

Abstract
Compelling evidence have demonstrated that bulk tumors can arise from a unique subset of cells commonly termed “cancer stem cells” that has been proposed to be a strong driving force of tumorigenesis and a key mechanism of therapeutic resistance. Recent advances in epigenomics have illuminated key mechanisms by which epigenetic regulation contribute to cancer progression. In this review, we present a discussion of how deregulation of various epigenetic pathways can contribute to cancer initiation and tumorigenesis, particularly with respect to maintenance and survival of cancer stem cells. This information, together with several promising clinical and preclinical trials of epigenetic modulating drugs, offer new possibilities for targeting cancer stem cells as well as improving cancer therapy overall.
Keywords: Cancer stem cells, Epigenetics, Histone methylation, Histone methyltransferase, DNA methylation, Signaling pathway
Background
Advances in genomic and epigenomic research has shaped our understanding of cancer over the past two decades. Rather than merely a perpetuating mass of dysregulated cells growing in an uncontrolled manner, cancer is also defined by the dynamic genetic and epigenetic alterations that contribute to cancer initiation and progression. Since epigenetic changes such as DNA methylation and histone modifications are crucial factors in developmental programming of stem cells to specific lineages of cellular and tissue differentiation, aberrant epigenetic alterations may transform normal stem cells to cancer stem cells with the loss of differentiation capacity and the acquisition of stem-like characteristics. More importantly, epigenetic mechanisms have been shown to be implicated in the observed variability of treatment response. For instance, a small subset of cells has been shown to be resistant to drug therapy in a variety of cancers such as melanoma, gastric, colon and lung cancers as a result of aberrant expression of key epigenetic modifiers. In this review, we will focus our discussion on the epigenetic regulation of CSCs and their impact on tumor-initiation, progression and response to therapies. We will also discuss recent advances in using epigenetic therapy to target cancer stem cells.
Main text
Cancer stem cells (CSCs)
Cancer stem cells (CSCs) define a small, unique subset of cells with self-renewal ability and the capacity to generate the different cell types that constitute the whole tumor [1]. These cells are termed CSCs because of their “stem-like” properties commonly shared with normal tissue stem cells. Such properties include extensive self-renewal ability (symmetrical and asymmetrical) and differentiation capacity. It should be noted that a general capacity to differentiate is not a mandatory feature of CSCs and that the ability of CSCs to differentiate and repopulate the cell types found in the original tumor is of greater significance. More importantly, CSCs should demonstrate potent tumor-initiation capacity. This property is usually demonstrated by injecting limited number of CSCs into an orthotopic in vivo environment to generate the bulk tumor. Nevertheless, the concept of CSC is of significant importance as it highlights the need to eradicate the CSC populations to achieve an effective cure.
The first clear evidence of CSCs being a key tumor-initiating subset of cancer cells was demonstrated in acute myeloid leukemia (AML) where prospective CSCs were isolated using cell surface markers that identify normal haematopoietic stem cells and evaluated for their tumor-initiating properties [2, 3]. Since then, similar identifications of tumor-initiating populations have been identified in multiple solid tumors that includes brain, breast, liver, ovary, prostate, lung, melanoma and colon cancers, by using different cell surface markers or through side population (SP) analysis [4–11]. For instance, in malignant glioma and medulloblastoma tumors, a putative neural stem cell marker, CD133 has been demonstrated to be adequate and essential to initiate and recapitulate the tumor upon xenotransplantation into immune-compromised mice [5]. However, this finding has been disputed as tumors can also arise from CD133-negative cells in a subset of glioma tumors [12]. In addition, CD133 surface marker expression has been demonstrated to change according to disease state and progression, further complicating its role as a bona fide CSC marker in brain tumors [13, 14]. In liver cancers such as hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (ICC), similar use of cell surface markers such as epithelial cell adhesion molecule (EpCAM), cytokeratin 19 (CK19), CD133, CD90, CD44, CD24, and CD13 has been applied to define a subpopulation of liver cancer cells as CSCs [15]. Importantly, it has recently been shown that these CSC markers are not specific to liver CSCs, and that distinct populations of liver CSCs express different surface markers possibly due to the strong intra- and inter-heterogeneity and varied etiology of liver cancer [16]. As a result, CSC studies have begun to move away from the reliance of cell surface markers to identify tumor-initiating cells and have begun to identify other complementary methods of measuring the functional activities of CSCs that may serve to identify CSCs as well as the molecular mechanisms that regulate CSCs [17].
Currently, the central theme of the CSC model is the ability of a subset of cells at the apex of the hierarchy to propagate tumors and promote tumor progression as compared to the non-tumorigenic cells within the bulk tumor. One of the gold standards to functionally identify CSCs is the capacity of these cells to regenerate a phenotypic copy of the original tumor in an orthotopic transplantation model. Non-CSCs, by definition, lack this ability and fail to generate tumors in the transplantation model. It is important to note that the CSC hierarchy model may not be ubiquitous for all cancers and that some tumorigenic cells are common in certain cancers. It is also important to note that such transplantation assays measure the tumorigenic potential of the cells to form tumors and not their actual fate. For example, alterations in tumorigenic assays carried out by Quintana and colleagues showed that CSC frequency could be increased by changing several experimental parameters such as the use of extracellular matrix (ECM) in the form of matrigel, prolonging the duration for tumor formation, and varying the severity of immune-compromised mice used [18]. This study highlighted that the tumor-initiating capacity may be an artificial consequence of the conditions employed in xenograft mouse models.
While analyzing CSC surface marker expression in primary tumors has been often performed to study the clinical impact of CSCs on tumor progression, more often than not, this has resulted in ambiguous data possibly due to the fact that CSC properties that sustain the primary tumor phenotype are defined by more than just specific marker expression [19, 20]. Analysis of key signalling pathway activity that resembles those functioning in stem-like cells, is more likely to accurately interrogate the clinical contribution of CSCs. An example of such studies was carried out by Lim et al. in BRCA1 mutation-associated breast tumors, where the authors prospectively isolated distinct subpopulations of normal and tumorigenic epithelial cells from BRCA1 mutation heterozygous individuals and found that luminal progenitors were highly represented in BRCA1 mutation-associated breast tumors, more than the stem cell population [21]. This suggests that luminal progenitors are more likely the cells-of-origin for BRCA1 mutation-associated breast tumors, which was later confirmed in a transgenic mouse model study carried out by Molyneux and colleagues [22]. These studies highlight the predictive capability of gene expression mapping of pathway activation rather than specific marker identity. In a separate study, John Dick and colleagues demonstrated that tumor-initiating AML stem cells contribute to disease progression and patient survival outcome, underscoring the importance of functionally defining the CSCs [23]. More importantly, the contribution of CSCs, with preferential activation of core stem cell programs, to patient survival outcome has been demonstrated. The study by Shats et al. showed that a stemness gene signature derived from embryonic stem cells (ESCs) could predict a breast cancer patient cohort sensitive to drugs linked to this signature using a Connectivity Map [24], demonstrating the clinical contribution of CSCs to patient outcome [25]. Collectively, these studies highlight that CSCs that perpetuate tumors are not merely defined by surface marker expression, but more importantly and more accurately by their gene expression profiles and consequent pathway activations.
Epigenetics: normal and cancer stem cells
Epigenetic regulation of the genome is one of the primary means by which genetic code is altered to control cellular developmental hierarchies. Epigenetic mechanisms such as histone modifications, DNA methylation, chromatin remodelling and even changes in noncoding RNAs including miRNAs together govern the epigenome landscape that dictate the outcome of cell fate specification without changes to the DNA sequences. Such changes in the genome is important during normal mammalian development and ESCs differentiation [26]. Importantly, gene expression profiles change during cellular differentiation according to not only a network of transcription factors but also the “epigenomic landscape” of the cell. For the purpose of this review, we will focus our discussions on two primary mechanisms of epigenetic regulation: histone modifications and DNA methylation.
Histone methylation occurs predominantly on lysine (K) and arginine (R) residues and these methylation marks serve as docking sites for histone readers [27]. Both lysine and arginine methylation can occur on both histones and non-histone proteins. The highly conserved histone lysine methylation occurs at three different levels: mono-, di-, and tri-methylation. Such modifications are commonly associated with gene activation or repression, depending on the target histone modification. For instance, histone H3 lysine 4 (H3K4), histone H3 lysine 36 (H3K36), and histone H3 lysine 79 (H3K79) are associated with gene activation whereas histone H3 lysine 9 (H3K9), histone H3 lysine 27 (H3K27) and histone H4 lysine 20 (H4K20) are associated with gene repression. The N-terminal tails of histones frequently undergo other post-translational modifications, which play significant roles in various DNA-templated processes, including transcription [28]. Hence, aberrations in histone modifications can lead to deregulated gene expression as seen in various human disease and malignancies.
DNA methyltransferases (DNMTs) are a class of enzymes involved in transferring a methyl group from S-adenosyl methionine (SAM) to cytosine bases of CpG dinucleotides at gene promoters and regulatory regions [29]. CpG dinucleotides are concentrated in short CpG-rich regions known commonly as “CpG islands”. In humans, CpG islands occupy about 60% of the gene promoters. CpG promoter islands can be methylated during development that results in long-term gene silencing. One classic example of such naturally occurring CpG methylation is the X-chromosome inactivation and the imprinted genes. DNA hypermethylation has also been associated with the silencing of tumor suppressor genes as well as differentiation genes in various cancers [30]. The reduced expression of these genes may then contribute to the formation of CSCs within tumor cell populations [31, 32]. Indeed, the importance of DNA methylation in maintaining CSC properties have been reported in leukemic, lung and colon stem cells [33–35]. The accumulation of epigenetic abnormalities has been suggested to be an early event that predisposes these tumor cells to acquire further mutations and genomic instability. This is supported by the fact that epigenetic machinery is crucial for the maintenance of normal stem and progenitor cells and that any epigenetic deregulation can lead to accumulation of cells with increased stemness properties and self-renewal ability, thus giving rise to CSCs.
Key CSC pathways regulated by epigenetic mechanisms
Wnt/β-catenin signaling pathway
The canonical Wnt/β-catenin signaling pathway mediates gene activation through the transcription factor β-catenin. In the absence of Wnt signaling, cytoplasmic β-catenin is inactivated by a degradation complex comprising Adenomatous polyposis coli (APC), Axin, glycogen synthase kinase 3 beta (GSK-3β), and casein kinase 1 (CK1). Phosphorylation by GSK-3β targets β-catenin for ubiquitination and subsequent proteasomal degradation. Upon Wnt ligand binding to Frizzled receptors, the degradation complex is inactivated via low density lipoprotein receptor-related protein 5/6 (LDR5/6) and Dishevelled, allowing for stabilisation of β-catenin. Accumulated β-catenin then translocates into the nucleus, where it associates with T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors to induce transcription of Wnt target genes such as CCND1 and MYC. The Wnt/β-catenin pathway has important functions in normal tissue development and maintenance, as well as in self-renewal and differentiation of CSCs [36, 37]. In fact, the Wnt/β-catenin pathway has been found to be aberrantly activated in a variety of cancers, either via genetic alterations, such as mutations in CTNNB1, APC and AXIN genes [38–40], or through epigenetic modulation.
DNA methylation has been linked to aberrant Wnt/β-catenin pathway activation through the enhanced promoter methylation and subsequent silencing of various Wnt inhibitors such as Wnt inhibitory factor 1 (WIF-1), AXIN2, Secreted frizzled-related protein 1 (SFRP-1), and Dickkopf-related protein 1 (DKK1) in breast and colorectal cancers [41–43]. In gastric cancer, Yoda et al. showed that aberrant methylation of Wnt negative regulators, including DKK3, Naked cuticle homolog 1 (NKD1) and SFRP1, could lead to activation of Wnt/β-catenin pathway [44]. Deregulation of Wnt/β-catenin pathway in cancer is also mediated by aberrant histone modifications. Decreased acetylation of H3K16 and increased H3K27 trimethylation along with recruitment of Sirtuin 1 (SirT1), enhancer of zeste homolog 2 (EZH2) and suppressor of zeste 12 protein homolog (Suz12) (components of polycomb repressor complex 2, PCR2) to the promoter of DKK1 inhibited the expression of the DKK1 Wnt antagonist (Fig. 1) [45]. In colorectal cancer, Dishevelled-binding antagonist of beta-catenin 3 (DACT3), an antagonist of Dishevelled, was found to be regulated by bivalent histone modifications—activating H3K4me3 and repressive H3K27me3 histone marks—at its locus [46]. This bivalent histone state was associated with decreased DACT3 expression in colorectal cancer cell lines [46]. In addition, methylation of H3K4 at the regulatory element of DKK1 marks the site for binding by the transcription factor Achaete-scute family BHLH transcription factor 1 (ASCL1), resulting in a repressed chromatin configuration [47]. ASCL1-mediated inhibition of DKK1 consequently leads to activation of Wnt signaling, and ASCL1 was found to be crucial for glioblastoma CSC maintenance and tumorigenicity [47–49].
Fig. 1.
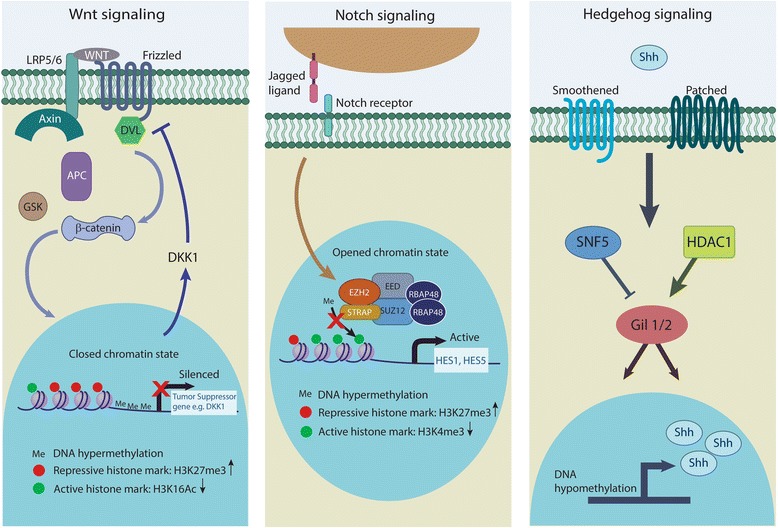
Regulation of key cancer stem cell signaling pathways by epigenetic mechanisms. Wnt/β-catenin signaling can be enhanced by decreased expression of the DKK1 inhibitor through promoter hypermethylation and increased H3K27me3 and decreased H3K16 acetylation marks. Notch signaling target genes such as Hes1 and Hes5 can be activated by inhibition of H3K27 inhibitory methylation mark at their promoter region by STRAP. Hedgehog signaling pathway can be activated in CSCs epigenetically by Shh promoter hypomethylation and increase HDAC1 expression. Epigenetic deregulation of CSC-related signaling pathways allows cancer cells to acquire self-renewal ability and drug resistance properties
Besides alterations in DNA and histones, non-coding RNAs have also been found to act as epigenetic modulators of Wnt/β-catenin signaling. Wang et al. demonstrated that long non-coding RNA of transcription factor 7 (lncTCF7), which is highly upregulated in liver CSCs, is able to induce TCF7 expression by recruiting the Switch/sucrose non-fermentable (SWI/SNF) chromatin remodelling complex to its promoter [50]. This subsequently activates the Wnt pathway, leading to self-renewal of liver CSCs and tumor propagation.
Hedgehog signaling pathway
The Hedgehog (Hh) signaling pathway plays important roles in guiding cell fate during embryonic development and in maintaining adult tissue homeostasis [51, 52]. It also functions in regulating stem and progenitor cell proliferation and maintenance in several tissues [53]. In the absence of sonic hedgehog ligand (Shh), the Patched receptor (PTCH1) prevents activation of Smoothened (SMO), allowing Gli proteins to be sequestered by suppressor of fused homolog (SUFU) and kinesin family member 7 (Kif7). Upon Shh binding to PTCH1, SMO is activated and mediates Hh signaling transduction via release of Gli proteins, which then enter the nucleus and act as transcription factors. Gli1 activates transcription of Hh target genes, Gli2 can both activate and repress transcription, while Gli3 functions as a transcriptional repressor.
The Hh signaling has been implicated in tumorigenesis in various tissues [54]. In basal cell carcinoma (BCC), upregulation of Hh signaling in stem cells in the interfollicular epidermis [55] or within the hair follicle [56] has been reported to contribute to tumor formation. In medulloblastomas, granule neuron progenitors/precursors (GNPs) in the cerebellum that have constitutively active Hh signaling have been identified as cells-of-origin of the tumor [54, 57, 58].
The Hh pathway is activated by genetic mutations in both BCC and medulloblastoma. However, epigenetic mechanisms also play a role in modulating the expression and function of Hh pathway components in various tumors. The chromatin remodelling protein SNF5 directly interacts with Hh signaling effector Gli1 to downregulate expression of Hh target genes. SNF5 is a member of the SWI-SNF complex and inhibits gene expression by altering chromatin structure at Gli1-regulated promoters, which includes genes such as Ptch1 and Gli1 itself. Hence, inactivation of SNF5 would contribute to aberrant Hh signaling activity as seen in human malignant rhabdoid tumors [59–61].
In addition, histone deacetylases are also involved in regulating Gli protein function. Gli1 and Gli2 proteins require deacetylation by HDAC1 to be transcriptionally active, and they, in turn, can induce HDAC1 expression through a positive autoregulatory loop [62]. This mechanism is inhibited by E3-ubiquitin ligase complex (comprising Cullin3 and renin, REN)-mediated degradation of HDAC1. However, REN is often deleted in human medulloblastoma [63], resulting in increased levels of HDAC1 and Gli1, and subsequent deregulation of Hh signaling in neural progenitors and tumor cells [62].
Hh pathway can also be epigenetically regulated by aberrant DNA methylation. Studies have found that hypomethylation of Shh promoter leads to enhanced expression of Shh ligand in breast and gastric cancers (Fig. 1) [64, 65]. Indeed, Duan and colleagues reported that promoter hypomethylation allowed nuclear factor kappa b (NF-κB) to bind and activate transcription of Shh, resulting in overexpression of the ligand [66]. Consequently, the upregulation of Hh signaling was able to promote self-renewal and invasiveness in breast cancer cells [66].
Notch signaling pathway
Notch is a transmembrane receptor involved in cell contact-dependent signaling [67]. Binding of ligands Jagged1/2 or Delta1-4 triggers cleavage of Notch intracellular domain (NICD) by γ-secretase and its release into the cytoplasm [68]. NICD then translocates into the nucleus, where it interacts with recombination signal binding protein for immunoglobulin kappa J region (RBPJ-κ) to transcriptionally induce expression of Notch target genes, such as MYC and HES1 [69]. In the inactive state, RBPJ-κ recruits co-repressor complexes to suppress Notch target genes [70].
Notch signaling is an evolutionarily conserved pathway that has important roles in development of various tissues and organs [71]. It also regulates cell proliferation and differentiation across a wide range of cell types and during different stages of cell lineage progression [69]. Furthermore, Notch pathway modulates stem cell differentiation and self-renewal. Importantly, Notch signaling has been shown to be crucial for survival of neural stem cells (NSCs) [72]. In murine intestinal stem cells, loss of B-lymphoma Mo-MLV insertion region 1 homolog (Bmi1), a target of Notch signaling, decreases proliferation and induces cellular differentiation into goblet cells [73]. Deregulation of Notch pathway has been implicated in various tumors such as prostate cancer, breast cancer, lung cancer, colorectal cancer and haematological malignancies [74–77]. Recent studies have also reported the role of Notch signaling in breast, colon and oesophageal CSCs [78–80].
Epigenetic modifications affecting various components of the Notch pathway have been found to cause aberrations in Notch signaling activity. Overexpression of Notch ligand Jagged2 in multiple myeloma has been associated with enhanced histone acetylation at the JAGGED2 promoter region [81]. Nuclear co-repressors such as nuclear receptor co-repressor 2 (SMRT) normally recruit HDACs to promoter regions to regulate gene expression. However, in multiple myeloma, the decreased levels of nuclear co-repressor SMRT reduces HDAC recruitment to JAGGED2 promoter, resulting in increased transcription of the Notch ligand and subsequent activation of Notch signaling [81]. In addition, Jin et al. reported that serine-threonine kinase receptor-associated protein (STRAP) promotes stemness in colorectal cancer-initiating cells via modulating the Notch pathway [80]. They found that STRAP interacts with EZH2 and SUZ12 of PRC2 complex, inhibiting histone methylation of H3K27 on HES1 and HES5 promoters, leading to gene activation (Fig. 1). This was concordant with the finding that both genes had increased activating (H3K4me3) and decreased repressive (H3K27me3) histone marks in wild-type (WT) cells as compared to STRAP knockdown (KD) cells. Moreover, ectopically expressed HES1 or HES5 was able to rescue the stemness phenotype in STRAP KD cells [80], further demonstrating the significance of Notch signaling in regulating stemness potential in CSCs.
Epigenetic regulation of metastasis and chemoresistance pathways
During tumor progression, metastasis of tumor cells has been linked to the induction of epithelial-to-mesenchymal transition (EMT). EMT is a multi-step process that results in decreased cell-cell adhesion, loss of cell polarity, increased cell motility, and gain of invasive mesenchymal properties [82, 83]. There is evidence that activation of EMT can confer cells with CSC and tumor-initiating properties [84, 85]. It was reported that EMT induction in both immortalised and transformed human mammary epithelial cells resulted in increased expression of CSC markers and mammosphere formation. Moreover, stem-like cells of mammary carcinomas were also found to express markers of EMT [85]. The relationship between EMT and acquisition of stem-like properties in tumor cells suggests that stemness properties may help increase the chances of disseminated tumor cells to successfully metastasize to distant sites [70].
Several signaling pathways involved in embryonic development, such as Wnt, Hedgehog and Notch, have been identified to regulate the EMT process [86, 87]. The transforming growth factor-β (TGF-β) family of cytokines are also known inducers of EMT [88, 89]. Hence, deregulation of these pathways and proteins could activate aberrant EMT induction, resulting in tumor metastasis and contribute to poorer patient prognosis. A hallmark of EMT is the loss of membrane protein E-cadherin, which functions in maintaining cell-cell adhesion [90–92]. Loss of E-cadherin can arise from mutations in its encoding gene CDH1, or via mechanisms that regulate its expression and function, including transcriptional repressors Twist-related protein 1 (TWIST1), Snail family zinc finger 1 (SNAIL), Zinc finger E-box-binding homeobox 1 (ZEB1) and Zinc finger E-box-binding homeobox 2 (ZEB2) [93]. Epigenetic mechanisms have also been found to play a dynamic role in silencing E-cadherin expression. For instance, DNA methylation of E-cadherin promoter helps to recruit HDACs to the site, leading to histone deacetylation and transcriptional silencing [94, 95]. In addition, histone methylation of CDH1 promoter by EZH2 and PRC2 complex, which is recruited by Snail1, also represses E-cadherin expression [96, 97].
Micro RNAs (miRNAs) that regulate the EMT pathway are epigenetically regulated as well. MiR-200 family members and miR-205 repress EMT and invasion by directly inhibiting transcription factors ZEB1 and ZEB2 [98–100]. Hence, inhibition of these miRNAs would result in increased EMT and metastasis. This is observed in high-grade breast cancers, whereby low levels of miR-200c is correlated with upregulation of EMT and stemness markers [101]. Silencing of miR-200c and miR-205 expression can also occur via enrichment of H3K27me3-mediated chromatin remodelling and DNA methylation, which leads to induction of EMT and CSC phenotype in immortalised human bronchial epithelial cells [102].
Studies have shown that cells with both CSC properties and EMT-like phenotype tend to be more resistant to chemotherapy drugs as compared to other cancer cell populations [103–105]. Arumugam et al. demonstrated that pancreatic cancer cell lines with EMT features were resistant to common chemotherapy drugs such as gemcitabine, 5-fluorouracil and cisplatin [106]. Moreover, cells that were resistant to gemcitabine expressed high ZEB1 and low E-cadherin, and acquired greater cell migration ability [106]. Indeed, these findings indicate that epigenetic modulations involved in the gain of CSC and EMT properties would most likely impact tumor cells’ response to therapy.
The increased drug resistance observed in CSCs is commonly mediated by enhanced expression of drug efflux transporters, such as ATP-binding cassette (ABC) family of transporters, which includes ATP-binding cassette sub-family G member 2 (ABCG2), multidrug resistance protein 1 (MDR1) and multidrug resistance-associated protein 1 (MRP1) [17, 107, 108]. These drug transporters utilise ATP in moving drugs out of the cell against its concentration gradient. The expression of these transporters are regulated by various mechanisms and pathways, and their deregulation would result in an enrichment of these proteins and drug efflux capability. Studies have shown that MRP1 expression can be upregulated by Notch signaling, and is responsible for drug resistance in CSCs [109, 110]. Expression of ABCG2 is upregulated upon enrichment of permissive histone modifications such as greater histone H3 acetylation, increased H3K4 tri-methylation and phosphorylation of H3S10, as well as decreased HDAC1 levels [111]. These histone marks along with decreased H3K9 tri-methylation allow RNA polymerase II and chromatin remodelling protein Brahma-related gene 1 (Brg1) to gain access to the promoter and activate transcription of ABCG2 [111]. Collectively, a complex network of signaling pathways that function in modulating the activity of normal stem cells can be susceptible to deregulation as a result of aberrant epigenetic modifications during the course of tumor formation. These abnormal alterations in key signaling pathways contribute to CSC proliferation and maintenance, as well as tumor progression and invasion. Hence, epigenetic regulation of these signalling pathways may serve as potential mechanisms for targeted therapy against CSCs.
Therapeutic intervention using epigenetic modifying drugs
As epigenetic mechanisms have important functions in modulating stem cell properties in cancer cells, targeting components of these epigenetic pathways would help in eradicating both CSCs and the bulk tumor population. Inhibitors of epigenetic modulatory enzymes such as HDACs and DNMTs have been widely studied and many are currently in clinical trials for treatment of a variety of cancers. In addition, deregulation of chromatin remodelling has been associated with tumorigenesis and tumor progression, thus making chromatin remodelling proteins viable targets for small molecule inhibitors as well. Indeed, many of these therapeutic strategies aim to induce differentiation of CSCs and to sensitise these cells to chemotherapy, with the ultimate goal of reducing tumor relapse and improving patient survival. Here, we review the development of various epigenetic therapies designed to target different components of the epigenetic machinery. A summary of these epigenetic drugs and their clinical status can be found in Table 1.
Table 1.
Epigenetic modulators in cancer
| Drug | Target | Clinical status | Indication | References |
|---|---|---|---|---|
| DNMT inhibitors | ||||
| Azacitidine | Inhibit DNMT (act as nucleoside analog) | FDA-approved | MDS | [277, 278] |
| Decitabine | Inhibit DNMT (act as nucleoside analog) | FDA-approved | MDS | [115, 277] |
| SGI-110 | Inhibit DMNT by incorporating into guanine nucleotide | Phase 3 (NCT02348489 Phase 1/2 (NCT01261312, NCT02197676) Phase 2 (NCT01752933) Phase 1/2 (NCT01696032) |
AML MDS, AML Advanced HCC Platinum-resistant recurrent ovarian cancer |
[279–281] |
| HDAC inhibitors | ||||
| Vorinostat | Inhibitor of Class I and II HDACs | FDA-approved | Cutaneous T cell lymphoma | [135] |
| Romidepsin | Inhibitor of Class I HDACs | FDA-approved | Cutaneous T cell lymphoma | [136] |
| Panobinostat | Pan-HDAC inhibitor | Phase 3 (NCT01034163) Phase 2/3 (NCT00425555) |
Hodgkin’s lymphoma Cutaneous T cell lymphoma |
[282, 283] |
| Entinostat | Inhibitor of Class I HDACs | Phase 2 (NCT00866333) Phase 1/2 (NCT01038778) |
Hodgkin’s lymphoma Clear cell renal cell carcinoma, metastatic renal cell cancer |
[284] |
| Belinostat | Inhibitor of Class I and II HDACs | Phase 2 (NCT00357032) Phase 2 (NCT00274651) Phase 2 (NCT00301756) |
Relapsed/refractory AML or older patients with newly diagnosed AML Recurrent/refractory cutaneous and peripheral T cell lymphomas Ovarian cancer |
[285–287] |
| Pracinostat | Inhibitor of Class I and II HDACs | Phase 2 (NCT01112384, NCT01075308) |
Translocation-associated recurrent/metastatic sarcomas, metastatic prostate cancer | [288, 289] |
| Givinostat | Inhibitor of Class I and II HDACs | Phase 2 (NCT01761968) |
Chronic myeloproliferative neoplasms | [290] |
| Valproic acid | Inhibitor of Class I and II HDACs | Phase 2 (NCT01900730) |
Breast cancer | [163, 291] |
| HMT inhibitors | ||||
| EPZ-5676 | Inhibit DOT1L methyltransferase (H3K79) activity by competing with SAM | Phase 1 (NCT02141828, NCT01684150) |
MLL-fusion leukemia, AML, acute lymphocytic/lymphoblastic leukemia, MDS, myeloproliferative disorders | [182] |
| DZNep | Inhibit HMT activity of EZH2 via inhibiting S-adenosylhomocysteine (SAH) hydrolase | Not in trial | Breast cancer, prostate cancer, glioblastoma multiforme (GBM) |
[193, 292, 293] |
| E7438 (EPZ-6438) | Inhibit HMT activity of EZH2 by competing with co-factor S-adenosyl-methionine (SAM) | Phase 2 (NCT02860286, NCT02601950) |
Malignant mesothelioma, rhabdoid tumors, synovial sarcoma, epitheloid sarcoma | [196] |
| GSK2816126 (GSK126) | Inhibit HMT activity of EZH2 by competing with co-factor S-adenosyl-methionine (SAM) | Phase 1 (NCT02082977) |
Relapsed/refractory DLBCL, transformed follicular lymphoma, multiple myeloma, non-Hodgkin’s lymphoma, solid tumors | [198, 199] |
| CPI-1205 | Inhibit HMT activity of EZH2 by competing with co-factor S-adenosyl-methionine (SAM) | Phase 1 (NCT02395601) |
B-cell lymphoma | [294] |
| Chaetocin | Inhibit SUV39H1 | Not in trial | HCC, multiple myeloma | [216, 218] |
| BIX01294 | Inhibit G9a (substrate-competitive) | Not in trial | Breast cancer, colon cancer | [209, 212] |
| UNC0638 | Inhibit G9a (substrate-competitive) | Preclinical | Breast cancer | [213] |
| UNC0642 | Inhibit G9a (substrate-competitive) | Preclinical | Pancreatic cancer | [214] |
| HDM inhibitors | ||||
| Tranylcypromine | Irreversible inhibitor of LSD1 | Phase 1 (NCT02273102, NCT02717884) Phase1/2 (NCT02261779) |
AML, MDS AML |
[226] |
| ORY-1001 | Irreversible inhibitor of LSD1 | Phase 1 | AML | [227] |
| GSK2879552 | Irreversible inhibition of LSD1 activity by modifying its cofactor FAD | Phase 1 (NCT02034123, NCT02177812) |
Small cell lung cancer, AML |
[231] |
| GSK-J4 | Inhibit UTX | Not in trial | T-cell acute lymphoblastic leukemia (T-ALL) | [295] |
| BET inhibitors | ||||
| I-BET762 (GSK525762A) | Interfere with binding of BET proteins to acetylated histones | Phase 1 (NCT01587703, NCT01943851) |
Solid tumors, relapsed refractory haematological malignancies | [243, 244] |
| JQ1 | Interfere with binding of BET proteins to acetylated histones (greatest specificity for BRD3 and BRD4) | Not in trial | NUT midline carcinoma (NMC), multiple myeloma, AML, Burkitt’s lymphoma, DLBCL | [239, 240, 296, 297] |
| I-BET151 (GSK1210151A) | Interfere with binding of BET proteins to acetylated histones | Not in trial | MLL fusion leukemia, medulloblastoma | [298, 299] |
| OTX015 | Interfere with binding of BET proteins to acetylated histones | Phase 1 (NCT01713582) |
Hematological malignancies | [248, 297] |
| CPI-203 | Interfere with binding of BET proteins to acetylated histones | Not in trial | Lymphoma | [300] |
| CPI-0610 | Interfere with binding of BET proteins to acetylated histones | Phase 1 (NCT02157636, NCT01949883, NCT02158858) |
Acute leukaemia, MDS, myelodysplastic/myeloproliferative neoplasms, lymphoma, multiple myeloma | [301] |
Targeting DNA methylation - DNA methyltransferase (DNMT) inhibitors
Inhibitors of DNA methylation were among the first epigenetic drugs tested for use in treatment of cancer [112]. The most widely studied DNMT inhibitors include azacitidine (5-azacitidine) and decitabine (5-aza-2’-deoxycytidine), which act as analogues of cytosine. These molecules get incorporated into DNA and covalently bond with DNA methyltransferase, thus preventing its function [113, 114] and leading to its degradation [115]. These drugs were initially used as cytotoxic chemotherapeutics in the late 1960s [116], but were found to be highly toxic [117–120]. Subsequently, recent studies have discovered that low doses of DNMT inhibitors had greater efficacy in sustaining decreased DNA methylation and associated re-expression of silenced genes in leukemic and epithelial tumor cells [121]. These lower doses were also able to reduce tumorigenicity and target CSC populations within the tumor. In lung cancer, Liu et al. showed that inhibition of DNMT1 was able to decrease proliferation and tumorigenic ability of lung CSCs [34].
Multiple studies have also demonstrated the role of DNMT inhibitors in differentiation therapy. Pinto et al. showed that azacitidine could induce primary AML cells from patients to differentiate into less or non-malignant cells [122, 123]. Prostate cancer derived-CSCs that were treated with decitabine showed decreased expression of stemness genes Octamer-binding transcription factor 4 (OCT40029 and Nanog homeobox (NANOG), leading to overall reduction in tumor growth [124]. In addition, low doses of SGI-110, a newer DNMT inhibitor, was recently reported to be capable of reprogramming ovarian CSCs to a more differentiated state [125]. Treatment with SGI-110 also decreased tumor-initiating ability and re-sensitized these cells to platinum, suggesting a potential use of DNMT inhibitors in combination with other chemotherapeutic agents in preventing recurrence of ovarian cancer [125]. Both azacitidine and decitabine have been approved by the FDA for treatment of myelodysplastic syndrome (MDS) [126]. Clinical trials for other indications such as AML and colorectal cancer are still ongoing. SGI-110 is also in phases of clinical trials for treatment of various cancers such as AML, MDS, liver cancer and platinum-resistant ovarian cancer.
Targeting histone deacetylation - Histone deacetylase(HDAC) inhibitors
An important histone tail modification is acetylation, which is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs are responsible for adding an acetyl group onto lysine residues of histone tail, which neutralizes the positive charge, resulting in a more “open” chromatin structure [127]. In contrast, HDACs remove the additional acetyl group, leading to increased binding affinity between DNA and histones, which is generally associated with gene repression [128]. Very often, deregulated gene silencing in cancers has been associated with aberrant histone deacetylation. For instance, in leukemia, this can be mediated through aberrant recruitment of HDACs by fusion proteins such as Acute myeloid leukemia protein 1 Eight twenty-one protein (AML1-ETO) and Promyelocytic leukemia protein retinoic acid receptor alpha (PML-RARα), which leads to abnormal gene silencing and subsequent leukemogenesis [129, 130]. Besides, HDACs can also acetylate non-histone proteins, including tumor suppressor p53 and oncogene B-cell lymphoma 2 (BCL2), resulting in inhibition of p53-dependent transcription [131] and upregulation of pro-survival protein, BCL2 [132]. Hence, the use of HDAC inhibitors in returning histone acetylation patterns to a normal state has been found to be effective in inducing apoptosis and differentiation as well as inhibit proliferation of tumor cells [129, 133]. These HDAC inhibitors can be divided mainly into two classes—the pan HDAC inhibitors and the class-specific inhibitors [134], and they all function via chelating the zinc atom in the active site of the enzyme [127].
Two HDAC inhibitors, vorinostat (subseroylanilide hydroxamic acid) and romidepsin (depsipeptide), have been approved for treatment of cutaneous T-cell lymphoma [135, 136]. Both drugs were found to produce durable response and efficacy in patients with cutaneous T-cell lymphoma in Phase 2 multi-center trials [135–138]. However, besides cutaneous T-cell lymphoma, monotherapy of vorinostat and romidepsin in treatment of various solid tumors have had little success in clinical trials [139–150]. Apart from these two compounds, many other HDAC inhibitors have also been developed and tested in clinical trials, the details of which have been well-reviewed elsewhere [115, 151–153]. Monotherapies of these compounds, including panobinostat [154, 155], entinostat [156, 157], belinostat [158, 159] and pracinostat (SB939) [160], are being tested against various haematological malignancies and solid tumors.
Another mechanism of action of HDAC inhibitors for cancer treatment is via differentiation or reprogramming of cancer cells. As therapy resistance is a major hurdle in cancer treatment and is often associated with CSCs and epigenetic control [161], HDAC inhibitors possess the ability to induce differentiation of CSCs from their quiescent state, thereby re-sensitising them to other chemotherapy agents. Valproic acid, an antiepileptic drug, has been found to be a powerful HDAC inhibitor [162]. Gottlicher et al. demonstrated that valproic acid could trigger differentiation of transformed hematopoietic progenitor cells and leukemic blasts from AML patients [162]. Furthermore, Travaglini et al. found that valproic acid was able to epigenetically reprogram breast cancer cells into a more “physiologic” phenotype, thus improving sensitivity to other forms of breast cancer therapy [163]. In addition, entinostat, a selective inhibitor of class I HDACs, was recently reported to reverse EMT phenotype and decrease the population of tumor-initiating cells in triple-negative breast cancer (TNBC) [164]. These tumor-initiating cells possessed CSC properties and were responsible for driving metastasis and drug resistance in TNBC, thus contributing to poor patient prognosis. Hence, this study demonstrated the utility of HDAC inhibitors in preventing CSC invasiveness and tumor metastasis. Overall, these studies demonstrate the potential use of epigenetic modulators towards the differentiation and therapeutic sensitization of CSCs.
Targeting histone methylation – Histone methyltransferase (HMT) inhibitors
A class of enzymes called histone lysine methyltransferases (HKMTs) mediate the addition of a methyl group to the nitrogen atom of the lysine side chain [165]. Despite catalysing a common chemical reaction, this family of HKMTs demonstrate large structural diversity of its active sites, allowing these enzymes to have high substrate specificity [127]. For example, DOT1L (KMT4) is a unique HKMT as it is currently the only known enzyme that methylates lysine 79 of histone H3 (H3K79) [166]. Similarly, methylation of H3K27 is only mediated by the catalytic subunit EZH2 (KMT6) of PRC2 [127]. In contrast, some methylation marks can be catalysed by several proteins, such as H3K9 methylation. These post-translational methylation of histones have important roles in regulation of gene expression, differentiation, DNA damage repair as well as in tumorigenesis [167, 168]. Aberrant histone methylation can be due to gene mutations, over-expression or deregulated control of epigenetic modulatory enzymes involved. Thus, HKMTs are potential therapeutic targets, and the structural differences between members of the family also enable greater selectivity in inhibition of these proteins by small molecule compounds [169].
HKMT inhibitors have only recently gained more attention as cancer therapeutics, resulting in a rapidly increasing number of these small molecule inhibitors being developed [170–172]. In fact, several DOT1 like histone H3K79 methyltransferase (DOT1L) and EZH2 inhibitors have progressed to being tested in clinical trials as cancer interventions [173]. H3K79 methylation by DOT1L is associated with transcriptional activation of genes under its regulation [174, 175], and overexpression or aberrant DOT1L activity has been found in cancer, such as leukemia with mixed lineage leukemia (MLL) gene translocation. The MLL fusion protein can recruit DOT1L into a transcription complex, which subsequently methylates H3K79 [176–180]. This leads to dysregulation and overexpression of many MLL-target genes, including Homeobox A9 (HoxA9) and Meis homeobox 1 (Meis1), which are key regulators of hematopoietic stem cell differentiation that contributes to leukemogenesis [165]. Therefore, DOT1L is an attractive target for therapy, resulting in the first selective DOT1L inhibitor EPZ-4777 to be synthesised with anti-tumor effects against murine models of MLL-rearranged leukemia [181]. Further optimisation of the drug led to the development of EPZ-5676, the first HKMT inhibitor to enter clinical trials. This compound has been shown to be highly potent and selective for DOT1L. Treatment with EPZ-5676 in a MLL-rearranged leukemia xenograft model showed durable and complete tumor regression [182]. EPZ-5676 is currently under clinical studies (Phase I) for MLL-fusion leukemia, AML, MDS and myeloproliferative disorders.
EZH2 is a member of PRC2, along with proteins embryonic ectoderm development protein (EED) and SUZ12, and is responsible for catalysing H3K27 mono-, di- and tri-methylation [183–185]. Overexpression of EZH2 has been found in various cancers of the breast, lung, prostate and haematological malignancies [186–191], and is associated with poor disease prognosis. Studies have also shown the role of EZH2 deregulation in tumor progression, metastasis [192, 193] and maintenance of CSC self-renewal properties [194]. In glioblastoma multiforme (GBM), inhibition of EZH2 by S-adenosylhomocysteine hydrolase (SAH) inhibitor 3-deazaneplanocin A (DZNep) was able to reduce self-renewal and tumor-initiating capabilities of GBM CSCs in vivo via affecting transcriptional regulation of oncogene MYC [193]. However, DZNep affects methylation of other histone residues [195], leading to the development of more specific EZH2 inhibitors. The earliest SAM-competitive and selective EZH2 inhibitor to advance into clinical trials for treatment of rhabdoid tumors and lymphomas is EPZ-6438 (E7438) [196, 197]. A more recent drug, GSK2816126 (GSK126) has also entered clinical studies for relapsed/refractory diffuse large B-cell lymphoma (DLBCL), multiple myeloma and transformed follicular lymphoma [198, 199]. Both drugs have shown high potency and selectivity in inhibiting tumor growth in preclinical studies [197, 198, 200].
H3K9 methyltransferases, such as euchromatic histone lysine methyltransferase 2 (G9a/EHMT2) and euchromatic histone lysine methyltransferase 1 (GLP/EHMT1), catalyse mono- and di-methylation of the lysine residue, while tri-methylation of H3K9 is mediated by Suppressor of variegation 3–9 homolog 1 (SUV39H1) and Suppressor of variegation 3–9 homolog 2 (SUV39H2) [201]. Upregulation of G9a activity has been linked to several types of cancer, including ovarian, lung, liver and bladder cancers [202–208]. Hence, several substrate-competitive inhibitors of these HKMTs have been developed. BIX-01294 is the first specific inhibitor of G9a and GLP, and studies have reported its ability to decrease H3K9me2 levels in mammalian cells [209–211]. Kim et al. reported that BIX-01294 was able to induce cell death in colon and breast cancer cells via EHMT dysfunction [212]. However, due to the increased toxicity levels of BIX-01294 at higher concentrations, the use of this drug is limited. This led to the recent development of a more potent, specific and selective EHMT inhibitor, UNC0638 that was found to decrease local H3K9me2 and DNA methylation levels [213]. Further development generated UNC0642, which possessed better pharmacokinetic properties and higher efficacy in inhibiting colony formation ability of pancreatic adenocarcinoma cells [214].
Methylation of H3K9 by SUV39H1 is associated with silencing of tumor suppressor genes, including E-cadherin and p15INK4B, in AML [215]. Overexpression of SUV39H1 has also been correlated with poor prognosis in multiple myeloma patients [216]. Treatment of multiple myeloma cells with chaetocin, a small molecule inhibitor of SUV39H1 showed anti-tumor effects at low doses of the drug [216]. Similarly, chaetocin was found to decrease H3K9me3 levels and induce differentiation of AML cells at non-toxic doses [217]. Furthermore, chaetocin was able to repress cell proliferation and induce apoptosis in hepatocellular carcinoma (HCC) cultures and xenografts [218], implying a potential tumorigenic role of EHMTs in HCC progression and development.
Targeting histone demethylation - Histone demethylase (HDM) inhibitors
Methylation of lysine on histones is also regulated by histone lysine demethylases (KDMs). This group of epigenetic erasers function in removing the methyl groups from lysine side chains on histones [219, 220]. As proper functioning of both HKMTs and KDMs is required to maintain stable histone methylation levels, small molecule inhibitors have also been developed to target KDMs. KDMs can be grouped into two families - the lysine-specific demethylase (LSD) family and Jumonji domain-containing (JmjC) family [221]. The LSD family are flavin adenine dinucleotide (FAD)-dependent amine oxidase that demethylates mono- and di-methyl lysine residues, while JmjC enzymes utilise 2-oxoglutarate and iron to oxidatively release methyl groups from all three methylation states at lysine residues [172, 222].
Upregulated expression of LSD1 (KDM1A) has been found in various human cancers, including AML, ovarian, lung, bladder and colorectal cancers [223–225]. Hence, small molecule inhibitors of LSD1 that target the enzyme cofactor FAD have been developed, the first of which is tranylcypromine [226]. Further studies have led to the synthesis of more selective derivatives of tranylcypromine, such as ORY-1001 [227] and GSK2879552 [228]. They function by irreversibly changing FAD, leading to the formation of a tetracyclic adduct [229]. LSD1 is important for normal hematopoiesis; loss of LSD1 has been found to inhibit differentiation and impair hematopoiesis [230]. This suggests a potential role of aberrant LSD1 activity in affecting stemness properties in tumor cells. The inhibitor ORY-1001 has been shown to decrease the population of AML stem cells and improve survival of mice with acute lymphoblastic leukemia (ALL) in preclinical studies [227, 228]. GSK2879552 has also been found to influence differentiation in small cell lung cancer (SCLC) [231]. These compounds are currently in phase 1 studies for relapsed or refractory AML (ORY-1001) and SCLC (GSK2879552).
Similarly, JmjC demethylases are amenable to pharmacological intervention as well. Ubiquitously transcribed tetratricopeptide repeat X chromosome (UTX), also known as KDM6A, is responsible for demethylating H3K27 [232–234], and loss of UTX activity has been found in multiple human malignancies, including multiple myeloma, esophageal squamous cell carcinoma and renal carcinoma [166]. However, no inhibitors of JmjC enzymes have advanced beyond biochemical studies [127]. Nevertheless, as UTX is a component of the mixed lineage leukemia protein 2 (MLL2) H3K4 methyltransferase complex, and interacts with SWI/SNF chromatin remodelling complex [235–237], it is still an important epigenetic target and its role in epigenetic modulation still warrants further study.
Targeting epigenetic readers – BET inhibitors
While epigenetic modulatory enzymes are obvious targets for therapy, epigenetic readers are also important components of the epigenetic machinery as they directly or indirectly regulate gene expression. One such group of readers called bromodomain and extra-terminal (BET) proteins modulate gene expression by recognising acetylated histones. Increased BET activities have been associated with NUT midline carcinoma (NMC), glioblastoma and various haematological malignancies, through aberrant transcription of disease-associated genes and oncogenes such as MYC [238]. Hence, BET proteins appear to be attractive therapeutic targets for controlling dysregulated gene expression.
JQ1 is a selective BET inhibitor of BRD family of proteins, including Bromodomain-containing protein 4 (BRD4) [239]. In preclinical studies, JQ1 was able to cause tumor regression in NMC mouse models, inhibit proliferation, induce apoptosis and differentiation in cancer cells [239–242]. Another BET inhibitor, I-BET762 (GSK525762A), functions by binding to the acetyl-binding pocket of BET proteins [243, 244]. Studies have shown that I-BET762 treatment was able to induce terminal differentiation of patient-derived malignant cells [245] and activate apoptosis in neuroblastoma and prostate cancer via inhibition of Myc-driven pathways [246, 247]. This compound is currently in phase I trials for solid tumors and relapsed or refractory haematological cancers.
OTX015 is another BET inhibitor that has progressed into clinical trials for various haematological malignancies. This compound has been found to possess anti-proliferative effects via directly influencing MYC expression and activity [248, 249]. Similarly, CPI-0610 has also entered clinical testing for lymphoma, multiple myeloma and myelodysplastic or myeloproliferative neoplasms. I-BET151 is a pan-BET inhibitor, similar to JQ1, and has been found to block proliferation and induce apoptosis in myeloma cells via repressing Myc activity [250]. Antitumor effects have also been observed in NMC, MLL, ALL, lung cancer and brain cancer [238].
Combination therapy with epigenetic modulators
While epigenetic drugs have been tested preclinically and clinically as single agents, further studies have revealed the increased efficacy of these drugs when used in combination with other therapies. One common combination of different epigenetic therapies is that of DNMT and HDAC inhibitors. Pathania et al. reported that combining azacitidine (DNMT inhibitor) and butyrate (HDAC inhibitor) was capable of significantly decreasing breast cancer CSC population [251]. In addition, combination of azacitidine and HDAC inhibitor entinostat at low doses in a phase I/II clinical trial showed sustained and favourable responses in treatment-resistant non-small cell lung cancer (NSCLC) patients [252]. Azacitidine and valproic acid co-treatment was also able to promote tumor regression in Patched mutant mouse models of medulloblastoma [253]. Besides DNMT-HDAC inhibitor combination therapy, studies have demonstrated synergistic effects of other epigenetic drug combinations. For example, inhibiting both EZH2 and G9a histone methyltransferases showed greater efficacy in blocking cell proliferation as compared to single drug treatment [254]. Furthermore, the DOT1L inhibitor EPZ-5676 could interact synergistically with DNA hypomethylating agents, such as azacitidine and decitabine, in MLL-rearranged leukemia cells [255].
In recent years, an increasing number of studies have reported the use of epigenetic drugs in combination with conventional chemotherapeutics, with underlying mechanisms of re-sensitising resistant CSCs to drug treatment, or to prime cancer cells for subsequent therapies [134, 256]. For example, low doses of SGI-110 (DNMT inhibitor) was found to drive ovarian CSCs towards a more differentiated phenotype and sensitise them to platinum treatment [125]. DOT1L inhibitor EPZ-5676 was also able to establish a chromatin state that enhanced the anti-tumor effects of cytarabine or daunorubicin in MLL-rearranged leukemia [255]. Moreover, pre-treatment with azacitidine was demonstrated to prime colon cancer cell lines to irinotecan therapy [257]. Indeed, various combinations have been tested in clinical trials with promising results on drug response and anti-tumor efficacy [258–261]. In addition to drug combination synergy, the method of delivery could also improve response to therapy. A recent paper by Li et al. showed that encapsulating decitabine and doxorubicin in nanoparticles was able to better target breast CSCs and inhibit tumor growth [262].
The use of immunotherapy in cancer has made significant progress over the past two decades, with several immunotherapy drugs being approved by the FDA for the treatment of cancer. These drugs function to overcome the mechanisms of immune tolerance that are employed by cancer cells to evade or limit the body’s immune response. These mechanisms include changes in antigen processing and presentation, creation of an immunosuppressive microenvironment, induction of T-cell death and activation of negative immune regulatory pathways [263]. One key receptor involved in the immunoinhibitory pathways is the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), which is expressed on the surface of immune cells and acts as an immune checkpoint. Studies have shown that targeting CTLA-4 receptor induced favourable responses in patients with advanced melanoma [264], and the FDA-approved CTLA-4 inhibitor, Ipilimumab, is now in clinical trials for prostate and lung cancers. Another immune checkpoint involved in tumor immune-resistance is the interaction between programmed cell death-1 (PD-1) and programmed death-ligand 1 (PD-L1) [265]. Specific targeting of PD-1 and PD-L1 has been clinically shown to be very effective in treatment of metastatic cancers and melanomas [266, 267].
However, as most of these immunotherapy strategies are mainly targeted at bulk tumors, which contain more differentiated cells with “differentiation antigens” [268], CSCs (which have a different set of tumor antigens) would not be successfully eradicated. Hence, more effective targeting of the CSC population can be achieved via CSC-specific immunologic approaches, or by combining immunotherapy with epigenetic therapies that induce CSC differentiation and alter surface protein expression. The latter approach would likely improve the overall antitumor efficacy as both CSC and bulk tumor populations can be targeted simultaneously. For instance, the use of DNA hypomethylating agent (5-aza-2’-deoxycytidine) in combination with anti-CTLA-4 monoclonal antibody in syngeneic transplantable murine models demonstrated significant reduction in tumor volumes as compared to single agent treatment alone [269]. The improved efficacy of this combination was attributed to the increased CD3+ T-cell infiltration in the combination cohort tumors and a sustained expression of cancer antigens and MHC proteins due to promoter demethylation. Furthermore, combinatorial drug treatment with immune checkpoint inhibitors (anti-CTLA-4 and anti-PD-1) and epigenetic modulators (5-azacytidine and Entinostat) showed remarkable eradication of CT26 colorectal tumors and 4 T1 mammary tumors in more than 80% of the tumor-bearing mice [270]. Importantly, 4 T1 tumor-bearing mice that were given combinatorial treatment did not develop metastases as compared to mice given single agent treatment. These findings demonstrate that epigenetic drugs in combination with immunotherapy can enhance the reversal of immune tolerance in cancer cells, including CSCs.
Another way in which cancer cells evade cytotoxic T-cells is by down-regulating human leukocyte antigen (HLA) to avoid tumor antigen presentation [134]. Hypermethylation of HLA promoters was frequently observed in gastric cancer and esophageal squamous cell cancers [271, 272]. Treatment with DNMT and HDAC inhibitors were found to be capable of reversing this hypermethylation and increasing HLA expression [272–275], thus priming these cells for immunotherapy. In addition, Li et al. showed that azacitidine treatment was able to enhance immunomodulatory pathways, such as antigen processing/presentation and interferon signaling, in breast, colorectal and ovarian cancers [276]. These preclinical data highlight the promising potential of combining epigenetic and immunotherapies in improving cancer treatment efficacy, which will be verified in several ongoing clinical trials.
Conclusion
Our understanding of cancer has changed over the last decade with the advances in sequencing technologies and the deciphering of the human genome. It is now clear to us that the tumor genome is complex and heterogeneous and that tumors do not arise from a single clone with a single tumor genome. We have discussed several important aspects and examples of how epigenetic deregulation may drive or promote tumorigenesis and metastasis by alteration of key transcriptomic programs and signaling pathways, especially in CSCs. More importantly, we have provided several evidences that these epigenetic modifiers are targetable and many of these epigenetic modulating drugs have entered clinical trials, and some including azacitidine, decitabine, vorinostat and romidepsin have been approved for various indications by the FDA. We believe that the success of these epigenetic therapeutic trials will provide a promising path to follow.
Acknowledgements
We apologize for not reviewing numerous original and pertinent articles due to space limitations and the scope of this review. We would also like to thank Mr Wong Chun Xi (Mechanobiology Institute, Singapore) for his help in illustrating the figure in this manuscript.
Funding
The authors are supported by the National Research Foundation Cancer Science Institute of Singapore RCE Main Grant, Ministry of Education Academic Research Fund (MOE AcRF Tier 2 (MOE2015-T2-2-126, Seed Fund Grant T1-BSRG 2014–05). This work is funded by the NCIS Yong Siew Yoon Research Grant through donations from the Yong Loo Lin Trust.
Availability of data and materials
Not applicable.
Authors’ Contributions
TBT, JJL and EKC were major contributors in the writing of the manuscript. TBT and EKC designed the manuscript. EKC revised and edited the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- ABC
ATP-binding cassette
- ABCG2
ATP-binding cassette sub-family G member 2
- ALL
Acute lymphoblastic leukemia
- AML
Acute myeloid leukemia
- AML1-ETO
Acute myeloid leukemia protein 1 Eight twenty-one protein
- APC
Adenomatous polyposis coli
- ASCL1
Achaete-scute family BHLH transcription factor 1
- BCC
Basal cell carcinoma
- BCL2
B-cell lymphoma 2
- BET
Bromodomain and extra-terminal
- Bmi1
B-lymphoma Mo-MLV insertion region 1 homolog
- CK1
Casein kinase 1
- CK19
Cytokeratin 19
- CSC
Cancer stem cells
- DACT3
Polycomb repressor complex 2, PCR2 Dishevelled-binding antagonist of beta-catenin 3
- DKK1
Dickkopf-related protein 1
- DNMTs
DNA methyltransferases
- DZNep
3-deazaneplanocin A
- ECM
Extracellular matrix
- EED
Embryonic ectoderm development protein
- EMT
Epithelial-to-mesenchymal transition
- EpCAM
Epithelial cell adhesion molecule
- ESCs
Embryonic stem cells
- EZH2
Enhancer of zeste homolog 2
- FAD
Flavin adenine dinucleotide
- G9a/EHMT2
Euchromatic histone lysine methyltransferase 2
- GBM
Glioblastoma multiforme
- GLP/EHMT1
Euchromatic histone lysine methyltransferase 1
- GNPs
Granule neuron progenitors/precursors
- GSK-3β
Glycogen synthase kinase 3 beta
- H3K27
Histone H3 lysine 27
- H3K36
Histone H3 lysine 36
- H3K4
Histone H3 lysine 4
- H3K79
Histone H3 lysine 79
- H3K9
Histone H3 lysine 9
- H4K20
Histone H4 lysine 20
- HAT
Histone acetyltransferases
- HCC
Hepatocellular carcinoma
- HDAC
Histone deacetylase
- Hh
Hedgehog
- HKMT
Histone lysine methyltransferase
- HoxA9
Homeobox A9
- ICC
Intrahepatic cholangiocarcinoma
- JmjC
Jumonji domain-containing (JmjC)
- KDM
Histone lysine demethylase
- Kif7
Kinesin family member 7
- LDR5/6
Low density lipoprotein receptor-related protein 5/6
- LSD
Lysine-specific demethylase
- MDR1
Multidrug resistance protein 1
- Meis1
Meis homeobox 1
- miRNAs
Micro RNAs
- MLL
mixed lineage leukemia
- MLL2
Mixed lineage leukemia protein 2
- MRP1
Multidrug resistance-associated protein 1
- NF-κB
Nuclear factor kappa b
- NICD
Notch intracellular domain
- NKD1
Naked cuticle homolog 1
- NMC
NUT midline carcinoma
- NSCLC
non-small cell lung cancer
- NSCs
Neural stem cells
- OCT4
Octamer-binding transcription factor 4
- PML-RARα
Promyelocytic leukemia protein retinoic acid receptor alpha
- PTCH1
Patched receptor
- RBPJ-κ
Recombination signal binding protein for immunoglobulin kappa J region
- SAH
S-adenosylhomocysteine hydrolase
- SAM
S-adenosyl methionine
- SFRP-1
Secreted frizzled-related protein 1
- Shh
Sonic hedgehog ligand
- SirT1
Sirtuin 1
- SMO
Smoothened
- SMRT
Nuclear receptor co-repressor 2
- SNAIL
Snail family zinc finger 1
- SP
Side population
- STRAP
Serine-threonine kinase receptor-associated protein
- SUFU
Suppressor of fused homolog
- SUV39H1
Suppressor of variegation 3–9 homolog 1
- SUV39H2
Suppressor of variegation 3–9 homolog 2
- Suz12
Suppressor of zeste 12 protein homolog
- TCF/LEF
T-cell factor/lymphoid enhancer factor
- TGF-β
Transforming growth factor-β
- TNBC
Triple-negative breast cancer
- TWIST1
Twist-related protein 1
- UTX
Ubiquitously transcribed tetratricopeptide repeat, X chromosome (UTX)
- WIF-1
Wnt inhibitory factor 1
- ZEB1
Zinc finger E-box-binding homeobox 1
- ZEB2
Zinc finger E-box-binding homeobox 2
Contributor Information
Tan Boon Toh, Email: csittb@nus.edu.sg.
Jhin Jieh Lim, Email: csiljj@nus.edu.sg.
Edward Kai-Hua Chow, Phone: +65 6516-8707, Email: csikce@nus.edu.sg.
References
- 1.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 2.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 3.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 4.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 5.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 6.Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, Zhan Q, Jordan S, Duncan LM, Weishaupt C, et al. Identification of cells initiating human melanomas. Nature. 2008;451:345–349. doi: 10.1038/nature06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, Yan PS, Huang TH, Nephew KP. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68:4311–4320. doi: 10.1158/0008-5472.CAN-08-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO, Zheng BJ, Guan XY. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132:2542–2556. doi: 10.1053/j.gastro.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 10.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–10951. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 11.Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, Conticello C, Ruco L, Peschle C, De Maria R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504–514. doi: 10.1038/sj.cdd.4402283. [DOI] [PubMed] [Google Scholar]
- 12.Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, Aigner L, Brawanski A, Bogdahn U, Beier CP. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 13.Kemper K, Sprick MR, de Bree M, Scopelliti A, Vermeulen L, Hoek M, Zeilstra J, Pals ST, Mehmet H, Stassi G, Medema JP. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010;70:719–729. doi: 10.1158/0008-5472.CAN-09-1820. [DOI] [PubMed] [Google Scholar]
- 14.Zhou F, Cui C, Ge Y, Chen H, Li Q, Yang Z, Wu G, Sun S, Chen K, Gu J, et al. Alpha2,3-Sialylation regulates the stability of stem cell marker CD133. J Biochem. 2010;148:273–280. doi: 10.1093/jb/mvq062. [DOI] [PubMed] [Google Scholar]
- 15.Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, Jia H, Ye Q, Qin LX, Wauthier E, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012–1024. doi: 10.1053/j.gastro.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson GS, Hu Z, Duan W, Tian A, Wang XM, McLeod D, Lam V, George J, Qiao L. Efficacy of using cancer stem cell markers in isolating and characterizing liver cancer stem cells. Stem Cells Dev. 2013;22:2655–2664. doi: 10.1089/scd.2012.0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chow EK, Fan LL, Chen X, Bishop JM. Oncogene-specific formation of chemoresistant murine hepatic cancer stem cells. Hepatology. 2012;56:1331–1341. doi: 10.1002/hep.25776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, Piccirillo S, Vescovi AL, DiMeco F, Olivi A, Eberhart CG. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25:2524–2533. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakariassen PO, Prestegarden L, Wang J, Skaftnesmo KO, Mahesparan R, Molthoff C, Sminia P, Sundlisaeter E, Misra A, Tysnes BB, et al. Angiogenesis-independent tumor growth mediated by stem-like cancer cells. Proc Natl Acad Sci U S A. 2006;103:16466–16471. doi: 10.1073/pnas.0607668103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, Asselin-Labat ML, Gyorki DE, Ward T, Partanen A, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 22.Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R, Mackay A, Grigoriadis A, Tutt A, Ashworth A, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010;7:403–417. doi: 10.1016/j.stem.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 23.Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, Poeppl A, Ling V, Beyene J, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17:1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 24.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 25.Shats I, Gatza ML, Chang JT, Mori S, Wang J, Rich J, Nevins JR. Using a stem cell-based signature to guide therapeutic selection in cancer. Cancer Res. 2011;71:1772–1780. doi: 10.1158/0008-5472.CAN-10-1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 27.Stallcup MR. Role of protein methylation in chromatin remodeling and transcriptional regulation. Oncogene. 2001;20:3014–3020. doi: 10.1038/sj.onc.1204325. [DOI] [PubMed] [Google Scholar]
- 28.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 29.Wongtrakoongate P. Epigenetic therapy of cancer stem and progenitor cells by targeting DNA methylation machineries. World J Stem Cells. 2015;7:137–148. doi: 10.4252/wjsc.v7.i1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 2007;16 Spec No 1:R50–59. doi: 10.1093/hmg/ddm018. [DOI] [PubMed] [Google Scholar]
- 31.Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39:237–242. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Broske AM, Vockentanz L, Kharazi S, Huska MR, Mancini E, Scheller M, Kuhl C, Enns A, Prinz M, Jaenisch R, et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat Genet. 2009;41:1207–1215. doi: 10.1038/ng.463. [DOI] [PubMed] [Google Scholar]
- 34.Liu CC, Lin JH, Hsu TW, Su K, Li AF, Hsu HS, Hung SC. IL-6 enriched lung cancer stem-like cell population by inhibition of cell cycle regulators via DNMT1 upregulation. Int J Cancer. 2015;136:547–559. doi: 10.1002/ijc.29033. [DOI] [PubMed] [Google Scholar]
- 35.Morita R, Hirohashi Y, Suzuki H, Takahashi A, Tamura Y, Kanaseki T, Asanuma H, Inoda S, Kondo T, Hashino S, et al. DNA methyltransferase 1 is essential for initiation of the colon cancers. Exp Mol Pathol. 2013;94:322–329. doi: 10.1016/j.yexmp.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 36.Hoffmeyer K, Raggioli A, Rudloff S, Anton R, Hierholzer A, Del Valle I, Hein K, Vogt R, Kemler R. Wnt/beta-catenin signaling regulates telomerase in stem cells and cancer cells. Science. 2012;336:1549–1554. doi: 10.1126/science.1218370. [DOI] [PubMed] [Google Scholar]
- 37.Myant KB, Cammareri P, McGhee EJ, Ridgway RA, Huels DJ, Cordero JB, Schwitalla S, Kalna G, Ogg EL, Athineos D, et al. ROS production and NF-kappaB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell. 2013;12:761–773. doi: 10.1016/j.stem.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov. 2006;5:997–1014. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- 39.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 40.Lindvall C, Bu W, Williams BO, Li Y. Wnt signaling, stem cells, and the cellular origin of breast cancer. Stem Cell Rev. 2007;3:157–168. doi: 10.1007/s12015-007-0025-3. [DOI] [PubMed] [Google Scholar]
- 41.Klarmann GJ, Decker A, Farrar WL. Epigenetic gene silencing in the Wnt pathway in breast cancer. Epigenetics. 2008;3:59–63. doi: 10.4161/epi.3.2.5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, Pretlow TP, Yang B, Akiyama Y, Van Engeland M, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36:417–422. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]
- 43.Koinuma K, Yamashita Y, Liu W, Hatanaka H, Kurashina K, Wada T, Takada S, Kaneda R, Choi YL, Fujiwara SI, et al. Epigenetic silencing of AXIN2 in colorectal carcinoma with microsatellite instability. Oncogene. 2006;25:139–146. doi: 10.1038/sj.onc.1209009. [DOI] [PubMed] [Google Scholar]
- 44.Yoda Y, Takeshima H, Niwa T, Kim JG, Ando T, Kushima R, Sugiyama T, Katai H, Noshiro H, Ushijima T. Integrated analysis of cancer-related pathways affected by genetic and epigenetic alterations in gastric cancer. Gastric Cancer. 2015;18:65–76. doi: 10.1007/s10120-014-0348-0. [DOI] [PubMed] [Google Scholar]
- 45.Hussain M, Rao M, Humphries AE, Hong JA, Liu F, Yang M, Caragacianu D, Schrump DS. Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer Res. 2009;69:3570–3578. doi: 10.1158/0008-5472.CAN-08-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang X, Tan J, Li J, Kivimae S, Yang X, Zhuang L, Lee PL, Chan MT, Stanton LW, Liu ET, et al. DACT3 is an epigenetic regulator of Wnt/beta-catenin signaling in colorectal cancer and is a therapeutic target of histone modifications. Cancer Cell. 2008;13:529–541. doi: 10.1016/j.ccr.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rheinbay E, Suva ML, Gillespie SM, Wakimoto H, Patel AP, Shahid M, Oksuz O, Rabkin SD, Martuza RL, Rivera MN, et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 2013;3:1567–1579. doi: 10.1016/j.celrep.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H, Huang H, Xue J, Liu M, Wang Y, et al. FoxM1 promotes beta-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell. 2011;20:427–442. doi: 10.1016/j.ccr.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng H, Ying H, Wiedemeyer R, Yan H, Quayle SN, Ivanova EV, Paik JH, Zhang H, Xiao Y, Perry SR, et al. PLAGL2 regulates Wnt signaling to impede differentiation in neural stem cells and gliomas. Cancer Cell. 2010;17:497–509. doi: 10.1016/j.ccr.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y, He L, Du Y, Zhu P, Huang G, Luo J, Yan X, Ye B, Li C, Xia P, et al. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell. 2015;16:413–425. doi: 10.1016/j.stem.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 51.Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 52.Ma J, Meng Y, Kwiatkowski DJ, Chen X, Peng H, Sun Q, Zha X, Wang F, Wang Y, Jing Y, et al. Mammalian target of rapamycin regulates murine and human cell differentiation through STAT3/p63/Jagged/Notch cascade. J Clin Invest. 2010;120:103–114. doi: 10.1172/JCI37964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432:324–331. doi: 10.1038/nature03100. [DOI] [PubMed] [Google Scholar]
- 54.Teglund S, Toftgard R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta. 1805;2010:181–208. doi: 10.1016/j.bbcan.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 55.Youssef KK, Van Keymeulen A, Lapouge G, Beck B, Michaux C, Achouri Y, Sotiropoulou PA, Blanpain C. Identification of the cell lineage at the origin of basal cell carcinoma. Nat Cell Biol. 2010;12:299–305. doi: 10.1038/ncb2031. [DOI] [PubMed] [Google Scholar]
- 56.Sellheyer K. Basal cell carcinoma: cell of origin, cancer stem cell hypothesis and stem cell markers. Br J Dermatol. 2011;164:696–711. doi: 10.1111/j.1365-2133.2010.10158.x. [DOI] [PubMed] [Google Scholar]
- 57.Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, Schuller U, Machold R, Fishell G, Rowitch DH, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008;14:135–145. doi: 10.1016/j.ccr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999;59:74–79. [PubMed] [Google Scholar]
- 60.Sevenet N, Sheridan E, Amram D, Schneider P, Handgretinger R, Delattre O. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet. 1999;65:1342–1348. doi: 10.1086/302639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- 62.Canettieri G, Di Marcotullio L, Greco A, Coni S, Antonucci L, Infante P, Pietrosanti L, De Smaele E, Ferretti E, Miele E, et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010;12:132–142. doi: 10.1038/ncb2013. [DOI] [PubMed] [Google Scholar]
- 63.Di Marcotullio L, Ferretti E, De Smaele E, Argenti B, Mincione C, Zazzeroni F, Gallo R, Masuelli L, Napolitano M, Maroder M, et al. REN(KCTD11) is a suppressor of Hedgehog signaling and is deleted in human medulloblastoma. Proc Natl Acad Sci U S A. 2004;101:10833–10838. doi: 10.1073/pnas.0400690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cui W, Wang LH, Wen YY, Song M, Li BL, Chen XL, Xu M, An SX, Zhao J, Lu YY, et al. Expression and regulation mechanisms of Sonic Hedgehog in breast cancer. Cancer Sci. 2010;101:927–933. doi: 10.1111/j.1349-7006.2010.01495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang LH, Choi YL, Hua XY, Shin YK, Song YJ, Youn SJ, Yun HY, Park SM, Kim WJ, Kim HJ, et al. Increased expression of sonic hedgehog and altered methylation of its promoter region in gastric cancer and its related lesions. Mod Pathol. 2006;19:675–683. doi: 10.1038/modpathol.3800573. [DOI] [PubMed] [Google Scholar]
- 66.Duan ZH, Wang HC, Zhao DM, Ji XX, Song M, Yang XJ, Cui W. Cooperatively transcriptional and epigenetic regulation of sonic hedgehog overexpression drives malignant potential of breast cancer. Cancer Sci. 2015;106:1084–1091. doi: 10.1111/cas.12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling--are we there yet? Nat Rev Drug Discov. 2014;13:357–378. doi: 10.1038/nrd4252. [DOI] [PubMed] [Google Scholar]
- 68.Kopan R. Notch signaling. Cold Spring Harb Perspect Biol. 2012;4. [DOI] [PMC free article] [PubMed]
- 69.Andersson ER, Sandberg R, Lendahl U. Notch signaling: simplicity in design, versatility in function. Development. 2011;138:3593–3612. doi: 10.1242/dev.063610. [DOI] [PubMed] [Google Scholar]
- 70.Munoz P, Iliou MS, Esteller M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol Oncol. 2012;6:620–636. doi: 10.1016/j.molonc.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu J, Sato C, Cerletti M, Wagers A. Notch signaling in the regulation of stem cell self-renewal and differentiation. Curr Top Dev Biol. 2010;92:367–409. doi: 10.1016/S0070-2153(10)92012-7. [DOI] [PubMed] [Google Scholar]
- 72.Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, Rueger MA, Bae SK, Kittappa R, McKay RD. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006;442:823–826. doi: 10.1038/nature04940. [DOI] [PubMed] [Google Scholar]
- 73.Lopez-Arribillaga E, Rodilla V, Pellegrinet L, Guiu J, Iglesias M, Roman AC, Gutarra S, Gonzalez S, Munoz-Canoves P, Fernandez-Salguero P, et al. Bmi1 regulates murine intestinal stem cell proliferation and self-renewal downstream of Notch. Development. 2015;142:41–50. doi: 10.1242/dev.107714. [DOI] [PubMed] [Google Scholar]
- 74.Yuan X, Wu H, Han N, Xu H, Chu Q, Yu S, Chen Y, Wu K. Notch signaling and EMT in non-small cell lung cancer: biological significance and therapeutic application. J Hematol Oncol. 2014;7:87. doi: 10.1186/s13045-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ichida JK, Tcw J, Williams LA, Carter AC, Shi Y, Moura MT, Ziller M, Singh S, Amabile G, Bock C, et al. Notch inhibition allows oncogene-independent generation of iPS cells. Nat Chem Biol. 2014;10:632–639. doi: 10.1038/nchembio.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zou J, Li P, Lu F, Liu N, Dai J, Ye J, Qu X, Sun X, Ma D, Park J, Ji C. Notch1 is required for hypoxia-induced proliferation, invasion and chemoresistance of T-cell acute lymphoblastic leukemia cells. J Hematol Oncol. 2013;6:3. doi: 10.1186/1756-8722-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gallahan D, Callahan R. The mouse mammary tumor associated gene INT3 is a unique member of the NOTCH gene family (NOTCH4) Oncogene. 1997;14:1883–1890. doi: 10.1038/sj.onc.1201035. [DOI] [PubMed] [Google Scholar]
- 78.D’Angelo RC, Ouzounova M, Davis A, Choi D, Tchuenkam SM, Kim G, Luther T, Quraishi AA, Senbabaoglu Y, Conley SJ, et al. Notch reporter activity in breast cancer cell lines identifies a subset of cells with stem cell activity. Mol Cancer Ther. 2015;14:779–787. doi: 10.1158/1535-7163.MCT-14-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang Z, Da Silva TG, Jin K, Han X, Ranganathan P, Zhu X, Sanchez-Mejias A, Bai F, Li B, Fei DL, et al. Notch signaling drives stemness and tumorigenicity of esophageal adenocarcinoma. Cancer Res. 2014;74:6364–6374. doi: 10.1158/0008-5472.CAN-14-2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jin L, Vu TT, Datta PK. Abstract 1709: STRAP mediates the stemness of human colorectal cancer cells by epigenetic regulation of Notch pathway. Cancer Res. 2016;76:1709. doi: 10.1158/1538-7445.AM2016-1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ghoshal P, Nganga AJ, Moran-Giuati J, Szafranek A, Johnson TR, Bigelow AJ, Houde CM, Avet-Loiseau H, Smiraglia DJ, Ersing N, et al. Loss of the SMRT/NCoR2 corepressor correlates with JAG2 overexpression in multiple myeloma. Cancer Res. 2009;69:4380–4387. doi: 10.1158/0008-5472.CAN-08-3467. [DOI] [PubMed] [Google Scholar]
- 82.Heerboth S, Housman G, Leary M, Longacre M, Byler S, Lapinska K, Willbanks A, Sarkar S. EMT and tumor metastasis. Clin Transl Med. 2015;4:6. doi: 10.1186/s40169-015-0048-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 84.Morel A-P, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of Breast Cancer Stem Cells through Epithelial-Mesenchymal Transition. PLoS One. 2008;3:e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dissanayake SK, Wade M, Johnson CE, O’Connell MP, Leotlela PD, French AD, Shah KV, Hewitt KJ, Rosenthal DT, Indig FE, et al. The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J Biol Chem. 2007;282:17259–17271. doi: 10.1074/jbc.M700075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vincan E, Barker N. The upstream components of the Wnt signalling pathway in the dynamic EMT and MET associated with colorectal cancer progression. Clin Exp Metastasis. 2008;25:657–663. doi: 10.1007/s10585-008-9156-4. [DOI] [PubMed] [Google Scholar]
- 88.Massague J. TGFbeta in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 90.Dohadwala M, Yang SC, Luo J, Sharma S, Batra RK, Huang M, Lin Y, Goodglick L, Krysan K, Fishbein MC, et al. Cyclooxygenase-2-dependent regulation of E-cadherin: prostaglandin E(2) induces transcriptional repressors ZEB1 and snail in non-small cell lung cancer. Cancer Res. 2006;66:5338–5345. doi: 10.1158/0008-5472.CAN-05-3635. [DOI] [PubMed] [Google Scholar]
- 91.Dumont N, Wilson MB, Crawford YG, Reynolds PA, Sigaroudinia M, Tlsty TD. Sustained induction of epithelial to mesenchymal transition activates DNA methylation of genes silenced in basal-like breast cancers. Proc Natl Acad Sci U S A. 2008;105:14867–14872. doi: 10.1073/pnas.0807146105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ, Thilaganathan N, Du L, Zhang Y, Pertsemlidis A, Kurie JM. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009;23:2140–2151. doi: 10.1101/gad.1820209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Avgustinova A, Benitah SA. The epigenetics of tumour initiation: cancer stem cells and their chromatin. Curr Opin Genet Dev. 2016;36:8–15. doi: 10.1016/j.gde.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 94.Koizume S, Tachibana K, Sekiya T, Hirohashi S, Shiraishi M. Heterogeneity in the modification and involvement of chromatin components of the CpG island of the silenced human CDH1 gene in cancer cells. Nucleic Acids Res. 2002;30:4770–4780. doi: 10.1093/nar/gkf593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Y, Shang Y. Epigenetic control of epithelial-to-mesenchymal transition and cancer metastasis. Exp Cell Res. 2013;319:160–169. doi: 10.1016/j.yexcr.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 96.Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani RS, Tomlins SA, Mehra R, Laxman B, Cao X, Yu J, et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene. 2008;27:7274–7284. doi: 10.1038/onc.2008.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Herranz N, Pasini D, Diaz VM, Franci C, Gutierrez A, Dave N, Escriva M, Hernandez-Munoz I, Di Croce L, Helin K, et al. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol Cell Biol. 2008;28:4772–4781. doi: 10.1128/MCB.00323-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 99.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, Yu WH, Rehman SK, Hsu JL, Lee HH, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317–323. doi: 10.1038/ncb2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tellez CS, Juri DE, Do K, Bernauer AM, Thomas CL, Damiani LA, Tessema M, Leng S, Belinsky SA. EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells. Cancer Res. 2011;71:3087–3097. doi: 10.1158/0008-5472.CAN-10-3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shah AN, Summy JM, Zhang J, Park SI, Parikh NU, Gallick GE. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann Surg Oncol. 2007;14:3629–3637. doi: 10.1245/s10434-007-9583-5. [DOI] [PubMed] [Google Scholar]
- 104.Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, Ali S, Abbruzzese JL, Gallick GE, Sarkar FH. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009;69:2400–2407. doi: 10.1158/0008-5472.CAN-08-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shukla S, Meeran SM. Epigenetics of cancer stem cells: Pathways and therapeutics. Biochim Biophys Acta. 1840;2014:3494–3502. doi: 10.1016/j.bbagen.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 106.Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey DJ, Choi W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820–5828. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wu CP, Calcagno AM, Ambudkar SV. Reversal of ABC drug transporter-mediated multidrug resistance in cancer cells: evaluation of current strategies. Curr Mol Pharmacol. 2008;1:93–105. doi: 10.2174/1874467210801020093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang X, Low XC, Hou W, Abdullah LN, Toh TB. Mohd Abdul Rashid M, Ho D, Chow EK: Epirubicin-adsorbed nanodiamonds kill chemoresistant hepatic cancer stem cells. ACS Nano. 2014;8:12151–12166. doi: 10.1021/nn503491e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cho S, Lu M, He X, Ee PL, Bhat U, Schneider E, Miele L, Beck WT. Notch1 regulates the expression of the multidrug resistance gene ABCC1/MRP1 in cultured cancer cells. Proc Natl Acad Sci U S A. 2011;108:20778–20783. doi: 10.1073/pnas.1019452108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kim B, Stephen SL, Hanby AM, Horgan K, Perry SL, Richardson J, Roundhill EA, Valleley EM, Verghese ET, Williams BJ, et al. Chemotherapy induces Notch1-dependent MRP1 up-regulation, inhibition of which sensitizes breast cancer cells to chemotherapy. BMC Cancer. 2015;15:634. doi: 10.1186/s12885-015-1625-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.To KK, Polgar O, Huff LM, Morisaki K, Bates SE. Histone modifications at the ABCG2 promoter following treatment with histone deacetylase inhibitor mirror those in multidrug-resistant cells. Mol Cancer Res. 2008;6:151–164. doi: 10.1158/1541-7786.MCR-07-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123:8–13. doi: 10.1002/ijc.23607. [DOI] [PubMed] [Google Scholar]
- 114.Juttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci U S A. 1994;91:11797–11801. doi: 10.1073/pnas.91.25.11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ghoshal K, Datta J, Majumder S, Bai S, Kutay H, Motiwala T, Jacob ST. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol. 2005;25:4727–4741. doi: 10.1128/MCB.25.11.4727-4741.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 116.Sorm F, Vesely J. the activity of a new antimetabolite, 5-azacytidine, against lymphoid leukaemia in ak mice. Neoplasma. 1964;11:123–130. [PubMed] [Google Scholar]
- 117.Abele R, Clavel M, Dodion P, Bruntsch U, Gundersen S, Smyth J, Renard J, van Glabbeke M, Pinedo HM. The EORTC Early Clinical Trials Cooperative Group experience with 5-aza-2′-deoxycytidine (NSC 127716) in patients with colo-rectal, head and neck, renal carcinomas and malignant melanomas. Eur J Cancer Clin Oncol. 1987;23:1921–1924. doi: 10.1016/0277-5379(87)90060-5. [DOI] [PubMed] [Google Scholar]
- 118.Cowan LA, Talwar S, Yang AS. Will DNA methylation inhibitors work in solid tumors? A review of the clinical experience with azacitidine and decitabine in solid tumors. Epigenomics. 2010;2:71–86. doi: 10.2217/epi.09.44. [DOI] [PubMed] [Google Scholar]
- 119.Clavel M, Monfardini S, Fossa S, Smyth J, Renard J, Kaye SB. 5-Aza-2′-deoxycytidine (NSC 127716) in non-seminomatous testicular cancer. Phase II from the EORTC Early Clinical Trials Cooperative Group and Genito-Urinary Group. Ann Oncol. 1992;3:399–400. doi: 10.1093/annonc/3.suppl_5.iii. [DOI] [PubMed] [Google Scholar]
- 120.Stadler WM, Margolin K, Ferber S, McCulloch W, Thompson JA. A phase II study of depsipeptide in refractory metastatic renal cell cancer. Clin Genitourin Cancer. 2006;5:57–60. doi: 10.3816/CGC.2006.n.018. [DOI] [PubMed] [Google Scholar]
- 121.Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21:430–446. doi: 10.1016/j.ccr.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pinto A, Zagonel V, Attadia V, Bullian PL, Gattei V, Carbone A, Monfardini S, Colombatti A. 5-Aza-2′-deoxycytidine as a differentiation inducer in acute myeloid leukaemias and myelodysplastic syndromes of the elderly. Bone Marrow Transplant. 1989;4(Suppl 3):28–32. [PubMed] [Google Scholar]
- 123.Zagonel V, Lo Re G, Marotta G, Babare R, Sardeo G, Gattei V, De Angelis V, Monfardini S, Pinto A. 5-Aza-2′-deoxycytidine (Decitabine) induces trilineage response in unfavourable myelodysplastic syndromes. Leukemia. 1993;7(Suppl 1):30–35. [PubMed] [Google Scholar]
- 124.Tian J, Lee SO, Liang L, Luo J, Huang CK, Li L, Niu Y, Chang C. Targeting the unique methylation pattern of androgen receptor (AR) promoter in prostate stem/progenitor cells with 5-aza-2′-deoxycytidine (5-AZA) leads to suppressed prostate tumorigenesis. J Biol Chem. 2012;287:39954–39966. doi: 10.1074/jbc.M112.395574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang Y, Cardenas H, Fang F, Condello S, Taverna P, Segar M, Liu Y, Nephew KP, Matei D. Epigenetic targeting of ovarian cancer stem cells. Cancer Res. 2014;74:4922–4936. doi: 10.1158/0008-5472.CAN-14-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kaminskas E, Farrell AT, Wang YC, Sridhara R, Pazdur R. FDA drug approval summary: azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist. 2005;10:176–182. doi: 10.1634/theoncologist.10-3-176. [DOI] [PubMed] [Google Scholar]
- 127.Finley A, Copeland RA. Small molecule control of chromatin remodeling. Chem Biol. 2014;21:1196–1210. doi: 10.1016/j.chembiol.2014.07.024. [DOI] [PubMed] [Google Scholar]
- 128.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/bj20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Johnstone RW, Licht JD. Histone deacetylase inhibitors in cancer therapy: is transcription the primary target? Cancer Cell. 2003;4:13–18. doi: 10.1016/S1535-6108(03)00165-X. [DOI] [PubMed] [Google Scholar]
- 130.Gelmetti V, Zhang J, Fanelli M, Minucci S, Pelicci PG, Lazar MA. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol Cell Biol. 1998;18:7185–7191. doi: 10.1128/MCB.18.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408:377–381. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- 132.Duan H, Heckman CA, Boxer LM. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol Cell Biol. 2005;25:1608–1619. doi: 10.1128/MCB.25.5.1608-1619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Federico M, Bagella L. Histone deacetylase inhibitors in the treatment of hematological malignancies and solid tumors. J Biomed Biotechnol. 2011;2011:475641. doi: 10.1155/2011/475641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Juo YY, Gong XJ, Mishra A, Cui X, Baylin SB, Azad NS, Ahuja N. Epigenetic therapy for solid tumors: from bench science to clinical trials. Epigenomics. 2015;7:215–235. doi: 10.2217/epi.14.73. [DOI] [PubMed] [Google Scholar]
- 135.Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, Frankel SR, Chen C, Ricker JL, Arduino JM, Duvic M. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:3109–3115. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- 136.Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, Zain J, Prince HM, Leonard JP, Geskin LJ, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27:5410–5417. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Thaler F. Current trends in the development of histone deacetylase inhibitors: a review of recent patent applications. Pharm Pat Anal. 2012;1:75–90. doi: 10.4155/ppa.11.3. [DOI] [PubMed] [Google Scholar]
- 138.Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, Scarisbrick J, Reddy S, Robak T, Becker JC, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28:4485–4491. doi: 10.1200/JCO.2010.28.9066. [DOI] [PubMed] [Google Scholar]
- 139.Vansteenkiste J, Van Cutsem E, Dumez H, Chen C, Ricker JL, Randolph SS, Schoffski P. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Invest New Drugs. 2008;26:483–488. doi: 10.1007/s10637-008-9131-6. [DOI] [PubMed] [Google Scholar]
- 140.Luu TH, Morgan RJ, Leong L, Lim D, McNamara M, Portnow J, Frankel P, Smith DD, Doroshow JH, Wong C, et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: a California Cancer Consortium study. Clin Cancer Res. 2008;14:7138–7142. doi: 10.1158/1078-0432.CCR-08-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Woyach JA, Kloos RT, Ringel MD, Arbogast D, Collamore M, Zwiebel JA, Grever M, Villalona-Calero M, Shah MH. Lack of therapeutic effect of the histone deacetylase inhibitor vorinostat in patients with metastatic radioiodine-refractory thyroid carcinoma. J Clin Endocrinol Metab. 2009;94:164–170. doi: 10.1210/jc.2008-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Modesitt SC, Sill M, Hoffman JS, Bender DP. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2008;109:182–186. doi: 10.1016/j.ygyno.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 143.Blumenschein GR, Jr, Kies MS, Papadimitrakopoulou VA, Lu C, Kumar AJ, Ricker JL, Chiao JH, Chen C, Frankel SR. Phase II trial of the histone deacetylase inhibitor vorinostat (Zolinza, suberoylanilide hydroxamic acid, SAHA) in patients with recurrent and/or metastatic head and neck cancer. Invest New Drugs. 2008;26:81–87. doi: 10.1007/s10637-007-9075-2. [DOI] [PubMed] [Google Scholar]
- 144.Traynor AM, Dubey S, Eickhoff JC, Kolesar JM, Schell K, Huie MS, Groteluschen DL, Marcotte SM, Hallahan CM, Weeks HR, et al. Vorinostat (NSC# 701852) in patients with relapsed non-small cell lung cancer: a Wisconsin Oncology Network phase II study. J Thorac Oncol. 2009;4:522–526. doi: 10.1097/JTO.0b013e3181952478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS, Reilly JF, Loboda A, Nebozhyn M, Fantin VR, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. 2009;27:2052–2058. doi: 10.1200/JCO.2008.19.0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Schrump DS, Fischette MR, Nguyen DM, Zhao M, Li X, Kunst TF, Hancox A, Hong JA, Chen GA, Kruchin E, et al. Clinical and molecular responses in lung cancer patients receiving Romidepsin. Clin Cancer Res. 2008;14:188–198. doi: 10.1158/1078-0432.CCR-07-0135. [DOI] [PubMed] [Google Scholar]
- 147.Otterson GA, Hodgson L, Pang H, Vokes EE. Cancer, Leukemia Group B: Phase II study of the histone deacetylase inhibitor Romidepsin in relapsed small cell lung cancer (Cancer and Leukemia Group B 30304) J Thorac Oncol. 2010;5:1644–1648. doi: 10.1097/JTO.0b013e3181ec1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Iwamoto FM, Lamborn KR, Kuhn JG, Wen PY, Yung WK, Gilbert MR, Chang SM, Lieberman FS, Prados MD, Fine HA. A phase I/II trial of the histone deacetylase inhibitor romidepsin for adults with recurrent malignant glioma: North American Brain Tumor Consortium Study 03–03. Neuro Oncol. 2011;13:509–516. doi: 10.1093/neuonc/nor017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Whitehead RP, Rankin C, Hoff PM, Gold PJ, Billingsley KG, Chapman RA, Wong L, Ward JH, Abbruzzese JL, Blanke CD. Phase II trial of romidepsin (NSC-630176) in previously treated colorectal cancer patients with advanced disease: a Southwest Oncology Group study (S0336) Invest New Drugs. 2009;27:469–475. doi: 10.1007/s10637-008-9190-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Molife LR, Attard G, Fong PC, Karavasilis V, Reid AH, Patterson S, Riggs CE, Jr, Higano C, Stadler WM, McCulloch W, et al. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC) Ann Oncol. 2010;21:109–113. doi: 10.1093/annonc/mdp270. [DOI] [PubMed] [Google Scholar]
- 151.Ahuja N, Easwaran H, Baylin SB. Harnessing the potential of epigenetic therapy to target solid tumors. J Clin Invest. 2014;124:56–63. doi: 10.1172/JCI69736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 154.Tarhini AA, Zahoor H, McLaughlin B, Gooding WE, Schmitz JC, Siegfried JM, Socinski MA, Argiris A. Phase I trial of carboplatin and etoposide in combination with panobinostat in patients with lung cancer. Anticancer Res. 2013;33:4475–4481. [PMC free article] [PubMed] [Google Scholar]
- 155.Cassier PA, Lefranc A, Amela EY, Chevreau C, Bui BN, Lecesne A, Ray-Coquard I, Chabaud S, Penel N, Berge Y, et al. A phase II trial of panobinostat in patients with advanced pretreated soft tissue sarcoma. A study from the French Sarcoma Group. Br J Cancer. 2013;109:909–914. doi: 10.1038/bjc.2013.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Witta SE, Jotte RM, Konduri K, Neubauer MA, Spira AI, Ruxer RL, Varella-Garcia M, Bunn PA, Jr, Hirsch FR. Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J Clin Oncol. 2012;30:2248–2255. doi: 10.1200/JCO.2011.38.9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, Cruickshank S, Miller KD, Lee MJ, Trepel JB. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. 2013;31:2128–2135. doi: 10.1200/JCO.2012.43.7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yeo W, Chung HC, Chan SL, Wang LZ, Lim R, Picus J, Boyer M, Mo FK, Koh J, Rha SY, et al. Epigenetic therapy using belinostat for patients with unresectable hepatocellular carcinoma: a multicenter phase I/II study with biomarker and pharmacokinetic analysis of tumors from patients in the mayo phase II consortium and the cancer therapeutics research group. J Clin Oncol. 2012;30:3361–3367. doi: 10.1200/JCO.2011.41.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Dizon DS, Blessing JA, Penson RT, Drake RD, Walker JL, Johnston CM, Disilvestro PA, Fader AN. a phase ii evaluation of belinostat and carboplatin in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal carcinoma: a gynecologic oncology group study. Gynecol Oncol. 2012;125:367–371. doi: 10.1016/j.ygyno.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zorzi AP, Bernstein M, Samson Y, Wall DA, Desai S, Nicksy D, Wainman N, Eisenhauer E, Baruchel S. A phase I study of histone deacetylase inhibitor, pracinostat (SB939), in pediatric patients with refractory solid tumors: IND203 a trial of the NCIC IND program/C17 pediatric phase I consortium. Pediatr Blood Cancer. 2013;60:1868–1874. doi: 10.1002/pbc.24694. [DOI] [PubMed] [Google Scholar]
- 161.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. Embo j. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Travaglini L, Vian L, Billi M, Grignani F, Nervi C. Epigenetic reprogramming of breast cancer cells by valproic acid occurs regardless of estrogen receptor status. Int J Biochem Cell Biol. 2009;41:225–234. doi: 10.1016/j.biocel.2008.08.019. [DOI] [PubMed] [Google Scholar]
- 164.Schech A, Kazi A, Yu S, Shah P, Sabnis G. Histone Deacetylase Inhibitor Entinostat Inhibits Tumor-Initiating Cells in Triple-Negative Breast Cancer Cells. Mol Cancer Ther. 2015;14:1848–1857. doi: 10.1158/1535-7163.MCT-14-0778. [DOI] [PubMed] [Google Scholar]
- 165.Song Y, Wu F, Wu J. Targeting histone methylation for cancer therapy: enzymes, inhibitors, biological activity and perspectives. J Hematol Oncol. 2016;9:49. doi: 10.1186/s13045-016-0279-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Popovic R, Licht JD. Emerging epigenetic targets and therapies in cancer medicine. Cancer Discov. 2012;2:405–413. doi: 10.1158/2159-8290.CD-12-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov. 2009;8:724–732. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- 168.Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol. 2007;14:1008–1016. doi: 10.1038/nsmb1337. [DOI] [PubMed] [Google Scholar]
- 169.Basavapathruni A, Jin L, Daigle SR, Majer CR, Therkelsen CA, Wigle TJ, Kuntz KW, Chesworth R, Pollock RM, Scott MP, et al. Conformational adaptation drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L. Chem Biol Drug Des. 2012;80:971–980. doi: 10.1111/cbdd.12050. [DOI] [PubMed] [Google Scholar]
- 170.Liu Y, Liu K, Qin S, Xu C, Min J. Epigenetic targets and drug discovery: part 1: histone methylation. Pharmacol Ther. 2014;143:275–294. doi: 10.1016/j.pharmthera.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 171.Liu K, Liu Y, Lau JL, Min J. Epigenetic targets and drug discovery Part 2: Histone demethylation and DNA methylation. Pharmacol Ther. 2015;151:121–140. doi: 10.1016/j.pharmthera.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 172.McGrath J, Trojer P. Targeting histone lysine methylation in cancer. Pharmacol Ther. 2015;150:1–22. doi: 10.1016/j.pharmthera.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 173.Copeland RA, Moyer MP, Richon VM. Targeting genetic alterations in protein methyltransferases for personalized cancer therapeutics. Oncogene. 2013;32:939–946. doi: 10.1038/onc.2012.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008;28:2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mueller D, Bach C, Zeisig D, Garcia-Cuellar MP, Monroe S, Sreekumar A, Zhou R, Nesvizhskii A, Chinnaiyan A, Hess JL, Slany RK. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood. 2007;110:4445–4454. doi: 10.1182/blood-2007-05-090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet. 2007;16:92–106. doi: 10.1093/hmg/ddl444. [DOI] [PubMed] [Google Scholar]
- 178.Zhang W, Xia X, Reisenauer MR, Hemenway CS, Kone BC. Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCalpha in an aldosterone-sensitive manner. J Biol Chem. 2006;281:18059–18068. doi: 10.1074/jbc.M601903200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, Zhang Y. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 180.Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, Xia X, Jesneck J, Bracken AP, Silverman LB, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14:355–368. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, Allain CJ, Klaus CR, Raimondi A, Scott MP, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122:1017–1025. doi: 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 184.Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O’Connor MB, Kingston RE, Simon JA. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/S0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 185.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wang X, Zhao H, Lv L, Bao L, Wang X, Han S. Prognostic Significance of EZH2 Expression in Non-Small Cell Lung Cancer: A Meta-analysis. Sci Rep. 2016;6:19239. doi: 10.1038/srep19239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647:21–29. doi: 10.1016/j.mrfmmm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 189.Li Z, Wang Y, Qiu J, Li Q, Yuan C, Zhang W, Wang D, Ye J, Jiang H, Yang J, Cheng J. The polycomb group protein EZH2 is a novel therapeutic target in tongue cancer. Oncotarget. 2013;4:2532–2549. doi: 10.18632/oncotarget.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 191.Weikert S, Christoph F, Kollermann J, Muller M, Schrader M, Miller K, Krause H. Expression levels of the EZH2 polycomb transcriptional repressor correlate with aggressiveness and invasive potential of bladder carcinomas. Int J Mol Med. 2005;16:349–353. [PubMed] [Google Scholar]
- 192.Shi J, Wang E, Zuber J, Rappaport A, Taylor M, Johns C, Lowe SW, Vakoc CR. The Polycomb complex PRC2 supports aberrant self-renewal in a mouse model of MLL-AF9;Nras(G12D) acute myeloid leukemia. Oncogene. 2013;32:930–938. doi: 10.1038/onc.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Suva ML, Riggi N, Janiszewska M, Radovanovic I, Provero P, Stehle JC, Baumer K, Le Bitoux MA, Marino D, Cironi L, et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. 2009;69:9211–9218. doi: 10.1158/0008-5472.CAN-09-1622. [DOI] [PubMed] [Google Scholar]
- 194.Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;20:1123–1136. doi: 10.1101/gad.381706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, Marquez VE, Jones PA. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8:1579–1588. doi: 10.1158/1535-7163.MCT-09-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110:7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014;13:842–854. doi: 10.1158/1535-7163.MCT-13-0773. [DOI] [PubMed] [Google Scholar]
- 198.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, 3rd, Diaz E, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 199.Chen YT, Zhu F, Lin WR, Ying RB, Yang YP, Zeng LH. The novel EZH2 inhibitor, GSK126, suppresses cell migration and angiogenesis via down-regulating VEGF-A. Cancer Chemother Pharmacol. 2016;77:757–765. doi: 10.1007/s00280-016-2990-1. [DOI] [PubMed] [Google Scholar]
- 200.Van Aller GS, Pappalardi MB, Ott HM, Diaz E, Brandt M, Schwartz BJ, Miller WH, Dhanak D, McCabe MT, Verma SK, et al. Long residence time inhibition of EZH2 in activated polycomb repressive complex 2. ACS Chem Biol. 2014;9:622–629. doi: 10.1021/cb4008748. [DOI] [PubMed] [Google Scholar]
- 201.Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, Shinkai Y. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002;16:1779–1791. doi: 10.1101/gad.989402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Huang J, Dorsey J, Chuikov S, Perez-Burgos L, Zhang X, Jenuwein T, Reinberg D, Berger SL. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J Biol Chem. 2010;285:9636–9641. doi: 10.1074/jbc.M109.062588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kondo Y, Shen L, Ahmed S, Boumber Y, Sekido Y, Haddad BR, Issa JP. Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS One. 2008;3:e2037. doi: 10.1371/journal.pone.0002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kondo Y, Shen L, Suzuki S, Kurokawa T, Masuko K, Tanaka Y, Kato H, Mizuno Y, Yokoe M, Sugauchi F, et al. Alterations of DNA methylation and histone modifications contribute to gene silencing in hepatocellular carcinomas. Hepatol Res. 2007;37:974–983. doi: 10.1111/j.1872-034X.2007.00141.x. [DOI] [PubMed] [Google Scholar]
- 205.Chen MW, Hua KT, Kao HJ, Chi CC, Wei LH, Johansson G, Shiah SG, Chen PS, Jeng YM, Cheng TY, et al. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 2010;70:7830–7840. doi: 10.1158/0008-5472.CAN-10-0833. [DOI] [PubMed] [Google Scholar]
- 206.Cho HS, Kelly JD, Hayami S, Toyokawa G, Takawa M, Yoshimatsu M, Tsunoda T, Field HI, Neal DE, Ponder BA, et al. Enhanced expression of EHMT2 is involved in the proliferation of cancer cells through negative regulation of SIAH1. Neoplasia. 2011;13:676–684. doi: 10.1593/neo.11512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hua KT, Wang MY, Chen MW, Wei LH, Chen CK, Ko CH, Jeng YM, Sung PL, Jan YH, Hsiao M, et al. The H3K9 methyltransferase G9a is a marker of aggressive ovarian cancer that promotes peritoneal metastasis. Mol Cancer. 2014;13:189. doi: 10.1186/1476-4598-13-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Bai K, Cao Y, Huang C, Chen J, Zhang X, Jiang Y. Association of Histone Methyltransferase G9a and Overall Survival After Liver Resection of Patients With Hepatocellular Carcinoma With a Median Observation of 40 Months. Medicine (Baltimore) 2016;95:e2493. doi: 10.1097/MD.0000000000002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kubicek S, O’Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, Rea S, Mechtler K, Kowalski JA, Homon CA, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 210.Trojer P, Zhang J, Yonezawa M, Schmidt A, Zheng H, Jenuwein T, Reinberg D. Dynamic Histone H1 Isotype 4 Methylation and Demethylation by Histone Lysine Methyltransferase G9a/KMT1C and the Jumonji Domain-containing JMJD2/KDM4 Proteins. J Biol Chem. 2009;284:8395–8405. doi: 10.1074/jbc.M807818200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Maze I, Covington HE, 3rd, Dietz DM, LaPlant Q, Renthal W, Russo SJ, Mechanic M, Mouzon E, Neve RL, Haggarty SJ, et al. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 2010;327:213–216. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kim Y, Kim YS, Kim DE, Lee JS, Song JH, Kim HG, Cho DH, Jeong SY, Jin DH, Jang SJ, et al. BIX-01294 induces autophagy-associated cell death via EHMT2/G9a dysfunction and intracellular reactive oxygen species production. Autophagy. 2013;9:2126–2139. doi: 10.4161/auto.26308. [DOI] [PubMed] [Google Scholar]
- 213.Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, Wigle TJ, Dimaggio PA, Wasney GA, Siarheyeva A, et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol. 2011;7:566–574. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Liu F, Barsyte-Lovejoy D, Li F, Xiong Y, Korboukh V, Huang XP, Allali-Hassani A, Janzen WP, Roth BL, Frye SV, et al. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J Med Chem. 2013;56:8931–8942. doi: 10.1021/jm401480r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Lakshmikuttyamma A, Scott SA, DeCoteau JF, Geyer CR. Reexpression of epigenetically silenced AML tumor suppressor genes by SUV39H1 inhibition. Oncogene. 2010;29:576–588. doi: 10.1038/onc.2009.361. [DOI] [PubMed] [Google Scholar]
- 216.Devin J, Viziteu E, Herviou L, Seckinger A, Camille G, Goldschmidt H, Vincent L, Pasero P, Hose D, Klein B, Moreaux J. Inhibition of SUV39H Methyltransferase As a Potent Therapeutic Target in Multiple Myeloma. Blood. 2015;126:1771. [Google Scholar]
- 217.Lai YS, Chen JY, Tsai HJ, Chen TY, Hung WC. The SUV39H1 inhibitor chaetocin induces differentiation and shows synergistic cytotoxicity with other epigenetic drugs in acute myeloid leukemia cells. Blood Cancer J. 2015;5:e313. doi: 10.1038/bcj.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Chiba T, Saito T, Yuki K, Zen Y, Koide S, Kanogawa N, Motoyama T, Ogasawara S, Suzuki E, Ooka Y, et al. Histone lysine methyltransferase SUV39H1 is a potent target for epigenetic therapy of hepatocellular carcinoma. Int J Cancer. 2015;136:289–298. doi: 10.1002/ijc.28985. [DOI] [PubMed] [Google Scholar]
- 219.Dimitrova E, Turberfield AH, Klose RJ. Histone demethylases in chromatin biology and beyond. EMBO Rep. 2015;16:1620–1639. doi: 10.15252/embr.201541113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hojfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov. 2013;12:917–930. doi: 10.1038/nrd4154. [DOI] [PubMed] [Google Scholar]
- 221.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11:384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 222.Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 223.Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, Ciceri F, Blaser JG, Greystoke BF, Jordan AM, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21:473–487. doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 224.Konovalov S, Garcia-Bassets I. Analysis of the levels of lysine-specific demethylase 1 (LSD1) mRNA in human ovarian tumors and the effects of chemical LSD1 inhibitors in ovarian cancer cell lines. J Ovarian Res. 2013;6:75. doi: 10.1186/1757-2215-6-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Lynch JT, Harris WJ, Somervaille TC. LSD1 inhibition: a therapeutic strategy in cancer? Expert Opin Ther Targets. 2012;16:1239–1249. doi: 10.1517/14728222.2012.722206. [DOI] [PubMed] [Google Scholar]
- 226.Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, Klein HU, Popescu AC, Burnett A, Mills K, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012;18:605–611. doi: 10.1038/nm.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Maes T, Tirapu I, Mascaró C, Ortega A, Estiarte A, Valls N, Castro-Palomino J, Arjol CB, Kurz G. Preclinical characterization of a potent and selective inhibitor of the histone demethylase KDM1A for MLL leukemia. J Clin Oncol. 2013;31:e13543. [Google Scholar]
- 228.Mohammad H, Smitheman K, Cusan M, Liu Y, Pappalardi M, Federowicz K, Van Aller G, Kasparec J, Tian X, Suarez D, et al. Inhibition Of LSD1 As a Therapeutic Strategy For The Treatment Of Acute Myeloid Leukemia. Blood. 2013;122:3964. doi: 10.1182/blood-2013-05-506212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Culhane JC, Cole PA. LSD1 and the chemistry of histone demethylation. Curr Opin Chem Biol. 2007;11:561–568. doi: 10.1016/j.cbpa.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Sprussel A, Schulte JH, Weber S, Necke M, Handschke K, Thor T, Pajtler KW, Schramm A, Konig K, Diehl L, et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia. 2012;26:2039–2051. doi: 10.1038/leu.2012.157. [DOI] [PubMed] [Google Scholar]
- 231.Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, Schneck JL, Carson JD, Liu Y, Butticello M, et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell. 2015;28:57–69. doi: 10.1016/j.ccell.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 232.Lan F, Bayliss PE, Rinn JL, Whetstine JR, Wang JK, Chen S, Iwase S, Alpatov R, Issaeva I, Canaani E, et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 2007;449:689–694. doi: 10.1038/nature06192. [DOI] [PubMed] [Google Scholar]
- 233.Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449:731–734. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- 234.Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, Shiekhattar R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318:447–450. doi: 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- 235.Issaeva I, Zonis Y, Rozovskaia T, Orlovsky K, Croce CM, Nakamura T, Mazo A, Eisenbach L, Canaani E. Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol Cell Biol. 2007;27:1889–1903. doi: 10.1128/MCB.01506-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Cho YW, Hong T, Hong S, Guo H, Yu H, Kim D, Guszczynski T, Dressler GR, Copeland TD, Kalkum M, Ge K. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem. 2007;282:20395–20406. doi: 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Miller SA, Mohn SE, Weinmann AS. Jmjd3 and UTX play a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression. Mol Cell. 2010;40:594–605. doi: 10.1016/j.molcel.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Fu LL, Tian M, Li X, Li JJ, Huang J, Ouyang L, Zhang Y, Liu B. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget. 2015;6:5501–5516. doi: 10.18632/oncotarget.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Feng Q, Zhang Z, Shea MJ, Creighton CJ, Coarfa C, Hilsenbeck SG, Lanz R, He B, Wang L, Fu X, et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014;24:809–819. doi: 10.1038/cr.2014.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Rajagopalan V, Vaidyanathan M, Janardhanam VA, Bradner JE. Pre-clinical analysis of changes in intra-cellular biochemistry of glioblastoma multiforme (GBM) cells due to c-Myc silencing. Cell Mol Neurobiol. 2014;34:1059–1069. doi: 10.1007/s10571-014-0083-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Gosmini R, Nguyen VL, Toum J, Simon C, Brusq JM, Krysa G, Mirguet O, Riou-Eymard AM, Boursier EV, Trottet L, et al. The discovery of I-BET726 (GSK1324726A), a potent tetrahydroquinoline ApoA1 up-regulator and selective BET bromodomain inhibitor. J Med Chem. 2014;57:8111–8131. doi: 10.1021/jm5010539. [DOI] [PubMed] [Google Scholar]
- 245.Garnier JM, Sharp PP, Burns CJ. BET bromodomain inhibitors: a patent review. Expert Opin Ther Pat. 2014;24:185–199. doi: 10.1517/13543776.2014.859244. [DOI] [PubMed] [Google Scholar]
- 246.Wyce A, Degenhardt Y, Bai Y, Le B, Korenchuk S, Crouthame MC, McHugh CF, Vessella R, Creasy CL, Tummino PJ, Barbash O. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget. 2013;4:2419–2429. doi: 10.18632/oncotarget.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Wyce A, Ganji G, Smitheman KN, Chung CW, Korenchuk S, Bai Y, Barbash O, Le B, Craggs PD, McCabe MT, et al. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLoS One. 2013;8:e72967. doi: 10.1371/journal.pone.0072967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Herait PE, Berthon C, Thieblemont C, Raffoux E, Magarotto V, Stathis A, Thomas X, Leleu X, Gomez-Roca C, Odore E, et al. Abstract CT231: BET-bromodomain inhibitor OTX015 shows clinically meaningful activity at nontoxic doses: interim results of an ongoing phase I trial in hematologic malignancies. Cancer Res. 2014;74:CT231. doi: 10.1158/1538-7445.AM2014-CT231. [DOI] [Google Scholar]
- 249.Bonetti P, Ponzoni M, Tibiletti MG, Stathis A, Heirat P, Zucca E, Bertoni F. 528 The BRD-inhibitor OTX015 Shows Pre-clinical Activity in Diffuse Large B-cell Lymphoma (DLBCL). Eur J Cancer. 2012;48:163.
- 250.Chaidos A, Caputo V, Gouvedenou K, Liu B, Marigo I, Chaudhry MS, Rotolo A, Tough DF, Smithers NN, Bassil AK, et al. Potent antimyeloma activity of the novel bromodomain inhibitors I-BET151 and I-BET762. Blood. 2014;123:697–705. doi: 10.1182/blood-2013-01-478420. [DOI] [PubMed] [Google Scholar]
- 251.Pathania R, Ramachandran S, Mariappan G, Thakur P, Shi H, Choi J-H, Manicassamy S, Kolhe R, Prasad PD, Sharma S, et al. Combined inhibition of DNMT and HDAC blocks the tumorigenicity of cancer stem-like cells and attenuates mammary tumor growth. Cancer Res. 2016;76:3224–3235. doi: 10.1158/0008-5472.CAN-15-2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, Sebree R, Rodgers K, Hooker CM, Franco N, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1:598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ecke I, Petry F, Rosenberger A, Tauber S, Monkemeyer S, Hess I, Dullin C, Kimmina S, Pirngruber J, Johnsen SA, et al. Antitumor effects of a combined 5-aza-2’deoxycytidine and valproic acid treatment on rhabdomyosarcoma and medulloblastoma in Ptch mutant mice. Cancer Res. 2009;69:887–895. doi: 10.1158/0008-5472.CAN-08-0946. [DOI] [PubMed] [Google Scholar]
- 254.Curry E, Green I, Chapman-Rothe N, Shamsaei E, Kandil S, Cherblanc FL, Payne L, Bell E, Ganesh T, Srimongkolpithak N, et al. Dual EZH2 and EHMT2 histone methyltransferase inhibition increases biological efficacy in breast cancer cells. Clin Epigenetics. 2015;7:84. doi: 10.1186/s13148-015-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Klaus CR, Iwanowicz D, Johnston D, Campbell CA, Smith JJ, Moyer MP, Copeland RA, Olhava EJ, Scott MP, Pollock RM, et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J Pharmacol Exp Ther. 2014;350:646–656. doi: 10.1124/jpet.114.214577. [DOI] [PubMed] [Google Scholar]
- 256.Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54:716–727. doi: 10.1016/j.molcel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Vatapalli RJ, Guzzetta AA, Fu T, Syed LH, Kwak R, Ahuja N. Improving Sensitivity to Irinotecan Using 5-Azacytidine in Colon Cancer Cell Lines. J Surgical Res. 2013;179:251–252.
- 258.Fu S, Hu W, Iyer R, Kavanagh JJ, Coleman RL, Levenback CF, Sood AK, Wolf JK, Gershenson DM, Markman M, et al. Phase 1b-2a study to reverse platinum resistance through use of a hypomethylating agent, azacitidine, in patients with platinum-resistant or platinum-refractory epithelial ovarian cancer. Cancer. 2011;117:1661–1669. doi: 10.1002/cncr.25701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Appleton K, Mackay HJ, Judson I, Plumb JA, McCormick C, Strathdee G, Lee C, Barrett S, Reade S, Jadayel D, et al. Phase I and pharmacodynamic trial of the DNA methyltransferase inhibitor decitabine and carboplatin in solid tumors. J Clin Oncol. 2007;25:4603–4609. doi: 10.1200/JCO.2007.10.8688. [DOI] [PubMed] [Google Scholar]
- 260.Matei D, Fang F, Shen C, Schilder J, Arnold A, Zeng Y, Berry WA, Huang T, Nephew KP. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res. 2012;72:2197–2205. doi: 10.1158/0008-5472.CAN-11-3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Ramaswamy B, Fiskus W, Cohen B, Pellegrino C, Hershman DL, Chuang E, Luu T, Somlo G, Goetz M, Swaby R, et al. Phase I-II study of vorinostat plus paclitaxel and bevacizumab in metastatic breast cancer: evidence for vorinostat-induced tubulin acetylation and Hsp90 inhibition in vivo. Breast Cancer Res Treat. 2012;132:1063–1072. doi: 10.1007/s10549-011-1928-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Li SY, Sun R, Wang HX, Shen S, Liu Y, Du XJ, Zhu YH, Jun W. Combination therapy with epigenetic-targeted and chemotherapeutic drugs delivered by nanoparticles to enhance the chemotherapy response and overcome resistance by breast cancer stem cells. J Control Release. 2015;205:7–14. doi: 10.1016/j.jconrel.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 263.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Blank C, Gajewski TF, Mackensen A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol Immunother. 2005;54:307–314. doi: 10.1007/s00262-004-0593-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Pan Q, Li Q, Liu S, Ning N, Zhang X, Xu Y, Chang AE, Wicha MS. Concise Review: Targeting Cancer Stem Cells Using Immunologic Approaches. Stem Cells. 2015;33:2085–2092. doi: 10.1002/stem.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Covre A, Coral S, Nicolay H, Parisi G, Fazio C, Colizzi F, Fratta E, Di Giacomo AM, Sigalotti L, Natali PG, Maio M. Antitumor activity of epigenetic immunomodulation combined with CTLA-4 blockade in syngeneic mouse models. Oncoimmunology. 2015;4:e1019978. doi: 10.1080/2162402X.2015.1019978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, Diaz LA, Jr, Papadopoulos N, Kinzler KW, Vogelstein B, Zhou S. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. 2014;111:11774–11779. doi: 10.1073/pnas.1410626111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Qifeng S, Bo C, Xingtao J, Chuanliang P, Xiaogang Z. Methylation of the promoter of human leukocyte antigen class I in human esophageal squamous cell carcinoma and its histopathological characteristics. J Thorac Cardiovasc Surg. 2011;141:808–814. doi: 10.1016/j.jtcvs.2010.04.031. [DOI] [PubMed] [Google Scholar]
- 272.Ye Q, Shen Y, Wang X, Yang J, Miao F, Shen C, Zhang J. Hypermethylation of HLA class I gene is associated with HLA class I down-regulation in human gastric cancer. Tissue Antigens. 2010;75:30–39. doi: 10.1111/j.1399-0039.2009.01390.x. [DOI] [PubMed] [Google Scholar]
- 273.Kitamura H, Torigoe T, Asanuma H, Honma I, Sato N, Tsukamoto T. Down-regulation of HLA class I antigens in prostate cancer tissues and up-regulation by histone deacetylase inhibition. J Urol. 2007;178:692–696. doi: 10.1016/j.juro.2007.03.109. [DOI] [PubMed] [Google Scholar]
- 274.Magner WJ, Kazim AL, Stewart C, Romano MA, Catalano G, Grande C, Keiser N, Santaniello F, Tomasi TB. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol. 2000;165:7017–7024. doi: 10.4049/jimmunol.165.12.7017. [DOI] [PubMed] [Google Scholar]
- 275.Mora-Garcia Mde L, Duenas-Gonzalez A, Hernandez-Montes J, De la Cruz-Hernandez E, Perez-Cardenas E, Weiss-Steider B, Santiago-Osorio E, Ortiz-Navarrete VF, Rosales VH, Cantu D, et al. Up-regulation of HLA class-I antigen expression and antigen-specific CTL response in cervical cancer cells by the demethylating agent hydralazine and the histone deacetylase inhibitor valproic acid. J Transl Med. 2006;4:55. doi: 10.1186/1479-5876-4-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R, Topper MJ, Luo J, Connolly RM, Azad NS, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014;5:587–598. doi: 10.18632/oncotarget.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours--lessons from the past. Nat Rev Clin Oncol. 2013;10:256–266. doi: 10.1038/nrclinonc.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Silverman LR, Fenaux P, Mufti GJ, Santini V, Hellstrom-Lindberg E, Gattermann N, Sanz G, List AF, Gore SD, Seymour JF. Continued azacitidine therapy beyond time of first response improves quality of response in patients with higher-risk myelodysplastic syndromes. Cancer. 2011;117:2697–2702. doi: 10.1002/cncr.25774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Griffiths EA, Choy G, Redkar S, Taverna P, Azab M, Karpf AR. SGI-110: DNA Methyltransferase Inhibitor Oncolytic. Drugs Future. 2013;38:535–543. [PMC free article] [PubMed] [Google Scholar]
- 280.Issa JP, Roboz G, Rizzieri D, Jabbour E, Stock W, O’Connell C, Yee K, Tibes R, Griffiths EA, Walsh K, et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015;16:1099–1110. doi: 10.1016/S1470-2045(15)00038-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Fang F, Munck J, Tang J, Taverna P, Wang Y, Miller DF, Pilrose J, Choy G, Azab M, Pawelczak KS, et al. The novel, small-molecule DNA methylation inhibitor SGI-110 as an ovarian cancer chemosensitizer. Clin Cancer Res. 2014;20:6504–6516. doi: 10.1158/1078-0432.CCR-14-1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Younes A, Sureda A, Ben-Yehuda D, Zinzani PL, Ong TC, Prince HM, Harrison SJ, Kirschbaum M, Johnston P, Gallagher J, et al. Panobinostat in patients with relapsed/refractory Hodgkin’s lymphoma after autologous stem-cell transplantation: results of a phase II study. J Clin Oncol. 2012;30:2197–2203. doi: 10.1200/JCO.2011.38.1350. [DOI] [PubMed] [Google Scholar]
- 283.Duvic M, Dummer R, Becker JC, Poulalhon N, Ortiz Romero P, Grazia Bernengo M, Lebbe C, Assaf C, Squier M, Williams D, et al. Panobinostat activity in both bexarotene-exposed and -naive patients with refractory cutaneous T-cell lymphoma: results of a phase II trial. Eur J Cancer. 2013;49:386–394. doi: 10.1016/j.ejca.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 284.Batlevi CL, Kasamon Y, Bociek RG, Lee P, Gore L, Copeland A, Sorensen R, Ordentlich P, Cruickshank S, Kunkel L, et al. ENGAGE- 501: phase II study of entinostat (SNDX-275) in relapsed and refractory Hodgkin lymphoma. Haematologica. 2016;101:968–975. doi: 10.3324/haematol.2016.142406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Kirschbaum MH, Foon KA, Frankel P, Ruel C, Pulone B, Tuscano JM, Newman EM. A phase 2 study of belinostat (PXD101) in patients with relapsed or refractory acute myeloid leukemia or patients over the age of 60 with newly diagnosed acute myeloid leukemia: a California Cancer Consortium Study. Leuk Lymphoma. 2014;55:2301–2304. doi: 10.3109/10428194.2013.877134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.O’Connor OA, Horwitz S, Masszi T, Van Hoof A, Brown P, Doorduijn J, Hess G, Jurczak W, Knoblauch P, Chawla S, et al. Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J Clin Oncol. 2015;33:2492–2499. doi: 10.1200/JCO.2014.59.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Mackay HJ, Hirte H, Colgan T, Covens A, MacAlpine K, Grenci P, Wang L, Mason J, Pham PA, Tsao MS, et al. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. Eur J Cancer. 2010;46:1573–1579. doi: 10.1016/j.ejca.2010.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Chu QS, Nielsen TO, Alcindor T, Gupta A, Endo M, Goytain A, Xu H, Verma S, Tozer R, Knowling M, et al. A phase II study of SB939, a novel pan-histone deacetylase inhibitor, in patients with translocation-associated recurrent/metastatic sarcomas-NCIC-CTG IND 200dagger. Ann Oncol. 2015;26:973–981. doi: 10.1093/annonc/mdv033. [DOI] [PubMed] [Google Scholar]
- 289.Eigl BJ, North S, Winquist E, Finch D, Wood L, Sridhar SS, Powers J, Good J, Sharma M, Squire JA, et al. A phase II study of the HDAC inhibitor SB939 in patients with castration resistant prostate cancer: NCIC clinical trials group study IND195. Invest New Drugs. 2015;33:969–976. doi: 10.1007/s10637-015-0252-4. [DOI] [PubMed] [Google Scholar]
- 290.Rambaldi A, Dellacasa CM, Finazzi G, Carobbio A, Ferrari ML, Guglielmelli P, Gattoni E, Salmoiraghi S, Finazzi MC, Di Tollo S, et al. A pilot study of the Histone-Deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol. 2010;150:446–455. doi: 10.1111/j.1365-2141.2010.08266.x. [DOI] [PubMed] [Google Scholar]
- 291.Mawatari T, Ninomiya I, Inokuchi M, Harada S, Hayashi H, Oyama K, Makino I, Nakagawara H, Miyashita T, Tajima H, et al. Valproic acid inhibits proliferation of HER2-expressing breast cancer cells by inducing cell cycle arrest and apoptosis through Hsp70 acetylation. Int J Oncol. 2015;47:2073–2081. doi: 10.3892/ijo.2015.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, Karuturi RK, Tan PB, Liu ET, Yu Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–1063. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Crea F, Hurt EM, Mathews LA, Cabarcas SM, Sun L, Marquez VE, Danesi R, Farrar WL. Pharmacologic disruption of Polycomb Repressive Complex 2 inhibits tumorigenicity and tumor progression in prostate cancer. Mol Cancer. 2011;10:40. doi: 10.1186/1476-4598-10-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Bradley WD, Arora S, Busby J, Balasubramanian S, Gehling VS, Nasveschuk CG, Vaswani RG, Yuan CC, Hatton C, Zhao F, et al. EZH2 inhibitor efficacy in non-Hodgkin’s lymphoma does not require suppression of H3K27 monomethylation. Chem Biol. 2014;21:1463–1475. doi: 10.1016/j.chembiol.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 295.Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488:404–408. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ., 3rd Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777–790. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Long J, Li B, Rodriguez-Blanco J, Pastori C, Volmar CH, Wahlestedt C, Capobianco A, Bai F, Pei XH, Ayad NG, Robbins DJ. The BET bromodomain inhibitor I-BET151 acts downstream of smoothened protein to abrogate the growth of hedgehog protein-driven cancers. J Biol Chem. 2014;289:35494–35502. doi: 10.1074/jbc.M114.595348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Moros A, Rodriguez V, Saborit-Villarroya I, Montraveta A, Balsas P, Sandy P, Martinez A, Wiestner A, Normant E, Campo E, et al. Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia. 2014;28:2049–2059. doi: 10.1038/leu.2014.106. [DOI] [PubMed] [Google Scholar]
- 301.Albrecht BK, Gehling VS, Hewitt MC, Vaswani RG, Cote A, Leblanc Y, Nasveschuk CG, Bellon S, Bergeron L, Campbell R, et al. Identification of a Benzoisoxazoloazepine Inhibitor (CPI-0610) of the Bromodomain and Extra-Terminal (BET) Family as a Candidate for Human Clinical Trials. J Med Chem. 2016;59:1330–1339. doi: 10.1021/acs.jmedchem.5b01882. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.