

Abstract
The mouse has been a major model system for biomedical research. Gene editing technologies will further increase its importance for studying the human condition and human diseases.

Subject Categories: Methods & Resources, Molecular Biology of Disease, S&S: Technology
The usefulness of a specific technology often hits a ceiling based on technical limitations. Then, a single advance, frequently orthogonal to the core methodology, dramatically expands the utility of this technology. The effectiveness of early surgeons wielding a scalpel was severely limited by how much a patient could withstand the pain of an operation. Anesthesia was the core discovery that permitted surgery to become a consistently effective medical intervention. Molecular cloning was a powerful technology to understand the importance of genes in biology, but it had limited utility in population genetics and in medicine, because of the arduous requirements for cloning a single gene. The invention of polymerase chain reaction dramatically expanded the utility of molecular biology into high‐throughput sequencing, epidemiology, forensics, diagnostics, and gene therapy. We believe that the advent of powerful gene editing technologies such as the CRISPR/CAS system will be this transformative technology for murine genetics.
The mouse as experimental model
The subsequent massive sequencing efforts to define the mutational spectrum of human diseases have uncovered a bewildering number of genetic changes. It is now recognized that most diseases are genetically complex and that their dissection requires the enrollment of many hundreds of thousands of individuals to achieve adequate statistical power to ensure the validity of an association. This purely statistical approach is clearly impracticable, given the many variants and mutations that are being uncovered with each sequence‐based study. Thus, the translation of human genome research into health care heavily relies on appropriate organismal models that accurately reflect the human condition. This is where the laboratory mouse (Mus musculus) comes in. For many diseases that require an intact organ system in an intact animal—such as neurological disorders, renal disease, complex cardiopulmonary syndromes, and aging—the mouse has been a primary experimental model for mammalian biology since the turn of the last century.
… the translation of human genome research into healthcare heavily relies on appropriate organismal models that accurately reflect the human condition.
Its utility was originally limited to studying observable transmissible phenomena, such as the generation of tumors from retroviruses, or the heredity of visually detectable traits. But as their genes were mapped, the usefulness of the mouse for assigning a range of phenotypes to specific gene mutations made it a powerful model system for mammalian genetics. This was significantly expanded when transgenesis and homologous recombination allowed the creation of new genetic models to directly test gene function. With each development, the importance of the mouse as a biological model of the human condition increased. Recently, however, some have claimed that the experimental mouse has approached an asymptote in its utility in biomedical research. Already, there have been criticisms that the mouse is not an appropriate model system for biomedical validation of therapeutic agents 1, 2. The solution is either to abandon the mouse for a better, yet cost‐effective, model system, or to improve it as the premier model system for mammalian genetics. There is now evidence that the latter will be the likely scenario and that modern gene editing technologies, especially the CRISPR/Cas system, will be the primary driver.
The impact of CRISPR/Cas
Though there are a number of CRISPR (clustered regularly interspaced short palindromic repeats) systems, the type II CRISPR/Cas9 system is the current workhorse for advanced gene editing 3. Cas9 cuts one or both strands of DNA at precise genomic locations as directed by a guide RNA. Once DNA double‐strand breaks are made, the cell's repair machinery takes over to mend broken ends. DNA repair is executed by one of the two major repair mechanisms: error‐prone non‐homologous end joining (NHEJ) or high‐fidelity homology‐directed repair (HDR). NHEJ provides a quick fix by ligating two loose ends together but often generates deletions or insertions. HDR requires a homologous repair template, such as the sister chromatid, a homologous chromosome, or a piece of DNA with homologous sequences delivered to the cell. This enables insertion of a new or redesigned fragment of DNA without leaving any undesired footprint behind. Though off‐target alterations can appear, these are not frequent and have been further mitigated by careful selection of guide RNAs and the use of recombinant Cas9 proteins.
It is this combination of precisely and efficiently placing the DNA break and providing the desired repair template that makes CRISPR/Cas9 genome editing superior to the traditional recombination approach that relied on a very rare recombination event in embryonic stem (ES) cells (Fig 1). Another advantage over conventional gene targeting is the time frame from concept to actual mouse model. Before a correctly edited ES cell clone is produced, it can take many months to generate the construct, and target and validate selected clones. By contrast, CRISPR guide RNAs and repair templates can be easily designed and synthetized in a few days. ES cell injection into blastocyst embryos generates chimeric mice that need further breeding to transmit the targeted allele to next generation. However, transmission rates vary depending on the quality of ES cells and may require multiple attempts or chimeras to produce heterozygous mutants, further delaying the process. In contrast, injection of CRISPR reagents into one‐cell embryos can directly generate heterozygotes for breeding or even homozygous mutants for early assessment of the phenotype. Although in many cases editing in zygotes produces a mosaic founder, the transmission rates are often much higher than in the case of chimeras from traditional methods.
Figure 1. Conventional gene targeting in ES cells versus CRISPR/Cas9‐mediated gene editing in zygotes.
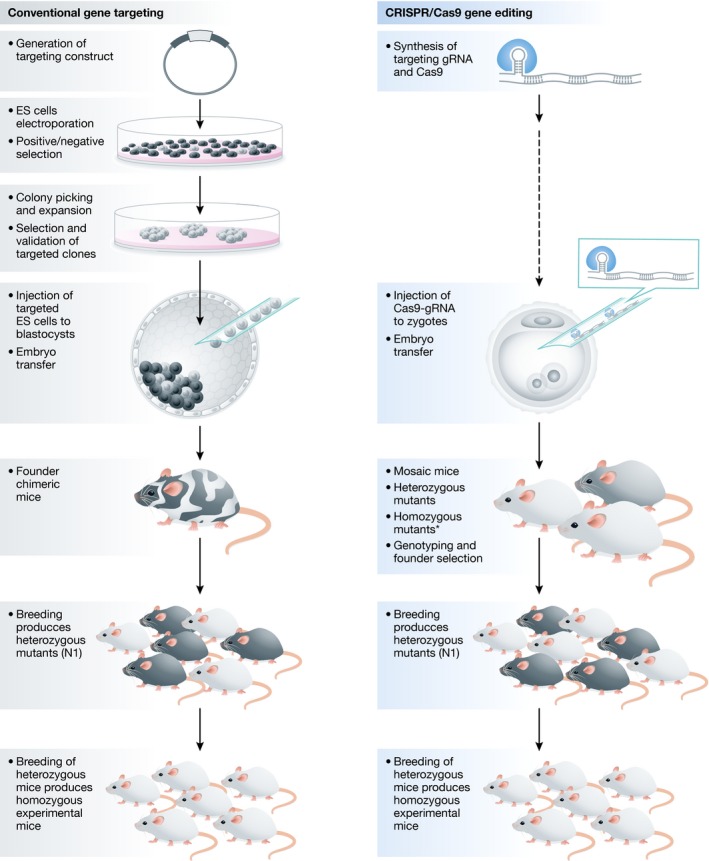
Generation of genetically engineered mouse models via ES cell targeting can take an average of 1 year and involves many intermediate steps. In contrast, CRISPR/Cas9 simplifies and accelerates the production of mice carrying the desired mutation (N1). This is achieved by eliminating the need to generate targeted ES cell line. In some cases, CRISPR/Cas9‐mediated embryo editing also produces homozygous mutants, which can add the additional benefit of preliminary phenotype assessment.
Whereas multiple changes could not be accomplished by standard approaches except by stacking knock‐ins through serial crosses or tedious manipulations in ES cells, CRISPR can achieve simultaneous engineering of multiple loci even for closely linked genes, which is almost impossible to achieve by breeding two knock‐in lines. Classical gene engineering by homologous recombination requires manipulation in established ES cell lines, which limits the strains in which the initial engineering takes place, and often leaves foreign genetic material behind such as LoxP sites or selection cassettes. Moreover, if no robust mouse ES cells exist for a preferred background strain, the process would need to perform gene targeting on a strain for which mouse ES cells exist, followed by backcrosses to the desired background. A minimum of five backcrosses and one additional year would be required to develop a mouse with the desired mutation on the preferred genetic background. This can be drastically accelerated by using CRISPR gene modifications directly into the preferred genetic background.
Since the CRISPR/Cas technologies are in their early childhood, neither their full potential nor their theoretical limits can be foreseen. But in its current iteration, CRISPR has already significantly reduced the time and cost to creating an engineered mouse (Table 1). The technology still has limitations: As the complexity of the targeted mutation increases from inducing a disruptive deletion to inserting genetic material of increasing size, the time and cost advantage of CRISPR diminishes. Still, the range of strains that can be manipulated, and the simplicity of developing the construct, especially for gene disruptions, has made CRISPR the preferred methodology for gene engineering in the mouse.
Table 1.
The percentage cost savings and the timelines using the CRISPR/Cas9 method compared with traditional homologous recombination technology in murine ES cells
| mES cells | CRISPR (in zygotes) | |||||
|---|---|---|---|---|---|---|
| conventional gene targeting | indel (small deletion/insertion) | deletion | precise editing (SNP) | conditional KO (LoxP, FRT) | Kl (reporter) | |
| % cost savings | baseline | 81% | 68% | 59% | 38% | 41% |
| avg. timelines | 12 months | 6.5 months | 7 months | 7 months | 8 months | 8 months |
These estimates include project initiation through the N1 stage, at which germline transmission has been achieved and a genetically modified mouse strain has been established. The major difference between the costs and timelines reflects the necessity of performing electroporation, selection, clone picking, and the subsequent screening in mES cells for traditional gene targeting. Downstream phases (injections and subsequent breeding) are similar with respect to the timelines. Note that for conventional targeting for which there are no murine ES cells for the desired background strain, the project timeline will increase by an additional 6–18 months, depending on the extent of backcrossing required to move the mutation to the desired background strain.
The recent reports of cytoplasmic delivery of gRNAs and Cas9 RNAs through electroporation demonstrated the potential to significantly increase the throughput of manipulating zygotes. Implementation of this advance could reduce the need for trained microinjectionists to serially inject single‐cell embryos. This would parallelize the process by orders of magnitude. Whenever a process undergoes quantum jumps in efficiency through parallelization, unique applications emerge. PCR is a great example: While its initial use was massively amplifying a genetic fragment, its miniaturization and parallelization gave rise to next‐generation sequencing. We can expect similar innovations with CRISPR for whole organism engineering in the future.
What scientific vistas will CRISPR open for mouse genetics? Immediately, we see four: creation of complete allelic series of a genetic disease; experimental validation of the phenotypic output of combinatorial gene variations in the whole mammalian organism; the progressive reshaping of the whole mouse genome so entire systems resemble the human—the “sapienization” of the mouse; and a comprehensive understanding of genetic background and its effect on a Mendelian disease mutation. Together, these comprise the next‐generation mouse models that our students will be exploiting.
Modeling the genetic diversity of a disease
Creation of complete allelic series of a genetic disorder starts with identifying candidate mutations for a particular disease, anywhere from autism, to idiopathic cardiomyopathies and epilepsies, to Alzheimer's disease. In each case, tens to hundreds of different mutations of individual genes and of tens to hundreds of genes comprise the causative genetic universe of each syndrome. Additionally, each of these diseases is best represented by output from an intact organism as opposed to cell cultures. In amyotrophic lateral sclerosis (ALS), our knowledge was restricted for many years to a handful of genes from families of known genetic inheritance. These familial forms of ALS accounted for only 10% of the patient population. With NGS, the identification of causative genes for ALS has increased dramatically. There are currently more than 20 genes known to be associated with ALS, over seven of which have been identified in the last 2–3 years. Using CRISPR technology, pathogenic variants are now being created in mice. Previously, genetic engineering was restricted to a single variant owing to high cost, but it is now feasible to create patient‐specific mutations in different functional domains of the protein.
For rare and orphan diseases, NGS has pointed to specific gene diagnoses, where none existed before. Mice are not only critical to understanding the pathophysiology of the disease, but also play an important role in therapeutic strategies, many of which can be specific to a given mutation. For spinal muscular atrophy (SMA), a motor neuron disease that controls voluntary muscle movement in children, a number of SMA mouse models were critical in assessing therapies for exon inclusion, RNA stabilization, and gene therapy. Other mouse models were instrumental to inform clinical trials on the therapeutic window for treatment 4, and still other models helped understanding the possible benefits of therapies for patients in more advanced disease states.
With the power of CRISPR‐driven model creation, it is conceivable that the entire universe of mutations cause a disease can be recapitulated in a series of mice
Indeed, it is rare that any one mouse model will provide us with all the information we need to understand disease mechanism and test therapeutics. To this end, the ease by which we can now genetically engineer mutations in mice allows us to develop the preclinical resources needed for precision medicine strategies. With the power of CRISPR‐driven model creation, it is conceivable that the entire universe of mutations that cause a disease can be recapitulated in a series of mice. The range of phenotypic diversity will allow for a true representation of the heterogeneity of the human disease so as to avoid the “overfitting” of treatments in preclinical studies performed on only one mutation in one mouse strain. These advanced models will facilitate the identification of drugs that only work in one or a few such representations of the disease. The limited response to a drug in a predictable subset of disease‐causing mutations would actually be considered a success in the paradigm of precision medical treatments; that is, that response heterogeneity can be explained by genetic heterogeneity.
Assessing combinatorial gene effects
Experimental validation of the phenotypic output of combinatorial gene variations in the whole mammalian organism is perhaps one of the more elusive goals in systems biology. It has so far been limited to cell culture and single‐cell organisms such as yeast, and the relevant diseases elucidated by these cellular screens have been largely related to cancer since it is primarily a disease of a cell. For disorders involving the development or function of an organ, the mouse has again been the favored genetic model. However, the genetic crosses necessary to generate double and triple mutations in one animal are arduous and costly and therefore have been very limited.
With CRISPR, moderate scans of synthetic phenotypes—the most obvious being lethality—can be performed on a whole mammalian organism for the first time. The first phase of this approach has been pursued by the International Mouse Phenotyping Consortium and uncovered 410 lethal genes during the production of the first 1,751 unique gene knockouts 5. Embryonic lethality of likely pairs of non‐lethal gene knockouts can now be “scanned” by stacking second mutations introduced by CRISPR into a viable knockout. This approach can theoretically be performed in vitro through microinjection or electroporation of multiple gRNAs and Cas9 RNAs into single‐cell embryos and screening for severe perturbations of early embryonic formation. Already, the multiplex introduction of 13 CRISPR/gRNAs against tumor suppressor genes (TSGs) into the pancreatic cells of intact mice susceptible to pancreatic cancers showed that the tumors that arose in these transfected animals had a minimum of 47% (6/13) mutations in specific tumor suppressors 6. Not only did this experiment show the TSG mutational load that seemed necessary for pancreatic cancer formation, but it also pinpointed which combinations were sufficient to cause cancer.
In germline gene engineering, it is imaginable that, if three to five mutations can be generated in one mouse (as has been claimed 7), crosses between these combinatorial models would generate animals with combinations of up to 6–10 mutations. Assuming the mutated genes are located sufficiently distant in the genome to allow independent segregation, different mutational loads less than the theoretical maximum will be represented in the offspring. Of course, there are limitations to the number and scope of mice and genes, but the idea of examining the effects of combinations of ten candidate genes is tantalizing for systems geneticists.
The “sapienization” of the mouse
The mouse is the most practicable mammalian genetic model system because of cost, the deep knowledge of its physiology, and the facile ability to manipulate its genome. But the theoretical limitations of mouse models are obvious: After more than 65 million years of evolutionary separation, the mouse is at a systems level different from the human despite the high homology between the mouse and the human genes. Nonetheless, the biological principles uncovered by research in murine systems have largely been representative of human biology.
… exchanging mouse genes for their human homologues have rendered the resultant mouse models more useful to test immunological, viral, and pharmacological outcomes
The challenge is that the expectations for medical relevancy of these model systems are now much higher, as translational impact has become de rigueur in biological research. Precision medicine requires model systems that can accurately reflect the diverse genotype–phenotype associations seen in patients. However, there are many examples where the evolutionary distance between mouse and human is the major experimental hurdle to optimizing the mouse as a model system for human biology. For example, cytokines GM‐CSF and Kit Ligand, though highly homologous between the human and the mouse, are sufficiently diverged such that the human and mouse proteins are not functionally interchangeable. This has limited the utility of immunodeficient mouse models to engraft a more complete human immune system. The solution to this problem has been to genetically replace the murine version of the cytokine with the human homologue. The expression of human cytokines in “humanized” immunodeficient mice has increased engraftment and function of human hematopoietic stem cells. Similarly, exchanging mouse genes for their human homologues has rendered the resultant mouse models more useful to test immunological, viral, and pharmacological outcomes. It is this progressive conversion of mouse systems to resemble the Homo sapiens genome that we call sapienization.
Perhaps the most impressive example of sapienization is the conversion of mouse immunoglobulin‐producing genes to their human counterparts to generate human monoclonal antibodies. Dubbed HumAb mice, the first iterations, disrupted the endogenous mouse immunoglobulin genes and randomly inserted the human transgene. This was found to be lacking because of immunodeficiency in the engineered mice owing to the incompatibility of the human constant regions with the murine B‐cell receptor‐signaling complex. By selectively replacing the murine germline variable region with the human variable region sequences in situ, but leaving the native mouse constant regions intact, the resultant mice exhibit a normal functioning humoral system but produce antibodies with human variable region sequences 8. These mice have become invaluable in speeding the generation of therapeutic monoclonal antibodies.
In these examples, the reengineering of functional portions of the mouse genome resulted in highly valuable tools for drug discovery, either as a platform for testing or as a generator of therapeutic drugs. The crafting of new discovery tools in mice will be greatly facilitated by the CRISPR systems. Not only will the speed of model creation be accelerated, but the ability to stack successive genomic changes on preexisting ones allowing for complex reengineering of the mouse genome will become practicable.
A comprehensive understanding of genetic backgrounds
Inbred mice are effective models for identifying genes that contribute to simple Mendelian traits. However, commonly used inbred mouse strains have been only modestly successful in mirroring complex human traits due to their limited genetic diversity. Experimental approaches in the mouse have not kept pace with rapid developments in human genetic studies, and new strategies for complex trait dissection in model organisms are needed.
To address this shortcoming, interbred panels of mouse strains resulting in cohorts with high genetic diversity have been specifically designed for gene association studies with the requisite statistical power and resolution for mapping complex traits. Genetically defined mouse models have advantages over other model organisms and cell‐based assays, because they provide well‐defined experimental systems in the context of an intact living mammal, under controlled environmental conditions and unlimited access to tissues. Of the extant mouse resources, the Hybrid Diversity Mouse Panel (HMDP), initiated at UCLA in 2010, which comprises 100 inbred strains, has yielded linkage data involving clinical, expression, proteomic, and metabolic traits.
… obtaining an existing model off‐the‐shelf bypasses the long and costly exercise of turning a new allele into a useful mouse model, and improves reproducibility through standardization
Further efforts to improve diversity and increase the precision of mouse genetic resources led to the creation of the Collaborative Cross (CC). CC strains exhibit a genetic structure more representative of the genetic variation present in the human population. It has the essential characteristics of an ideal experimental population: genome‐wide causative variants with unique strain distribution patterns, reproducible populations supporting cumulative data integration, and large populations providing statistical support for systems networks. The Diversity Outbred (DO) population, a newer mouse resource derived from partially inbred CC strains, offers an allelic diversity that is much greater than in either HMDP or CC, such that each animal has a high degree of heterozygosity and carries a unique combination of alleles 9.
When CRISPR technologies are coupled with high throughput gRNA/Cas RNA delivery systems, it is conceivable to create the same mutation replicating a Mendelian disorder in a large number of CC strains, or even DO embryos. This would permit the mapping of genetic modifiers across the entire diversity landscape of the mouse. While the prospect of targeted mutagenesis across mouse diversity panels was heretofore untenable using classic ESC‐based mutagenesis, CRISPR technology adds dramatic power to classic screening approaches for genetic modifiers.
Challenges from other model systems
With their rapid reproductive cycles and large litter sizes, mice have historically dominated the field of transgenesis despite the inherent impediments of mammalian homologous recombination. CRISPR has now been successfully applied to produce rat strains carrying alterations in multiple genes. Although they are an excellent model system for physiology, rats are considerably more expensive to maintain and age slowly and are unlikely to displace the mouse with its established databases, rich mutant resources, and diverse, well‐characterized genetic backgrounds.
CRISPR technology also promises to overcome many of the technological barriers to the generation of targeted mutants in larger species such as non‐human primates. A limited number of genetically modified primates (mainly macaques and marmosets) have already been developed as models of CNS disorders that affect higher cognitive functions. However, primate models present considerable hurdles, with relatively diverse genetic backgrounds, low efficiency of CRISPR germline modification, long generation periods, and singleton births. Most experiments will necessarily be carried out on a handful of founder animals, which challenges the consistency in measuring phenotypes and the design of appropriately powered studies. Ethical considerations, as well as more logistical barriers, such as transport and cost, are likely to limit the adoption of the primate as a widely used genetic system. The mouse, as a model system for human disease, will not go away.
What does it mean for mouse facilities?
With the spread of CRISPR expertise, one could argue that repositories will no longer be needed since everyone can make their own models. This was the case for cDNA resources once PCR enabled ready recreation of any defined DNA sequence. However, the germline transmission of a newly engineered allele is only the first step in the complex process of producing a model of human disease, or a useful tool for mechanistic investigation. New alleles need careful quality assurance, while the presumed model needs to be validated through in‐depth phenotyping. Thus, while local self‐creation of models might be the modus operandi for a few centers or laboratories, obtaining an existing model off‐the‐shelf bypasses the long and costly exercise of turning a new allele into a useful mouse model and improves reproducibility through standardization.
Moreover, as the relevance of mouse diversity becomes more widespread, scientists will want to start with existing models on defined genetic backgrounds and engineer better ones in different genetic backgrounds, or with preexisting engineered genetic cassettes. This also requires access to repository resources. At the Jackson Laboratory, which hosts a major mouse repository of more than 9,000 distinct lines, we believe that CRISPR/Cas9 will enable the generation of greater diversity of new models, rather than duplication of prior efforts, and that the role of repositories may be increasingly important, especially for combinatorial gene models. The analogy with libraries is pertinent. When books were few, only a few would own these books, but with a plethora of books published, public libraries became a necessity even for the wealthy.
CRISPR has lowered the hurdle for any investigator to create a mouse model. But the true limiting factor will be the availability and the quality of mouse phenotyping facilities to analyze the physiological outputs. They are expensive to maintain and currently not structured for high‐throughput screening. These factors strongly suggest that the next generation of mouse experiments will be characterized by greater numbers and complexity of the animal models used. In this setting, very few facilities can mount the requisite scale of operations to match the need. We therefore believe that the establishment of national laboratories to support the scientific community for large‐scale preclinical experimentation will be necessary. Akin to the Department of Energy's (DOE) national laboratories hosting linear accelerators, these National Laboratories for Model Systems can conduct consensus‐driven experiments critical to answering biomedical questions. The prototype for mouse studies may be the Interventions Testing Program (ITP; http://www.nia.nih.gov/research/dab/interventions-testing-program-itp), in which drugs are tested for longevity effects 10. Such facilities will be cost‐effective because of economies of scale and can host visiting scholars to conduct specialized parts of collective research programs.
With refinement of available genetic backgrounds and increasing sophistication in physiological characterization, we predict that CRISPR technology will reinstate the mouse as the premier experimental system to model the human condition. However, at a nascent phase of development, this technology has already reshaped the conduct of functional genomic sciences.
Sidebar A: Further Reading.
Articles describing the range of genetic mutations found in any human disease
Taylor JC, Martin HC, Lise S, Broxholme J, Cazier JB, Rimmer A, Kanapin A, Lunter G, Fiddy S, Allan C et al (2015) Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet 47: 717–726
White MA, Sreedharan J (2016) Amyotrophic lateral sclerosis: recent genetic highlights. Curr Opin Neurol 29: 557–564
Mouse models of human disease
Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH (2005) SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full‐length SMN. Hum Mol Genet 14: 845–857
Gogliotti RG, Cardona H, Singh J, Bail S, Emery C, Kuntz N, Jorgensen M, Durens M, Xia B, Barlow C et al (2013) The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Hum Mol Genet 22: 4084–40101
Brehm MA, Wiles MV, Greiner DL, Shultz LD (2014) Generation of improved humanized mouse models for human infectious diseases. J Immunol Methods 410: 3–17
Boverhof DR, Chamberlain MP, Elcombe CR, Gonzalez FJ, Heflich RH, Hernández LG, Jacobs AC, Jacobson‐Kram D, Luijten M, Maggi A et al (2011) Transgenic animal models in toxicology: historical perspectives and future outlook. Toxicol Sci 121: 207–233
Mestas J, Hughes CC (2004) Of mice and not men: differences between mouse and human immunology. J Immunol 172: 2731–2738
Ghazalpour A, Rau CD, Farber CR, Bennett BJ, Orozco LD, van Nas A, Pan C, Allayee H, Beaven SW, Civelek M et al (2012) Hybrid mouse diversity panel: a panel of inbred mouse strains suitable for analysis of complex genetic traits. Mamm Genome 23: 680–692
Technologies using the mouse
Lonberg N (2005) Human antibodies from transgenic animals. Nat Biotechnol 23: 1117–1125
Qin W, Dion SL, Kutny PM, Zhang Y, Cheng AW, Jillette NL, Malhotra A, Geurts AM, Chen YG, Wang H (2015) Efficient CRISPR/Cas9‐mediated genome editing in mice by zygote electroporation of nuclease. Genetics 200: 423–430
Churchill GA, Gatti DM, Munger SC, Svenson KL (2012) The Diversity outbred mouse population. Mamm Genome 23: 713–718
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald‐Smith GP, Gao H, Hennessy L et al (2013) Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA 110: 3507–3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cavanaugh SE, Pippin JJ, Barnard ND (2014) Animal models of Alzheimer disease: historical pitfalls and a path forward. ALTEX 31: 279–302 [DOI] [PubMed] [Google Scholar]
- 3. Singh P, Schimenti JC, Bolcun‐Filas E (2015) A mouse geneticist's practical guide to CRISPR applications. Genetics 199: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lutz CM, Kariya S, Patruni S, Osborne MA, Liu D, Henderson CE, Li DK, Pellizzoni L, Rojas J, Valenzuela DM et al (2011) Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J Clin Invest 121: 3029–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, Meehan TF, Weninger WJ, Westerberg H, Adissu H et al (2016) High‐throughput discovery of novel developmental phenotypes. Nature 537: 508–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maresch R, Mueller S, Veltkamp C, Öllinger R, Friedrich M, Heid I, Steiger K, Weber J, Engleitner T, Barenboim M et al (2016) Multiplexed pancreatic genome engineering and cancer induction by transfection‐based CRISPR/Cas9 delivery in mice. Nat Commun 7: 10770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R (2013) One‐step generation of mice carrying mutations in multiple genes by CRISPR/Cas‐mediated genome engineering. Cell 153: 910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Macdonald LE, Karow M, Stevens S, Auerbach W, Poueymirou WT, Yasenchak J, Frendewey D, Valenzuela DM, Giallourakis CC, Alt FW et al (2014) Precise and in situ genetic humanization of 6 Mb of mouse immunoglobulin genes. Proc Natl Acad Sci USA 111: 5147–5152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chick JM, Munger SC, Simecek P, Huttlin EL, Choi K, Gatti DM, Raghupathy N, Svenson KL, Churchill GA, Gygi SP (2016) Defining the consequences of genetic variation on a proteome‐wide scale. Nature 534: 500–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nadon N, Strong R, Miller RA, Nelson J, Javors M, Sharp ZD, Peralba JM, Harrison D (2008) Design of aging intervention studies: the NIA interventions testing program. Age 30: 187–199 [DOI] [PMC free article] [PubMed] [Google Scholar]