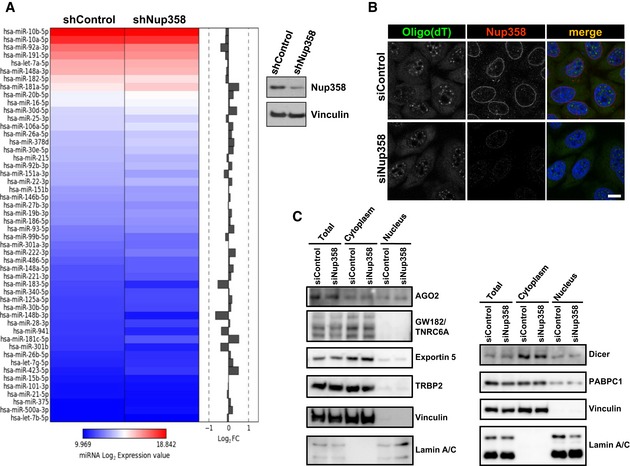
- HEK293T cells were transfected with shRNA control (shControl) or shRNA against Nup358 (shNup358). Heatmap shows relative levels of 50 most abundant miRNAs detected between shControl and shNup358‐transfected HEK293T cells. Western blot (right panel) shows the extent of Nup358 depletion. Vinculin was used as loading control.
- HeLa cells were transfected with control (siControl) or Nup358 (siNup358)‐specific siRNA. Cells were fixed and stained for mRNA using labeled oligo(dT) (red) and for Nup358 (green) using specific antibodies. DNA was stained with Hoechst 33342 (blue, merge). Scale bar, 10 μm.
- HeLa cells were subjected to control (siControl) or Nup358 (siNup358) depletion and the nucleo‐cytoplasmic fractionation was performed and analyzed for different proteins as indicated. Vinculin and lamin A/C were used as cytoplasmic and nuclear markers, respectively. Right and left panels represent two independent fractionations.
As compared to control cells, Nup358‐depleted cells did not show any detectable change in the relative distribution of mRNAs between the nucleus and the cytoplasm as assessed by oligo(dT) staining (Fig EV3B). Moreover, the total level and the nucleo‐cytoplasmic distribution of specific proteins involved in miRNA pathway were largely unaltered by Nup358 depletion (Fig EV3C). Together, these results indicated that the observed impairment of miRNA function in the absence of Nup358 might not be due to any indirect effect caused by defects in miRNA export and/or processing, mRNA export, or nucleo‐cytoplasmic distribution of key proteins involved in miRNA pathway.
Nup358 is not required for miRISC formation, but is essential for association of target mRNA with miRISC
As the mature miRNA levels were mostly unaffected when Nup358 was depleted, we examined whether this nucleoporin is involved in loading of miRNAs onto AGO proteins. Towards addressing this, AGO2 immunoprecipitation was performed in control or Nup358 siRNA‐treated cells, and the immunoprecipitates were analyzed for the presence of specific miRNAs by qPCR. Two abundant miRNAs in HeLa cells (let‐7a and miR‐17) showed no significant difference in the extent of their association with AGO2 in the absence or presence of Nup358 (Fig 3B). These results suggested that Nup358 is not involved in loading of miRNA onto AGO2 or miRISC formation.
We next investigated whether Nup358 depletion caused any effect on the interaction of target mRNA with the miRISC. To test this, we immunoprecipitated endogenous AGO2 from HeLa cells and examined for the presence of RL‐3xBulge mRNA (a miRNA target that has three imperfect let‐7a binding sites) using qPCR. Similar to Dicer‐ and AGO2‐depleted conditions, Nup358 knockdown significantly impaired the ability of miRISC to associate with the RL‐3xBulge target mRNA (Fig 3B). Furthermore, we analyzed the association of two validated endogenous mRNA targets with AGO2 63, in the absence and presence of Nup358. In Nup358‐depleted cells, binding of AGO2 to Serbp1 and Dnajb11 mRNAs was significantly reduced as compared to that of GAPDH mRNA (Fig 3C). Consistent with a role for Nup358 in miRNA pathway, the protein levels of established miRNA targets such as Ras 64 and c‐Myc 65, 66 were found to be increased in Nup358‐depleted cells (Fig 3D). Collectively, these results indicated that Nup358 is involved in the coupling of target mRNA with miRISC.
To verify this further, we utilized a previously reported artificial AGO2 tethering assay that bypasses the requirement of miRNA for suppression of the target mRNA 67. The suppression of AGO2‐tethered mRNA, however, is dependent on GW182 and downstream effectors 68. We reasoned that if Nup358 plays a role in the coupling of miRNA with target mRNA, it should be dispensable in a condition where suppression of target mRNA occurs independent of miRNA. Our results showed that Nup358 depletion did not have a specific effect on the ability of tethered AGO2 to suppress the reporter RNA (Fig EV4). This outcome is consistent with the finding that Nup358 is essential for association of target mRNA with miRISC, but is not required once the target mRNA is directly made to interact with the AGO protein. The results also suggested that Nup358 depletion does not cause any general effect on miRNA pathway downstream to miRNA–mRNA association.
Figure EV4. Nup358 depletion does not affect repression of mRNAs tethered with AGO2.
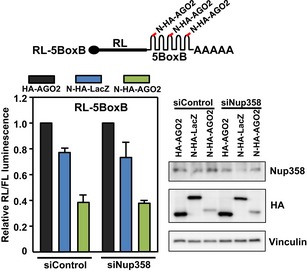
HEK293T cells were sequentially transfected with control (siControl) or Nup358 siRNA (siNup358), followed by co‐transfection of pRL‐5BoxB construct (a representative schematic is shown on top) with FL as internal control, and HA‐AGO2, N‐HA‐LacZ, or N‐HA‐AGO2, as indicated. Left panel: Graph represents RL/FL luminescence normalized to HA‐AGO2 transfected samples for control and Nup358 knockdown, separately. RL/FL luminescence ratio was calculated. Data are presented as mean ± SD (n = 3). Western blot (right panel) shows the extent of Nup358 knockdown in cells transfected with indicated siRNA and expression constructs, using specific antibodies. Vinculin was used as loading control.
RNA‐binding zinc finger (ZnF) domains of Nup358 are dispensable for miRNA function
Our results suggested that Nup358 could play a role in the coupling of target mRNA with miRISC. Human Nup358 contains eight ZnF domains in the middle region (Fig EV5A), and previous studies have shown these domains to directly bind the signal sequence coding regions present in a subset of mRNAs encoding secretory proteins 69. Also, RanBP2‐type ZnF domains are shown to be present in some RNA binding proteins and contribute to RNA binding 70. We were interested to examine whether ZnF domains are required for Nup358's function in mRNA–miRISC coupling process. Toward this, let‐7a miRNA activity was monitored using reporter assays in Nup358‐depleted HeLa cells after expressing a Nup358 deletion mutant that was devoid of ZnF domains. The results showed that the mutant protein was capable of almost completely rescuing the de‐repression caused by Nup358 depletion (Fig EV5B), supporting the conclusion that the RNA‐binding ZnF domains are dispensable for Nup358's function in miRNA pathway.
Figure EV5. The middle region of Nup358, consisting of the RNA‐binding RanBP2‐type zinc fingers, is dispensable for miRNA function.
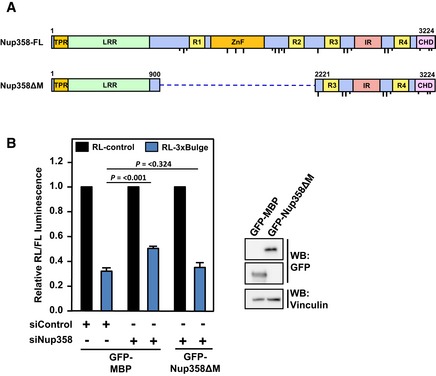
- The domain architecture of human full‐length (FL) Nup358. TPR, tetratricopeptide repeat; LRR, leucine‐rich region; R1–R4, RanGTP‐binding domain; ZnF, zinc finger domains; IR, internal repeats; CHD, cyclophilin‐homology domain. Dotted line indicates the deleted region in Nup358ΔM.
- HeLa cells were transfected with control (siControl) or Nup358 (siNup358)‐specific siRNAs. Subsequently, RL‐3xBulge or RL‐3xBulge‐mut reporter construct was transfected along with GFP‐maltose binding protein (MBP, control) or GFP‐Nup358ΔM construct. The dual‐luciferase assay was performed to monitor the miRNA activity. FL was used as a transfection control. Left panel: RL/FL luminescence ratio was calculated. Data are presented as mean ± SD (n = 3), P‐values were calculated using Student's t‐test. Right panel: Western blot showing relative expression of indicated constructs in HEK293T cells using anti‐GFP antibodies.
Nup358 interacts with AGO2 and GW182
Based on the finding that Nup358 functions in coupling the target mRNA to miRISC, we tested whether Nup358 interacts with components of miRISC. We immunoprecipitated endogenous Nup358 from HeLa cells and analyzed the immunoprecipitate for the presence of AGO2 and GW182. The co‐immunoprecipitation assay suggested that Nup358 associated with both AGO2 and GW182 in vivo (Fig 3E). This interaction was found to be independent of RNA, as RNase A treatment did not alter the association of Nup358 with AGO2 or GW182 considerably (Fig 3E). Under the same condition, as previously shown, the interaction of AGO2 with PABPC1 was found to be RNA‐dependent 63 (Fig 3E).
We further tested whether Nup358 associates with miRISC and target mRNAs in vivo. HeLa cells were transfected with RL‐3xBulge (let‐7a target), and RNA–protein cross‐linking was performed using formaldehyde. The cell lysates were subjected to immunoprecipitation using IgG control or Nup358‐specific antibodies, and the immunoprecipitates were analyzed for the presence of let‐7a by qPCR. As compared to IgG control, Nup358 immunoprecipitate had significantly higher levels of let‐7a miRNA (Fig 3F). Moreover, the ectopically expressed (RL‐3xBulge) and endogenous (Dnajb11) mRNA targets were also enriched in Nup358 immunoprecipitate (Fig 3F). Collectively, these results suggested that Nup358 associates with the protein and RNA components of the miRNA‐induced silencing complex in vivo.
Nup358 interacts with AGO proteins through the IR region
Nup358 is a large nucleoporin with multiple domains (Fig 4A). We wished to characterize the interaction between Nup358 and AGO proteins in detail. To delineate the region in Nup358 that is involved in the interaction with AGO, GFP‐tagged N‐terminal (Nup358‐N), middle (Nup358‐M), or C‐terminal region (Nup358‐C) of Nup358 was expressed along with HA‐AGO2 in HEK293T cells. Co‐immunoprecipitation assays revealed that AGO2 specifically interacted with all three fragments of Nup358, and more prominently with the C‐terminal fragment (Fig 4B). Further, we proceeded to identify and characterize the minimum region in Nup358‐C required for interaction with AGO2. Experiments with deletion constructs of Nup358‐C indicated that the IR region was sufficient to mediate the interaction with AGO2 (Fig 4C). Consistent with this, a deletion mutant of Nup358‐C lacking the IR region failed to interact with AGO2 (Fig 4D).
Figure 4. AGO proteins interact with IR region of Nup358.
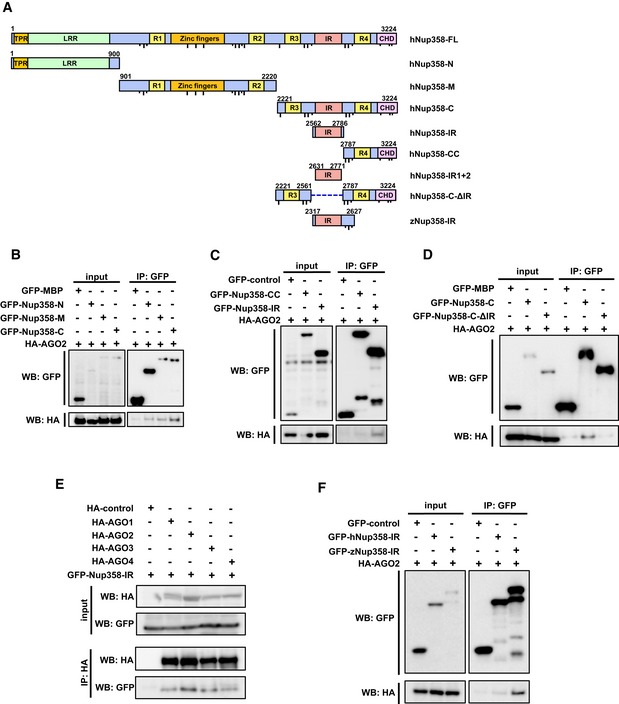
- Schematic diagram representing the domains and constructs of human (h) and zebrafish (z) Nup358 used in this study. TPR, tetratricopeptide repeat; LRR, leucine‐rich region; R1–R4, RanGTP‐binding domain; ZnF, zinc finger domains; IR, internal repeats; CHD, cyclophilin‐homology domain. Dotted line indicates the deleted region in the indicated construct. FG and F × FG sequence positions are represented as short and long black lines, respectively. Amino acid positions are indicated in numbers. FL, full‐length; N, amino‐terminal region; M, middle region; C, carboxy‐terminal region.
- HEK293T cells were co‐transfected with GFP‐maltose binding protein (MBP) control or GFP‐tagged version of indicated Nup358 fragments along with HA‐AGO2. Immunoprecipitation (IP) was performed using GFP antibodies and the presence of AGO2 was detected by Western blotting (WB) using HA antibodies.
- Cells were transfected with indicated constructs and IP and WB were performed as described for (B).
- HEK293T cells transfected with GFP‐MBP (control), GFP‐Nup358‐C, or GFP‐Nup358‐CΔIR (a mutant devoid of IR region) and HA‐AGO2. IP and WB analyses were performed as indicated.
- Cells were transfected with HA‐tagged version of indicated AGO subfamily member and GFP‐Nup358‐IR. IP and WB analyses were performed using indicated antibodies.
- Lysates from cells expressing GFP‐control, GFP‐human (h) Nup358‐IR, or GFP‐zebrafish (z) Nup358‐IR and HA‐AGO2 were immunoprecipitated using GFP antibodies, and the immunoprecipitates were analyzed for the presence of HA‐AGO2 by Western blotting.
To examine the conservation of interaction between Nup358 and AGO subfamily proteins, GFP‐IR was co‐expressed with HA‐tagged AGO1–4 in HEK293T cells. Co‐immunoprecipitation assay confirmed that IR region physically associated with all four AGO proteins (Fig 4E). Moreover, a fragment encompassing the IR region derived from zebrafish Nup358 also showed specific interaction with human AGO2 (Fig 4F). Collectively, these data demonstrated that IR provides a conserved region for interaction with AGO subclade of proteins.
SIM is a conserved motif for AGO interaction
As the results suggested that IR region of Nup358 is involved in binding to AGO proteins, we investigated this molecular interaction in detail. Nup358‐IR possesses two internal repeats, each of them having a SIM‐binding and a Ubc9‐binding region (Fig 5A) 50. Interestingly, we found that both IR1 and IR2 could independently interact with AGO2 in a SIM‐dependent manner (Fig 5B). However, as known earlier, IR1, and not IR2, specifically interacted with endogenous SUMOylated RanGAP and Ubc9 50, 51. Also, deletion of SIM1 from IR1 abrogated the interaction with SUMO~RanGAP, but not with Ubc9 (Fig 5B). To test whether SIM is sufficient for AGO interaction, we co‐expressed HA‐AGO2 with GFP‐SIM1 or GFP‐SIM2. Co‐immunoprecipitation assay clearly indicated that SIM1 and SIM2 independently were capable of interacting with AGO2, but not with SUMO~RanGAP or Ubc9 (Fig 5C).
Figure 5. SIM is the minimum region in Nup358‐IR required for binding to AGO2.
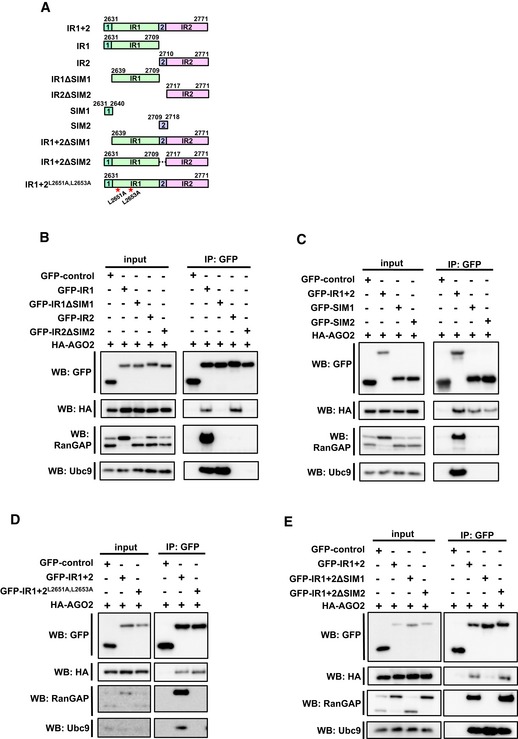
- Schematic representation of human Nup358‐IR region and the constructs used in this study. IR, internal repeats; SIM, SUMO‐interacting motif; 1, 2, SIM1 and SIM2. Amino acids substituted in Ubc9 mutant are indicated with red asterisks.
- HEK293T cells were transfected with indicated constructs, and immunoprecipitation (IP) was performed using GFP‐specific antibodies and probed for HA‐AGO2 by Western blotting (WB) using HA antibodies. Endogenous RanGAP and Ubc9 were probed with specific antibodies.
- Lysates prepared from cells expressing the indicated constructs were subjected to IP and WB using indicated antibodies. The presence of endogenous RanGAP and Ubc9 was determined by WB.
- GFP‐control, GFP‐IR1 + 2 wild type, or mutants were co‐expressed with HA‐AGO and IP and WB analyses were performed to detect the interaction using indicated antibodies.
- Lysates prepared from cells expressing the indicated proteins were immunoprecipitated with GFP‐specific antibodies, and the presence of specific proteins in the immunoprecipitates was detected by WB with indicated antibodies.
It has been shown previously that Ubc9 preferably binds to IR1 and stabilizes the interaction between IR1 and SUMO~RanGAP 50, 51. We wished to test whether Ubc9 is required for the interaction between IR (IR1 + 2) and AGO2. Interestingly, a mutant of IR that is defective in binding to Ubc9 (IR1 + 2L2651A,L2653A) 37 still efficiently associated with AGO2, whereas as expected, it failed to interact with SUMO~RanGAP (Fig 5D). Moreover, deletion of SIM1, but not SIM2, from IR disrupted its interaction with AGO2, indicating that in the context of intact IR, AGO2 interaction is mainly dependent on SIM1 (Fig 5E). As expected, SIM1 deletion also impaired the ability of IR to associate with SUMO~RanGAP, and not with Ubc9 (Fig 5E). Taken together, these results suggested that AGO‐IR interaction requires neither Ubc9 nor SUMO~RanGAP, and SIM is the minimum region in Nup358 required for the interaction with AGO2.
Earlier studies indicated that within Nup358‐SIM1, the hydrophobic amino acids V2632, I2634, and L2635 contribute to SUMO binding 48. We found that substituting these residues with alanine compromised the SIM's ability to bind AGO protein (Fig 6A), indicating that the binding mode of SIM with SUMO and AGO proteins could be similar. The finding that both SIM1 and SIM2 from Nup358‐IR independently interacted with AGO2 prompted us to investigate whether SIMs derived from other SUMO‐interacting proteins are capable of binding to AGO. Toward this, GFP‐fused SIMs from PIAS1 and TTRAP (Fig 6B) were individually co‐expressed with HA‐AGO2 in HEK293T cells and assessed for their ability to interact with AGO2 by co‐immunoprecipitation assays. It has already been shown that PIAS1‐SIM interacts with both SUMO1 and SUMO2, whereas TTRAP‐SIM shows greater preference for SUMO2 43. We found that irrespective of the proteins from which they were derived, both the SIMs interacted with AGO2 (Fig 6B). Moreover, SIM could associate with both AGO1 and AGO2 (Fig 6C). Collectively, these results indicated that SIM provides a binding platform for conserved interaction with AGO subfamily of proteins.
Figure 6. SIM provides a conserved platform for interaction with AGO family of proteins.
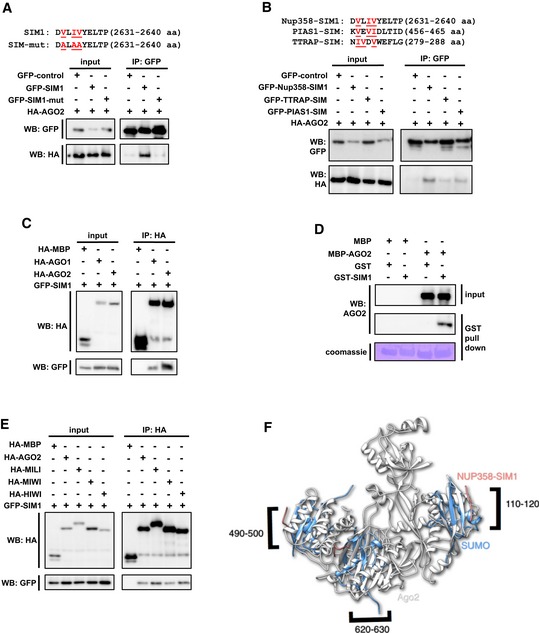
- Top panel: depiction of the amino acid sequence corresponding to the Nup358‐SIM1 region and substitutions introduced in the SIM1 mutant. Bottom panel: HEK293T cells were co‐transfected with GFP‐control, GFP‐SIM1, or GFP‐SIM1‐mut along with HA‐AGO2 and immunoprecipitation (IP) was performed with GFP‐specific antibodies, and Western analysis (WB) of the input lysate and immunoprecipitates was carried out using indicated antibodies.
- Top panel: depiction of the amino acid sequence corresponding to the SIMs in the indicated proteins used in this study. Hydrophobic residues involved in SUMO binding are indicated in red. Bottom panel: Constructs expressing GFP‐control or GFP‐fused SIMs of indicated proteins were co‐transfected with HA‐AGO2 in HEK293T cells, and IP and WB were performed to determine the extent of interaction.
- HA‐MBP‐control, HA‐AGO1, or HA‐AGO2 was co‐expressed with GFP‐SIM1 and the cell lysates were subjected to IP with HA antibodies and WB analysis with indicated antibodies to detect the presence of proteins in the immunoprecipitates.
- SIM1 directly interacts with AGO2. Bacterial lysates expressing MBP control or MBP‐AGO2 and GST or GST‐SIM1 were mixed and GST pull‐down assay was performed. The presence of proteins in the pull‐down samples was analyzed by WB using specific antibodies. The extent of GST pull‐down was monitored by Coomassie staining of the membrane (Coomassie).
- SIM1 interacts with PIWI subfamily of proteins. HEK293T cells were co‐transfected with indicated constructs expressing HA‐tagged version of PIWI proteins and GFP‐SIM1. IP was performed using HA antibodies. The immunoprecipitates were probed for the presence of GFP‐SIM1 using GFP‐specific antibodies by WB.
- Putative SIM‐binding regions in AGO2. Three distinct superimpositions of AGO2 (gray) and SUMO (blue), in complex with Nup358‐SIM1(red), are shown in cartoon representation rendered with Chimera 89. The residue numbers on the AGO2 that could be the putative SIM‐binding regions are labeled. To identify possible Nup358 binding sites on AGO2, the 3D structure of AGO2 (PDB ID: 4W5O) 88 was compared with that of SUMO (PDB ID: 1Z5S) 47.
To examine whether SIM directly binds to AGO proteins, we resorted to bacterially expressed recombinant proteins. Mixed bacterial lysates expressing maltose binding protein (MBP, control) or MBP‐AGO2 along with GST (control) or GST‐SIM1 were used for performing GST pull‐down assays. The results suggested that GST‐SIM1 specifically interacted with AGO2 in vitro, indicating that SIM can directly bind to AGO2 (Fig 6D). Collectively, these experiments suggested that SIM provides a direct binding platform for AGO proteins.
Nup358 contains five potential SIMs as predicted by the GPS‐SUMO program at medium SUMO interaction threshold values (http://sumosp.biocuckoo.org) 71. These include one SIM in the N‐terminal region (Nup358‐N), two within the middle region (Nup358‐M), and two in the C‐terminal region (Nup358‐C) (Appendix Fig S1). The presence of SIMs in all the three regions provides an explanation for the association of all these fragments with AGO2 (Fig 4B).
SIM can bind to PIWI clade of proteins
As PIWI and AGO subfamilies of proteins share similarity in domain architecture and functions in terms of small RNA‐mediated gene silencing 4, we tested whether SIM could also bind to PIWI proteins. To address this, co‐immunoprecipitation assay was performed using HEK293T cells co‐expressing GFP‐SIM1 and HA‐tagged MBP (control), AGO2, MILI, MIWI, or HIWI proteins. The results clearly indicated that PIWI clade proteins, similar to AGO subfamily members, specifically interacted with SIM (Fig 6E). We conclude that SIM provides a general binding platform for interaction with AGO family of proteins.
Potential SIM‐binding sites in AGO2
The finding that AGO proteins bind to SIM raised the possibility that AGO has SUMO‐like domains found to be present in a few proteins 72, 73. However, analysis suggested that such domains are absent in AGO proteins. The other possibility included that there could be regions in AGO proteins that are structurally similar to the regions in SUMO that are involved in interaction with SIM 74. Using a recently developed algorithm, CLICK, probable regions on AGO2 were recognized by structural similarity to the SUMO regions that interact with SIM 75, 76. This structural analysis identified three distinct regions on AGO2 (Fig 6F). The structures of SUMO and AGO2 in the superimposed regions were between 70 and 87% geometrically similar with RMSD values ranging from 1.6 to 1.9 Å. The predicted binding sites comprised amino acids belonging to the N, MID, and PIWI domains of AGO2. These analyses point to the presence of multiple putative SIM‐binding sites in AGO proteins.
Ectopically Nup358‐SIM1 functionally interferes with miRNA pathway
As SIM was identified as the minimum region in Nup358 required for binding to AGO proteins, we overexpressed GFP‐MBP (control), GFP‐Nup358‐SIM1, or GFP‐Nup358‐SIM1‐mutant (defective in interaction with AGO) in HeLa cells along with RL‐3xBulge reporter to monitor the let‐7a miRNA activity 61. SIM1, but not SIM1 mutant, specifically interfered with the miRNA‐mediated suppression of the reporter RNA (Fig 7A), indicating that Nup358‐SIM1 has the ability to act in a dominant‐negative fashion, possibly by interfering with interaction of endogenous Nup358 with AGO proteins.
Figure 7. Functional relevance of the AGO‐interacting motif in miRNA pathway.
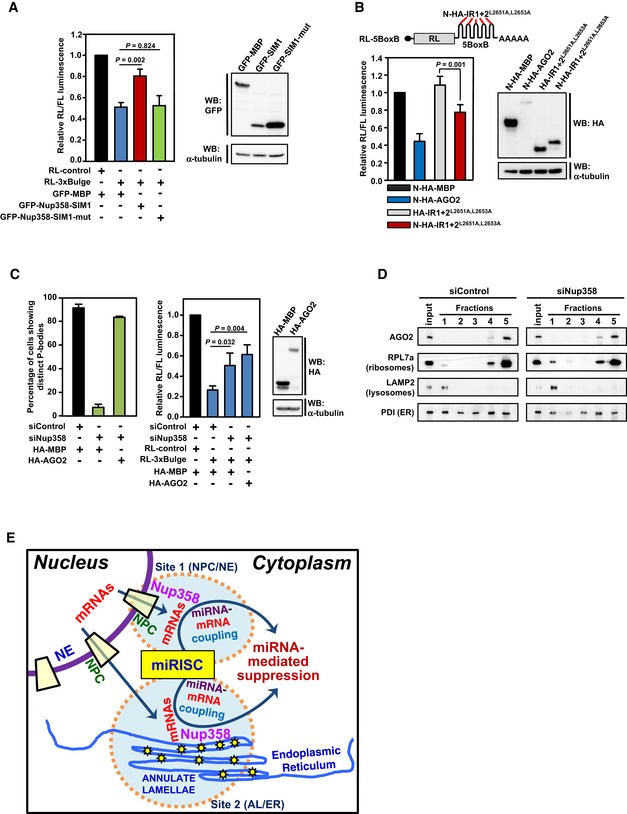
- Left panel: HeLa cells were co‐transfected with GFP‐MBP (control), GFP‐SIM1, or GFP‐SIM1‐mut (AGO interaction‐defective) along with RL‐control or RL‐3xBulge. Firefly luciferase was used as transfection control. Dual‐luciferase assay was performed. RL/FL luminescence ratio was plotted after normalization with GFP‐control. Data are presented as mean ± SD (n = 3), P‐values were calculated using Student's t‐test. Right panel: Western blots showing the expression of transfected constructs using indicated antibodies. α‐Tubulin was used as loading control.
- Top panel: Reporter construct used in this study 61. Bottom left panel: HEK293T cells were co‐transfected with indicated expression constructs and the RL reporter. Firefly luciferase (FL) was used as transfection control. Dual‐luciferase assay was performed. The ratio of RL/FL luminescence was plotted after normalization with N‐HA‐MBP control. Data are presented as mean ± SD (n = 3), and P‐value was calculated using Student's t‐test. Bottom right panel: Western blot for monitoring the expression levels of indicated proteins using antibodies. α‐Tubulin was used as loading control.
- Ectopic expression of AGO2 restores P body formation, but fails to rescue the impairment in miRNA function in Nup358‐depleted cells. HeLa cells were transfected with control (siControl) or Nup358 (siNup358)‐specific siRNA, followed by indicated constructs. Left panel: Quantitative representation showing the number of cells showing microscopically distinct P bodies in different indicated conditions. The data were obtained from three independent experiments, and in each experiment, 100 cells were counted from different fields for the presence of intact P bodies and expressed as percentage. Middle panel: HeLa cells were transfected with indicated siRNAs, followed by expression constructs and miRNA RL reporters. FL was used as transfection control. The ratio of RL/FL luminescence was calculated. Data are presented as mean ± SD (n = 3), and P‐values were obtained using Student's t‐test. Right panel: Western blot analysis for the relative expression of indicated constructs in HEK293T cells. α‐Tubulin was used as loading control.
- HeLa cells were transfected with siControl or siNup358 and subjected to membrane flotation assay. Fractions were collected and analyzed for the presence of indicated proteins by Western blotting.
- Model for the role of Nup58 in miRNA‐mediated translational suppression. Exported mRNAs could be sorted for miRNA‐mediated suppression at two potential sites in the cytoplasm: site 1—the nuclear pore complex (NPC) as a part of nuclear envelope (NE); and/or site 2—annulate lamellae (AL) as a part of endoplasmic reticulum (ER). Nup358, a component of the NPC and AL, plays an important role in the coupling of target mRNA with miRISC.
Artificial tethering of AGO proteins to the 3′‐UTR of reporter mRNA has been shown to suppress the reporter mRNA 67. We wished to test whether Nup358‐IR could be tethered to mRNAs to suppress their expression, presumably by recruiting AGO proteins. As we found that IR1 + 2 mutant (IR1 + 2L2651A,L2653A) was incapable of binding to endogenous SUMO~RanGAP or Ubc9, but retained the ability to bind to AGO proteins (Fig 5D), we used this mutant in tethering studies. HEK293T cells were transfected with Nλ‐peptide‐HA‐tagged IR1 + 2L2651A,L2653A (N‐HA‐IR1 + 2L2651A,L2653A) along with a reporter construct engineered to express the Renilla luciferase mRNA with five BoxB hairpins at its 3′‐UTR, which provides binding site for N‐HA‐tagged proteins 67. In addition to N‐HA‐MBP, HA‐IR1 + 2L2651A,L2653A that is incapable of binding to BoxB hairpin was used as control. The results from tethering assays clearly suggested that N‐HA‐IR1 + 2L2651A,L2653A could significantly suppress the expression of the reporter mRNA (Fig 7B). Collectively, these results support the notion that IR region, and particularly SIM, acts functionally as an AGO‐interacting motif.
In Nup358‐depleted cells, exogenous expression of AGO2 restores P body formation but does not rescue the defect in miRNA pathway
Previous studies have suggested that P bodies are the sites at which miRNA‐mediated suppression/degradation of the target mRNA could occur 5, 6. However, other studies indicated that microscopically visible P body formation is a consequence of miRNA‐mediated repression 60. Therefore, it may be possible that Nup358 participates in miRNA pathway by playing a role at the level of formation and/or functioning of P bodies. Interestingly, exogenous expression of HA‐AGO2, but not HA‐MBP control, restored P body formation in Nup358‐depleted cells to a level almost comparable to that in the control siRNA‐treated cells (Fig 7C). Next, we tested whether ectopic expression of AGO2 is sufficient to revert back the miRNA‐mediated de‐repression caused by Nup358 depletion. HeLa cells were treated with control or Nup358‐specific siRNA and subsequently assessed for let‐7‐mediated repression of the RL‐3xBulge reporter mRNA in the presence of ectopically expressed HA‐MBP (control) or HA‐AGO2. Interestingly, expression of AGO2 did not rescue the impairment of miRNA pathway caused by Nup358 knockdown (Fig 7C). Collectively, this suggested that restoration of P body assembly in Nup358‐depleted cells was not sufficient to rescue the miRNA‐mediated suppression defect. Although the mechanism by which AGO2 expression induced P body formation in Nup358 knockdown cells is not clear, the results supported a functional role for Nup358 in miRNA pathway upstream to P body assembly.
Nup358 depletion does not affect localization of AGO2 to rough ER
Previously, it has been shown that miRNA‐loaded AGO2 localizes to the rough ER 11. To investigate whether Nup358 plays a role in targeting AGO2 to the rER, membrane flotation assay was performed using cells that were treated with control or Nup358 siRNA. The fractionation was verified by using PDI (ER lumen marker), RPL7a (ribosome marker), and LAMP2 (lysosome marker). The results suggested that Nup358 depletion did not affect the overall localization of AGO2 to rER (Fig 7D), indicating that it may not function in targeting AGO proteins to the ER.
Based on the data presented here, we propose the following model (Fig 7E). Exported mRNAs could be sorted at the NE (NPC) and at the ER (AL) for determination of their eventual fate in the cytoplasm. Nup358, as a component of NPC and AL, is involved in the coupling of target mRNA with miRISC to bring about miRNA‐mediated suppression. We propose that Nup358 could essentially exert its function both at the NE and ER. The findings thereby also indicate a specific role of ER in coordinating the cytoplasmic events involved in miRNA‐mediated gene silencing pathway.
Discussion
Here, we show that Nup358 plays an important role in miRNA‐mediated translational suppression, by aiding in the coupling of target mRNA with miRISC in vivo. Consistent with this function, we find that Nup358 physically interacts with the protein and RNA components of the miRNA‐induced silencing complex. Further characterization of Nup358–AGO interaction revealed the previously characterized SIM as a conserved motif for interaction with AGO family of proteins as well. The functional and evolutionary significance of the finding that the same motif performs binding to AGO and SUMO proteins deserves further investigation. Furthermore, our results with tethering and dominant‐negative experiments support the idea that SIM‐mediated interaction of Nup358 with AGO proteins is important for miRNA function.
Where does the coupling of miRISC with target mRNA occur? Recent studies indicated a role for ER in this process 10, 11. Our live imaging studies clearly show an intimate interaction of mRNP granules such as P bodies and SGs with cytoplasmic AL and NE. We also found that some of the AL could originate from the NE. Although untested, it is possible that AL could carry mRNAs into the cytoplasm, where the fate of these mRNAs could be regulated. The results reported here point to a potential role of AL as platforms for the regulation of exported and/or cytoplasmic mRNAs by coordinating with machineries involved in mRNA remodeling in the cytoplasm. Although a robust, dynamic association of cytoplasmic AL with the mRNP granules was evident from our live imaging studies, we are unsure whether the coupling occurs only at the AL. Further studies are required to delineate the contribution of NPC‐ and AL‐associated Nup358 in this process. Understanding the mechanistic details by which Nup358 gets targeted to these structures would help address this question conclusively.
Interestingly, exogenous expression of AGO2 could almost completely restore P body formation in Nup358‐deficient cells, but failed to rescue the impaired miRNA function (Fig 7C), arguing against an indirect role for Nup358 in miRNA pathway through maintenance of P body integrity. These data strengthen the conclusion that Nup358 plays a role in the coupling of mRNA target with miRISC, and P body disruption observed in Nup358‐depleted cells could be a consequence of impaired miRNA function. Furthermore, the observation that overexpression of Nup358 enhances miRNA function (Fig EV2C and D) indicates it to be a rate‐limiting factor regulating miRNA pathway in mammalian cells.
Whether Nup358–AGO2 interaction increases the quantitative efficiency of AGO2 to associate with the target mRNA through compartmentalization or it affects miRISC–mRNA interaction at the molecular level is unclear. This interaction, however, does not seem to be essential for targeting the miRNA‐loaded AGO2 to the rER (Fig 7D). It is possible that Nup358 acts as a scaffold for mediating the coupling of the target RNA with the miRISC in the context of rER. This could involve interaction of Nup358 with target RNA as well as with the miRISC components AGO and GW182. Interestingly, we have observed that depletion of Nup358 affects target RNA suppression mediated by of miRNA (Fig 2B–D) and not when AGO2 is artificially tethered to the target RNA (Fig EV4). How does Nup358 participate in the coupling of mRNA with AGO protein (or miRISC) only in case of miRNA‐mediated suppression? One of the possibilities is that Nup358–AGO interaction is required for a conformational change in AGO protein that favors its binding to target RNA and/or facilitates the miRNA–mRNA base pairing within the RNA‐binding groove. The extensive interaction between Nup358 and AGO would help achieve this.
A recent study suggested that Nup358 is important for translational activation of a set of mRNAs encoding secretory proteins 69. The authors have shown that Nup358 interacts with mRNAs through the ZnF domains and potentiates their translation. However, the ZnF domains appear to be dispensable for the Nup358's function in miRNA pathway. Combined with the earlier report, our results suggest that Nup358 might play a general role in determining the cytoplasmic fate of mRNAs (translational activation or suppression), possibly involving different mechanisms.
Our findings show that SUMO~RanGAP interacts with the same region (SIM1) to which AGO also binds. Association of SUMO~RanGAP with the IR region could be competed with increasing levels of AGO2 in cells, indicating that the binding of AGO and SUMO~RanGAP is mutually exclusive (Appendix Fig S2). However, the binding of SUMO~RanGAP and Ubc9 with IR appears to be rather stronger and AGO2 does not completely compete out this interaction. Understanding the functional relevance of mutually exclusive Nup358 complexes, containing either SUMO~RanGAP or AGO proteins, requires further investigation. This could spatially and temporally regulate aspects of Nup358 function within the cell. Intriguingly, the binding of AGO proteins to IR region might influence RanGAP's function and Nup358's SUMO E3 ligase activity.
Previous studies have shown that human AGO2 gets SUMOylated at lysine 402 77, 78. Consistent with this, we also found that the lysine residue 402, but not 266 or 693, of human AGO2 is modified by SUMO (Appendix Fig S3A). Interestingly, similar to wild type, SUMO‐defective mutant (K402R) of AGO2 retained the ability to associate with IR region (Appendix Fig S3B), indicating that the binding of AGO2 to SIM is not dependent on SUMO modification. Moreover, we noticed that unmodified AGO is capable of binding to IR/SIM as revealed by our co‐immunoprecipitation assays.
Does AGO bind to IR/SIM as a potential substrate for SUMO modification? We believe it to be unlikely because of the following reasons. Firstly, previous studies have shown that IR acts as an E3 ligase by not directly binding to the substrate, rather binding to SUMO~Ubc9 thioester to enhance the Ubc9 (E2) activity for SUMOylation 47, 53. Secondly, among the four AGO subclade members, only AGO1 and AGO2 have the conserved lysine residue (K402 in AGO2) that undergoes SUMOylation 78. In spite of this, we found all AGO proteins to have the ability to efficiently bind to IR. Thirdly, binding of SIM to SUMO (as a part of SUMO~Ubc9 thioester) is required for the IR to act as an E3 ligase 50, 79, and due to the mutually exclusive nature of interaction between SUMO and AGO with SIM, AGO binding to the SIM would in principle affect IR's E3 ligase activity.
It is worth noting that many RNP structures are found either to be associated with the NE or to be present in close proximity to the nucleus in different organisms. For example, many germ cell RNP granules are present in the perinuclear region, often adjacent to NE or NPCs, and genome‐wide screenings have identified some nucleoporins in having roles in the assembly and function of these structures 80, 81, 82, 83. The physical proximity of these RNP granules to NE or NPCs could also help coupling the exported RNAs to machineries involved in RNA regulation in the cytoplasm.
Is the AL‐RNP granule association conserved across evolution? In earlier studies, AL are shown to associate with MEX‐3‐positive RNP granules in the arrested C. elegans oocytes 21. Interestingly, depletion of npp‐9, the Nup358 homolog in C. elegans, led to disruption of MEX‐3 granules, indicating a role for this nucleoporin in the mRNP assembly 21. In another study, it was shown that P granules in C. elegans are present in the perinuclear region, often adjacent to NPC clusters enriched in npp‐9 84. This nuclear association was implicated to play a role in collection of mRNAs by P granule components prior to their release into the cytoplasm. The nucleoporin npp‐9 also was shown to physically interact with P granules that were released into the cytoplasm 84. However, the functional role of npp‐9 in this context is currently unknown and requires further investigation.
In summary, we have elucidated a novel function for Nup358 in coupling miRISC with the target mRNAs, and a possible role for ER (AL) in this process. Further studies on the interaction between Nup358 and AGO proteins revealed that the previously characterized SUMO interaction motif acts as a binding motif for AGO family of proteins as well. These findings provide a framework not only for exploring the mechanistic details of how Nup358 functions in miRNA pathway, but also to understand the role of ER (AL) in miRNA‐mediated suppression.
Materials and Methods
Cell culture, treatments, and transfection
HeLa S3, HEK293T, and COS‐7 cells were grown in Dulbecco's modified Eagle's medium (Invitrogen) with 10% fetal bovine serum (v/v) (Invitrogen) and 10 μg/ml ciprofloxacin antibiotics in a humidified incubator at 37°C under 5% CO2. For induction of SGs, cells were grown to 60–80% confluency and were subjected to 0.5 mM sodium arsenite treatment for 30 min.
In each well of 24‐well plates, 8 × 104 cells were seeded on glass coverslip (for immunofluorescence), or 3 × 105 cells in each well of 6‐well plates or 1.2 × 106 cells in each 100‐mm dish (for immunoprecipitation). After 12 h, cells were transfected using Lipofectamine 2000 (Invitrogen) for HeLa or polyethyleneimine, linear (PEI, MW‐25,000; Polysciences Inc.) for HEK293T and COS‐7 cells, following the manufacturer's instructions.
For siRNA transfections, Lipofectamine RNAiMax (Invitrogen) was used according to the manufacturer's instructions. Unless otherwise indicated, all siRNAs were used at a final concentration of 40 nM. The siRNAs were synthesized (Dharmacon) against the following target sequences for different genes: siNup358‐A (5′ GGTGAAGATGGATGGAATA 3′), siNup358‐B (5′ GGTGTGAAATAAAAGTTTA 3′), siNup358‐C (5′ GGACAGTGGGATTGT AGTG 3′), siControl (5′ TTCTCCGAACGTGTCACGT 3′), siNup214 (5′ TCAAATA CCTCTAACCTAT 3′), siDicer (5′ TTGTTGCGAGGTTGATTCT 3′), and siAGO2 (CAGAGT CCCGTGTGTCCTA 3′). siNup358‐A was used as siNup358 in every experiment, unless mentioned otherwise. siNup358‐B is directed against 3′‐UTR of human Nup358 and was used for rescue experiments.
Plasmid constructs
GFP‐tagged versions of full‐length (FL) and fragments of human Nup358 (GFP‐Nup358‐N, GFP‐Nup358‐M and GFP‐Nup358‐C) have been described earlier 31. GFP‐Nup358‐CC (2,787–3,224) and GFP‐Nup358‐IR (2,652–2,786) were generated from GFP‐BPC by deleting required 5′ and 3′ regions using appropriate restriction sites and end‐filling. GFP BPC∆IR was generated by replacing the SpeI/KpnI fragment with specific annealed oligos to delete amino acids 2,562–2,786 and to keep the reading frame intact. pEGFP‐Nup358ΔM was generated by removing the ApaI fragment corresponding to Nup358‐M in pEGFP‐Nup358‐FL, and replacing it with two annealed oligos (5′ CAA GCT TCG CGG CCC GCC CCG GGC C 3′ and 5′ CGG GGC GGG CCG CGA AGC TTG GGC C 3′) to keep N‐terminal and C‐terminal regions of Nup358 in frame. For generation of deletion mutants of IR, specific primers were used to amplify regions corresponding to 2,631–2,771 (IR1 + 2), 2,631–2,709 (IR1), 2,710–2,771 (IR2), 2,639–2,771 (IR1 + 2∆SIM1), 2,639–2,709 (IR1∆SIM1), and 2,717–2,771 (IR2∆SIM2) using pEGFP‐Nup358‐IR as the template. The PCR products were cloned at EcoRI/SalI sites of pEGFP‐C2 (Clontech). Zebrafish Nup358‐IR was amplified using specific primers from a cDNA sample prepared from zebrafish embryos. The region corresponding to 2,317–2,627 amino acids of zebrafish Nup358 was PCR‐amplified and cloned into pEGFP‐C2 at EcoRI site. To generate 2,631–2,709 + 2,717–2,711 (IR1 + 2∆SIM2) mutant, appropriate primers were used to PCR‐amplify the entire pEGFP‐Nup358‐IR construct devoid of SIM2 region. The PCR product was self‐ligated to obtain the deletion mutant. The resultant construct was verified by sequencing.
The mutant GFP‐IR1 + 2L2651A,L2653A was generated using a similar strategy as described above, except that the template used for PCR amplification was HA‐RanBP2 (2,553–2,838)L2651A,L2653A (a kind gift from Jan van Deursen, Mayo Clinic College of Medicine, USA) 37. To obtain N‐HA‐IR1 + 2L2651A,L2653A, the PCR product obtained by using appropriate primers and GFP‐IR1 + 2L2651A,L2653A as the template was cloned at the EcoRI/EcoRV sites of pCI‐neo‐N‐HA vector. To obtain HA‐IR1 + 2L2651A,L2653A, EcoRI/XhoI fragment from GFP‐IR1 + 2L2651A,L2653A was subcloned at respective sites in pcDNA‐HA vector. For generating Nup358‐SIM1 (2,631–2,640), Nup358‐SIM2 (2,709–2,718), PIAS1‐SIM (456–465), and TTRAP‐SIM (279–288), annealed oligos containing these coding sequences with EcoR1/Xho1 overhangs were cloned into pEGFP‐C2 (Clontech) at EcoRI/SalI sites. HA‐AGO2 was generated by subcloning hAGO2 open reading frame (ORF) from pCMV‐SPORT‐hAGO2 (a gift from Shigeyuki Yokoyama, RIKEN Genomic Sciences Center, Yokohama, Japan) into pcDNA‐HA vector using appropriate restriction sites. To generate HA‐MBP control, MBP ORF was PCR‐amplified from pMAL‐p2 (New England Biolabs) as the template, and the product was cloned at EcoRI/SmaI sites of pCI‐neo‐N‐HA. A similar strategy was used to clone MBP into pEGFP‐C2 vector to generate GFP‐MBP control. For in vitro interactions, pMAL‐AGO2 was generated by cloning the BamH1 fragment having AGO2 ORF from pCMV‐SPORT‐hAGO2 into pMAL‐c2 (New England Biolabs). To generate GST‐SIM1, Nup358‐SIM1 oligos were annealed and cloned into EcoRI/XhoI sites of pGEX‐6P1 vector (GE healthcare). HA‐AGO2 mutants—K693R, K402R, and K266R—were generated by PCR‐based method using pcDNA‐HA‐AGO2 as a template and verified by sequencing. pEGFP‐SUMO1G and pEGFP‐SUMO1GG were generated by PCR‐amplifying the SUMO ORF using specific primers and subcloning into pEGFP‐C1 (Clontech) at KpnI and SmaI sites.
For generation of shNup358, the oligos (forward: 5′ GATCCCCGGTGAAGATGGATGGAATATTCAAGAGATATTCCATCCATCTTCACCTTTTTGGAAA 3′; reverse: 5′ AGCTTTTCCAAAAAGGTGAAGATGGATGGAATATCTCTTGAATATTCCATCCATCTTCACCGGG 3′) were annealed and cloned into pSUPER‐EGFP (a gift from Ian Macara, Vanderbilt University Medical Center, TN, USA) at BglII‐HindIII sites. pSUPER‐EGFP vector was used as control.
siRNA‐resistant GFP‐Nup358ΔM was generated for rescue experiments used in Fig EV5. The siRNA target sequence in Nup358 was changed from 5′ GGTGAAGATGGATGGAATA 3′ to 5′ GGCGAGGACGGGTGGAACA 3′, which results in no change in amino acids. Underlined nucleotides are the ones that were changed. The construct was verified by sequencing.
The constructs, RL‐control, RL‐3xBulge, RL‐5BoxB reporters, N‐HA‐AGO1, N‐HA‐AGO2, N‐HA‐AGO3, N‐HA‐AGO4, N‐HA‐LacZ, HA‐MILI, HA‐MIWI, and HA‐HIWI were kind gifts from Ramesh Pillai, EMBL Grenoble, France. pCMV‐FL‐miR30 (P) and pSUPER miR30 constructs were kind gifts from Bryan Cullen (Duke University Medical Center, USA). FL constructs containing wild‐type (HMGA2‐wt) and mutant (HMGA2‐mut) HMGA2 3′‐UTR were kind gifts from Gunter Meister (Universität Regensburg, Germany) 63.
Immunofluorescence microscopy
For immunofluorescence analysis, cells grown on coverslips were fixed using chilled methanol for 5 min. Cells were quickly washed with 0.1% Triton X‐100 in Tris‐buffered saline (TBS) and then incubated with indicated primary antibodies diluted in TBS containing 2% normal horse serum (Vector Laboratories) for 30 min at room temperature (RT). Cells were washed three times with TBS, followed by addition of fluorescently conjugated secondary antibodies and incubation for 30 min at RT. Hoechst 33342 dye (Sigma) was used to stain the DNA, which was added to the secondary antibody solution. Cells were again washed three times with TBS, and the coverslips were mounted on glass slides using Vectashield mounting medium (Vector Laboratories). To avoid dehydration, coverslips were sealed using nail polish, and were later observed under microscope. Images were captured with Leica TCS SP5 or Zeiss 510 Meta laser‐scanning confocal microscope using a Plan Apochromat 63×/1.4 NA oil immersion objective. Images were processed further in Adobe Photoshop CS2. The microscope has been regularly calibrated for alignment/chromatic aberration between two channels, as evident from the intensity profiles obtained using TetraSpeck beads (Appendix Fig S4).
The following antibodies and dilutions were used for immunofluorescence studies: rabbit anti‐Nup358 (1:1,000) generated in the lab 25, mouse anti‐Dcp1a (1:1,000; Sigma, WH0055802M6), rabbit anti‐Dcp1a (1:2,000; a kind gift from Jens Lykke‐Andersen, University of California, San Diego, USA), mouse anti‐RanGAP1 (1:500; Santa Cruz, 28322), rabbit anti‐Nup214 (1:100; Abcam, ab70497), goat anti‐eIF3η (1:500; Santa Cruz, sc‐16377), mouse anti‐Nup62 (1:100; BD, 610497), mouse anti‐protein disulfide isomerase (PDI) (1:1,500; Abcam, ab5484), and mouse anti‐Nup153 (a generous gift from Katharine Ullman, University of Utah, USA) antibodies. The following fluorescently labeled secondary antibodies were used: Alexa Fluor 350, 488, 568, or 594 (1:1,000; Invitrogen).
Live cell imaging
GFP‐Nup358 construct has been described earlier 26. COS7 cells were plated onto 24‐mm coverslips (approximately 2 × 105 cells). After 12 h, cells were co‐transfected with indicated constructs, and after 36 h, coverslips were assembled on Attofluor Cell Chamber (Invitrogen) for live cell imaging in DMEM without phenol red. For analyzing the GFP‐Nup358‐positive AL dynamics (Fig 1C and Movies EV1 and EV2), the expressing COS‐7 cell was imaged for 60 min at 1‐min interval, using Zeiss LSM 780 confocal microscope. To analyze P body–Nup358 or SG–Nup358 association (Movies EV3 and EV4), live imaging was performed for 30 min, at 1‐min interval, using a laser‐scanning confocal microscope (TCS SP5; Leica) with a Plan Apochromat 63× objective (1.4 NA, oil).
For live cell imaging of Nup358 with P bodies or SGs, COS‐7 cells were co‐transfected with GFP‐Nup358 and RFP‐Dcp1a (P bodies) (a gift from Ken Fujimura, University of Tokyo, Japan) or RFP‐G3BP1 (SGs). The construct RFP‐G3BP1 was generated by subcloning human G3BP1‐S149A mutant that constitutively induces SGs upon expression 85, from pEGFP‐G3BP1‐S149A (a generous gift from Jamal Tazi, Institut de Génétique Moléculaire de Montpellier, France) to pDsRed‐Express‐C1 (Clontech) using appropriate restriction sites.
Immunoprecipitation and Western blotting
For co‐immunoprecipitation of AGO2 and GW182 with Nup358, HeLa cells were washed with ice‐cold TBS, scrapped using a cell scrapper, and resuspended in chilled lysis buffer (25 mM Tris–HCl, pH 7.4, 150 mM KCl, 0.5% NP‐40, 2 mM EDTA) supplemented with 7.5 mM NaF, 0.75 mM sodium orthovanadate, 1 mM PMSF, protease inhibitor cocktail (Roche), leupeptin 50 μg/ml, aprotinin 5 μg/ml, and pepstatin 2 μg/ml. To make a clear lysate, mild sonication was given (three pulses at 30% amplitude) and repeated 3–4 times depending upon the initial cell volume. Lysate was centrifuged in Eppendorf centrifuge (5417 R) to remove cellular debris at ∼10,600 g for 10 min at 4°C and was further pre‐cleared by incubating with protein A–Sepharose beads (Invitrogen). Starting material was prepared by mixing 1/10th volume of the pre‐cleared lysate with equal volume 3× SDS–PAGE loading dye and heated at 95°C for 5 min. In the meantime, protein A–Sepharose beads were bound with indicated antibodies by incubating in TBS for 1 h 30 min at 4°C on a rotospin (Bangalore Genei). Antibody‐bound beads were then washed with lysis buffer twice, and the pre‐cleared lysate was incubated for 2 h at 4°C on rotospin. The immunoprecipitates were then washed with lysis buffer twice, followed by a final wash with TBS before eluting in SDS–PAGE loading dye. The immunoprecipitates were separated on SDS–PAGE and transferred to PVDF membrane (Millipore) using semi‐dry transfer apparatus (Bio‐Rad or GE). For Western blotting, PVDF membranes were incubated with primary antibody in 1% BSA in TBS with 0.1% Tween‐20 (TBST) for 2 h at RT or overnight at 4°C. Membranes were washed thrice with TBST for 3 min each and incubated with HRP‐conjugated secondary antibodies in 1% BSA in TBST for 1 h. Membranes were washed thrice with TBST and developed using ECL Plus Western Detection Kit (GE Healthcare or Thermo Scientific) following the manufacturer's instructions. The images were acquired using ImageQuant LAS 4000 (GE Healthcare). EZview™ Red Anti‐HA Affinity beads (Sigma) and rabbit GFP antibody 86‐bound protein A beads (Invitrogen) were used for immunoprecipitation of HA‐ and GFP‐tagged proteins, respectively.
For analyzing the RNA dependence of protein interactions, cell lysis and immunoprecipitations were performed using 100 mM Tris–HCl, pH 7.4, 150 mM NaCl, and 0.05% NP‐40 supplemented with 7.5 mM NaF, 1 mM PMSF, PIC (Roche), and 80 U/ml of murine RNase inhibitor (New England Biolabs). The immunoprecipitates were washed with lysis buffer once and divided equally into two. One of the samples was incubated with TBS and the other one with TBS containing RNase A (100 μg/ml) at 25°C for 10 min. The IP samples were eluted in SDS loading dye and analyzed by Western blotting.
For monitoring the SUMOylation of AGO2, the immunoprecipitation protocol was similar to that used for co‐immunoprecipitation as mentioned above, except that 20 mM N‐ethylmaleimide (NEM) was used in TBS and lysis buffer (20 mM HEPES, pH 8.0, 10 mM KCl, 1 mM MgCl2, 20% glycerol, and 0.01% Triton X‐100).
The following antibodies and dilutions were used for Western blotting: mouse anti‐Nup358 (1:1,000; Santa Cruz Biotechnology Inc., sc‐74518), rabbit anti‐Dicer (1:1,500; Santa Cruz Biotechnology Inc., sc‐30226), rabbit anti‐Nup214 (1:1,000; Bethyl Laboratories, A300‐717A), mouse anti‐GFP (1:8,000; Santa Cruz Biotechnology Inc., sc‐9996), mouse anti‐vinculin (1:10,000; Sigma, V9131), mouse anti‐tubulin (1:10,000; Sigma, T5168), rabbit anti‐TNRC6A/GW182 (1:500; Bethyl Laboratories, A302‐329A), mouse anti‐HA (1:5,000; Covance Research Products, MMS‐101R), rabbit anti‐AGO2 (1:1,000; Cell Signaling Technology, #2897), rat anti‐AGO2 (1:1,000; Millipore, MABE253), rabbit RPL7a (1:80,000; Abcam, ab70753), rabbit anti‐PDI (1:2,000; Santa Cruz Technology, sc‐20132), mouse anti‐LAMP2 (1:1,000; BD Bioscience, 555803), rabbit anti‐exportin‐5 (1:3,000; Santa Cruz Biotechnology Inc., sc‐66885), mouse anti‐PABP (1:5,000; Abcam, ab6125), rabbit anti‐TRBP2 (1:3,000; Abcam, ab42018), mouse anti‐lamin A/C (1:3,000; Santa Cruz Biotechnology Inc., sc‐7292), mouse anti‐RanGAP1 (1:5,000; Santa Cruz Biotechnology, sc‐28322), mouse anti‐Ubc9 (1:10,000; BD Biosciences, 610748), mouse anti‐AGO2 (1:1,000; Sigma‐Aldrich, WH0027161M1), rabbit anti‐Ras (1:10,000; Abcam, ab52939), mouse anti‐c‐Myc (1:1,000; Santa Cruz Biotechnology Inc., sc‐40), mouse anti‐PABPC1 (1:3,000; Abcam, ab6125), donkey anti‐rabbit IgG‐HRP (1:10,000; GE Healthcare, NA‐934), sheep anti‐mouse IgG‐HRP (1:10,000; GE Healthcare, NA‐931), goat anti‐rat IgG‐HRP (1:10,000; GE Healthcare, NA935V), and HRP‐rec‐protein A (1:10,000; Invitrogen, 101123). Rabbit anti‐GFP (1:10,000) and rabbit anti‐Nup358 (1:3,000) antibodies have been described earlier 25, 86.
Dual‐luciferase reporter assay
For dual‐luciferase reporter assay, 4 × 104 HeLa cells were seeded in each well of a 24‐well plate, 12 h prior to siRNA transfection. Control and indicated siRNAs were transfected at a working concentration of 40 nM (unless otherwise mentioned), and cells were incubated for 36 h. Cells were then subjected to a second round of transfection with reporter DNA (20 ng of indicated RL reporter, and 100 ng of pcDNA‐FL as internal control) and incubated for 24–36 h. Cells were lysed in passive lysis buffer and the reporter assay was performed using Dual‐Luciferase assay system (Promega) in Glomax Multi Detection System (Promega). Relative luciferase activity was calculated; values obtained from control and specific knockdown conditions were individually normalized with respect to RL‐control value of each set and plotted with arbitrary units.
For rescue experiments with full‐length Nup358 expression (Fig EV2D), HeLa cells were transfected with 20 nM control (siControl) or Nup358‐specific siRNA (siNup358‐A) for 36 h. Later, reporter constructs (20 ng of RL‐control or RL‐3xBulge reporter and 100 ng of pcDNA‐FL as internal control) along with GFP or GFP‐Nup358 construct (500 ng/well in a 24‐well plate) were transfected for 36 h, before dual‐luciferase assay was performed. For rescue experiments with Nup358ΔM (Fig EV5), HeLa cells were transfected with 40 nM of control or Nup358‐specific siRNA for 60 h, and later, the reporters (20 ng of RL‐3xBulge‐mutant‐control or RL‐3xBulge reporter, and 100 ng of pcDNA‐FL as internal control) were co‐transfected with indicated plasmid GFP‐MBP or GFP‐Nup358ΔM (100 ng/well in a 24‐well plate). GFP‐Nup358ΔM construct was generated by replacing the middle ApaI fragment with a designed annealed pair of oligos to keep the N‐terminal and C‐terminal regions in frame. GFP‐MBP was generated by PCR‐amplifying the ORF for MBP using pMAL‐p2 (New England Biolabs) as the template and cloning the product into pEGFP‐C2 at appropriate restriction sites. Dual‐luciferase assay was performed after 36 h.
For miR30 reporter system, 8 × 104 HEK293T cells were seeded 12 h prior to siRNA transfection. Forty‐eight hours after siRNA transfection, the following constructs were transfected: pCMV‐FL‐miR30 (P) reporter (50 ng) with pSUPER‐control or pSUPER miR30 constructs (2 ng) and RL as internal control (1 ng). The experiment was terminated 12 h after reporter transfection. Relative luciferase activity (FL/RL) was calculated and normalized with respect to FL‐control value of each set and plotted. For HMGA2 3′‐UTR‐containing FL‐reporter (wt and mut) assays, the protocol remained similar to that for RL‐3xBulge, except that 10 ng of HMGA2‐wt or HMGA2‐mut along with 1 ng of RL‐control was transfected and the relative FL/RL activity was measured 63.
Dominant‐negative effect of Nup358‐SIM1 region on miRNA pathway was assessed as described below. For dual‐luciferase reporter assay, 8 × 104 HeLa cells were seeded in each well of a 24‐well plate, 12 h prior to transfection. Cells were transfected with reporter DNA (25 ng of indicated Renilla luciferase (RL)‐reporter and 100 ng of pcDNA‐firefly luciferase (FL) as internal control) and plasmid DNA (500 ng of GFP‐MBP or GFP‐SIM1 or 130 ng of GFP‐SIM1 mutant with 370 ng of fill‐in DNA) for 36 h. Transfection was performed using Lipofectamine 2000 (Invitrogen). Dual‐luciferase assay was performed as described earlier. Relative luciferase (RL/FL ratio) activity was calculated; values obtained were individually normalized with respect to RL‐control value of each set and plotted.
Tethering assay was performed in HEK293T cells. 4 × 104 cells were seeded in each well of a 24‐well plate. Control or Nup358 siRNA was transfected for 48 h. Then, 25 ng of RL‐5BoxB reporter and 50 ng of pcDNA‐FL were transfected as internal control. The reporters (500 ng) were co‐transfected with HA‐AGO2, N‐HA‐AGO2, or N‐HA‐LacZ. Assay was performed after 24 h of transfection. In some tethering assays, HA‐IR1 + 2, N‐HA‐IR1 + 2, or N‐HA‐IR1 + 2L2651A,L2653A was used.
Tethering assay using Nup358‐IR1 + 2 was performed in HEK293T cells as described below. 1 × 105 cells were seeded in each well of a 24‐well plate. Then, 25 ng of RL‐5BoxB reporter and 100 ng of pcDNA‐FL were transfected as internal control. The reporters were co‐transfected with N‐HA‐MBP, N‐HA‐AGO2, HA‐IR1 + 2L2651A,L2653A, or N‐HA‐IR1 + 2L2651A,L2653A (500 ng each) for 24 h. Polyethyleneimine (Polysciences Inc.) was used as transfecting reagent. Cells were washed with 1× TBS once and lysed in passive lysis buffer, and dual‐luciferase assay was performed. Relative luciferase activity (RL/FL ratio) was calculated, and values obtained were individually normalized with respect to the RL‐control value with N‐HA‐MBP and plotted.
RNA isolation
RNA was isolated from cell lysates using TRIzol (Invitrogen). Three volumes of TRIzol was added to one volume of the lysate and incubated for 15 min at RT. To this, 1/5th volume of chloroform was added and rigorously agitated for homogenous mixing. The mixture was then incubated on ice for 5 min and centrifuged at ∼15,200 g for 12 min using Eppendorf centrifuge (5417 R). Upper aqueous layer was aspirated into another tube without disturbing the interface. An equal volume of isopropanol was added and incubated for 1 h at −20°C. The tube was then centrifuged at ∼15,200 g for 12 min. The RNA pellet obtained was washed with 70% ethanol twice, followed by a brief centrifugation. The remaining ethanol was removed and the pellet was left at RT for air‐drying. The pellet was then dissolved in autoclaved glass distilled water, heated at 65°C for 5 min, and stored at −80°C. In case of RNA isolation for miRNA qPCR, heating step was not included.
RNA immunoprecipitation
To analyze the relative association of mRNA with AGO2, HeLa cells were grown in 100‐mm culture dishes and were transfected with control, AGO2, Dicer, and Nup358 siRNA and incubated for 36 h. Further, cells were transfected with RL‐3xBulge construct and incubated for 24 h. For RNA immunoprecipitation analysis, cells were washed with ice‐cold 1× PBS, removed using a cell scrapper, and resuspended in chilled NET2 buffer (100 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.05% NP‐40) supplemented with 7.5 mM NaF, 1 mM PMSF, PIC (Roche), and 100 U/ml of murine RNase inhibitor. To make a clear lysate, mild sonication was given, three pulses at 30% amplitude, and repeated thrice. Lysate was centrifuged to clear cellular debris at ∼15,200 g for 10 min at 4°C. Lysate was further pre‐cleared by incubating the supernatant with protein G–Sepharose beads (Invitrogen) for 1 h at 4°C. A part of the pre‐cleared lysate was taken out as starting material for RNA isolation and Western analysis. For AGO2 immunoprecipitation, mouse anti‐AGO2 (Sigma, WH0027161M1) or rat anti‐AGO2 (Sigma, SAB4200085) antibodies were used. Mouse IgG (Vector Laboratories) and rat IgG (BD Pharmingen) were used as respective control IgGs. In the meantime, tRNA‐saturated beads were prepared by incubating protein A–Sepharose beads with yeast tRNA (Invitrogen), herring sperm DNA (Sigma), and glycogen (Sigma) in NET2 buffer for 90 min at 4°C. Finally, tRNA‐saturated beads were incubated with the lysate containing antibodies and incubated for 1 h at 4°C. Beads were then washed 5–7 times with NET2 buffer and the RNA was isolated from beads using TRIzol as mentioned earlier, except that the RNA precipitation was performed for 4 h at −20°C. RNA samples were further used for cDNA synthesis, followed by real‐time PCR analysis as described below.
For assessing the extent of AGO2 association with endogenous target mRNAs (Serbp1 or DnaJB1), the same procedure was followed, except that the cells were not transfected with RL‐3xBulge construct. The mRNAs in the AGO2‐IP were quantified using specific primers described earlier 63.
For analyzing the association of miRNA and mRNA with Nup358, HeLa cells were transfected with the miRNA reporter construct RL‐3xBulge. Twenty hours post‐transfection, cells were washed thrice with PBS and then cross‐linked with 1% formaldehyde for 10 min. Glycine was added to a final concentration of 0.25 M for neutralization for 5 min. The cells were washed with PBS three times and lysed in RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% NP‐40, 0.1% SDS, 0.5% sodium deoxycholate) containing PIC (Roche) and 100 U/ml of murine RNase inhibitor). Following a mild sonication, the lysate was centrifuged at 12,000 g for 10 min. The supernatant was then incubated with rabbit IgG (control) or Nup358‐specific antibodies for 45 min at 4°C. In the meanwhile, protein A–Sepharose beads were coated with herring serum DNA, tRNA, and glycogen for 30 min. The lysate was then incubated with coated beads for 45 min at 4°C. The immunoprecipitate was given five washes with RIPA buffer containing 1 M NaCl. Delinking was done with 1% SDS, 10 mM DTT, 5 mM EDTA, and 50 mM Tris, pH 7.4, for 1 h at 70°C, followed by proteinase K treatment (10 μg/ml) for 45 min at 55°C. RNA was isolated using TRIzol method as mentioned above, and RNA precipitation was done overnight in the presence of glycogen. cDNA synthesis for miRNA and mRNA and the respective qPCRs were done using the protocols mentioned below.
cDNA synthesis and semi‐quantitative PCR
Total RNA isolated was subjected to DNase I treatment before cDNA synthesis. One microgram total RNA, 1 μl 10× DNase I reaction buffer, and 1 μl DNase I, Amp grade 1 U/μl (Invitrogen), were mixed and the volume was made up to 10 μl as per the manufacturer's instructions and incubated for 20–30 min at 37°C. DNase I was inactivated by addition of 1 μl of 25 mM EDTA solution and incubating the mixture for 10 min at 65°C. Reaction was scaled up as per requirement. cDNA synthesis was performed with Oligo(dT)20 primers or random primers using SuperScript III First‐Strand Synthesis kit (Invitrogen) as per the manufacturer's instructions. The synthesized cDNA was further diluted and used as template for PCR amplification.
Real‐time quantitative PCR
Isolated RNA from input, control, or AGO2 immunoprecipitate was treated with DNase I, and cDNA synthesis was performed as described above. To assess the specific association of miRNA target (RL‐3xBulge) with AGO2, quantitative PCR (qPCR) was performed with the following primers on a Rotor‐Gene Q (Qiagen) using the SYBR Select Master Mix (Invitrogen). GAPDH was used as negative control. The primers used were Renilla luciferase (forward: 5′ CGAGCACCAAGACAAGATCA 3′; reverse: 5′ GTAGGCAGCGAACTCCTCAG 3′) and GAPDH (forward: 5′ GATTCCACCCATGGCAAATTC 3′; reverse: 5′ AGCATCGCCCCACTTGATT 3′).
For relative association of let‐7a and miR‐17 with AGO2, total RNA isolated from input, control, or AGO2 immunoprecipitate was used. Association of U6 RNA with AGO2 was monitored to assess the extent of non‐specificity. Using TaqMan MicroRNA Reverse Transcription Kits (Invitrogen, 4427975; ID: 000377, 002308, 001973), the RNA was converted to cDNA with specific microRNA primers supplied with specific kits. qPCR mix was prepared according to the TaqMan Small RNA Assay (Invitrogen) protocol, and the PCR was performed on a Rotor‐Gene Q (Qiagen). A similar protocol was followed for isolation and quantitation of let‐7a in control and Nup358‐depleted HeLa cells.
Oligo(dT) staining
HeLa cells seeded on coverslips were fixed with 4% paraformaldehyde in 1× TBS for 15 min at RT. Chilled methanol was added to the cells and incubated for 5 min. This was followed by incubation of the coverslips with 2× SSC at RT for 10 min. Further, hybridization chamber was used to carry out the hybridization process. In the process, moistened strips of filter paper were kept on the edges of the stage to avoid the coverslips from drying out. The hybridization mix was made up of the following components: 40% formamide, 10% dextran sulfate, 0.1 mg herring sperm DNA, and 5 ng/μl of FAM oligo(dT) in a 2× SSC solution. One hundred microliters of the hybridization mix was added per coverslip. Hybridization was carried out at 37°C for 3 h. Further, the coverslips were washed twice with 2× SSC followed by one wash with 0.2× SSC. Followed by three washes with 1× TBS, the cells were incubated with Hoechst 33342 diluted in 1× TBS. Cells were then washed with 1× TBS and coverslips were mounted using Vectashield mounting medium (Vector Laboratories) and observed under microscope.
Nucleo‐cytoplasmic fractionation
Control and Nup358 siRNA‐transfected HeLa cells were resuspended in cytoplasmic extraction buffer (20 mM HEPES, pH 8.0, 10 mM KCl, 1 mM MgCl2, 20% glycerol, 0.2% Triton X‐100). Cells were lysed by gently pipetting for 45 min at an interval of 15 min. Lysate was centrifuged with Eppendorf centrifuge (5417 R) at ∼15,200 g for 20 min. Supernatant was collected as cytoplasmic fraction and the pellet was further processed for nuclear fraction after washing three times with 1× TBS. Nuclear lysis buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 2 mM EGTA, 2 mM DTT, 1% Triton X‐100) was added to the pellet and then sonicated for 5 s with a pulse of 1 s on and 2 s off at 30% amplitude using Vibra‐Cell sonicator (Sonics & Materials Inc.). Lysis was carried out for 45 min by sonicating the samples at an interval of 15 min. Lysate was then centrifuged at ∼15,200 g at 4°C for 20 min. The supernatant was collected as nuclear fraction. The proteins were separated by SDS–PAGE and analyzed by Western blotting. Vinculin and lamin A/C were used as markers for cytoplasmic and nuclear fractions, respectively. Cytoplasmic and nuclear lysates were mixed in equal proportion to make total protein fraction. For total fraction, double the volume of lysate was loaded for analysis.
Membrane flotation assay
Membrane flotation assay was carried out as described earlier 11, 87. Briefly, 6 × 106 HeLa cells were seeded in each 60‐mm dish. Twelve hours later, cells were transfected with 40 nM of control or Nup358 siRNA for 72 h. Cells in each dish were washed twice with ice‐cold 1× PBS and lysed in 1 ml of hypotonic buffer [10 mM Tris, pH 8, 10 mM KCl, 1.5 mM MgCl2, 10 mM DTT, and EDTA‐free protease inhibitor cocktail (Roche)] by passing through a 23‐gauge needle 15 times. Lysates were centrifuged at 1,000 g for 5 min at 4°C. The resultant post‐nuclear supernatant (446 μl) was mixed with 72% (w/v) sucrose made in hypotonic buffer (1.78 ml) and overlaid with 55% (w/v) sucrose (2.23 ml), followed by 0.54 ml of 10% (w/v) sucrose (0.54 ml). Ultracentrifugation was performed at 140,000 g (4°C) using MLS‐50 rotor for 12 h. After centrifugation, 1‐ml fractions were collected from top of the gradient. To each fraction, 5 ml of hypotonic buffer was initially added and mixed, followed by 1.5 ml of 100% TCA. Proteins were precipitated by centrifugation at 1,400 g (4°C) for 5 min. The protein pellets were washed four times with ice‐cold acetone and air‐dried before resuspending in 25 μl of NP‐40 lysis buffer. After addition of 25 μl of 3× SDS loading dye, the samples were heated at 95°C for 5 min and subjected to SDS–PAGE for Western analysis.
Northern blotting
For Northern blotting, 10 μg of total RNA was resolved on a 12% urea–PAGE and electro‐transferred to Hybond N+ nylon membrane (GE Healthcare). The membrane was UV‐cross‐linked and washed for 1 h at 65°C in prewash buffer (0.1× SSC, 0.1% SDS). Prehybridization was performed at 42°C in hybridization solution (10× Denhardt's solution, 6× SSC, 0.1% SDS) for 6 h. Body‐labeled oligonucleotides complementary to miRNAs were used as probes for Northern blotting. Radiolabeled probes were heat‐denatured at 65°C for 5 min and added to the hybridization solution and kept for overnight incubation at 42°C. The membrane was washed twice for 10 min at 25°C in wash solution (6× SSC, 0.1% SDS) and was exposed to X‐ray film and analyzed by autoradiography.
miRNA deep sequencing
HEK293T cells were transfected with control or Nup358 shRNA. Total RNA was isolated and miRNA deep sequencing was performed at Genotypic Technology, Bengaluru, India. Small RNA libraries for sequencing were constructed according to the Illumina TruSeq Small RNA preparation Guide. One microgram of total RNA was used as the starting material. Briefly, 3′ adaptors were ligated to the specific 3′OH group of small RNAs followed by 5′ adaptor ligation. The ligated products were reverse‐transcribed with SuperScript II reverse transcriptase. The cDNA was enriched by PCR and cleaned using polyacrylamide gel electrophoresis. The library was size selected in the range of 140–180 bp followed by overnight gel elution and precipitation using glycogen, 3 M sodium acetate, and absolute ethanol. The precipitate was resuspended in resuspension buffer. The prepared library was quantified using Nanodrop and Qubit Fluorometer and validated for quality by running an aliquot on High Sensitivity Bioanalyzer Chip (Agilent). The libraries were sequenced for 54SE small RNA sequencing in GAIIX (Illumina). The relative expression of miRNAs was analyzed by GeneSpring NGS software. The values were log‐transformed and the heatmap was generated using Matplotlib. The corresponding fold change has been represented by bar graphs.
Bacterial expression of proteins and pull‐down assays
GST‐control (pGEX‐6P1), GST‐Nup358‐SIM1, MBP (pMAL‐c2), or pMAL‐hAGO2 was transformed in Escherichia coli BL21 pLysS strain and induced with 0.5 mM IPTG at 18°C for 6 h. Cells were lysed in lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.05% NP‐40) supplemented with 7.5 mM NaF, 0.75 mM sodium orthovanadate, 1 mM PMSF, protease inhibitor cocktail (PIC, Roche), leupeptin (50 μg/ml), aprotinin (5 μg/ml), and pepstatin (2 μg/ml) and lysozyme (200 μg/ml). The indicated lysates were mixed and GST pull‐down assay was performed. The proteins were eluted in 3× SDS–PAGE loading dye and Western blot analysis was performed as described above.
Structural analysis
To identify possible Nup358 binding sites on AGO2, we compared the 3D structure of AGO2 (PDB ID: 4W5O) 88 with that of SUMO (PDB ID: 1Z5S) 47. The chosen SUMO structure was in complex with the SIM1 of Nup358. Structural superimpositions were performed using the CLICK program 75, 76 that identifies similar sub‐structures regardless of topology. All superimpositions were carried out with full‐length AGO2 and a truncated SUMO molecule comprising the residues 20–54 and 86–91. These regions on SUMO consisted of regular secondary structures that were most proximal to the bound Nup358. On superimposing the AGO2 with SUMO structures, the position of the Nup358 was transferred onto the former. Of the 93 different superimpositions obtained, 46 cases had collisions between Nup358‐SIM1 and the AGO2 molecule. Therefore, these superimpositions were deemed unphysical and omitted from further consideration. The remaining 47 alignments were clustered by spatial proximity to one another, and one structure was chosen to represent each cluster (Fig 6F).
Statistical analysis
The experiments were independently repeated at least three times (n = 3), and the values are expressed as mean ± SD. P‐values were calculated using Student's t‐test (SigmaStat2.03). P‐value ≤ 0.05 was considered statistically significant. Graphs were plotted using SigmaPlot8.0.
Author contributions
MRS conducted most of the experiments regarding Nup358's role in miRNA pathway, SG mostly worked on Nup358–AGO interaction, and DK mainly contributed to the characterization of AL and its association with mRNP granules. MA, MH, SKY, AS, IM, PD, SD, and PKS supported design and execution of some of the experiments. YR, MSM, and PG contributed to identification of putative SIM‐binding regions in AGO2. VS contributed to experimental design and provided scientific advice. JJ contributed to overall design and supervision and along with other authors wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Review Process File
Acknowledgements
We thank Ramesh Pillai (EMBL Grenoble, France), Gunter Meister (Universität Regensburg, Germany), Bryan Cullen (Duke University Medical Center, USA), Ken Fujimura (University of Tokyo, Japan), Jamal Tazi (Institut de Génétique Moléculaire de Montpellier, France), Shigeyuki Yokoyama (RIKEN Genomic Sciences Center, Yokohama, Japan), Katharine Ullman (University of Utah, USA), Mary Dasso (NICHD, NIH, USA), and Ian Macara (Vanderbilt University Medical Center, TN, USA) for sharing reagents. We are grateful to Joseph and Seshadri lab members for scientific discussions and helpful suggestions. We thank Shekhar Mande (NCCS, Pune) and Radha Chauhan (NCCS, Pune) for insightful discussions on AGO, SUMO, and SIM structures. Deepa Subramanyam (NCCS, Pune) and Jyotsna Dhawan (CCMB, Hyderabad) are acknowledged for critical reading of the manuscript. Financial support from Council of Scientific and Industrial Research, Government of India, to M.R. Sahoo, D. Khuperkar, S. Gaikwad, S.K. Yadav, A. Singh, I. Magre, and P. Deshmukh, from Department of Biotechnology, Government of India, to M. Ashok, M. Helen, Y. Ramtirtha and S. Dhanvijay, and from INSPIRE (DST) and IYBA (DBT) fellowships to P. Gayathri is gratefully acknowledged. MSM would like to acknowledge the Wellcome trust‐DBT India alliance for a senior fellowship. We thank Pankhuri Vyas for generating pSUPER Nup358 shRNA clones, Pravin Sawale for generating GFP‐SUMO1G and GFP‐SUMO1GG clones, and Ashwini Atre (NCCS, Pune) and Richa Ricky (IISER, Pune) for the help with microscopy. We are grateful to Gaurang Mahajan (NCCS, Pune) for helping with miRNA analysis. The work was partly supported by intramural funding from NCCS and a grant (EMR/2014/001092) from Department of Science and Technology, Government of India.
EMBO Reports (2017) 18: 241–263
References
- 1. Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Friedman RC, Farh KK, Burge CB, Bartel DP (2009) Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ha M, Kim VN (2014) Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15: 509–524 [DOI] [PubMed] [Google Scholar]
- 4. Meister G (2013) Argonaute proteins: functional insights and emerging roles. Nat Rev Genet 14: 447–459 [DOI] [PubMed] [Google Scholar]
- 5. Liu J, Rivas FV, Wohlschlegel J, Yates JR III, Parker R, Hannon GJ (2005) A role for the P‐body component GW182 in microRNA function. Nat Cell Biol 7: 1261–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sen GL, Blau HM (2005) Argonaute 2/RISC resides in sites of mammalian mRNA decay known as cytoplasmic bodies. Nat Cell Biol 7: 633–636 [DOI] [PubMed] [Google Scholar]
- 7. El Shami M, Pontier D, Lahmy S, Braun L, Picart C, Vega D, Hakimi MA, Jacobsen SE, Cooke R, Lagrange T (2007) Reiterated WG/GW motifs form functionally and evolutionarily conserved ARGONAUTE‐binding platforms in RNAi‐related components. Genes Dev 21: 2539–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Till S, Lejeune E, Thermann R, Bortfeld M, Hothorn M, Enderle D, Heinrich C, Hentze MW, Ladurner AG (2007) A conserved motif in Argonaute‐interacting proteins mediates functional interactions through the Argonaute PIWI domain. Nat Struct Mol Biol 14: 897–903 [DOI] [PubMed] [Google Scholar]
- 9. Siomi MC, Sato K, Pezic D, Aravin AA (2011) PIWI‐interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol 12: 246–258 [DOI] [PubMed] [Google Scholar]
- 10. Li S, Liu L, Zhuang X, Yu Y, Liu X, Cui X, Ji L, Pan Z, Cao X, Mo B et al (2013) MicroRNAs inhibit the translation of target mRNAs on the endoplasmic reticulum in Arabidopsis . Cell 153: 562–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stalder L, Heusermann W, Sokol L, Trojer D, Wirz J, Hean J, Fritzsche A, Aeschimann F, Pfanzagl V, Basselet P et al (2013) The rough endoplasmatic reticulum is a central nucleation site of siRNA‐mediated RNA silencing. EMBO J 32: 1115–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hetzer MW, Wente SR (2009) Border control at the nucleus: biogenesis and organization of the nuclear membrane and pore complexes. Dev Cell 17: 606–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cronshaw JM, Krutchinsky AN, Zhang W, Chait BT, Matunis MJ (2002) Proteomic analysis of the mammalian nuclear pore complex. J Cell Biol 158: 915–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoelz A, Debler EW, Blobel G (2011) The structure of the nuclear pore complex. Annu Rev Biochem 80: 613–643 [DOI] [PubMed] [Google Scholar]
- 15. Chatel G, Fahrenkrog B (2012) Dynamics and diverse functions of nuclear pore complex proteins. Nucleus 3: 162–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chow KH, Factor RE, Ullman KS (2012) The nuclear envelope environment and its cancer connections. Nat Rev Cancer 12: 196–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kessel RG (1992) Annulate lamellae: a last frontier in cellular organelles. Int Rev Cytol 133: 43–120 [DOI] [PubMed] [Google Scholar]
- 18. Meier E, Miller BR, Forbes DJ (1995) Nuclear pore complex assembly studied with a biochemical assay for annulate lamellae formation. J Cell Biol 129: 1459–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wischnitzer S (1970) The annulate lamellae. Int Rev Cytol 27: 65–100 [DOI] [PubMed] [Google Scholar]
- 20. Onischenko EA, Gubanova NV, Kieselbach T, Kiseleva EV, Hallberg E (2004) Annulate lamellae play only a minor role in the storage of excess nucleoporins in Drosophila embryos. Traffic 5: 152–164 [DOI] [PubMed] [Google Scholar]
- 21. Patterson JR, Wood MP, Schisa JA (2011) Assembly of RNP granules in stressed and aging oocytes requires nucleoporins and is coordinated with nuclear membrane blebbing. Dev Biol 353: 173–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Asally M, Yasuda Y, Oka M, Otsuka S, Yoshimura SH, Takeyasu K, Yoneda Y (2011) Nup358, a nucleoporin, functions as a key determinant of the nuclear pore complex structure remodeling during skeletal myogenesis. FEBS J 278: 610–621 [DOI] [PubMed] [Google Scholar]
- 23. Cho KI, Yi H, Desai R, Hand AR, Haas AL, Ferreira PA (2009) RANBP2 is an allosteric activator of the conventional kinesin‐1 motor protein, KIF5B, in a minimal cell‐free system. EMBO Rep 10: 480–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Joseph J, Tan SH, Karpova TS, McNally JG, Dasso M (2002) SUMO‐1 targets RanGAP1 to kinetochores and mitotic spindles. J Cell Biol 156: 595–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Joseph J, Liu ST, Jablonski SA, Yen TJ, Dasso M (2004) The RanGAP1‐RanBP2 complex is essential for microtubule‐kinetochore interactions in vivo. Curr Biol 14: 611–617 [DOI] [PubMed] [Google Scholar]
- 26. Joseph J, Dasso M (2008) The nucleoporin Nup358 associates with and regulates interphase microtubules. FEBS Lett 582: 190–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murawala P, Tripathi MM, Vyas P, Salunke A, Joseph J (2009) Nup358 interacts with APC and plays a role in cell polarization. J Cell Sci 122: 3113–3122 [DOI] [PubMed] [Google Scholar]
- 28. Pichler A, Gast A, Seeler JS, Dejean A, Melchior F (2002) The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 108: 109–120 [DOI] [PubMed] [Google Scholar]
- 29. Prunuske AJ, Liu J, Elgort S, Joseph J, Dasso M, Ullman KS (2006) Nuclear envelope breakdown is coordinated by both Nup358/RanBP2 and Nup153, two nucleoporins with zinc finger modules. Mol Biol Cell 17: 760–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Salina D, Enarson P, Rattner JB, Burke B (2003) Nup358 integrates nuclear envelope breakdown with kinetochore assembly. J Cell Biol 162: 991–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vyas P, Singh A, Murawala P, Joseph J (2013) Nup358 interacts with Dishevelled and aPKC to regulate neuronal polarity. Biol Open 2: 1270–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Frohnert C, Hutten S, Walde S, Nath A, Kehlenbach RH (2014) Importin 7 and Nup358 promote nuclear import of the protein component of human telomerase. PLoS One 9: e88887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hamada M, Haeger A, Jeganathan KB, van Ree JH, Malureanu L, Walde S, Joseph J, Kehlenbach RH, van Deursen JM (2011) Ran‐dependent docking of importin‐beta to RanBP2/Nup358 filaments is essential for protein import and cell viability. J Cell Biol 194: 597–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hutten S, Flotho A, Melchior F, Kehlenbach RH (2008) The Nup358‐RanGAP complex is required for efficient importin alpha/beta‐dependent nuclear import. Mol Biol Cell 19: 2300–2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hutten S, Walde S, Spillner C, Hauber J, Kehlenbach RH (2009) The nuclear pore component Nup358 promotes transportin‐dependent nuclear import. J Cell Sci 122: 1100–1110 [DOI] [PubMed] [Google Scholar]
- 36. Walde S, Thakar K, Hutten S, Spillner C, Nath A, Rothbauer U, Wiemann S, Kehlenbach RH (2012) The nucleoporin Nup358/RanBP2 promotes nuclear import in a cargo‐ and transport receptor‐specific manner. Traffic 13: 218–233 [DOI] [PubMed] [Google Scholar]
- 37. Dawlaty MM, Malureanu L, Jeganathan KB, Kao E, Sustmann C, Tahk S, Shuai K, Grosschedl R, van Deursen JM (2008) Resolution of sister centromeres requires RanBP2‐mediated SUMOylation of topoisomerase IIalpha. Cell 133: 103–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Klein UR, Haindl M, Nigg EA, Muller S (2009) RanBP2 and SENP3 function in a mitotic SUMO2/3 conjugation‐deconjugation cycle on Borealin. Mol Biol Cell 20: 410–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sakin V, Richter SM, Hsiao HH, Urlaub H, Melchior F (2015) Sumoylation of the GTPase ran by the RanBP2 SUMO E3 ligase complex. J Biol Chem 290: 23589–23602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gareau JR, Lima CD (2010) The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol 11: 861–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Y, Dasso M (2009) SUMOylation and deSUMOylation at a glance. J Cell Sci 122: 4249–4252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Flotho A, Melchior F (2013) Sumoylation: a regulatory protein modification in health and disease. Annu Rev Biochem 82: 357–385 [DOI] [PubMed] [Google Scholar]
- 43. Hecker CM, Rabiller M, Haglund K, Bayer P, Dikic I (2006) Specification of SUMO1‐ and SUMO2‐interacting motifs. J Biol Chem 281: 16117–16127 [DOI] [PubMed] [Google Scholar]
- 44. Mahajan R, Delphin C, Guan T, Gerace L, Melchior F (1997) A small ubiquitin‐related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell 88: 97–107 [DOI] [PubMed] [Google Scholar]
- 45. Matunis MJ, Wu J, Blobel G (1998) SUMO‐1 modification and its role in targeting the Ran GTPase‐activating protein, RanGAP1, to the nuclear pore complex. J Cell Biol 140: 499–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Saitoh H, Sparrow DB, Shiomi T, Pu RT, Nishimoto T, Mohun TJ, Dasso M (1998) Ubc9p and the conjugation of SUMO‐1 to RanGAP1 and RanBP2. Curr Biol 8: 121–124 [DOI] [PubMed] [Google Scholar]
- 47. Reverter D, Lima CD (2005) Insights into E3 ligase activity revealed by a SUMO‐RanGAP1‐Ubc9‐Nup358 complex. Nature 435: 687–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Song J, Durrin LK, Wilkinson TA, Krontiris TG, Chen Y (2004) Identification of a SUMO‐binding motif that recognizes SUMO‐modified proteins. Proc Natl Acad Sci USA 101: 14373–14378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Namanja AT, Li YJ, Su Y, Wong S, Lu J, Colson LT, Wu C, Li SS, Chen Y (2012) Insights into high affinity small ubiquitin‐like modifier (SUMO) recognition by SUMO‐interacting motifs (SIMs) revealed by a combination of NMR and peptide array analysis. J Biol Chem 287: 3231–3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tatham MH, Kim S, Jaffray E, Song J, Chen Y, Hay RT (2005) Unique binding interactions among Ubc9, SUMO and RanBP2 reveal a mechanism for SUMO paralog selection. Nat Struct Mol Biol 12: 67–74 [DOI] [PubMed] [Google Scholar]
- 51. Werner A, Flotho A, Melchior F (2012) The RanBP2/RanGAP1*SUMO1/Ubc9 complex is a multisubunit SUMO E3 ligase. Mol Cell 46: 287–298 [DOI] [PubMed] [Google Scholar]
- 52. Gareau JR, Reverter D, Lima CD (2012) Determinants of small ubiquitin‐like modifier 1 (SUMO1) protein specificity, E3 ligase, and SUMO‐RanGAP1 binding activities of nucleoporin RanBP2. J Biol Chem 287: 4740–4751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pichler A, Knipscheer P, Saitoh H, Sixma TK, Melchior F (2004) The RanBP2 SUMO E3 ligase is neither HECT‐ nor RING‐type. Nat Struct Mol Biol 11: 984–991 [DOI] [PubMed] [Google Scholar]
- 54. Pemberton LF, Paschal BM (2005) Mechanisms of receptor‐mediated nuclear import and nuclear export. Traffic 6: 187–198 [DOI] [PubMed] [Google Scholar]
- 55. Stewart M (2007) Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol 8: 195–208 [DOI] [PubMed] [Google Scholar]
- 56. Swaminathan S, Kiendl F, Korner R, Lupetti R, Hengst L, Melchior F (2004) RanGAP1*SUMO1 is phosphorylated at the onset of mitosis and remains associated with RanBP2 upon NPC disassembly. J Cell Biol 164: 965–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cordes VC, Reidenbach S, Rackwitz HR, Franke WW (1997) Identification of protein p270/Tpr as a constitutive component of the nuclear pore complex‐attached intranuclear filaments. J Cell Biol 136: 515–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wu X, Kasper LH, Mantcheva RT, Mantchev GT, Springett MJ, van Deursen JM (2001) Disruption of the FG nucleoporin NUP98 causes selective changes in nuclear pore complex stoichiometry and function. Proc Natl Acad Sci USA 98: 3191–3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke‐Andersen J, Fritzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P (2005) Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol 169: 871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Eulalio A, Behm‐Ansmant I, Schweizer D, Izaurralde E (2007) P‐body formation is a consequence, not the cause, of RNA‐mediated gene silencing. Mol Cell Biol 27: 3970–3981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, Filipowicz W (2005) Inhibition of translational initiation by Let‐7 MicroRNA in human cells. Science 309: 1573–1576 [DOI] [PubMed] [Google Scholar]
- 62. Zeng Y, Yi R, Cullen BR (2003) MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc Natl Acad Sci USA 100: 9779–9784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Weinmann L, Hock J, Ivacevic T, Ohrt T, Mutze J, Schwille P, Kremmer E, Benes V, Urlaub H, Meister G (2009) Importin 8 is a gene silencing factor that targets argonaute proteins to distinct mRNAs. Cell 136: 496–507 [DOI] [PubMed] [Google Scholar]
- 64. Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ (2005) RAS is regulated by the let‐7 microRNA family. Cell 120: 635–647 [DOI] [PubMed] [Google Scholar]
- 65. Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, Elble R, Watabe K, Mo YY (2009) p53 represses c‐Myc through induction of the tumor suppressor miR‐145. Proc Natl Acad Sci USA 106: 3207–3212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sampson VB, Rong NH, Han J, Yang Q, Aris V, Soteropoulos P, Petrelli NJ, Dunn SP, Krueger LJ (2007) MicroRNA let‐7a down‐regulates MYC and reverts MYC‐induced growth in Burkitt lymphoma cells. Cancer Res 67: 9762–9770 [DOI] [PubMed] [Google Scholar]
- 67. Pillai RS, Artus CG, Filipowicz W (2004) Tethering of human Ago proteins to mRNA mimics the miRNA‐mediated repression of protein synthesis. RNA 10: 1518–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Braun JE, Huntzinger E, Izaurralde E (2013) The role of GW182 proteins in miRNA‐mediated gene silencing. Adv Exp Med Biol 768: 147–163 [DOI] [PubMed] [Google Scholar]
- 69. Mahadevan K, Zhang H, Akef A, Cui XA, Gueroussov S, Cenik C, Roth FP, Palazzo AF (2013) RanBP2/Nup358 potentiates the translation of a subset of mRNAs encoding secretory proteins. PLoS Biol 11: e1001545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nguyen CD, Mansfield RE, Leung W, Vaz PM, Loughlin FE, Grant RP, Mackay JP (2011) Characterization of a family of RanBP2‐type zinc fingers that can recognize single‐stranded RNA. J Mol Biol 407: 273–283 [DOI] [PubMed] [Google Scholar]
- 71. Zhao Q, Xie Y, Zheng Y, Jiang S, Liu W, Mu W, Liu Z, Zhao Y, Xue Y, Ren J (2014) GPS‐SUMO: a tool for the prediction of sumoylation sites and SUMO‐interaction motifs. Nucleic Acids Res 42: W325–W330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Novatchkova M, Bachmair A, Eisenhaber B, Eisenhaber F (2005) Proteins with two SUMO‐like domains in chromatin‐associated complexes: the RENi (Rad60‐Esc2‐NIP45) family. BMC Bioinformatics 6: 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Urulangodi M, Sebesta M, Menolfi D, Szakal B, Sollier J, Sisakova A, Krejci L, Branzei D (2015) Local regulation of the Srs2 helicase by the SUMO‐like domain protein Esc2 promotes recombination at sites of stalled replication. Genes Dev 29: 2067–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kerscher O (2007) SUMO junction‐what's your function? New insights through SUMO‐interacting motifs. EMBO Rep 8: 550–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nguyen MN, Tan KP, Madhusudhan MS (2011) CLICK–topology‐independent comparison of biomolecular 3D structures. Nucleic Acids Res 39: W24–W28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nguyen MN, Madhusudhan MS (2011) Biological insights from topology independent comparison of protein 3D structures. Nucleic Acids Res 39: e94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Josa‐Prado F, Henley JM, Wilkinson KA (2015) SUMOylation of Argonaute‐2 regulates RNA interference activity. Biochem Biophys Res Commun 464: 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sahin U, Lapaquette P, Andrieux A, Faure G, Dejean A (2014) Sumoylation of human Argonaute 2 at lysine‐402 regulates its stability. PLoS One 9: e102957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Truong K, Su Y, Song J, Chen Y (2011) Entropy‐driven mechanism of an E3 ligase. Biochemistry 50: 5757–5766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schisa JA (2012) New insights into the regulation of RNP granule assembly in oocytes. Int Rev Cell Mol Biol 295: 233–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Updike DL, Strome S (2009) A genomewide RNAi screen for genes that affect the stability, distribution and function of P granules in Caenorhabditis elegans . Genetics 183: 1397–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Updike DL, Hachey SJ, Kreher J, Strome S (2011) P granules extend the nuclear pore complex environment in the C. elegans germ line. J Cell Biol 192: 939–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Voronina E, Seydoux G, Sassone‐Corsi P, Nagamori I (2011) RNA granules in germ cells. Cold Spring Harb Perspect Biol 3: a002774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sheth U, Pitt J, Dennis S, Priess JR (2010) Perinuclear P granules are the principal sites of mRNA export in adult C. elegans germ cells. Development 137: 1305–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tourriere H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, Tazi J (2003) The RasGAP‐associated endoribonuclease G3BP assembles stress granules. J Cell Biol 160: 823–831 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 86. Sahoo PK, Murawala P, Sawale PT, Sahoo MR, Tripathi MM, Gaikwad SR, Seshadri V, Joseph J (2012) Wnt signalling antagonizes stress granule assembly through a Dishevelled‐dependent mechanism. Biol Open 1: 109–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Tahbaz N, Kolb FA, Zhang H, Jaronczyk K, Filipowicz W, Hobman TC (2004) Characterization of the interactions between mammalian PAZ PIWI domain proteins and Dicer. EMBO Rep 5: 189–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schirle NT, Sheu‐Gruttadauria J, MacRae IJ (2014) Structural basis for microRNA targeting. Science 346: 608–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Review Process File