

Abstract
Genetically engineered mouse models (GEMMs) have contributed significantly to the field of cancer research. In contrast to cancer cell inoculation models, GEMMs develop de novo tumors in a natural immune‐proficient microenvironment. Tumors arising in advanced GEMMs closely mimic the histopathological and molecular features of their human counterparts, display genetic heterogeneity, and are able to spontaneously progress toward metastatic disease. As such, GEMMs are generally superior to cancer cell inoculation models, which show no or limited heterogeneity and are often metastatic from the start. Given that GEMMs capture both tumor cell‐intrinsic and cell‐extrinsic factors that drive de novo tumor initiation and progression toward metastatic disease, these models are indispensable for preclinical research. GEMMs have successfully been used to validate candidate cancer genes and drug targets, assess therapy efficacy, dissect the impact of the tumor microenvironment, and evaluate mechanisms of drug resistance. In vivo validation of candidate cancer genes and therapeutic targets is further accelerated by recent advances in genetic engineering that enable fast‐track generation and fine‐tuning of GEMMs to more closely resemble human patients. In addition, aligning preclinical tumor intervention studies in advanced GEMMs with clinical studies in patients is expected to accelerate the development of novel therapeutic strategies and their translation into the clinic.
Keywords: cancer, genetically engineered mouse models, metastasis, therapy, tumor microenvironment
Subject Categories: Cancer
Glossary
- Allotransplantation
Transplantation of established mouse cancer cell lines in immunoproficient mice with the same genetic background.
- Cancer immunotherapy
Therapeutic strategies aimed at harnessing the patient's immune system to attack cancer. For example, immune checkpoint blockade interferes with negative regulatory molecules of T‐cell activation.
- Cancer‐associated fibroblasts (CAFs)
Fibroblasts that reside in the tumor microenvironment.
- Clinically overt metastatic disease
Metastatic disease that affects multiple organs, eventually leading to organ failure and death.
- Conventional GEMM
Oncogene‐bearing transgenic mice (aka oncomice) and mice carrying germline mutations in tumor suppressor genes (TSGs).
- Cre‐ERT
A fusion protein in which Cre‐recombinase is fused to a mutated hormone‐binding domain of the estrogen receptor (ERT). Administration of the estrogen analogue tamoxifen leads to post‐translational activation of Cre‐recombinase activity and excision of the target gene flanked by loxP sites.
- Cre‐loxP
A site‐specific recombination system that allows for Cre‐recombinase‐mediated deletion of genes flanked by loxP recombination sites. Expression of Cre‐recombinase can be induced in a tissue‐restricted manner.
- CRISPR/Cas9
A genome editing system that enables induction of DNA double‐strand breaks (DSBs) at defined genomic locations by directing the Cas9 nuclease to a predefined genomic locus using single‐guide RNAs (sgRNAs). DSB repair by non‐homologous end‐joining or homologous recombination (in the presence of an oligonucleotide) will lead to gene knockout or modification, respectively.
- Epithelial‐to‐mesenchymal transition (EMT)
A process by which epithelial cells lose polarity and cell–cell adhesion, and gain mesenchymal‐like migratory properties.
- Extracellular matrix (ECM)
The non‐cellular component present within all tissues and organs, which provides essential physical scaffolding for cellular structures and has biochemical and mechanical properties important for tissue development and homeostasis.
- Flp‐FRT
A site‐specific recombination system that allows for Flp‐recombinase‐mediated deletion of genes flanked by FRT recombination sites.
- GEMM‐ESC
A technique for rapid introduction of additional genetic modifications and subsequent production of chimeric mice from embryonic stem cells (ESCs) derived from existing GEMMs.
- Germline GEMMs
Mouse models carrying genetically engineered alleles (transgenes, conventional knockout/knock‐in alleles, or loxP/FRT‐flanked conditional alleles) in all cells including the germline.
- Next‐generation GEMMs
Mouse models that are genetically engineered to accurately mimic sporadic human cancer.
- Non‐germline GEMMs
Mouse models carrying genetically engineered alleles in somatic cells but not in germline cells. These mouse models include chimeric mice derived from genetically engineered (GEMM‐derived) ESCs and mice produced by CRISPR/Cas9‐mediated somatic gene editing.
- Oncogene addiction
Tumors display “oncogene addiction” when they are highly dependent on a single oncogene for their growth and maintenance.
- Oncomouse
Mouse with transgenic expression of a specific oncogene under control of a tissue‐specific promoter.
- Patient‐derived tumor xenografts (PDTX)
Mouse models based on transplantation and serial propagation of fresh human tumor biopsies in immunodeficient mice.
- Tumor microenvironment (TME)
The cellular environment in which tumor cells reside. The tumor microenvironment is composed of different populations of stromal cells, including endothelial cells, fibroblasts, extracellular matrix, and immune cells.
- Xenotransplantation
Transplantation of human tumor cells or tissue in immunocom‐promised mice.
Introduction
Despite the fact that survival rates of cancer patients have improved over the last decades, we are still facing numerous challenges in the clinic. One of the major problems is the development of drug resistance. Monotherapy with targeted anti‐cancer agents or chemotherapeutics invariably results in drug resistance caused by de novo mutations or outgrowth of pre‐existing therapy‐resistant clones within heterogeneous tumors. Moreover, after apparently successful treatments, small numbers of drug‐tolerant tumor cells can survive and remain dormant for extended periods of time and eventually relapse to form recurrent disease that can be phenotypically different from the original tumor (Kottke et al, 2013; Blatter & Rottenberg, 2015). Another major challenge is metastatic disease, which accounts for over ninety percent of cancer‐related deaths (Weigelt et al, 2005). These secondary tumors are often unresponsive to therapy and are at present mostly incurable. Encouraging advancements have been made with cancer immunotherapy, aimed at harnessing the patient's immune system to attack cancer. However, even though durable responses are observed in some cases, a large proportion of cancer patients does not show clinical benefit (Sharma & Allison, 2015).
Successful treatment of cancer requires a multidisciplinary approach in which different strategies such as surgery, irradiation, cytotoxic therapy, and immunotherapy are combined. To design such combinations, it is critical to improve our insights into the cancer cell‐intrinsic and cell‐extrinsic mechanisms underlying tumor development, metastasis, and therapy responsiveness. To find the most effective treatment for different cancer types, we heavily rely on preclinical research in animal models. Despite successful validation of novel anti‐cancer therapies in conventional preclinical mouse models based on xenotransplantation of established human cancer cell lines or allotransplantation of mouse tumor cell lines, the majority of the phase 3 clinical trials fail (Reichert & Wenger, 2008). The overall poor clinical predictability of these conventional in vivo tumor models emphasizes the need for more advanced preclinical in vivo models with better predictive power. Until fairly recently, progress in the field was hampered by the poor availability of preclinical models that closely recapitulate the natural course of human cancer. However, recent technological developments have led to fast‐track generation of sophisticated mouse models that more closely mimic human cancer in terms of genetic composition, interactions of cancer cells with their tumor microenvironment, drug response, and resistance. These next‐generation genetically engineered mouse models (GEMMs) are of great importance to improve our understanding of the complex mechanisms underlying cancer biology, and are anticipated to improve translation of new therapeutic strategies into the clinic (Fig 1)—ultimately leading to increased survival of cancer patients. This review describes the evolution and recent technological advances of mouse model engineering, and the applications of the resulting models in basic and translational oncology research.
Figure 1. Applications of GEMMs in basic cancer research and translational oncology.

Evolution of mouse cancer modeling
Since the advent of tumor transplantation models in nude mice 50 years ago, advances in mouse genome engineering have led to the generation of various types of transplantation‐based and genetically engineered tumor models to study cancer biology (Fig 2, Table 1). Here, we give an overview of mouse models that are most commonly used in cancer research.
Figure 2. Schematic overview of transplantation‐based mouse models and germline GEMMs.

(A) Cancer cell line transplantation models are based on (orthotopic) inoculation of cultured human or mouse cancer cells in immunodeficient or syngeneic mice, respectively. (B) Patient‐derived tumor xenograft models or GEMM‐derived tumor allograft models are based on direct (orthotopic) implantation of human or mouse tumor fragments in immunodeficient or syngeneic mice, respectively. (C) In oncomice, de novo tumorigenesis is induced by transgenic expression of an oncogene (ONC) from a tissue‐specific promoter. (D) In tumor suppressor gene (TSG) knockout mice, de novo tumorigenesis is induced by germline inactivation of a TSG. (E, F) In conditional GEMMs, de novo formation of sporadic tumors is induced by tissue‐specific Cre‐loxP‐mediated inactivation of conditional TSG alleles (E) and/or activation of conditional oncogenes (F). Tissue‐specific expression of the Cre‐recombinase is achieved by crossbreeding with Cre transgenic mice, tamoxifen‐inducible Cre‐ERT transgenic mice, or by local administration of Cre‐encoding lenti‐ or adenoviruses. (G, H) Oncogene addiction can be studied using GEMMs with tamoxifen‐ or doxycycline‐inducible gene expression. (G) Administration of tamoxifen to transgenic mice carrying an oncogene‐ERT (ONC‐ERT) fusion will induce tumors that may undergo no, temporal, or durable regression upon tamoxifen withdrawal. (H) Similar studies can be performed by administration of doxycycline to bi‐transgenic mice with tissue‐specific expression of the reverse tetracycline‐controlled transactivator (rtTA) and carrying an oncogene or shRNA under transcriptional control of a tetracycline response element (TRE).
Table 1.
Advantages and disadvantages of different mouse cancer models
| De novo carcinogenesis | Natural immunocompetent TME | Spontaneous metastasis | Cancer gene validation | Genetic validation drug targets | Preclinical drug testing | Drug resistance mechanisms | Immunotherapy | |
|---|---|---|---|---|---|---|---|---|
| Allograft cell line inoculation | No | Maybe | Model‐dependent | Yes | Yes | Yes | Yes | Yes |
| Xenograft cell line inoculationa | No | No | Model‐dependent | Yes | Yes | Yes | Maybe | No |
| Patient‐derived tumor xenograft (PDTX)a | No | No | Model‐dependent | No | No | Yes | Yes | No |
| Oncomouse | Yes | Yes | Model‐dependent | Yes | No | Yes | Yes | Yes |
| Cre‐LoxP | Yes | Yes | Model‐dependent | Yes | No | Yes | Yes | Yes |
| Tet‐on/off TetO‐Cre | Yes | Yes | Model‐dependent | Yes | Yes | Yes | Yes | Yes |
| GEMM‐ESC | Yes | Yes | Model‐dependent | Yes | Yes | Yes | Yes | Yes |
| CRISPR/Cas9 | Yes | Yes, when used in Cas9‐tolerant hosts | Model‐dependent | Yes | Yes | Yes | Yes | Expected |
These cancer models require the use of immunodeficient hosts.
Cancer cell line transplantation models
Mouse models based on xenografting human cancer cell lines or allografting mouse tumor cells are the most commonly used in vivo tumor models in cancer research. These transplantation models allow for rapid testing of potential cancer‐ and metastasis‐related genes and are often used for preclinical drug testing. For example, xenotransplantation studies provided insights into the mechanisms underlying intrinsic resistance of colorectal cancer (CRC) to vemurafenib. The results of this study led to the initiation of a clinical trial in which CRC patients were treated with a combination therapy targeting both BRAF V600E and EGFR (Prahallad et al, 2012), illustrating the usefulness of xenotransplantation models in establishing novel combinatorial treatment strategies. Moreover, xenograft studies identified distinct gene expression signatures that mediate organ‐specific patterns of metastatic colonization (Kang et al, 2003; Minn et al, 2005; Bos et al, 2009). In addition, these models revealed that disseminated breast cancer cells reside adjacent to blood vessels, which might provide a niche regulating dormancy of disseminated cancer cells (Ghajar et al, 2013). Furthermore, many of the fundamental insights into anti‐tumor immunity, T‐cell tolerance mechanisms, and immune‐escape routes of tumors come from in vivo studies with cell line allograft models (Leach et al, 1996). These findings have laid the foundation for the currently ongoing cancer immunotherapy revolution.
Nevertheless, as cancer cell lines contain multiple mutations from the start and acquire additional aberrations when cultured in vitro for extended periods of time, these inoculation models do not reflect the morphology and genetic heterogeneity of human cancers, and are therefore mostly poor predictors of clinical response. While allografting of mouse cancer cell lines can be performed in immunoproficient hosts, xenotransplantation of cell lines must be performed in immunocompromised mice to prevent rejection, which makes them less suitable to study the roles of the immune system in tumor development and therapy response.
Patient‐derived tumor xenografts
Patient‐derived tumor xenografts (PDTX) are derived from fresh human tumor biopsies that are transplanted in immunodeficient mice. Unlike cell line transplantation models, PDTX tumors maintain the molecular, genetic, and histological heterogeneity as observed in cancer patients, even after serial passaging in mice (Hidalgo et al, 2014). Therefore, PDTX models can be valuable tools to define personalized medicine as was demonstrated by preclinical drug screening in PDTX models of non‐small‐cell lung cancer (NSCLC) (Fichtner et al, 2008; Merk et al, 2009, 2011), breast cancer (Marangoni et al, 2007), melanoma (Kemper et al, 2015; Girotti et al, 2016), prostate cancer (Yoshida et al, 2005; Qu et al, 2014), and colorectal cancer (Bertotti et al, 2011, 2015; Bardelli et al, 2013; Kavuri et al, 2015; Misale et al, 2015; Zanella et al, 2015). For small‐cell lung cancer (SCLC), it was shown that circulating tumor cells (CTCs) from blood can be used to establish CTC‐derived explant (CDX) models that mirror the donor patient's response to platinum and etoposide (Hodgkinson et al, 2014). Large‐scale studies are now carried out using PDTX models to predict responses of clinical drug candidates. Approximately 1,000 PDTX models were established with a diverse panel of mutations, and subsequently used for in vivo compound screens, yielding correlations between drug response and tumor genotype that were both reproducible and clinically translatable (Gao et al, 2015). In a recent study using PDTX models of triple‐negative breast cancer, single‐cell gene expression analysis revealed that early‐stage metastatic cells express distinct signatures enriched in stem‐like genes, identifying novel potential drug targets to tackle metastatic breast cancer (Lawson et al, 2015).
Unfortunately, a major obstacle of PDTX modeling is the disappointing take rate of various tumor types, such as estrogen receptor‐positive breast cancer and prostate cancer (Landis et al, 2013; Lawrence et al, 2015). In addition, PDTX modeling must be performed in immunocompromised mice, thereby circumventing the natural anti‐ and pro‐tumor activity provided by the adaptive immune system. Given the complex crosstalk between adaptive immune components, the innate immune system, and cancer cells, it is important to realize that PDTX models can provide clinically valuable data, albeit in the absence of the influential adaptive immune system. Current efforts to generate humanized mice by engrafting immunodeficient mice with human CD34+ hematopoietic stem cells or precursor cells have shown remarkable progress (Drake et al, 2012; Holzapfel et al, 2015). Although reconstitution of immune cells from specific lineages remains challenging, the introduction of transgenes encoding human cytokines, chemokines, and growth factors can support the development of human myeloid cells in mice. To support development of HLA‐restricted T cells, recipient immunodeficient mice can be further optimized by transgenic expression of human HLA molecules and deficiency of mouse MHC class‐I and class‐II molecules. While the limited availability of hematopoietic donor stem cells (obtained from umbilical cord blood or fetal liver) and the relatively high costs of these models are potential disadvantages, humanized mouse models could provide a useful platform for preclinical evaluation of immunotherapeutics.
Modeling de novo cancer in genetically engineered mice
In the early 1980s, the first cloned cancer genes were introduced into the genome of transgenic mice, which were termed oncomice (Hanahan et al, 2007). The first oncomouse was a GEMM with transgenic expression of a specific activated oncogene (v‐HRas) under control of a mammary‐specific promoter (MMTV), making the mouse prone to developing mammary tumors (Sinn et al, 1987). The first oncomice led to great excitement in the cancer research community as they provided unambiguous proof for the hypothesis that oncogene expression in normal cells could lead to tumor formation (Brinster et al, 1984; Hanahan, 1985; Lacey et al, 1986; Sinn et al, 1987; Rüther et al, 1989). With the development of gene‐targeting technology in 1992, also cancer predisposition in tumor suppressor gene (TSG) knockout mice could be studied (Finlay, 1992).
Though oncomice and TSG knockout mice have provided a wealth of knowledge, they also have their limitations. Given that transgenes are expressed in all cells of a particular tissue and TSGs in knockout mice are inactivated in all cells of the animal, these models fail to mimic sporadic cancers in which accumulation of genetic events in a single cell results in tumorigenesis in an otherwise healthy organ. To circumvent this, more sophisticated mouse models are currently available that allow somatic inactivation of tumor suppressors or activation of (mutant) oncogenes in conditional GEMMs (Jonkers & Berns, 2002). One of the first examples is the generation of a mouse colorectal cancer model using Cre‐loxP‐mediated somatic inactivation of Apc. With this technique, any gene flanked by loxP recombination sites will be deleted after activation of the Cre‐recombinase. APC loss in intestinal epithelial cells was sporadically induced through adenovirus‐mediated delivery of Cre‐recombinase, resulting in the rapid onset of colorectal adenomas that shared many features with adenomas in familial adenomatous polyposis coli (FAP) patients (Shibata et al, 1997). By introducing mutations associated with a specific type of cancer, one can generate mouse models that closely mimic the histopathological, molecular, and clinical features of tumors in patients (Frese & Tuveson, 2007; Walrath et al, 2010).
Induction of somatic mutations at a chosen time and in a specific tissue can be achieved by using Cre‐ERT fusion proteins, in which a mutated hormone‐binding domain of the estrogen receptor (ERT) is fused to the Cre‐recombinase. Cre‐ERT is an inducible Cre‐recombinase: administration of the estrogen analogue tamoxifen leads to post‐translational activation of Cre‐recombinase activity and excision of the target gene flanked by loxP sites. Hence, mice with (tissue‐specific) expression of Cre‐ERT allow for spatiotemporally controlled Cre‐mediated genomic recombination upon administration of tamoxifen (Vooijs et al, 2001).
Even though the Cre‐loxP system can be applied to alter the expression of more than one gene, it does so simultaneously, and therefore does not fully mimic the sequential accumulation of mutations during multistep carcinogenesis. Recently, an inducible dual‐recombinase system was developed which combines Flp‐FRT and Cre‐loxP recombination systems, allowing sequential genetic manipulation of gene expression by two independent recombination systems (Schönhuber et al, 2014). This approach allows for (i) independent targeting of tumor cell autonomous and non‐autonomous pathways/processes, (ii) sequential induction of mutations to model human multistep carcinogenesis, and (iii) genetic validation of therapeutic targets in autochthonous tumors.
Speeding up and fine‐tuning mouse cancer modeling
While GEMMs have proven to be valuable tools for cancer research, there are still aspects that can be improved. A major limitation of germline GEMMs is that development and validation of these models is time‐consuming, laborious, and expensive. This is exemplified when a novel germline mutation has to be introduced in an existing multi‐allelic mouse model, as this requires extensive breeding. The rapidly increasing number of mutations identified in cancer sequencing studies calls for novel mouse modeling strategies that enable accelerated in vivo evaluation of candidate cancer genes and patient‐relevant mutations in known cancer genes in non‐germline GEMMs (Fig 3).
Figure 3. Schematic overview of non‐germline GEMMs.

(A) Embryonic stem cell (ESC)‐derived non‐germline GEMMs. ESCs from wild‐type mice or established GEMMs can be used to introduce single or multiple mutations using CRISPR/Cas9‐based gene editing and/or mutant alleles using recombinase‐mediated cassette exchange (RMCE). The resulting ESCs can be injected into host blastocysts, which are implanted into pseudo‐pregnant females to produce chimeric mice. (B, C) CRISPR/Cas9‐based non‐germline GEMMs. CRISPR/Cas9‐mediated in situ gene editing can be achieved by local administration of sgRNA‐encoding lentiviruses in transgenic mice with tissue‐specific Cas9 expression (B), or by local administration of lentiviruses that encode both Cas9 and sgRNA in wild‐type mice (C). The latter approach may require immunodeficient or Cas9‐tolerant mice to avoid Cas9‐specific immune responses.
Embryonic stem cell‐based mouse cancer models
To speed up the generation of novel GEMMs of human cancer, embryonic stem cells (ESCs) can be genetically altered and used to produce cohorts of non‐germline GEMMs (Heyer et al, 2010). An alternative approach is the recently developed GEMM‐ESC strategy, which employs ESCs that are derived from existing (multi‐allelic) GEMMs. These GEMM‐derived ESCs can be used for rapid introduction of additional genetic modifications and subsequent production of chimeric mice that show the same characteristics as the established GEMM but now contain the additional genetic modification (Huijbers et al, 2011, 2014). For example, the GEMM‐ESC strategy was used to introduce the Met proto‐oncogene in a GEMM of BRCA1‐associated breast cancer, which yielded a novel mouse model of BRCA1‐deficient metaplastic breast cancer (Henneman et al, 2015). Whereas BRCA1‐deficient mouse mammary carcinomas showed high sensitivity to the clinical PARP inhibitor olaparib, BRCA1‐deficient metaplastic mammary tumors showed intrinsic resistance.
In vivo RNA interference
RNA interference (RNAi) by short hairpin RNAs (shRNAs) allows reversible silencing of gene expression without modifying the genome, and therefore, it can be used as an alternative to homologous recombination‐based gene inactivation approaches. RNAi‐based genetic screens have proven to be powerful tools to rapidly identify and validate cancer genes. In vivo RNAi screens have been successfully used to identify novel TSGs in mouse models of hepatocellular carcinoma and lymphoma (Hemann et al, 2003; Zender et al, 2008; Bric et al, 2009), and to identify genes involved in resistance to the tyrosine kinase inhibitor sorafenib in liver cancer (Rudalska et al, 2014). Moreover, the development of systems for doxycycline‐inducible shRNA expression in transgenic mice allows reversible expression of shRNAs in a time‐ and tissue‐specific manner (Dickins et al, 2007; Premsrirut et al, 2011). Using the latter approach, Dow et al (2015b) have shown that shRNA‐mediated APC suppression in the presence of Kras and Trp53 mutations induces intestinal carcinomas, which undergo sustained regression upon restoration of APC expression by turning off shRNA expression.
Genome editing using CRISPR/Cas9 technology
In the past decades, additional approaches for genome editing have been developed such as Zinc‐finger nucleases (ZFNs) and transcription‐activator‐like effector nucleases (TALENs) (Li et al, 2011; Wefers et al, 2013). These approaches have now been outperformed by the development of CRISPR/Cas9 systems for genome editing (Cong et al, 2013), which have revolutionized biological research over the past 3 years and are considered the biggest game changer since PCR. The CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 system was first discovered as a prokaryotic immune system that confers resistance to foreign genetic elements, but soon thereafter has been exploited to achieve gene editing (Ishino et al, 1987; Mojica et al, 2000; Jansen et al, 2002). By using appropriate single‐guide RNAs (sgRNAs), the Cas9 nuclease can be directed to any genomic locus, where it induces double‐stranded cleavage of matching target DNA sequences, leading to gene knockout (Cong et al, 2013). The CRISPR/Cas9 system can also be used to introduce defined mutations or loxP/FRT recombination sites, by simply co‐introducing oligonucleotides that can serve as a template for repair of the Cas9‐induced break (Yang et al, 2014).
CRISPR/Cas9 technology seems the system of choice for rapid cancer modeling in mice, as it has proven to be an efficient gene‐targeting strategy with the potential for multiplexed genome editing (Sánchez‐Rivera & Jacks, 2015). Virtually, all (combinations of) genetic alterations found in human tumors can now be rapidly introduced in the mouse germline, including (conditional) gene deletions (Wang et al, 2013; Yang et al, 2013), point mutations (Wang et al, 2013), and translocations (Blasco et al, 2014; Choi & Meyerson, 2014; Torres et al, 2014). Other groups have successfully used CRISPR/Cas9 technology for somatic editing of oncogenes and TSGs in mice. These efforts have led to a new generation of non‐germline models of hepatocellular carcinoma (Xue et al, 2014; Weber et al, 2015), lung cancer (Platt et al, 2014; Sánchez‐Rivera et al, 2014), brain cancer (Zuckermann et al, 2015), pancreatic cancer (Chiou et al, 2015; Maresch et al, 2016), and breast cancer (Annunziato et al, 2016).
The CRISPR/Cas9 system has recently been modified to induce target gene repression (CRISPRi) or activation (CRISPRa) (Gilbert et al, 2013). These modified systems may be used to generate mice with inducible and reversible activation of oncogenes and/or inactivation of TSGs. Though extremely powerful, CRISPR/Cas9‐based systems for in vivo gene editing may also have certain drawbacks. For example, current CRISPR/Cas9 strategies are not suited to validate the oncogenic potential of putative oncogenes. To this end, CRISPRa‐based systems may be used to activate transcription of target genes (Braun et al, 2016). Moreover, somatic delivery of Cas9 may trigger Cas9‐specific immune responses resulting in clearance of Cas9‐expressing cells (Wang et al, 2015; Annunziato et al, 2016). To circumvent this issue, experiments should be performed in immunodeficient animals or mice that are engineered to develop immunological tolerance to Cas9. Finally, CRISPR/Cas9‐mediated genome editing may create unwanted off‐target mutations that may be circumvented by employing pairs of sgRNAs in mice with inducible expression of a Cas9n “nickase” variant that induces DNA single‐strand breaks (Dow et al, 2015a).
Fine‐tuning mouse cancer modeling with patient‐relevant alleles
Many cancer‐predisposing germline mutations and somatic mutations in human TSGs are missense or nonsense mutations that may result in the production of a mutant or truncated protein with residual activity. Such mutations are not adequately modeled in (conditional) knockout mice, in which deletion of one or more exons leads to complete loss of the protein. It is therefore essential to generate mouse models carrying patient‐relevant mutations to study their contribution to tumorigenesis and therapy response. Several studies have shown that patient‐relevant TSG mutations in mice induce different phenotypes compared to the null‐alleles. Compared to Trp53 knockouts, patient‐relevant Trp53 hotspot mutations in mice were shown to have enhanced oncogenic activity (Lang et al, 2004; Olive et al, 2004). Similarly, introduction of patient‐relevant Brca1 mutations in a conditional mouse model of BRCA1‐associated breast cancer showed that, in contrast to Brca1‐null tumors, mammary tumors with expression of Brca1 alleles harboring mutations in the RING domain readily acquired resistance to DNA‐damaging drugs due to residual activity of the RING‐less BRCA1 protein (Drost et al, 2011, 2016). Thus, by introducing specific somatic or germline mutations into GEMMs, the causal link between these mutations and therapy responsiveness can be determined.
Applications of GEMMs in basic cancer research
GEMMs of de novo tumorigenesis are the systems of choice for in vivo analysis of the cell‐intrinsic and cell‐extrinsic processes that contribute to cancer initiation, progression, and metastasis. Here, we discuss how GEMMs have contributed to advances in cancer biology.
Validation of candidate cancer genes
Given the growing number of candidate cancer genes that are identified in large‐scale tumor sequencing studies, there is a clear need for rapid in vivo strategies to validate these genes. Considering their speed and relative simplicity, GEMM‐ESC and CRISPR/Cas9 technologies are the methods of choice for fast‐track validation of candidate cancer genes. Especially, non‐germline models based on somatic CRISPR/Cas9‐mediated gene editing enable in vivo validation of (combinations of) candidate cancer genes in a truly high‐throughput manner, as was demonstrated in a mouse model for pancreatic cancer (Maresch et al, 2016). Here, transfection‐based multiplexed delivery of Cas9 and sgRNAs targeting 13 different cancer genes induced pancreatic cancer (PDAC) in the majority of mice. The PDACs displayed genome editing of over 60% of the target genes, indicating clonal expansion of CRISPR/Cas9‐induced driver mutations that induce cancer (Maresch et al, 2016). Likewise, GEMMs with doxycycline‐inducible Cas9 expression were employed to validate defined combinations of intestinal cancer genes, for example, Apc and Trp53 (Dow et al, 2015a). Besides modifying TSGs, CRISPR/Cas9 technology can be applied to validate the oncogenicity of chromosomal rearrangements, such as the Eml4‐Alk gene fusion observed in lung cancer (Maddalo et al, 2014).
Mouse models to study oncogene addiction
Some tumors are highly dependent on a single oncogene for their growth, a phenomenon called “oncogene addiction”. Conditional GEMMs are unsuitable models to determine oncogene addiction, as the genetic lesion is irreversible, and thus requires another layer of regulation. Oncogene‐ERT fusions can be employed to control oncogene expression; for example, Trp53 KI/KI mice in which both Trp53 alleles are replaced by a tamoxifen‐inducible Trp53‐ERT variant and were used to determine the therapeutic efficacy of p53 restoration in established tumors (Martins et al, 2006). P53 restoration in established Eμ‐Myc lymphomas triggered rapid apoptosis, which led to a significant increase in survival.
Also systems for doxycycline‐regulatable gene expression have been successfully used in GEMMs to turn oncogenes on to induce tumorigenesis; and off to determine how established tumors respond to oncogene inactivation (Gossen et al, 1995; Lewandoski, 2001). For example, continuous expression of a doxycycline‐inducible Myc transgene in hematopoietic cells resulted in the formation of malignant T‐cell lymphomas and acute myeloid leukemia that regressed upon de‐induction of Myc expression (Felsher & Bishop, 1999). The long‐term effects of temporal MYC de‐induction seem to differ between cancer types. For example, brief inactivation of MYC in osteogenic sarcomas resulted in sustained regression due to differentiation of sarcoma cells into mature osteocytes (Jain et al, 2002). In contrast, invasive liver cancers regressed after MYC inactivation, but residual tumor cells remained dormant and immediately restored their neoplastic features upon MYC reactivation (Shachaf et al, 2004).
Determining cells‐of‐origin of cancers
Identifying the cell‐of‐origin of malignancies may provide important information for the development of improved therapeutic strategies. Studies in GEMMs have successfully identified the cell‐of‐origin for several different cancer types. For example, the cell‐of‐origin of SCLC was determined by intratracheal injection of cell‐type‐restricted Adeno‐Cre viruses, to inactivate Trp53 and Rb1 in Clara, neuro‐endocrine (NE), and alveolar type 2 (SPC) cells, respectively. Trp53 and Rb1 inactivation in these specific cell types of the lung resulted in differences in tumor onset and tumor phenotype, and identified NE cells (and to a lesser extent SPC cells) as the cell‐of‐origin in SCLC (Sutherland et al, 2011). Cell‐of‐origin studies can also deliver surprising results, as was the case for BRCA1‐related basal‐like breast cancer. While BRCA1‐related basal‐like breast cancer was previously postulated to originate from basal epithelial stem cells, cell‐of‐origin studies in GEMMs revealed that in fact, luminal progenitors are the source of basal‐like tumors (Molyneux et al, 2010). Genetic aberrations, such as Pik3ca mutations, can have a profound effect on the stem cell pool, as was demonstrated recently by two independent laboratories. Expression of Pik3ca H1047R was shown to evoke dedifferentiation of lineage‐committed mammary epithelial cells into a multipotent stem‐like state (Koren et al, 2015; Van Keymeulen et al, 2015). Interestingly, the cell‐of‐origin of Pik3ca H1047R mammary tumors dictates their malignancy, highlighting the importance of pinpointing the cell‐of‐origin to improve specificity of anti‐cancer drugs and therapeutic outcome.
Studying the contribution of the tumor microenvironment
GEMMs have been indispensable in deciphering the contribution of tumor cell‐extrinsic factors such as cancer‐associated fibroblasts (CAFs) and immune cells to tumorigenesis. CAFs regulate deposition of extracellular matrix (ECM) and formation of basement membrane by synthesizing ECM components such as collagen, fibronectin, and laminin. Moreover, fibroblasts are a source of various soluble mediators including matrix metalloproteinases (MMPs), which enable ECM turnover, reinforcing their crucial role in maintaining ECM homeostasis (Kalluri & Zeisberg, 2006). Studies in GEMMs have demonstrated dual roles of fibroblasts in cancer. During malignant transformation of epithelial cells, CAFs can stimulate tumor progression by enhancing inflammation, angiogenesis, and ECM remodeling, as was demonstrated in the K14‐HPV16 squamous skin cancer model (Erez et al, 2010). In contrast, genetic in vivo depletion of CAFs was shown to accelerate tumor progression in two independent GEMMs of pancreatic cancer (Özdemir et al, 2014), suggesting a tumor‐restraining role for CAFs. The same controversy holds true for immune cells: originally, immune cells were thought to suppress tumorigenesis by attacking transformed cells; however, recent studies revealed that these cells could exert tumor‐promoting functions. The link between inflammation and cancer has been demonstrated in mouse models of several different cancer types (Greten et al, 2004; Pikarsky et al, 2004; DeNardo et al, 2009; Guerra et al, 2011; Jamieson et al, 2012; Pyonteck et al, 2013; Bald et al, 2014; Coffelt et al, 2015; Wculek & Malanchi, 2015). For example, in a mouse model of colitis‐associated cancer, genetic depletion of NF‐κB signaling in myeloid immune cells resulted in reduced tumor growth (Greten et al, 2004), demonstrating their tumor‐promoting function. Moreover, studies in the K14‐HPV16 model have shown that mast cells and bone marrow‐derived cells promote squamous skin cancer by activating angiogenesis and by reorganizing stromal architecture via MMP9 (Coussens et al, 1999, 2000). Using the same skin cancer model, chronic inflammation was found to promote de novo carcinogenesis in a B lymphocyte‐dependent manner (de Visser et al, 2005). Since then the tumor‐promoting roles of inflammation‐induced tumor‐associated macrophages (TAMs) (Lin et al, 2001; Noy & Pollard, 2014) and neutrophils (Jamieson et al, 2012; Bald et al, 2014; Coffelt et al, 2015, 2016; Wculek & Malanchi, 2015) have been described in several studies. For example, genetic ablation of colony‐stimulating factor 1 (CSF1), an important factor for macrophages, in the MMTV‐PyMT breast cancer mouse model delayed progression of mammary tumors to malignancy (Lin et al, 2001). Similarly, inhibition of CXCR2, a mediator of neutrophil‐migration, suppressed the formation of intestinal tumors in APC min/+ mice (Jamieson et al, 2012). Together, these studies emphasize that immune cells can act as co‐conspirators in tumor development and progression.
Deciphering spontaneous metastasis formation
Despite improved cancer treatment options, metastatic disease remains the primary cause of cancer‐related death. The metastatic cascade is a complex multi‐step process managed by a constant crosstalk between cancer cells and their microenvironment (Quail & Joyce, 2013; McAllister & Weinberg, 2014). Most preclinical metastasis research has been performed in cell line inoculation models, which do not recapitulate the metastatic process as it occurs in patients. GEMMs display de novo tumor progression and metastasis formation and are therefore indispensable for studying aspects of spontaneous metastasis formation that were unclear in the past (Fig 4). A potential drawback of GEMMs is that mice generally need to be sacrificed due to their primary tumor burden before macroscopic metastases have developed. This problem can be overcome by orthotopic transplantation of GEMM‐derived tumor fragments—which maintain the intratumoral heterogeneity of donor tumors—followed by surgical resection, allowing the development of clinically overt metastatic disease (Doornebal et al, 2013).
Figure 4. Applications of mouse models in metastasis research.

This overview summarizes the utility of different preclinical mouse models of experimental and spontaneous metastasis to study the different steps of the metastatic cascade. Conventional GEMMs represent oncomice and TSG knockout mice. Next‐generation GEMMs represent conditional mouse models that are genetically engineered to accurately mimic sporadic human cancer. For some models, the utility for studying specific steps in the metastatic cascade has yet to be determined, as indicated by a question mark. Moreover, several studies have shown that components of the adaptive immune system contribute to the various steps of the metastatic cascade. These aspects cannot be studied in models based on xenografting of human cancer cells or tumor fragments in immunodeficient hosts (indicated by an asterisk). To circumvent this, humanized mice can be used as hosts.
Several key aspects of metastasis have been discovered in GEMMs. For example, metastasis was originally believed to occur late in tumorigenesis. However, studies in BALB‐NeuT and MMTV‐PyMT mouse mammary tumor models revealed that transformed cells in early lesions are already capable of disseminating to bone marrow and lungs, and form micro‐metastasis (Hüsemann et al, 2008). Similarly, epithelial‐to‐mesenchymal transition (EMT) was thought to play a key role in tumor cell dissemination and metastasis. However, recent studies in GEMMs of pancreatic and breast cancer show that cancer cells retain their epithelial characteristics while colonizing metastatic sites, suggesting that EMT is not essential for metastasis formation in these models (Fischer et al, 2015; Zheng et al, 2015). Moreover, GEMMs have revolutionized the field by revealing the complex crosstalk between cancer cells and the immune system in metastasis formation. Several laboratories have shown that myeloid immune cells, such as macrophages and neutrophils, play key roles in promoting metastasis formation in different types of cancer (Lin et al, 2001; DeNardo et al, 2009; Bald et al, 2014; Coffelt et al, 2015; Wculek & Malanchi, 2015). Recently, we reported a mammary tumor‐induced systemic inflammatory state characterized by IL17‐producing γδ T cells and the subsequent expansion of immunosuppressive neutrophils that drives spontaneous metastasis formation in a GEMM of lobular breast cancer and a GEMM‐based transplantation model for spontaneous metastatic disease (Coffelt et al, 2015).
In conclusion, GEMMs have proven indispensable for understanding the complexity of metastasis and have challenged the current dogma that metastasis is a late‐stage cancer cell‐intrinsic process involving EMT. These findings may have important implications for treatment of metastatic cancer patients.
Applications of GEMMs in translational oncology
GEMMs of human cancer have been successfully used to validate candidate drug targets, assess therapy efficacy, and evaluate mechanisms of drug resistance. Since GEMMs develop de novo tumors in the context of an intact immune system, they are uniquely suited for investigating the potential of cancer immunotherapy. Close alignment of mouse and human studies can provide a platform that can aid in the development of novel treatment strategies for cancer patients (Fig 5). Below, we discuss how GEMMs can provide clinically relevant information for the design and development of novel anti‐cancer therapies.
Figure 5. Applications of mouse models in cancer drug development.
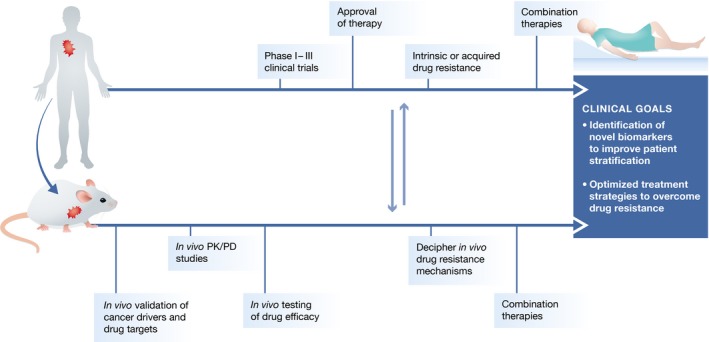
Development of novel treatment strategies in oncology requires preclinical studies in mouse cancer models to identify and validate novel cancer drivers and therapeutic targets, to determine in vivo drug pharmacokinetics and pharmacodynamics (PK/PD), and to evaluate in vivo anti‐cancer efficacy of novel therapeutics. When promising preclinical results are obtained, the tolerability and anti‐cancer efficacy of these drugs are evaluated in human patients in phase I–III clinical trials. A proportion of patients will show poor response due to intrinsic or acquired resistance, which may be studied mechanistically in preclinical mouse models to identify response biomarkers and combination therapies to prevent or overcome resistance. The close alignment of mouse studies and human clinical trials will lead to better patient stratification, identification of novel biomarkers, and development of optimal combination therapies, culminating in improved cancer patient care.
Validation of novel drug targets
Considering that not all cancer genes are essential for maintenance of established tumors, it is important to test whether reactivation of a TSG or down‐regulation of an oncogene results in durable regression of established tumors in a realistic preclinical setting, before drugs against these targets are developed. The relevance of oncogenes for tumor maintenance can be assessed in inducible mouse models in which oncogene expression can be de‐induced once tumors have developed. For example, de‐induction of oncogenic Pik3ca H1047R expression in a mouse model of breast cancer caused (partial) tumor regression demonstrating that these tumors are “addicted” to constitutively active PI3K signaling. However, most tumors eventually recurred due to Met or Myc amplifications, indicating that these genetic lesions may induce resistance to PI3K inhibitors (Liu et al, 2011). This example illustrates that preclinical studies in inducible GEMMs are not only useful for validating drug targets but also for identifying mechanisms underlying acquired drug resistance.
TSGs may sometimes also constitute valid drug targets. For example, p53 loss‐of‐function in cancer can result from dominant‐negative or inactivating mutations in the Trp53 gene or from amplification/overexpression of its specific inhibitors MDM2 and MDM4. Genetic studies in GEMMs with reversible inactivation of p53 have shown that restoration of p53 leads to rapid regression of established tumors (Martins et al, 2006; Ventura et al, 2007; Xue et al, 2007), providing strong rationale for designing anti‐cancer drugs that restore p53 function by inhibiting MDM2 (Vassilev et al, 2004) or by restoring wild‐type function to mutant p53 (Bykov et al, 2002). Similarly, GEMMs of colorectal cancer with inducible knockdown of APC showed that APC restoration initiates rapid and extensive tumor cell differentiation and sustained regression without relapse, providing in vivo validation of the WNT pathway as a therapeutic target for treatment of APC‐mutant colorectal cancers (Dow et al, 2015b).
Unraveling therapy response and resistance
To minimize the risk of failure of novel anti‐cancer therapeutics in clinical trials, preclinical evaluation of response and resistance in robust and predictive in vivo models is essential. Therapeutic responses of GEMMs to targeted therapy and conventional chemotherapy are very similar to those of human patients, as was assessed in GEMMs of Kras‐mutant lung cancer and pancreatic cancer (Singh et al, 2010). However, the differences in drug metabolism between mice and humans have to be taken into consideration. For example, the substrate specificity of the cytochrome P450 enzyme—which is involved in drug metabolism in the liver—is highly variable between species, an issue that could be overcome by humanizing mice (Scheer et al, 2012). Hence, preclinical drug efficacy studies in (humanized) GEMMs may advance the development of optimal (combinations of) anti‐cancer drugs to target specific tumors, and the identification of determinants of therapy response that may be used as predictive biomarkers for patient stratification. In addition, GEMMs may be used to identify mechanisms by which therapy‐sensitive tumors acquire drug resistance.
A clear example of a preclinical GEMM that has provided mechanistic insight into therapy response and resistance of BRCA1‐mutated breast cancer is the K14cre;Brca1 F/F ;Trp53 F/F (KB1P) mouse model. KB1P mice develop mammary tumors that mimic the histopathological features of human BRCA1‐mutated breast cancers as well as their hypersensitivity to platinum drugs and PARP inhibitors (Rottenberg et al, 2007, 2008). Clinical trials evaluated the PARP inhibitor olaparib for the treatment of ovarian, breast, and colorectal cancer (Lee et al, 2014). While olaparib did not seem promising in this diverse group of cancer patients, it did show significant responses in BRCA1‐mutation carriers, due to the synthetic lethal combination of PARP inhibition and BRCA1‐deficiency (Ledermann et al, 2012, 2014). BRCA1‐mutant cells are more vulnerable to PARP inhibition because the single‐strand DNA breaks induced by PARP inhibition, lead to double‐strand breaks during replication, which cannot be repaired by BRCA1‐deficient cells due to lack of homologous recombination. Based on promising results obtained in clinical trials (Ledermann et al, 2012, 2014), olaparib (trade name LynParza) was approved by the FDA in December 2014 for the treatment of patients with advanced BRCA1/2‐mutated ovarian cancer. Despite the good response of BRCA1/2‐mutated cancers to olaparib, acquired resistance is observed both in patients and GEMMs. Preclinical studies in KB1P mice revealed several mechanisms of resistance, such as elevated levels of drug efflux transporters and restoration of homologous recombination (Rottenberg et al, 2008; Jaspers et al, 2013; Henneman et al, 2015; Xu et al, 2015). These studies could aid in understanding clinical resistance and in designing improved treatment strategies for olaparib‐resistant patients in the clinic.
It is becoming clear that therapy response and resistance is not only influenced by tumor cell‐intrinsic factors but also by stromal factors such as fibroblasts and immune cells (Farmer et al, 2009; DeNardo et al, 2011; Shree et al, 2011; Acharyya et al, 2012; Nakasone et al, 2012; Boelens et al, 2014). This is illustrated by tumor intervention studies in a GEMM of PDAC, which showed that therapeutic inhibition of paracrine Sonic Hedgehog (SHH) signaling reduced desmoplastic tumor stroma and increased tumor vasculature, resulting in enhanced delivery of gemcitabine to tumors (Olive et al, 2009). However, the concept of targeting tumor stroma in PDAC has recently been challenged by two studies showing that stromal factors may suppress rather than promote PDAC growth, possibly by restraining tumor angiogenesis (Özdemir et al, 2014; Rhim et al, 2014). Together, these studies demonstrate that the contribution of the tumor microenvironment to therapy resistance may be more profound, but also more complex, than previously anticipated.
Cancer immunotherapy
Over the past decades, mechanistic insights into immune responses have culminated in therapeutic strategies that harness the patient's immune system to attack cancer. Recent clinical trials in patients with advanced melanoma and lung cancer confirm the remarkable potential of immune checkpoint blockade, including anti‐CTLA‐4 and anti‐PD‐1, to enhance effective anti‐tumor immunity and to improve survival in a proportion of the patients (Hodi et al, 2010; Topalian et al, 2012, 1). The basis of these clinical trials comes from several decades of fundamental research in experimental mouse models that have revealed the importance of CTLA‐4 and PD‐1 in restraining immune responses, as most clearly illustrated by the severe spontaneous autoimmunity phenotype in CTLA‐4‐deficient (Waterhouse et al, 1995) and to a milder extent in PD‐1‐deficient mice (Nishimura et al, 1999, 2001). CTLA‐4 blockade in mice bearing inoculated tumors enhances anti‐tumor T‐cell responses resulting in tumor rejection (Leach et al, 1996), illustrating that releasing the brake on T cells might be an interesting strategy to combat cancer. Nevertheless, a substantial proportion of patients do not respond to immunotherapy, and the current challenge is to understand why.
Currently, the majority of immunological studies are performed in tumor transplantation models, but we foresee a growing role for GEMMs. Several studies in GEMMs illustrate that during de novo carcinogenesis, T cells fail to respond due to tumor‐induced tolerance mechanisms (Willimsky & Blankenstein, 2005; Garbe et al, 2006; DuPage et al, 2011). Strikingly, transplantation of GEMM‐derived tumor cells in immunodeficient mice resulted in rapid tumor growth, while wild‐type mice rejected these tumors (Willimsky & Blankenstein, 2005; Garbe et al, 2006; DuPage et al, 2011), demonstrating that the cancer cells did not lose their immunogenicity and T cells are still able to recognize and attack them; however, they fail to do so in a host bearing de novo tumors.
Tumors are often characterized by chronic inflammation, which induces local and systemic immunosuppression that is unfavorable for T cells to perform their effector function (Mitchem et al, 2013; Ruffell et al, 2014; Coffelt et al, 2015). Moreover, tumors often show dysfunctional dendritic cells, which results in impaired T‐cell priming. For example, MMTV‐PyMT mammary tumors display dendritic cells that are potent activators of anti‐tumor T cells, but these cells are outcompeted by the overabundant presence of macrophages preventing proper T‐cell activation (Engelhardt et al, 2012; Broz et al, 2014). Recent studies have demonstrated that boosting dendritic cell function (Broz et al, 2014; Ruffell et al, 2014; Salmon et al, 2016; Sánchez‐Paulete et al, 2016) or blocking myeloid cell‐induced immunosuppression (Highfill et al, 2014; Zhu et al, 2014) improves the anti‐tumor efficacy of immune checkpoint blockade. Thus, patients that show resistance to T‐cell boosting immunotherapy might show improved clinical benefit when treatment is combined with compounds that either target immunosuppression or enhance T‐cell priming.
Immunotherapy studies in GEMMs require a different approach compared to inoculation models. Considering that tumors in GEMMs develop de novo, individual mice—like patients—have a unique set of tumor antigens. Consequently, heterogeneous responses are expected, which warrants identification of molecular differences between responsive and non‐responsive tumors, and may yield biomarkers that can predict clinical benefit. However, for most GEMM‐derived tumors, the identity of expressed tumor antigens that could be recognized by T cells is unknown. To circumvent this, clinically relevant tumor antigens could be introduced by genetic engineering to allow tracking of tumor‐specific T‐cell responses. For example, the introduction of tumor‐specific antigens in GEMMs with low immunogenic tumors, such as sarcomas and lung cancer, increased the immunogenicity of these tumors and resulted in a potent, but transient, anti‐tumor T‐cell response (DuPage et al, 2011, 2012). The initial anti‐tumor T‐cell response was quickly followed by regulatory T‐cell‐mediated immunosuppression (Joshi et al, 2015). Thus, these models can aid ongoing and future research to unravel the complex mechanisms underlying immune evasion (DuPage & Jacks, 2013), and may ultimately lead to novel (combination) strategies to improve cancer immunotherapy.
Co‐clinical trials in GEMMs
Recently, a “co‐clinical trial” paradigm has been developed in which preclinical trials in GEMMs are run in parallel with human clinical trials to predict therapeutic response (Clohessy & Pandolfi, 2015). This strategy was successfully used to identify genetic determinants of androgen deprivation resistance in prostate cancer, as well as novel combination therapies to overcome castration resistance (Lunardi et al, 2013). Similarly, co‐clinical trials in GEMMs of NSCLC showed that Kras/Lkb1‐mutant lung tumors are more resistant to combination therapy with docetaxel and the MEK inhibitor selumetinib than Kras‐ or Kras/p53‐mutant tumors, highlighting LKB1 as a potential determinant of resistance in clinical trials with this combination therapy (Chen et al, 2012). These studies demonstrate that preclinical efficacy studies in GEMMs of human cancer may identify novel biomarkers and (combination) therapies that can be validated in concurrent human clinical trials or used to optimize the design of future trials.
Concluding remarks and future perspectives
Many anti‐cancer drugs in clinical trials do not live up to the high expectations raised by preceding preclinical studies. How can we improve the predictive power of preclinical studies in the oncology arena? Most importantly, the preclinical tumor model of choice should reflect the human disease as faithfully as possible. To achieve this, it is important that preclinical mouse models capture both the intrinsic and extrinsic properties of cancer. First, preclinical models should contain the patient‐specific mutations that initiated the malignancy, and harbor the genetic variation as seen in patient populations. In addition, de novo tumorigenesis should occur in the natural microenvironment reflecting the crosstalk of cancer cells with the tumor microenvironment (including infiltrating immune cells, fibroblasts, and the lymphatic and blood vasculature) as observed in human cancer. It is also important to realize that the majority of patients who are enrolled in clinical trials have already developed advanced metastatic disease. Preclinical drug efficacy studies should therefore be preferably performed in mice that reflect the disease stage of the patients for which the therapy is intended. On a similar note, patients enrolled in clinical trials are frequently heavily pre‐treated, which is likely to negatively affect therapy outcome. Preclinical studies performed in treatment‐naive animals may thus overestimate therapy efficacy. On the other hand, treatments that are unsuccessful in heavily pre‐treated patients with advanced disease might still be beneficial for treatment‐naive patients with less advanced disease.
Current advances in genetic engineering allow for fast‐track generation and genetic fine‐tuning of mouse models that develop de novo cancer, which incorporates both cancer cell‐intrinsic and cell‐extrinsic properties of specific patient cohorts. We anticipate that these next‐generation GEMMs and GEMM‐based orthotopic transplantation models for spontaneous metastatic disease are currently the best available models to faithfully recapitulate human cancer. These models provide valuable tools to study the mechanisms underlying complex processes such as cancer initiation, organ‐specific metastasis formation, and the involvement of tumor microenvironment. But, more importantly for cancer patients, these models will provide better insights into (immune) therapy responsiveness and resistance, and disease recurrence. It is expected that preclinical efficacy studies with novel anti‐cancer drugs in next‐generation GEMMs will provide enhanced predictivity for their clinical efficacy, and thus accelerate the design and clinical implementation of novel anti‐cancer strategies that will improve cancer patient care.
Conflict of interest
The authors declare that they have no conflict of interest.
Pending issues.
Understanding of tumor cell‐intrinsic and cell‐extrinsic mechanisms underlying cancer and metastasis development, and therapy resistance.
Development of multidisciplinary therapeutic strategies including conventional anti‐cancer drugs and immunotherapy to successfully fight disseminated cancer.
Reduction in time and costs to generate next‐generation genetically engineered mouse models that closely recapitulate human cancer.
Close alignment of preclinical mouse studies and human clinical trials to improve cancer patient care.
Acknowledgements
Research in the laboratory of J.J. is supported by the Dutch Cancer Society (grants 5220, 6532, 7048, 7589, 7835, and 7877), the Netherlands Organization for Scientific Research (NWO; Cancer Genomics Netherlands [CGCNL], Cancer Systems Biology Center [CSBC], Zenith 93512009, and VICI 91814643), Worldwide Cancer Research (grant 14‐0288), the European Union Seventh Framework Program (EurocanPlatform project 260791 and Infrafrontier‐I3 project 312325), the European Research Council (ERC Synergy grant 319661: CombatCancer), and a National Roadmap grant for Large‐Scale Research Facilities from NWO. Research in the laboratory of K.E.d.V. is supported by the European Research Council (ERC Consolidator grant 615300: InflaMet), the European Union Seventh Framework Program (MCA‐ITN 317445: TIMCC), and the Beug Foundation for Metastasis Research. K.E.d.V. is an EMBO Young Investigator.
EMBO Mol Med (2017) 9: 137–153
See the Glossary for abbreviations used in this article.
Contributor Information
Martine H van Miltenburg, Email: m.v.miltenburg@nki.nl.
Jos Jonkers, Email: j.jonkers@nki.nl.
References
- Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, Manova‐Todorova K, Leversha M, Hogg N, Seshan VE et al (2012) A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 150: 165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato S, Kas SM, Nethe M, Yücel H, Del Bravo J, Pritchard C, Bin Ali R, van Gerwen B, Siteur B, Drenth AP et al (2016) Modeling invasive lobular breast carcinoma by CRISPR/Cas9‐mediated somatic genome editing of the mammary gland. Genes Dev 30: 1470–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bald T, Quast T, Landsberg J, Rogava M, Glodde N, Lopez‐Ramos D, Kohlmeyer J, Riesenberg S, van den Boorn‐Konijnenberg D, Hömig‐Hölzel C et al (2014) Ultraviolet‐radiation‐induced inflammation promotes angiotropism and metastasis in melanoma. Nature 507: 109–113 [DOI] [PubMed] [Google Scholar]
- Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, Sartore‐Bianchi A, Scala E, Cassingena A, Zecchin D et al (2013) Amplification of the MET receptor drives resistance to anti‐EGFR therapies in colorectal cancer. Cancer Discov 3: 658–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, Corà D, Di Nicolantonio F, Buscarino M, Petti C et al (2011) A molecularly annotated platform of patient‐derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab‐resistant colorectal cancer. Cancer Discov 1: 508–523 [DOI] [PubMed] [Google Scholar]
- Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, Sausen M, Phallen J, Hruban CA, Tokheim C et al (2015) The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 526: 263–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco RB, Karaca E, Ambrogio C, Cheong T‐C, Karayol E, Minero VG, Voena C, Chiarle R (2014) Simple and rapid in vivo generation of chromosomal rearrangements using CRISPR/Cas9 technology. Cell Rep 9: 1219–1227 [DOI] [PubMed] [Google Scholar]
- Blatter S, Rottenberg S (2015) Minimal residual disease in cancer therapy – Small things make all the difference. Drug Resist Updat 21–22: 1–10 [DOI] [PubMed] [Google Scholar]
- Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, Azzam DJ, Twyman‐Saint Victor C, Wiemann BZ, Ishwaran H et al (2014) Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 159: 499–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos PD, Zhang XH‐F, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA et al (2009) Genes that mediate breast cancer metastasis to the brain. Nature 459: 1005–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun CJ, Bruno PM, Horlbeck MA, Gilbert LA, Weissman JS, Hemann MT (2016) Versatile in vivo regulation of tumor phenotypes by dCas9‐mediated transcriptional perturbation. Proc Natl Acad Sci USA 113: E3892–E3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bric A, Miething C, Bialucha CU, Scuoppo C, Zender L, Krasnitz A, Xuan Z, Zuber J, Wigler M, Hicks J et al (2009) Functional identification of tumor suppressor genes through an in vivo RNA interference screen in a mouse lymphoma model. Cancer Cell 16: 324–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL, Chen HY, Messing A, van Dyke T, Levine AJ, Palmiter RD (1984) Transgenic mice harboring SV40 T‐antigen genes develop characteristic brain tumors. Cell 37: 367–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL et al (2014) Dissecting the tumor myeloid compartment reveals rare activating antigen‐presenting cells critical for T cell immunity. Cancer Cell 26: 638–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bykov VJN, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG, Selivanova G (2002) Restoration of the tumor suppressor function to mutant p53 by a low‐molecular‐weight compound. Nat Med 8: 282–288 [DOI] [PubMed] [Google Scholar]
- Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T, Liu Y, Tupper T, Ouyang J, Li J et al (2012) A murine lung cancer co‐clinical trial identifies genetic modifiers of therapeutic response. Nature 483: 613–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou S‐H, Winters IP, Wang J, Naranjo S, Dudgeon C, Tamburini FB, Brady JJ, Yang D, Grüner BM, Chuang C‐H et al (2015) Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9‐mediated somatic genome editing. Genes Dev 29: 1576–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi PS, Meyerson M (2014) Targeted genomic rearrangements using CRISPR/Cas technology. Nat Commun 5: 3728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clohessy JG, Pandolfi PP (2015) Mouse hospital and co‐clinical trial project‐from bench to bedside. Nat Rev Clin Oncol 12: 491–498 [DOI] [PubMed] [Google Scholar]
- Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau C‐S, Verstegen NJM, Ciampricotti M, Hawinkels LJAC, Jonkers J et al (2015) IL‐17‐producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522: 345–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffelt SB, Wellenstein MD, de Visser KE (2016) Neutrophils in cancer: neutral no more. Nat Rev Cancer 16: 431–446 [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Raymond WW, Bergers G, Laig‐Webster M, Behrendtsen O, Werb Z, Caughey GH, Hanahan D (1999) Inflammatory mast cells up‐regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev 13: 1382–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Tinkle CL, Hanahan D, Werb Z (2000) MMP‐9 supplied by bone marrow‐derived cells contributes to skin carcinogenesis. Cell 103: 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, Coussens LM (2009) CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16: 91–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA et al (2011) Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 1: 54–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickins RA, McJunkin K, Hernando E, Premsrirut PK, Krizhanovsky V, Burgess DJ, Kim SY, Cordon‐Cardo C, Zender L, Hannon GJ et al (2007) Tissue‐specific and reversible RNA interference in transgenic mice. Nat Genet 39: 914–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doornebal CW, Klarenbeek S, Braumuller TM, Klijn CN, Ciampricotti M, Hau C‐S, Hollmann MW, Jonkers J, de Visser KE (2013) A preclinical mouse model of invasive lobular breast cancer metastasis. Cancer Res 73: 353–363 [DOI] [PubMed] [Google Scholar]
- Dow LE, Fisher J, O'Rourke KP, Muley A, Kastenhuber ER, Livshits G, Tschaharganeh DF, Socci ND, Lowe SW (2015a) Inducible in vivo genome editing with CRISPR‐Cas9. Nat Biotechnol 33: 390–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow LE, O'Rourke KP, Simon J, Tschaharganeh DF, van Es JH, Clevers H, Lowe SW (2015b) Apc restoration promotes cellular differentiation and reestablishes Crypt homeostasis in colorectal cancer. Cell 161: 1539–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake AC, Chen Q, Chen J (2012) Engineering humanized mice for improved hematopoietic reconstitution. Cell Mol Immunol 9: 215–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost R, Bouwman P, Rottenberg S, Boon U, Schut E, Klarenbeek S, Klijn C, van der Heijden I, van der Gulden H, Wientjens E et al (2011) BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer Cell 20: 797–809 [DOI] [PubMed] [Google Scholar]
- Drost R, Dhillon KK, van der Gulden H, van der Heijden I, Brandsma I, Cruz C, Chondronasiou D, Castroviejo‐Bermejo M, Boon U, Schut E et al (2016) BRCA1185delAG tumors may acquire therapy resistance through expression of RING‐less BRCA1. J Clin Invest 126: 2903–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, Crowley D, Chen J, Jacks T (2011) Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell 19: 72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T (2012) Expression of tumour‐specific antigens underlies cancer immunoediting. Nature 482: 405–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Jacks T (2013) Genetically engineered mouse models of cancer reveal new insights about the antitumor immune response. Curr Opin Immunol 25: 192–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, Egeblad M, Krummel MF (2012) Marginating dendritic cells of the tumor microenvironment cross‐present tumor antigens and stably engage tumor‐specific T cells. Cancer Cell 21: 402–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erez N, Truitt M, Olson P, Arron ST, Hanahan D (2010) Cancer‐associated fibroblasts are activated in incipient neoplasia to orchestrate tumor‐promoting inflammation in an NF‐kappaB‐dependent manner. Cancer Cell 17: 135–147 [DOI] [PubMed] [Google Scholar]
- Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Wirapati P, Becette V, André S, Piccart M, Campone M et al (2009) A stroma‐related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med 15: 68–74 [DOI] [PubMed] [Google Scholar]
- Felsher DW, Bishop JM (1999) Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell 4: 199–207 [DOI] [PubMed] [Google Scholar]
- Fichtner I, Rolff J, Soong R, Hoffmann J, Hammer S, Sommer A, Becker M, Merk J (2008) Establishment of patient‐derived non‐small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin Cancer Res 14: 6456–6468 [DOI] [PubMed] [Google Scholar]
- Finlay CA (1992) p53 loss of function: implications for the processes of immortalization and tumorigenesis. BioEssays 14: 557–560 [DOI] [PubMed] [Google Scholar]
- Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, Choi H, El Rayes T, Ryu S, Troeger J et al (2015) Epithelial‐to‐mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527: 472–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese KK, Tuveson DA (2007) Maximizing mouse cancer models. Nat Rev Cancer 7: 645–658 [DOI] [PubMed] [Google Scholar]
- Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, Zhang C, Schnell C, Yang G, Zhang Y et al (2015) High‐throughput screening using patient‐derived tumor xenografts to predict clinical trial drug response. Nat Med 21: 1318–1325 [DOI] [PubMed] [Google Scholar]
- Garbe AI, Vermeer B, Gamrekelashvili J, von Wasielewski R, Greten FR, Westendorf AM, Buer J, Schmid RM, Manns MP, Korangy F et al (2006) Genetically induced pancreatic adenocarcinoma is highly immunogenic and causes spontaneous tumor‐specific immune responses. Cancer Res 66: 508–516 [DOI] [PubMed] [Google Scholar]
- Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, Almeida D, Koller A, Hajjar KA, Stainier DYR et al (2013) The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 15: 807–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern‐Ginossar N, Brandman O, Whitehead EH, Doudna JA et al (2013) CRISPR‐mediated modular RNA‐guided regulation of transcription in eukaryotes. Cell 154: 442–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girotti MR, Gremel G, Lee R, Galvani E, Rothwell D, Viros A, Mandal AK, Lim KHJ, Saturno G, Furney SJ et al (2016) Application of sequencing, liquid biopsies, and patient‐derived xenografts for personalized medicine in melanoma. Cancer Discov 6: 286–299 [DOI] [PubMed] [Google Scholar]
- Gossen M, Freundlieb S, Bender G, Müller G, Hillen W, Bujard H (1995) Transcriptional activation by tetracyclines in mammalian cells. Science 268: 1766–1769 [DOI] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li Z‐W, Egan LJ, Kagnoff MF, Karin M (2004) IKKβ links inflammation and tumorigenesis in a mouse model of colitis‐associated cancer. Cell 118: 285–296 [DOI] [PubMed] [Google Scholar]
- Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernández‐Porras I, Cañamero M, Rodriguez‐Justo M, Serrano M, Barbacid M (2011) Pancreatitis‐induced inflammation contributes to pancreatic cancer by inhibiting oncogene‐induced senescence. Cancer Cell 19: 728–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D (1985) Heritable formation of pancreatic beta‐cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 315: 115–122 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Wagner EF, Palmiter RD (2007) The origins of oncomice: a history of the first transgenic mice genetically engineered to develop cancer. Genes Dev 21: 2258–2270 [DOI] [PubMed] [Google Scholar]
- Hemann MT, Fridman JS, Zilfou JT, Hernando E, Paddison PJ, Cordon‐Cardo C, Hannon GJ, Lowe SW (2003) An epi‐allelic series of p53 hypomorphs created by stable RNAi produces distinct tumor phenotypes in vivo. Nat Genet 33: 396–400 [DOI] [PubMed] [Google Scholar]
- Henneman L, van Miltenburg MH, Michalak EM, Braumuller TM, Jaspers JE, Drenth AP, de Korte‐Grimmerink R, Gogola E, Szuhai K, Schlicker A et al (2015) Selective resistance to the PARP inhibitor olaparib in a mouse model for BRCA1‐deficient metaplastic breast cancer. Proc Natl Acad Sci USA 112: 8409–8414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer J, Kwong LN, Lowe SW, Chin L (2010) Non‐germline genetically engineered mouse models for translational cancer research. Nat Rev Cancer 10: 470–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo M, Amant F, Biankin AV, Budinská E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Mælandsmo GM et al (2014) Patient‐derived xenograft models: an emerging platform for translational cancer research. Cancer Discov 4: 998–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, Kaplan RN, Mackall CL (2014) Disruption of CXCR2‐mediated MDSC tumor trafficking enhances anti‐PD1 efficacy. Sci Transl Med 6: 237ra67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkinson CL, Morrow CJ, Li Y, Metcalf RL, Rothwell DG, Trapani F, Polanski R, Burt DJ, Simpson KL, Morris K et al (2014) Tumorigenicity and genetic profiling of circulating tumor cells in small‐cell lung cancer. Nat Med 20: 897–903 [DOI] [PubMed] [Google Scholar]
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363: 711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzapfel BM, Wagner F, Thibaudeau L, Levesque J‐P, Hutmacher DW (2015) Concise review: humanized models of tumor immunology in the 21st century: convergence of cancer research and tissue engineering. Stem Cells 33: 1696–1704 [DOI] [PubMed] [Google Scholar]
- Huijbers IJ, Krimpenfort P, Berns A, Jonkers J (2011) Rapid validation of cancer genes in chimeras derived from established genetically engineered mouse models. BioEssays 33: 701–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijbers IJ, Bin Ali R, Pritchard C, Cozijnsen M, Kwon M‐C, Proost N, Song J‐Y, de Vries H, Badhai J, Sutherland K et al (2014) Rapid target gene validation in complex cancer mouse models using re‐derived embryonic stem cells. EMBO Mol Med 6: 212–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüsemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, Forni G, Eils R, Fehm T, Riethmüller G et al (2008) Systemic spread is an early step in breast cancer. Cancer Cell 13: 58–68 [DOI] [PubMed] [Google Scholar]
- Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169: 5429–5433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, Sundberg CD, Bishop JM, Felsher DW (2002) Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 297: 102–104 [DOI] [PubMed] [Google Scholar]
- Jamieson T, Clarke M, Steele CW, Samuel MS, Neumann J, Jung A, Huels D, Olson MF, Das S, Nibbs RJB et al (2012) Inhibition of CXCR2 profoundly suppresses inflammation‐driven and spontaneous tumorigenesis. J Clin Invest 122: 3127–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R, van Embden JDA, Gaastra W, Schouls LM (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43: 1565–1575 [DOI] [PubMed] [Google Scholar]
- Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A et al (2013) Loss of 53BP1 causes PARP inhibitor resistance in Brca1‐mutated mouse mammary tumors. Cancer Discov 3: 68–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers J, Berns A (2002) Conditional mouse models of sporadic cancer. Nat Rev Cancer 2: 251–265 [DOI] [PubMed] [Google Scholar]
- Joshi NS, Akama‐Garren EH, Lu Y, Lee D‐Y, Chang GP, Li A, DuPage M, Tammela T, Kerper NR, Farago AF et al (2015) Regulatory T cells in tumor‐associated tertiary lymphoid structures suppress anti‐tumor T cell responses. Immunity 43: 579–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6: 392–401 [DOI] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón‐Cardo C, Guise TA, Massagué J (2003) A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3: 537–549 [DOI] [PubMed] [Google Scholar]
- Kavuri SM, Jain N, Galimi F, Cottino F, Leto SM, Migliardi G, Searleman AC, Shen W, Monsey J, Trusolino L et al (2015) HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov 5: 832–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper K, Krijgsman O, Cornelissen‐Steijger P, Shahrabi A, Weeber F, Song J‐Y, Kuilman T, Vis DJ, Wessels LF, Voest EE et al (2015) Intra‐ and inter‐tumor heterogeneity in a vemurafenib‐resistant melanoma patient and derived xenografts. EMBO Mol Med 7: 1104–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Reavie L, Couto JP, De Silva D, Stadler MB, Roloff T, Britschgi A, Eichlisberger T, Kohler H, Aina O et al (2015) PIK3CA(H1047R) induces multipotency and multi‐lineage mammary tumours. Nature 525: 114–118 [DOI] [PubMed] [Google Scholar]
- Kottke T, Boisgerault N, Diaz RM, Donnelly O, Rommelfanger‐Konkol D, Pulido J, Thompson J, Mukhopadhyay D, Kaspar R, Coffey M et al (2013) Detecting and targeting tumor relapse by its resistance to innate effectors at early recurrence. Nat Med 19: 1625–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey M, Alpert S, Hanahan D (1986) Bovine papillomavirus genome elicits skin tumours in transgenic mice. Nature 322: 609–612 [DOI] [PubMed] [Google Scholar]
- Landis MD, Lehmann BD, Pietenpol JA, Chang JC (2013) Patient‐derived breast tumor xenografts facilitating personalized cancer therapy. Breast Cancer Res 15: 201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GA, Iwakuma T, Suh Y‐A, Liu G, Rao VA, Parant JM, Valentin‐Vega YA, Terzian T, Caldwell LC, Strong LC et al (2004) Gain of function of a p53 hot spot mutation in a mouse model of Li‐Fraumeni syndrome. Cell 119: 861–872 [DOI] [PubMed] [Google Scholar]
- Lawrence MG, Pook DW, Wang H, Porter LH, Frydenberg M, Kourambas J, Appu S, Poole C, Beardsley EK, Ryan A et al (2015) Establishment of primary patient‐derived xenografts of palliative TURP specimens to study castrate‐resistant prostate cancer. Prostate 75: 1475–1483 [DOI] [PubMed] [Google Scholar]
- Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang C‐Y et al (2015) Single‐cell analysis reveals a stem‐cell program in human metastatic breast cancer cells. Nature 526: 131–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach DR, Krummel MF, Allison JP (1996) Enhancement of antitumor immunity by CTLA‐4 blockade. Science 271: 1734–1736 [DOI] [PubMed] [Google Scholar]
- Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira‐Frommer R, Safra T et al (2012) Olaparib maintenance therapy in platinum‐sensitive relapsed ovarian cancer. N Engl J Med 366: 1382–1392 [DOI] [PubMed] [Google Scholar]
- Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira‐Frommer R, Safra T et al (2014) Olaparib maintenance therapy in patients with platinum‐sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 15: 852–861 [DOI] [PubMed] [Google Scholar]
- Lee J‐M, Ledermann JA, Kohn EC (2014) PARP Inhibitors for BRCA1/2 mutation‐associated and BRCA‐like malignancies. Ann Oncol 25: 32–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandoski M (2001) Conditional control of gene expression in the mouse. Nat Rev Genet 2: 743–755 [DOI] [PubMed] [Google Scholar]
- Li H, Haurigot V, Doyon Y, Li T, Wong SY, Bhagwat AS, Malani N, Anguela XM, Sharma R, Ivanciu L et al (2011) In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature 475: 217–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin EY, Nguyen AV, Russell RG, Pollard JW (2001) Colony‐stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med 193: 727–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Cheng H, Santiago S, Raeder M, Zhang F, Isabella A, Yang J, Semaan DJ, Chen C, Fox EA et al (2011) Oncogenic PIK3CA‐driven mammary tumors frequently recur via PI3K pathway‐dependent and PI3K pathway‐independent mechanisms. Nat Med 17: 1116–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunardi A, Ala U, Epping MT, Salmena L, Clohessy JG, Webster KA, Wang G, Mazzucchelli R, Bianconi M, Stack EC et al (2013) A co‐clinical approach identifies mechanisms and potential therapies for androgen deprivation resistance in prostate cancer. Nat Genet 45: 747–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han Y‐C, Ogrodowski P, Crippa A, Rekhtman N, de Stanchina E et al (2014) In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 516: 423–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangoni E, Vincent‐Salomon A, Auger N, Degeorges A, Assayag F, de Cremoux P, de Plater L, Guyader C, De Pinieux G, Judde J‐G et al (2007) A new model of patient tumor‐derived breast cancer xenografts for preclinical assays. Clin Cancer Res 13: 3989–3998 [DOI] [PubMed] [Google Scholar]
- Maresch R, Mueller S, Veltkamp C, Öllinger R, Friedrich M, Heid I, Steiger K, Weber J, Engleitner T, Barenboim M et al (2016) Multiplexed pancreatic genome engineering and cancer induction by transfection‐based CRISPR/Cas9 delivery in mice. Nat Commun 7: 10770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins CP, Brown‐Swigart L, Evan GI (2006) Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 127: 1323–1334 [DOI] [PubMed] [Google Scholar]
- McAllister SS, Weinberg RA (2014) The tumour‐induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol 16: 717–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merk J, Rolff J, Becker M, Leschber G, Fichtner I (2009) Patient‐derived xenografts of non‐small‐cell lung cancer: a pre‐clinical model to evaluate adjuvant chemotherapy? Eur J Cardiothorac Surg 36: 454–459 [DOI] [PubMed] [Google Scholar]
- Merk J, Rolff J, Dorn C, Leschber G, Fichtner I (2011) Chemoresistance in non‐small‐cell lung cancer: can multidrug resistance markers predict the response of xenograft lung cancer models to chemotherapy? Eur J Cardiothorac Surg 40: e29–e33 [DOI] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massagué J (2005) Genes that mediate breast cancer metastasis to lung. Nature 436: 518–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misale S, Bozic I, Tong J, Peraza‐Penton A, Lallo A, Baldi F, Lin KH, Truini M, Trusolino L, Bertotti A et al (2015) Vertical suppression of the EGFR pathway prevents onset of resistance in colorectal cancers. Nat Commun 6: 8305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica‐Worms D et al (2013) Targeting tumor‐infiltrating macrophages decreases tumor‐initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 73: 1128–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJ, Díez‐Villaseñor C, Soria E, Juez G (2000) Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol Microbiol 36: 244–246 [DOI] [PubMed] [Google Scholar]
- Molyneux G, Geyer FC, Magnay F‐A, McCarthy A, Kendrick H, Natrajan R, Mackay A, Grigoriadis A, Tutt A, Ashworth A et al (2010) BRCA1 basal‐like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 7: 403–417 [DOI] [PubMed] [Google Scholar]
- Nakasone ES, Askautrud HA, Kees T, Park J‐H, Plaks V, Ewald AJ, Fein M, Rasch MG, Tan Y‐X, Qiu J et al (2012) Imaging tumor‐stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell 21: 488–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura H, Nose M, Hiai H, Minato N, Honjo T (1999) Development of lupus‐like autoimmune diseases by disruption of the PD‐1 gene encoding an ITIM motif‐carrying immunoreceptor. Immunity 11: 141–151 [DOI] [PubMed] [Google Scholar]
- Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N et al (2001) Autoimmune dilated cardiomyopathy in PD‐1 receptor‐deficient mice. Science 291: 319–322 [DOI] [PubMed] [Google Scholar]
- Noy R, Pollard JW (2014) Tumor‐associated macrophages: from mechanisms to therapy. Immunity 41: 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T (2004) Mutant p53 gain of function in two mouse models of Li‐Fraumeni syndrome. Cell 119: 847–860 [DOI] [PubMed] [Google Scholar]
- Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D et al (2009) Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324: 1457–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özdemir BC, Pentcheva‐Hoang T, Carstens JL, Zheng X, Wu C‐C, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV et al (2014) Depletion of carcinoma‐associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25: 719–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich‐Pyest E, Urieli‐Shoval S, Galun E, Ben‐Neriah Y (2004) NF‐κB functions as a tumour promoter in inflammation‐associated cancer. Nature 431: 461–466 [DOI] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M et al (2014) CRISPR‐Cas9 knockin mice for genome editing and cancer modeling. Cell 159: 440–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R (2012) Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483: 100–103 [DOI] [PubMed] [Google Scholar]
- Premsrirut PK, Dow LE, Kim SY, Camiolo M, Malone CD, Miething C, Scuoppo C, Zuber J, Dickins RA, Kogan SC et al (2011) A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell 145: 145–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V et al (2013) CSF‐1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 19: 1264–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu S, Wang K, Xue H, Wang Y, Wu R, Liu C, Gao AC, Gout PW, Collins CC, Wang Y (2014) Enhanced anticancer activity of a combination of docetaxel and Aneustat (OMN54) in a patient‐derived, advanced prostate cancer tissue xenograft model. Mol Oncol 8: 311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Joyce JA (2013) Microenvironmental regulation of tumor progression and metastasis. Nat Med 19: 1423–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert JM, Wenger JB (2008) Development trends for new cancer therapeutics and vaccines. Drug Discov Today 13: 30–37 [DOI] [PubMed] [Google Scholar]
- Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW et al (2014) Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25: 735–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg S, Nygren AOH, Pajic M, van Leeuwen FWB, van der Heijden I, van de Wetering K, Liu X, de Visser KE, Gilhuijs KG, van Tellingen O et al (2007) Selective induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc Natl Acad Sci USA 104: 12117–12122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AOH, Zander SAL, Derksen PWB, de Bruin M, Zevenhoven J, Lau A et al (2008) High sensitivity of BRCA1‐deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA 105: 17079–17084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudalska R, Dauch D, Longerich T, McJunkin K, Wuestefeld T, Kang T‐W, Hohmeyer A, Pesic M, Leibold J, von Thun A et al (2014) In vivo RNAi screening identifies a mechanism of sorafenib resistance in liver cancer. Nat Med 20: 1138–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B, Chang‐Strachan D, Chan V, Rosenbusch A, Ho CMT, Pryer N, Daniel D, Hwang ES, Rugo HS, Coussens LM (2014) Macrophage IL‐10 blocks CD8+ T cell‐dependent responses to chemotherapy by suppressing IL‐12 expression in intratumoral dendritic cells. Cancer Cell 26: 623–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüther U, Komitowski D, Schubert FR, Wagner EF (1989) c‐fos expression induces bone tumors in transgenic mice. Oncogene 4: 861–865 [PubMed] [Google Scholar]
- Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova‐Acebes M, Khudoynazarova M, Agudo J, Tung N et al (2016) Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD‐L1 and BRAF inhibition. Immunity 44: 924–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez‐Paulete AR, Cueto FJ, Martínez‐López M, Labiano S, Morales‐Kastresana A, Rodríguez‐Ruiz ME, Jure‐Kunkel M, Azpilikueta A, Aznar MA, Quetglas JI et al (2016) Cancer immunotherapy with immunomodulatory anti‐CD137 and anti‐PD‐1 monoclonal antibodies requires BATF3‐dependent dendritic cells. Cancer Discov 6: 71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez‐Rivera FJ, Papagiannakopoulos T, Romero R, Tammela T, Bauer MR, Bhutkar A, Joshi NS, Subbaraj L, Bronson RT, Xue W et al (2014) Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 516: 428–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez‐Rivera FJ, Jacks T (2015) Applications of the CRISPR‐Cas9 system in cancer biology. Nat Rev Cancer 15: 387–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheer N, Kapelyukh Y, McEwan J, Beuger V, Stanley LA, Rode A, Wolf CR (2012) Modeling human cytochrome P450 2D6 metabolism and drug‐drug interaction by a novel panel of knockout and humanized mouse lines. Mol Pharmacol 81: 63–72 [DOI] [PubMed] [Google Scholar]
- Schönhuber N, Seidler B, Schuck K, Veltkamp C, Schachtler C, Zukowska M, Eser S, Feyerabend TB, Paul MC, Eser P et al (2014) A next‐generation dual‐recombinase system for time‐ and host‐specific targeting of pancreatic cancer. Nat Med 20: 1340–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD et al (2004) MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 431: 1112–1117 [DOI] [PubMed] [Google Scholar]
- Sharma P, Allison JP (2015) The future of immune checkpoint therapy. Science 348: 56–61 [DOI] [PubMed] [Google Scholar]
- Shibata H, Toyama K, Shioya H, Ito M, Hirota M, Hasegawa S, Matsumoto H, Takano H, Akiyama T, Toyoshima K et al (1997) Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science 278: 120–123 [DOI] [PubMed] [Google Scholar]
- Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, Bell‐McGuinn KM, Zabor EC, Brogi E, Joyce JA (2011) Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev 25: 2465–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Lima A, Molina R, Hamilton P, Clermont AC, Devasthali V, Thompson JD, Cheng JH, Bou Reslan H, Ho CCK et al (2010) Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat Biotechnol 28: 585–593 [DOI] [PubMed] [Google Scholar]
- Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P (1987) Coexpression of MMTV/v‐Ha‐ras and MMTV/c‐myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell 49: 465–475 [DOI] [PubMed] [Google Scholar]
- Sutherland KD, Proost N, Brouns I, Adriaensen D, Song J‐Y, Berns A (2011) Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 19: 754–764 [DOI] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al (2012) Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med 366: 2443–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres R, Martin MC, Garcia A, Cigudosa JC, Ramirez JC, Rodriguez‐Perales S (2014) Engineering human tumour‐associated chromosomal translocations with the RNA‐guided CRISPR‐Cas9 system. Nat Commun 5: 3964 [DOI] [PubMed] [Google Scholar]
- Van Keymeulen A, Lee MY, Ousset M, Brohée S, Rorive S, Giraddi RR, Wuidart A, Bouvencourt G, Dubois C, Salmon I et al (2015) Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 525: 119–123 [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C et al (2004) In vivo activation of the p53 pathway by small‐molecule antagonists of MDM2. Science 303: 844–848 [DOI] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T (2007) Restoration of p53 function leads to tumour regression in vivo. Nature 445: 661–665 [DOI] [PubMed] [Google Scholar]
- de Visser KE, Korets LV, Coussens LM (2005) De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell 7: 411–423 [DOI] [PubMed] [Google Scholar]
- Vooijs M, Jonkers J, Berns A (2001) A highly efficient ligand‐regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO Rep 2: 292–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walrath JC, Hawes JJ, Van Dyke T, Reilly KM (2010) Genetically engineered mouse models in cancer research. Adv Cancer Res 106: 113–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R (2013) One‐step generation of mice carrying mutations in multiple genes by CRISPR/Cas‐mediated genome engineering. Cell 153: 910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Mou H, Li S, Li Y, Hough S, Tran K, Li J, Yin H, Anderson DG, Sontheimer EJ et al (2015) Adenovirus‐mediated somatic genome editing of Pten by CRISPR/Cas9 in mouse liver in spite of Cas9‐specific immune responses. Hum Gene Ther 26: 432–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW (1995) Lymphoproliferative disorders with early lethality in mice deficient in Ctla‐4. Science 270: 985–988 [DOI] [PubMed] [Google Scholar]
- Wculek SK, Malanchi I (2015) Neutrophils support lung colonization of metastasis‐initiating breast cancer cells. Nature 528: 413–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J, Öllinger R, Friedrich M, Ehmer U, Barenboim M, Steiger K, Heid I, Mueller S, Maresch R, Engleitner T et al (2015) CRISPR/Cas9 somatic multiplex‐mutagenesis for high‐throughput functional cancer genomics in mice. Proc Natl Acad Sci USA 112: 13982–13987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wefers B, Panda SK, Ortiz O, Brandl C, Hensler S, Hansen J, Wurst W, Kühn R (2013) Generation of targeted mouse mutants by embryo microinjection of TALEN mRNA. Nat Protoc 8: 2355–2379 [DOI] [PubMed] [Google Scholar]
- Weigelt B, Peterse JL, van't Veer LJ (2005) Breast cancer metastasis: markers and models. Nat Rev Cancer 5: 591–602 [DOI] [PubMed] [Google Scholar]
- Willimsky G, Blankenstein T (2005) Sporadic immunogenic tumours avoid destruction by inducing T‐cell tolerance. Nature 437: 141–146 [DOI] [PubMed] [Google Scholar]
- Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M et al (2015) REV7 counteracts DNA double‐strand break resection and affects PARP inhibition. Nature 521: 541–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon‐Cardo C, Lowe SW (2007) Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445: 656–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, Cai W, Yang G, Bronson R, Crowley DG et al (2014) CRISPR‐mediated direct mutation of cancer genes in the mouse liver. Nature 514: 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R (2013) One‐step generation of mice carrying reporter and conditional alleles by CRISPR/Cas‐mediated genome engineering. Cell 154: 1370–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Jaenisch R (2014) Generating genetically modified mice using CRISPR/Cas‐mediated genome engineering. Nat Protoc 9: 1956–1968 [DOI] [PubMed] [Google Scholar]
- Yoshida T, Kinoshita H, Segawa T, Nakamura E, Inoue T, Shimizu Y, Kamoto T, Ogawa O (2005) Antiandrogen bicalutamide promotes tumor growth in a novel androgen‐dependent prostate cancer xenograft model derived from a bicalutamide‐treated patient. Cancer Res 65: 9611–9616 [DOI] [PubMed] [Google Scholar]
- Zanella ER, Galimi F, Sassi F, Migliardi G, Cottino F, Leto SM, Lupo B, Erriquez J, Isella C, Comoglio PM et al (2015) IGF2 is an actionable target that identifies a distinct subpopulation of colorectal cancer patients with marginal response to anti‐EGFR therapies. Sci Transl Med 7: 272ra12 [DOI] [PubMed] [Google Scholar]
- Zender L, Xue W, Zuber J, Semighini CP, Krasnitz A, Ma B, Zender P, Kubicka S, Luk JM, Schirmacher P et al (2008) An oncogenomics‐based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell 135: 852–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu C‐C, LeBleu VS, Kalluri R (2015) Epithelial‐to‐mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527: 525–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang‐Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG (2014) CSF1/CSF1R blockade reprograms tumor‐infiltrating macrophages and improves response to T‐cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 74: 5057–5069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckermann M, Hovestadt V, Knobbe‐Thomsen CB, Zapatka M, Northcott PA, Schramm K, Belic J, Jones DTW, Tschida B, Moriarity B et al (2015) Somatic CRISPR/Cas9‐mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat Commun 6: 7391 [DOI] [PMC free article] [PubMed] [Google Scholar]