

Abstract
Tumour progression is dependent on the interaction between tumour cells and cells of the surrounding microenvironment. The tumour is a dynamic milieu consisting of various cell types such as endothelial cells, fibroblasts, cells of the immune system and mesenchymal stem cells (MSCs). MSCs are multipotent stromal cells that are known to reside in various areas such as the bone marrow, fat and dental pulp. MSCs have been found to migrate towards inflammatory sites and studies have shown that they also migrate towards and incorporate into the tumour. The key question is how they interact there. MSCs may interact with tumour cells through paracrine signalling. On the other hand, MSCs have the capacity to differentiate to various cell types such as osteocytes, chondrocytes and adipocytes and it is possible that MSCs differentiate at the site of the tumour. More recently it has been shown that cross-talk between tumour cells and MSCs has been shown to increase metastatic potential and promote epithelial-to-mesenchymal transition. This review will focus on the role of MSCs in tumour development at various stages of progression from growth of the primary tumour to the establishment of distant metastasis.
Keywords: Mesenchymal stem cells, Tumour microenvironment, Cancer progression, Tumour metastasis, Tumour stroma
Background
It is now understood that tumour cells do not act alone. Cancer cells interact with their surrounding stroma and these interactions lead to an ‘activated state’ resulting in increased release of pro-inflammatory cytokines and growth factors [1]. The tumour is in a chronic state of inflammation and has been described as a ‘wound that never heals’ [2]. This inflammatory state drives the recruitment of responsive cell types such as macrophages, myeloid derived suppressor cells and mesenchymal stem cells (MSCs) [3–5]. Cross-talk between cancer cells and cells of the surrounding stroma promotes tumour progression and creates a dynamic extracellular matrix, favourable for the invasive tumour cell [6, 7].
The tumour stroma varies between each cancer type and the heterogeneous nature of the tumour makes it complicated to study. It is important to develop an understanding of what drives non-cancerous cells toward an activated state, what that activated state is and what it subsequently means for tumour cell progression.
MSCs are multipotent stem cells originally found to have the capacity to differentiate into the tri-lineages - osteoblasts, chondrocytes and adipocytes [8]. They are generally characterised by their tri-lineage differentiation capacity and by positivity for surface markers CD73, CD105 and CD90 [9]. More recent developments have revealed a wider range in differentiation potential such as differentiation to myocytes and neurons [10, 11]. They can be sourced from the bone marrow, adipose tissue and dental pulp [8, 12–14]. They are also found in circulation and are known to home to inflammatory sites [15]. Due to their capacity to home to injured tissue, research has suggested a reparative function for MSCs in multiple tissues including the lung [16], liver [17], brain [18] and heart [19].
MSCs reside in the bone marrow stroma alongside haematopoietic stem cells (HSCs), osteoblasts, osteoclasts, adipocytes, endothelial cells (ECs) and monocytes [20, 21]. MSCs may play a supportive role for HSCs and have previously been used to enhance long-term HSC engraftment in human transplantation [22, 23].
Knowledge of these characteristics as well as their differentiation capacity has caused excitement in the field of regenerative medicine and use of MSCs has potential for therapeutics in a range of fields such as cardiology, immunology and neurology. However, in the field of cancer research many studies suggest that MSC activity may contribute to poorer outcomes [24–27].
In recent studies, it has been shown that MSCs can also home to tumour sites and contribute to tumour growth and progression [26–29]. Analysis from human prostatectomies showed that MSCs represented 0.01–1.1% of total cells present in the prostate tumour [30]. MSCs have been found to increase the metastatic potential of tumour cells by promoting their motility and invasiveness as well as having a role in the creation of a metastatic niche at the secondary site [26, 31–33].
Main text
Mesenchymal stem cells at the primary tumour site
MSCs have been implicated in the promotion of tumour growth in numerous cancer types such as follicular lymphoma [24], head and neck carcinoma [25], glioma [34], breast [26], gastric [35], colon [36] and prostate cancer [27].
Karnoub and colleagues showed that co-injection of human bone marrow MSCs with only one of four breast cancer cell lines (MCF7) into mice led to accelerated tumour growth, however, co-injection with all cell lines (MDA-MB-231, HMLR, MDA-MB-435 and MCF7) led to increased metastasis [26]. Similarly, in a more recent study it was found that co-injection of human bone marrow MSCs with the triple negative inflammatory breast cancer cell line, SUM149, resulted in inhibited primary tumour growth but increased invasion and metastasis in mice [37]. These findings indicate a role for MSCs at the tumour site in the promotion of metastasis possibly through the induction of epithelial-to-mesenchymal transition (EMT) in primary tumour cells.
An increase in tumour growth was also found in mice following co-injection of human adipose tissue derived MSCs with the prostate cancer cell line MDA-PCa-118b [27]. In another study bone marrow MSCs were also found to stimulate the proliferation, migration and invasion of the prostate cancer cell line PC3 in vitro (see Fig. 1). This effect was inhibited by blocking transforming growth factor β (TGFβ) [38]. A similar study showed that TGFβ immunodepletion from oncostatin M treated human adipose tissue derived MSC conditioned media reduced the adhesion capacity of PC3 cells in vitro [39]. Like many growth factors and cytokines TGFβ plays a dual role in cancer. TGFβ can have a suppressive effect during the early initiating steps of carcinogenesis, acting as a tumour suppressor inhibiting cell proliferation, while in later stages it can induce epithelial to mesenchymal transition promoting the development of metastatic disease [40]. Of particular note is the dependency on stromal derived TGFβ for colorectal cancer metastasis initiation [41], and the association of stromal TGFβ expression with breast cancer outcome [42].
Fig. 1.
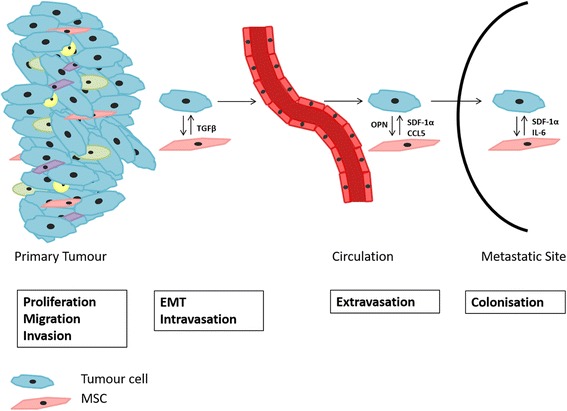
MSC and tumour cell interaction in cancer progression. MSCs have been shown to interact with tumour cells at the primary site and during metastatic colonisation in a manner that promotes cancer progression. MSCs have been shown to promote EMT in tumour cells through direct cell-cell contact, which could in part be due to TGFβ secretion [38, 82]. Additionally, tumour cell secretion of osteopontin (OPN) was found to induce MSC secretion of chemokine (C-C motif) ligand 5 (CCL5) stimulating breast cancer cell metastasis through interaction with the C-C chemokine receptor type 5 (CCR5) receptor [84]. Tumour cell migration towards and entry into the bone marrow metastatic site was shown to be mediated by stromal cell-derived factor 1 (SDF-1α) – a factor secreted by bone marrow MSCs – interaction with the C-X-C chemokine receptor type 4 (CXCR4) receptor expressed on breast and prostate tumour cells [33, 102, 103]
Some research groups have investigated the tumour promoting function of MSCs isolated from the tumour, arguably a more realistic approach to understanding the role of MSCs within the tumour microenvironment. Co-injection of MSCs isolated from human head and neck carcinoma [43], gastric cancer [25] and gliomas [34] with tumour cells into mouse models resulted in an increase in tumour growth and progression. Interestingly, Li and colleagues found that MSCs isolated from human gastric cancer tumours stimulated increased proliferation and migration of gastric cancer cell lines (BGC-823 and MKN-28) in vitro in comparison to bone marrow derived MSCs or MSCs isolated from non-cancerous adjacent tissue. They also found that they secreted more vascular endothelial growth factor (VEGF), macrophage inflammatory protein-2, TGF-β1, and the pro-inflammatory cytokines interleukin (IL)-6, and IL-8 while blockade of IL-8 attenuated the tumour promoting function of the gastric cancer MSCs [35].
From the studies described thus far, we can ascertain that MSCs are important players in the promotion of tumour growth and progression. Key thoughts to consider at this point would be whether naïve MSCs can induce such an effect upon arrival at the tumour through paracrine signalling and cell-cell contact, or do tumour microenvironment exposed MSCs transition to a determined ‘activated’ or reprogrammed state. The studies discussed above describing MSCs directly isolated from the tumour give evidence to the latter whereby tumour derived MSCs differed in activity to naïve bone marrow derived MSCs. Taking this into account, future studies should consider further investigation into the functional and molecular differences that occur in MSCs isolated from various tumour types. Are they functionally, morphologically and molecularly the same or does it depend on the tumour source?
Role in tumour suppression
In contrast to the research described above there is evidence to suggest that MSCs can also have an inhibitory effect on tumour growth. Suppression of tumour growth has been noted in breast cancer [44], Kaposi’s sarcoma [45], hepatoma [46] and melanoma [47] models. Human MSCs derived from the umbilical cord and adipose tissue were implanted into a breast cancer metastasis mouse model and found to inhibit metastasis to the lung and reduce tumour growth through poly (ADP-ribose) polymerase (PARP) and caspase-3 cleavage, which could in turn induce apoptosis [44]. However, MSCs derived from the bone marrow, adipose tissue and dental pulp are not functionally identical, therefore the studies using MSCs derived from other sources may not be replicated using bone marrow derived MSCs [48, 49].
MSCs are a heterogeneous population of cells containing subpopulations with differing differentiation capacities [50]. Moreover, MSCs were found to express embryonic stem cell or pluripotency markers which differed depending on the source. Bone marrow derived MSCs were found to express Oct4, Nanog, alkaline phosphatase and SSEA-4; adipose and dermis derived MSCs were found to express Oct4, Nanog, SOX2, alkaline phosphatase and SSEA-4; while heart MSCs were found to express Oct4, Nanog, SOX2 and SSEA-4 [51]. It is therefore relevant to consider the source of MSCs and the techniques used to isolate and characterise them in each study. Table 1 highlights the experimental methods used to identify MSCs in key studies described in this review. There is an apparent discrepancy between studies in the techniques used to isolate the MSCs, where only a portion used gradient centrifugation to separate a population of MSCs. Moreover, each study uses a different set of criteria to characterise the isolated population. Though the predominant positive markers used are CD105 and CD90, there is no overall consistency in molecular or phenotypic characterisation of the MSCs used in each study. Differences in isolation techniques and growth conditions can favour certain subpopulations and future research in this area should place emphasis on the methods for isolation and characterisation for increased clarification on the population of stromal cells used experimentally.
Table 1.
Isolation techniques and methods of characterisation used in a selection of studies
| Manuscript | Origin | Species | Isolation Technique | Characterisation | Tumour of Relevance | Tumour Function |
|---|---|---|---|---|---|---|
| Karnoub et al., 2007 [26] | Bone marrow (hip) | Human | Histopaque density centrifugation, bFGF supplemented, adherent to plastic | CD105+, CD45−/GlyA− | Breast | Promoting |
| Lacerda et al., 2015 [37] | Bone marrow | Human | Purchased from EMD Millipore (Billerica, MA, USA) (Part #SCC034, Lot N61710996) | Markers unspecified. Osteogenic, adipogenic and chondrogenic differentiation capacity. | Breast | Promoting |
| Ye et al., 2012 [38] | Bone marrow (iliac crest) | Human | Percoll gradient centrifugation, adherence to plastic | CD105+, CD90+, CD44+, CD29+, CD166+, HLA-ABC+, CD34−, CD14−, CD45− and HLA-DR−. Osteogenic and adipogenic differentiation capacity | Prostate | Promoting |
| Lee et al., 2013 [39] | Adipose tissue | Human | Adherence to plastic | CD105+, CD90+, CD44+, CD29+, CD73+, CD34−, CD45− and CD31− | Prostate | Promoting |
| Sun et al., 2009 [44] | Umbilical cord | Human | Ficoll density gradient centrifugation, adherence to plastic | CD105+, CD90+, CD44+, CD29+, CD73+, CD34−, CD45− and HLA-DR−. | Breast | Suppressive |
| Sun et al., 2009 [44] | Adipose tissue (mammary fat) | Human | Adherence to plastic | Characterised in a previous study: CD105+, CD90+, CD29+, CD34−, CD14−, CD45−, HLA-DR−and CD133−. | Breast | Suppressive |
| Otsu et al., 2009 [47] | Bone marrow | Rat and mouse | Adherence to plastic | CD90+, CD44+, CD29+, CD59+, CD54+, CD11b−, CD45− | Melanoma | Suppressive when administered at a 3:1 ratio with ECs. |
| Spaeth et al., 2009 [61] | Bone marrow | Human | Adherence to plastic | CD105+, CD90+, CD44+, CD146+, CD140b+, CD166+, CD31−, CD34−and CD45−. Osteogenic, adipogenic and mineralised cell differentiation capacity |
Transition to CAF following exposure to ovarian cancer ‘SKOV-3’ cells | Promoting following transition to CAF |
| Mishra et al., 2008 [70] | Bone marrow | Human | Ficoll gradient centrifugation, adherence to plastic | CD105+, CD90+, CD44+, HLA-ABC+, Stro1+, CD11b−, CD45− and HLA-DR−. Osteogenic, adipogenic and myogenic differentiation capacity | Transition to CAF following exposure to breast cancer ‘MDA-MB-231’ cells | Promoting following transition to CAF |
| Shangguan et al., 2012 [65] | Bone marrow | Human | Obtained from IH-supported MSC Distribution center in Texas A&M Health Science Center | CD105+, CD90+, CD44+, CD29+, CD49c+, CD49f+, CD59+, CD166+, CD34−, CD36−, CD117− and CD45−. Osteogenic, adipogenic and chondrogenic differentiation capacity | TGF-β dependent transition to CAF following exposure to breast cancer ‘MDA-MB-231’ cells | Promoting following transition to CAF |
Otsu et al. showed that murine bone marrow MSCs had a cytotoxic effect on the tumour in a melanoma mouse model through the release of reactive oxygen species when in contact with ECs present at the capillaries. This induced apoptosis of the ECs and reduced tumour growth. However, the cytotoxic effect of the MSC was only observed when implanted at high concentrations. MSCs seeded onto EC derived capillaries in matrigel evoked a cytotoxic effect at a EC:MSC ratio of 1:1 or 1:3. Cytotoxicity decreased when the MSC number was reduced by an order of magnitude [47] and given that in prostate cancer MSCs were only found to represent 0.01–1.1% of the tumour experiments using a high ratio of MSCs may not be reflective of the tumour microenvironment in vivo [30]. These results may explain the difference in outcome observed in studies showing tumour growth promotion by MSCs. Further investigation on the effect of dose on efficacy is warranted for any conclusions to be made, nonetheless, when examining the impact of MSC on tumour biology, the source and specific ratios of MSC to tumour cells reflective of the natural tumour environment is an important consideration.
Another explanation for the contrasting results is that like macrophages there is a polarisation of MSCs in response to secreted factors from the tumour that either drives the cells toward a tumour promoting or suppressive function. Tumour infiltrating macrophages can become induced by the stromal microenvironment and are referred to as tumour associated macrophages (TAMs) [52, 53]. Depending on the stimuli, macrophages can be polarised toward an M1 or M2 phenotype. The M1 phenotype can be induced by interferon gamma (IFN-γ) and lipopolysaccharides and have been shown to have cytotoxic effects on tumour cells. In contrast M2 macrophages are induced by IL-4, IL-13 and IL-10, promote wound healing and angiogenesis and are phenotypically similar to TAMs [52, 54–56].
MSCs were previously found to express toll-like receptor (TLR)- 1, 2, 3, 4, 5 and 6 and TLR-agonist interaction stimulated MSC migration and immunomodulatory factor secretion [57]. In particular LPS stimulation of TLR4 and Poly-IC stimulation of TLR3 resulted in enhanced phospho-IKKα/β and phospho-MAPK indicting that activation of TLR4 or TLR3 may regulate NFkB and/or MAPK signalling in MSCs. In particular IL-6 and IL-8 were highly induced upon TLR4 activation [57]. Interestingly, Waterman and colleagues proposed a polarisation of MSCs based on TLR signalling. They found functional differences between human bone marrow derived MSCs stimulated by either TLR4 or TLR3 and classified them as MSC1 and MSC2 respectively [58]. MSC1 cells were found to have an anti-tumour effect while MSC2 cells promoted tumour growth and metastasis [59]. Given that increased expression of both TLR3 and TLR4 in breast tumour epithelium is associated with increased risk of disease recurrence [60], and taken in the context of their anti-tumoural and pro-tumoural effects in MSCs [59], it is clear that targeting TLRs for the treatment of cancer is complex and its benefits may be dependent on the specific polarisation of MSCs and immune cells in the tumour microenvironment, in addition to the TLR expression patterns within the tumour epithelia in each individual patient.
Cancer associated fibroblasts: origins and characteristics
Cancer associated fibroblasts (CAFs) are a heterogeneous population of fibroblast-like cells with a tumour promoting function. The heterogeneity may be due to varying cell origins and the molecular constitution of tumour stroma from which the cell fate is determined. CAFs have been found to originate from bone marrow MSCs, fibroblasts and by transdifferentiation of epithelial and endothelial cells [61–63]. The mechanisms by which the cells differentiate or become ‘activated’ are largely unknown, however, exposure to TGF-β has been shown to induce the phenotypic changes regardless of cell origin [63–66].
MSCs as an origin for CAFs
Evidence to suggest CAFs can be derived from MSCs was found in in vivo studies whereby genetically tagged bone marrow derived cells, injected into mice, were found at the tumour site with myofibroblast morphology and expressing α smooth muscle actin (α-SMA) and the α1 chain of type I (pro)collagen [67–69]. A subsequent study in a murine ovarian carcinoma xenograft model, found that bone marrow derived MSCs engrafted at the tumour expressed CAF markers fibroblast activation protein, fibroblast specific protein 1, α-SMA and tenascin C (TN-C) [61].
Further evidence to support the hypothesis that CAFs can originate from MSCs comes from in vitro studies where MSCs are cultured long-term in tumour cell conditioned medium. In a study by Mishra et al. human MSCs were cultured for up to 30 days in the breast cancer cell line (MDA-MB-231) conditioned medium [70]. The resulting MSCs expressed increased levels of α-SMA, fibroblast specific protein 1 (FSP-1), SDF-1α and vimentin and stimulated tumour cell growth in both in vitro and in vivo models [70]. Long-term culture of human MSCs for 12–16 days in conditioned medium taken from ovarian cancer cell line, SKOV-3, induced the expression of CAF markers in MSCs and elevated secretion of IL-6, leading to increased tumour cell proliferation [61]. Interestingly, TGF-β may be involved in the transition as human bone marrow MSCs transduced with a lentiviral vector which inhibited TGF-β/smad signalling, expressed a decrease in CAF markers when conditioned for 10 days in tumour cell conditioned medium in comparison to naïve MSCs [65]. Furthermore, treatment of MSCs with the endoplasmic reticulum chaperone, GRP78, activated TGF-β/smad signalling and induced the transition to a CAF like phenotype [71]. Taken together, it is clear that TGF-β plays a major role in the transition from MSC to CAF, however it is unclear to what degree it affects the secretory profile of the cells and their functional characteristics. It is also interesting to note that the MSCs used in each of these studies are positive for the MSC markers CD105, CD90 and CD44, which allows a more robust interpretation of the findings (see Table 1).
On the other hand, it must be noted that MSCs and CAFs share many similarities. A study has shown that CAFs share many of the same surface markers as MSCs such as CD29, CD44, CD73, CD90, CD106 and CD117, and have the capacity to differentiate to osteocytes, chondrocytes and adipocytes, and express vimentin [72]. An interesting suggestion, which is discussed in more detail in a recent review by Kalluri, describes the idea that fibroblasts are resting mesenchymal cells that can be activated to become MSCs in response to certain stimuli [73]. Nonetheless, CAFs were found to have an increased proliferative capacity and secrete increased VEGF, TGF-β, IL-4, IL-10 and tumour necrosis factor-α (TNF-α) compared to MSCs [72]. This provides credibility to another proposal by Kalluri that resting fibroblasts are in fact MSCs that can be stimulated to an activated state such as what is described as a CAF or a cancer-associated MSC [73]. It could also be suggested that CAFs originate from a subpopulation of MSCs, a finding which could explain some of the shared characteristics. It was suggested in a review by Augsten that the term CAFs should be used to describe a heterogeneous population of fibroblasts that originate from different sources, reside in various tumour types but are not assigned a specific function. This suggestion borrows from previous literature describing macrophage polarisation where an F1 subtype would be associated with tumour suppressive properties and an F2 subtype would describe fibroblasts with tumour promoting effects [74].
Mesenchymal stem cells and metastasis
MSCs interact with cancer cells at multiple stages of cancer progression. At the primary tumour MSCs have been shown to drive tumour cells toward an invasive, pro-metastatic state. Human MSCs injected alone into mice with mammary carcinoma xenografts resulted in a 42% occurrence of metastatic lesions, compared with 17% in the control treated mice [75]. Similarly, human MSCs injected systemically into mice were found to migrate to the stroma of primary colon tumours as well as metastatic liver tumours [76]. Furthermore, co-culture of human bone marrow MSCs with MDA-MB-231 or MDA-MB-435 breast cancer cell lines 48 hours preceding injection resulted in enhanced metastasis in a mouse orthotopic implantation model, whereas the MSCs had no effect on metastasis without prior co-culture [77].
Tracking of MSCs using magnetic resonance imaging in a mouse xenograft model has shown that MSCs were more likely to home to the lung metastatic site than to the primary tumour [78]. A study suggests that tumour cells do not always leave the primary site as single cells but also as ‘heterotypic tumour fragments’ consisting of the metastatic cancer cells along with tumour stromal cells [32]. These clusters of cells were found to migrate to the metastatic site and promote tumour growth. Moreover, CAFs were found to migrate from the primary tumour to the lung metastatic site in mice [32]. Additionally, a study by Kaplan and colleagues using mouse models found that VEGF receptor (VEGFR1) expressing bone marrow derived cells migrated to and formed clusters in pre-metastatic sites before the arrival of tumour cells. Interestingly, blocking VEGFR1 function prevented cluster formation and metastasis [79]. These studies indicate a potential role for bone marrow derived cells in the creation and possibly the maintenance of a metastatic niche.
Role in the promotion of EMT
The presence of MSCs in the tumour stroma may stimulate EMT of the cancer cells. Research has shown that direct co-culture of breast or gastric cancer cells with human bone marrow derived MSCs resulted in the upregulation of EMT markers N-cadherin, vimentin, Twist and Snail and the downregulation of E-cadherin [80, 81]. Correspondingly, it was found that human MSCs pretreated with TNF-α and IFN-γ, secreted increased levels of TGF-β. Hepatocellular carcinoma cells grown in conditioned medium from the TNF-α and IFN-γ treated MSCs showed marked changes in molecular markers and functional characteristics associated with EMT, such as increased migration and invasion both in vitro and in vivo [82].
Role in the establishment of distant metastasis
A study by Karnoub and colleagues investigated the effect of MSCs on breast cancer cell motility and migration to the site of metastasis [26]. Human bone marrow derived MSCs were co-injected with the breast cancer cell line, MDA-MB-231, into mice. The chemokine CCL5 was secreted by MSCs, which in turn interacted with its receptor CCR5 on the breast cancer cells, resulting in increased metastasis to the lung [26]. Further strengthening these results, studies were published demonstrating the secretion of CCL5 by in vitro by human bone marrow derived MSCs in response to osteosarcoma cells [83] and breast cancer cells [84]. Additionally, it was found that the release of osteopontin (OPN) by tumour cells induced the production of CCL5 by MSCs, which in turn promoted CCR5 mediated breast cancer cell metastasis (see Fig. 1). Furthermore, MSCs isolated from the site of metastasis (the lung and liver) expressed the CAF markers α-SMA, SDF-1α, TN-C, MMP-2 and MMP-9 [84].
OPN is a chemoattractant with adhesive properties and can facilitate invasion through the binding of integrins, mainly αvβ1, αvβ3, αvβ5, αvβ6, α8β1 and α5β1, on many cell types [85–88]. Increased OPN levels were found to be correlated with prostate cancer progression and an indicator of the presence of distant metastases [89–92]. OPN deficient mice when injected with B16 melanoma cells developed decreased bone metastasis in comparison to wild-type mice [93]. OPN facilitates osteoclastogenesis by mediating osteoclast motility and anchorage to the bone mineral matrix [94–98]. Changes in OPN production within the bone marrow could therefore disrupt bone homeostasis as expression of OPN in breast cancer has been found to be associated with osteolytic bone metastasis [99, 100].
MSCs at the bone metastatic site
MSCs are bone marrow resident cells and given the poor prognosis in patients diagnosed with metastatic bone cancer, it is a key area in which to explore their role [101]. MSCs play a crucial supportive role for HSCs and their interaction with the surrounding microenvironment maintains a balance between bone formation and resorption. Given the plethora of studies showing the tumour promoting effect of MSC-tumour cell interaction, it is likely that tumour cell infiltration into the bone marrow will have a considerable impact on bone marrow homeostasis.
Entry of cancer cells into the bone marrow may be facilitated by MSCs through adherence of the metastatic cell to bone marrow ECs [33]. Several studies have found that the chemoattraction of tumour cells to the bone marrow is stimulated by bone marrow stromal cell production of SDF-1α (see Fig. 1) [33, 102, 103]. Prostate cancer cells were found to express the receptor CXCR4 and migrate and invade in response to SDF-1α [104, 105]. Human bone marrow derived MSCs were found to promote the transmigration of breast cancer cell lines (MCF7 and T47D) across bone marrow ECs [33]. Tac1 expression in the breast cancer cell lines was found to play a key role in bone marrow EC transmigration and the adherence of the metastatic cells to MSCs through the regulation of CXCR4 and SDF-1α production in the breast cancer cells [33].
Cells of the bone marrow including HSCs, megakaryocytes, macrophages and myeloid-derived suppressor cells have been implicated in developing a hospitable metastatic niche [106]. However, given the plasticity of MSCs and their role in bone remodelling it seems likely that the establishment of tumour cells within the bone marrow would result in cellular cross-talk that would disrupt bone homeostasis. Bone morphogenic protein-4 (BMP-4) within the bone marrow has been shown to stimulate the production of sonic hedgehog (SHH) in prostate cancer LNCaP cells which enhanced BMP-responsive reporter signalling in the mouse stromal cell line, MC3T3-E1, leading to increased osteoblastic differentiation [107].
An interesting study by Joseph et al. investigated the interaction between HSCs derived from the bone marrow of mice implanted with prostate cancer cell lines that formed either osteoblastic or osteolytic metastatic lesions. They found that HSCs derived from the mice with osteoblastic lesions stimulated osteoblastic differentiation of MSCs through BMP2 signalling, while HSCs derived from mice with osteolytic lesions enhanced the differentiation of mixed marrow mononuclear to osteoclasts through IL-6 signalling [108]. It is thought provoking research and the field would benefit from a similar study in which MSCs are isolated from both osteoclastic and osteoblastic metastatic lesions. A better understanding of the impact of tumour cell infiltration on the bone marrow resident cells could reveal better therapeutic targets. The other question is whether these effects are lasting, and if depletion of tumour cells from the metastatic site leaves behind a dysfunctional, destructive microenvironment.
IL-6 is a pro-inflammatory cytokine that is known to mediate cell proliferation, cell survival and lymphocyte differentiation [109]. IL-6 may have an important role in cross-talk within the tumour associated bone marrow microenvironment. Production of IL-6 in multiple myeloma by bone marrow stromal cells induces tumour cell adhesion and osteoclastogenesis [110, 111]. IL-6 secretion in MSCs was found to be stimulated by neuroblastoma cells within the bone marrow which in turn activated osteoclasts [112]. IL-6 was also found to act on neuroblastoma and multiple myeloma cells within the bone marrow by increasing cell proliferation and survival through activation of the signal transducer and activator of transcription 3 (STAT3) pathway [112, 113].
Conclusions
It is now understood that MSCs interact with and influence tumour cells at various stages of progression. It is not clear however, whether the effect is predominantly tumour promoting or suppressive. Explanations that could account for the conflicting results include differences in experimental design, the heterogeneity within the MSC population or varying responses dependent on the stimuli (explored more extensively in a review by Klopp et al. [114]). Nonetheless, there is extensive evidence to suggest that MSCs can promote tumour growth and drive metastatic progression. Despite this, MSCs are increasingly being studied for their potential in a range of different clinical therapies. It is therefore imperative to understand how they communicate with tumour cells and within the tumour stroma. Given the plasticity of MSCs, future research should consider whether they are reprogrammed at the site of the tumour or if they exert their effects solely through paracrine signalling and direct cell-cell contact. It would also be interesting to ascertain whether there are phenotypic differences in MSCs that are isolated from different tumour types and whether the MSC responds to the tumour according to its stage of progression.
Cancer therapies classically target tumour cells yet, what remains is an activated stroma that provides an encouraging microenvironment for any surviving tumour cells. Evidence to support this comes from studies in breast cancer in which stromal-related gene expression or gene signatures was predictive of clinical outcome [115, 116]. Moreover, pre-treatment of MSCs to concentrations of cisplatin which were toxic to breast cancer cells but not MSCs in vitro was found induce changes in kinase phosphorylation and increased cytokine production in the MSCs and co-culture with breast cancer cells lead to chemoresistance in the tumour cells [117]. It would therefore be of therapeutic interest to investigate the contribution of tumour stromal cells to cancer progression and their activity following cytotoxic treatment.
Acknowledgements
Not applicable.
Funding
Sarah Ridge was funded by a NUI Galway PhD Scholarship. Sharon Glynn was funded by the Irish Cancer Society (PCT13MCD). Francis Sullivan was funded by the Galway University Foundation.
Availability of data and materials
Not applicable.
Authors’ contributions
The initial draft of the paper was written by SMR. SAG revised and expanded the manuscript. SMR created the figure and designed the outline of the article. FJS proofread the manuscript. Each author has read and approved the final version of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable – review article.
Abbreviations
- BMP-4
Bone morphogenic protein-4
- CAF
Cancer associated fibroblast
- CCL5
Chemokine (C-C motif) ligand 5
- CCR5
C-C chemokine receptor type 5
- EC
Endothelial cell
- EMT
Epithelial-to-mesenchymal transition
- FSP1
Fibroblast specific protein 1
- HSC
Haematopoietic stem cell
- IFN-γ
Interferon gamma
- IL
Interleukin
- MSC
Mesenchymal stem cell
- OPN
Osteopontin
- SDF-1α
Stromal cell-derived factor 1
- SHH
Sonic hedgehog
- STAT3
Signal transducer and activator of transcription 3
- TAM
Tumour associated macrophage
- TGFβ
Transforming growth factor β
- TLR
Toll-like receptor
- TN-C
Tenascin C
- TNF-α
Tumour necrosis factor-α
- VEGF
Vascular endothelial growth factor
- αSMA
α smooth muscle actin
References
- 1.Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res. 2010;316(8):1324–31. doi: 10.1016/j.yexcr.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 2.Dvorak HF. Tumors: wounds that do not heal-redux. Cancer Immunol Res. 2015;3(1):1–11. doi: 10.1158/2326-6066.CIR-14-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71–8. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 4.Hall B, Andreeff M, Marini F. The participation of mesenchymal stem cells in tumor stroma formation and their application as targeted-gene delivery vehicles. Handb Exp Pharmacol. 2007;180:263–83. doi: 10.1007/978-3-540-68976-8_12. [DOI] [PubMed] [Google Scholar]
- 5.Young MR, Wright MA. Myelopoiesis-associated immune suppressor cells in mice bearing metastatic Lewis lung carcinoma tumors: gamma interferon plus tumor necrosis factor alpha synergistically reduces immune suppressor and tumor growth-promoting activities of bone marrow cells and diminishes tumor recurrence and metastasis. Cancer Res. 1992;52(22):6335–40. [PubMed] [Google Scholar]
- 6.Sato T, et al. Tumor-stromal cell contact promotes invasion of human uterine cervical carcinoma cells by augmenting the expression and activation of stromal matrix metalloproteinases. Gynecol Oncol. 2004;92(1):47–56. doi: 10.1016/j.ygyno.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 7.Sung SY, et al. Coevolution of prostate cancer and bone stroma in three-dimensional coculture: implications for cancer growth and metastasis. Cancer Res. 2008;68(23):9996–10003. doi: 10.1158/0008-5472.CAN-08-2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pittenger MF, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–7. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 9.Dominici M, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–7. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 10.Wakitani S, Saito T, Caplan AI. Myogenic cells derived from rat bone marrow mesenchymal stem cells exposed to 5-azacytidine. Muscle Nerve. 1995;18(12):1417–26. doi: 10.1002/mus.880181212. [DOI] [PubMed] [Google Scholar]
- 11.Kopen GC, Prockop DJ, Phinney DG. Marrow stromal cells migrate throughout forebrain and cerebellum, and they differentiate into astrocytes after injection into neonatal mouse brains. Proc Natl Acad Sci U S A. 1999;96(19):10711–6. doi: 10.1073/pnas.96.19.10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gronthos S, et al. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci U S A. 2000;97(25):13625–30. doi: 10.1073/pnas.240309797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang JI, et al. Rat extramedullary adipose tissue as a source of osteochondrogenic progenitor cells. Plast Reconstr Surg. 2002;109(3):1033–41. doi: 10.1097/00006534-200203000-00037. [DOI] [PubMed] [Google Scholar]
- 14.Friedenstein AJ, Chailakhjan RK, Lalykina KS. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Tissue Kinet. 1970;3(4):393–403. doi: 10.1111/j.1365-2184.1970.tb00347.x. [DOI] [PubMed] [Google Scholar]
- 15.Chamberlain G, et al. Concise review: mesenchymal stem cells: their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells. 2007;25(11):2739–49. doi: 10.1634/stemcells.2007-0197. [DOI] [PubMed] [Google Scholar]
- 16.Ortiz LA, et al. Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates its fibrotic effects. Proc Natl Acad Sci U S A. 2003;100(14):8407–11. doi: 10.1073/pnas.1432929100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato Y, et al. Human mesenchymal stem cells xenografted directly to rat liver are differentiated into human hepatocytes without fusion. Blood. 2005;106(2):756–63. doi: 10.1182/blood-2005-02-0572. [DOI] [PubMed] [Google Scholar]
- 18.Ji JF, et al. Interactions of chemokines and chemokine receptors mediate the migration of mesenchymal stem cells to the impaired site in the brain after hypoglossal nerve injury. Stem Cells. 2004;22(3):415–27. doi: 10.1634/stemcells.22-3-415. [DOI] [PubMed] [Google Scholar]
- 19.Wu GD, et al. Migration of mesenchymal stem cells to heart allografts during chronic rejection. Transplantation. 2003;75(5):679–85. doi: 10.1097/01.TP.0000048488.35010.95. [DOI] [PubMed] [Google Scholar]
- 20.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7(3):311–7. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 21.Chitteti BR, et al. Impact of interactions of cellular components of the bone marrow microenvironment on hematopoietic stem and progenitor cell function. Blood. 2010;115(16):3239–48. doi: 10.1182/blood-2009-09-246173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Almeida-Porada G, et al. Cotransplantation of human stromal cell progenitors into preimmune fetal sheep results in early appearance of human donor cells in circulation and boosts cell levels in bone marrow at later time points after transplantation. Blood. 2000;95(11):3620–7. [PubMed] [Google Scholar]
- 23.Maitra B, et al. Human mesenchymal stem cells support unrelated donor hematopoietic stem cells and suppress T-cell activation. Bone Marrow Transplant. 2004;33(6):597–604. doi: 10.1038/sj.bmt.1704400. [DOI] [PubMed] [Google Scholar]
- 24.Ame-Thomas P, et al. Human mesenchymal stem cells isolated from bone marrow and lymphoid organs support tumor B-cell growth: role of stromal cells in follicular lymphoma pathogenesis. Blood. 2007;109(2):693–702. doi: 10.1182/blood-2006-05-020800. [DOI] [PubMed] [Google Scholar]
- 25.Kansy BA, et al. The bidirectional tumor--mesenchymal stromal cell interaction promotes the progression of head and neck cancer. Stem Cell Res Ther. 2014;5(4):95. doi: 10.1186/scrt484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karnoub AE, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449(7162):557–63. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 27.Prantl L, et al. Adipose tissue-derived stem cells promote prostate tumor growth. Prostate. 2010;70(15):1709–15. doi: 10.1002/pros.21206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki K, et al. Mesenchymal stromal cells promote tumor growth through the enhancement of neovascularization. Mol Med. 2011;17(7–8):579–87. doi: 10.2119/molmed.2010.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kucerova L, et al. Tumor cell behaviour modulation by mesenchymal stromal cells. Mol Cancer. 2010;9:129. doi: 10.1186/1476-4598-9-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brennen WN, et al. Quantification of Mesenchymal Stem Cells (MSCs) at sites of human prostate cancer. Oncotarget. 2013;4(1):106–17. doi: 10.18632/oncotarget.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nabha SM, et al. Bone marrow stromal cells enhance prostate cancer cell invasion through type I collagen in an MMP-12 dependent manner. Int J Cancer. 2008;122(11):2482–90. doi: 10.1002/ijc.23431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duda DG, et al. Malignant cells facilitate lung metastasis by bringing their own soil. Proc Natl Acad Sci U S A. 2010;107(50):21677–82. doi: 10.1073/pnas.1016234107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corcoran KE, et al. Mesenchymal stem cells in early entry of breast cancer into bone marrow. PLoS One. 2008;3(6):e2563. doi: 10.1371/journal.pone.0002563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hossain A, et al. Mesenchymal stem cells isolated from human gliomas increase proliferation and maintain stemness of glioma stem cells through the IL-6/gp130/STAT3 pathway. Stem Cells. 2015;33(8):2400–15. doi: 10.1002/stem.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li W, et al. Gastric cancer-derived mesenchymal stem cells prompt gastric cancer progression through secretion of interleukin-8. J Exp Clin Cancer Res. 2015;34:52. doi: 10.1186/s13046-015-0172-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu W, et al. Mesenchymal stem cells derived from bone marrow favor tumor cell growth in vivo. Exp Mol Pathol. 2006;80(3):267–74. doi: 10.1016/j.yexmp.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 37.Lacerda L, et al. Mesenchymal stem cells mediate the clinical phenotype of inflammatory breast cancer in a preclinical model. Breast Cancer Res. 2015;17:42. doi: 10.1186/s13058-015-0549-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ye H, et al. Human bone marrow-derived mesenchymal stem cells produced TGFbeta contributes to progression and metastasis of prostate cancer. Cancer Invest. 2012;30(7):513–8. doi: 10.3109/07357907.2012.692171. [DOI] [PubMed] [Google Scholar]
- 39.Lee MJ, et al. Oncostatin M promotes mesenchymal stem cell-stimulated tumor growth through a paracrine mechanism involving periostin and TGFBI. Int J Biochem Cell Biol. 2013;45(8):1869–77. doi: 10.1016/j.biocel.2013.05.027. [DOI] [PubMed] [Google Scholar]
- 40.Costanza, B., et al. Stromal Modulators of TGF-beta in Cancer. J Clin Med. 2017. 6(1). [DOI] [PMC free article] [PubMed]
- 41.Calon A, et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22(5):571–84. doi: 10.1016/j.ccr.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richardsen E, et al. Immunohistochemical expression of epithelial and stromal immunomodulatory signalling molecules is a prognostic indicator in breast cancer. BMC Res Notes. 2012;5:110. doi: 10.1186/1756-0500-5-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim EK, et al. Endogenous gastric-resident mesenchymal stem cells contribute to formation of cancer stroma and progression of gastric cancer. Korean J Pathol. 2013;47(6):507–18. doi: 10.4132/KoreanJPathol.2013.47.6.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun B, et al. Therapeutic potential of mesenchymal stromal cells in a mouse breast cancer metastasis model. Cytotherapy. 2009;11(3):289–98. doi: 10.1080/14653240902807026. [DOI] [PubMed] [Google Scholar]
- 45.Khakoo AY, et al. Human mesenchymal stem cells exert potent antitumorigenic effects in a model of Kaposi’s sarcoma. J Exp Med. 2006;203(5):1235–47. doi: 10.1084/jem.20051921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qiao L, et al. Suppression of tumorigenesis by human mesenchymal stem cells in a hepatoma model. Cell Res. 2008;18(4):500–7. doi: 10.1038/cr.2008.40. [DOI] [PubMed] [Google Scholar]
- 47.Otsu K, et al. Concentration-dependent inhibition of angiogenesis by mesenchymal stem cells. Blood. 2009;113(18):4197–205. doi: 10.1182/blood-2008-09-176198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee RH, et al. Characterization and expression analysis of mesenchymal stem cells from human bone marrow and adipose tissue. Cell Physiol Biochem. 2004;14(4–6):311–24. doi: 10.1159/000080341. [DOI] [PubMed] [Google Scholar]
- 49.Wagner W, et al. Comparative characteristics of mesenchymal stem cells from human bone marrow, adipose tissue, and umbilical cord blood. Exp Hematol. 2005;33(11):1402–16. doi: 10.1016/j.exphem.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 50.Horwitz EM, et al. Clarification of the nomenclature for MSC: the international society for cellular therapy position statement. Cytotherapy. 2005;7(5):393–5. doi: 10.1080/14653240500319234. [DOI] [PubMed] [Google Scholar]
- 51.Riekstina U, et al. Embryonic stem cell marker expression pattern in human mesenchymal stem cells derived from bone marrow, adipose tissue, heart and dermis. Stem Cell Rev. 2009;5(4):378–86. doi: 10.1007/s12015-009-9094-9. [DOI] [PubMed] [Google Scholar]
- 52.Mantovani A, et al. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–55. doi: 10.1016/S1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 53.Sica A, Saccani A, Mantovani A. Tumor-associated macrophages: a molecular perspective. Int Immunopharmacol. 2002;2(8):1045–54. doi: 10.1016/S1567-5769(02)00064-4. [DOI] [PubMed] [Google Scholar]
- 54.Allavena P, et al. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol Rev. 2008;222:155–61. doi: 10.1111/j.1600-065X.2008.00607.x. [DOI] [PubMed] [Google Scholar]
- 55.Sica A, et al. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42(6):717–27. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 56.Solinas G, et al. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86(5):1065–73. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- 57.Tomchuck SL, et al. Toll-like receptors on human mesenchymal stem cells drive their migration and immunomodulating responses. Stem Cells. 2008;26(1):99–107. doi: 10.1634/stemcells.2007-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Waterman RS, et al. A new mesenchymal stem cell (MSC) paradigm: polarization into a pro-inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS One. 2010;5(4):e10088. doi: 10.1371/journal.pone.0010088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Waterman RS, Henkle SL, Betancourt AM. Mesenchymal stem cell 1 (MSC1)-based therapy attenuates tumor growth whereas MSC2-treatment promotes tumor growth and metastasis. PLoS One. 2012;7(9):e45590. doi: 10.1371/journal.pone.0045590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gonzalez-Reyes S, et al. Study of TLR3, TLR4 and TLR9 in breast carcinomas and their association with metastasis. BMC Cancer. 2010;10:665. doi: 10.1186/1471-2407-10-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Spaeth EL, et al. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One. 2009;4(4):e4992. doi: 10.1371/journal.pone.0004992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Evans RA, et al. TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp Cell Res. 2003;282(2):90–100. doi: 10.1016/S0014-4827(02)00015-0. [DOI] [PubMed] [Google Scholar]
- 63.Zeisberg EM, et al. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67(21):10123–8. doi: 10.1158/0008-5472.CAN-07-3127. [DOI] [PubMed] [Google Scholar]
- 64.Kojima Y, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107(46):20009–14. doi: 10.1073/pnas.1013805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shangguan L, et al. Inhibition of TGF-beta/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma-associated fibroblasts and abolishes their protumor effects. Stem Cells. 2012;30(12):2810–9. doi: 10.1002/stem.1251. [DOI] [PubMed] [Google Scholar]
- 66.Calon A, Tauriello DV, Batlle E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin Cancer Biol. 2014;25:15–22. doi: 10.1016/j.semcancer.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 67.Direkze NC, et al. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004;64(23):8492–5. doi: 10.1158/0008-5472.CAN-04-1708. [DOI] [PubMed] [Google Scholar]
- 68.Ishii G, et al. Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem Biophys Res Commun. 2003;309(1):232–40. doi: 10.1016/S0006-291X(03)01544-4. [DOI] [PubMed] [Google Scholar]
- 69.Direkze NC, et al. Bone marrow-derived stromal cells express lineage-related messenger RNA species. Cancer Res. 2006;66(3):1265–9. doi: 10.1158/0008-5472.CAN-05-3202. [DOI] [PubMed] [Google Scholar]
- 70.Mishra PJ, et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68(11):4331–9. doi: 10.1158/0008-5472.CAN-08-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peng Y, Li Z, Li Z. GRP78 secreted by tumor cells stimulates differentiation of bone marrow mesenchymal stem cells to cancer-associated fibroblasts. Biochem Biophys Res Commun. 2013;440(4):558–63. doi: 10.1016/j.bbrc.2013.09.108. [DOI] [PubMed] [Google Scholar]
- 72.Paunescu V, et al. Tumour-associated fibroblasts and mesenchymal stem cells: more similarities than differences. J Cell Mol Med. 2011;15(3):635–46. doi: 10.1111/j.1582-4934.2010.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–98. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 74.Augsten M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front Oncol. 2014;4:62. doi: 10.3389/fonc.2014.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Albarenque SM, Zwacka RM, Mohr A. Both human and mouse mesenchymal stem cells promote breast cancer metastasis. Stem Cell Res. 2011;7(2):163–71. doi: 10.1016/j.scr.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 76.Shinagawa K, et al. Mesenchymal stem cells enhance growth and metastasis of colon cancer. Int J Cancer. 2010;127(10):2323–33. doi: 10.1002/ijc.25440. [DOI] [PubMed] [Google Scholar]
- 77.Chaturvedi P, et al. Hypoxia-inducible factor-dependent breast cancer-mesenchymal stem cell bidirectional signaling promotes metastasis. J Clin Invest. 2013;123(1):189–205. doi: 10.1172/JCI64993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Loebinger MR, et al. Magnetic resonance imaging of mesenchymal stem cells homing to pulmonary metastases using biocompatible magnetic nanoparticles. Cancer Res. 2009;69(23):8862–7. doi: 10.1158/0008-5472.CAN-09-1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kaplan RN, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–7. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Martin FT, et al. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: stimulation of epithelial to mesenchymal transition (EMT) Breast Cancer Res Treat. 2010;124(2):317–26. doi: 10.1007/s10549-010-0734-1. [DOI] [PubMed] [Google Scholar]
- 81.Xue Z, et al. Mesenchymal stem cells promote epithelial to mesenchymal transition and metastasis in gastric cancer though paracrine cues and close physical contact. J Cell Biochem. 2015;116(4):618–27. doi: 10.1002/jcb.25013. [DOI] [PubMed] [Google Scholar]
- 82.Jing Y, et al. Mesenchymal stem cells in inflammation microenvironment accelerates hepatocellular carcinoma metastasis by inducing epithelial-mesenchymal transition. PLoS One. 2012;7(8):e43272. doi: 10.1371/journal.pone.0043272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu WT, et al. Human mesenchymal stem cells (hMSCs) target osteosarcoma and promote its growth and pulmonary metastasis. Cancer Lett. 2009;281(1):32–41. doi: 10.1016/j.canlet.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 84.Mi Z, et al. Osteopontin promotes CCL5-mesenchymal stromal cell-mediated breast cancer metastasis. Carcinogenesis. 2011;32(4):477–87. doi: 10.1093/carcin/bgr009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hu DD, et al. A biochemical characterization of the binding of osteopontin to integrins alpha v beta 1 and alpha v beta 5. J Biol Chem. 1995;270(44):26232–8. doi: 10.1074/jbc.270.44.26232. [DOI] [PubMed] [Google Scholar]
- 86.Liaw L, et al. The adhesive and migratory effects of osteopontin are mediated via distinct cell surface integrins. Role of alpha v beta 3 in smooth muscle cell migration to osteopontin in vitro. J Clin Invest. 1995;95(2):713–24. doi: 10.1172/JCI117718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Denda S, Reichardt LF, Muller U. Identification of osteopontin as a novel ligand for the integrin alpha8 beta1 and potential roles for this integrin-ligand interaction in kidney morphogenesis. Mol Biol Cell. 1998;9(6):1425–35. doi: 10.1091/mbc.9.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yokosaki Y, et al. The integrin alpha(9)beta(1) binds to a novel recognition sequence (SVVYGLR) in the thrombin-cleaved amino-terminal fragment of osteopontin. J Biol Chem. 1999;274(51):36328–34. doi: 10.1074/jbc.274.51.36328. [DOI] [PubMed] [Google Scholar]
- 89.Khodavirdi AC, et al. Increased expression of osteopontin contributes to the progression of prostate cancer. Cancer Res. 2006;66(2):883–8. doi: 10.1158/0008-5472.CAN-05-2816. [DOI] [PubMed] [Google Scholar]
- 90.Castellano G, et al. Activation of the osteopontin/matrix metalloproteinase-9 pathway correlates with prostate cancer progression. Clin Cancer Res. 2008;14(22):7470–80. doi: 10.1158/1078-0432.CCR-08-0870. [DOI] [PubMed] [Google Scholar]
- 91.Forootan SS, et al. Prognostic significance of osteopontin expression in human prostate cancer. Int J Cancer. 2006;118(9):2255–61. doi: 10.1002/ijc.21619. [DOI] [PubMed] [Google Scholar]
- 92.Ramankulov A, et al. Plasma osteopontin in comparison with bone markers as indicator of bone metastasis and survival outcome in patients with prostate cancer. Prostate. 2007;67(3):330–40. doi: 10.1002/pros.20540. [DOI] [PubMed] [Google Scholar]
- 93.Nemoto H, et al. Osteopontin deficiency reduces experimental tumor cell metastasis to bone and soft tissues. J Bone Miner Res. 2001;16(4):652–9. doi: 10.1359/jbmr.2001.16.4.652. [DOI] [PubMed] [Google Scholar]
- 94.Ishijima M, et al. Enhancement of osteoclastic bone resorption and suppression of osteoblastic bone formation in response to reduced mechanical stress do not occur in the absence of osteopontin. J Exp Med. 2001;193(3):399–404. doi: 10.1084/jem.193.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chellaiah MA, et al. Osteopontin deficiency produces osteoclast dysfunction due to reduced CD44 surface expression. Mol Biol Cell. 2003;14(1):173–89. doi: 10.1091/mbc.E02-06-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Reinholt FP, et al. Osteopontin--a possible anchor of osteoclasts to bone. Proc Natl Acad Sci U S A. 1990;87(12):4473–5. doi: 10.1073/pnas.87.12.4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ross FP, et al. Interactions between the bone matrix proteins osteopontin and bone sialoprotein and the osteoclast integrin alpha v beta 3 potentiate bone resorption. J Biol Chem. 1993;268(13):9901–7. [PubMed] [Google Scholar]
- 98.Yamate T, et al. Osteopontin expression by osteoclast and osteoblast progenitors in the murine bone marrow: demonstration of its requirement for osteoclastogenesis and its increase after ovariectomy. Endocrinology. 1997;138(7):3047–55. doi: 10.1210/endo.138.7.5285. [DOI] [PubMed] [Google Scholar]
- 99.Ibrahim T, et al. Expression of bone sialoprotein and osteopontin in breast cancer bone metastases. Clin Exp Metastasis. 2000;18(3):253–60. doi: 10.1023/A:1006754605901. [DOI] [PubMed] [Google Scholar]
- 100.Adwan H, Bauerle TJ, Berger MR. Downregulation of osteopontin and bone sialoprotein II is related to reduced colony formation and metastasis formation of MDA-MB-231 human breast cancer cells. Cancer Gene Ther. 2004;11(2):109–20. doi: 10.1038/sj.cgt.7700659. [DOI] [PubMed] [Google Scholar]
- 101.Coleman RE. Skeletal complications of malignancy. Cancer. 1997;80(8 Suppl):1588–94. doi: 10.1002/(SICI)1097-0142(19971015)80:8+<1588::AID-CNCR9>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 102.Cooper CR, et al. Stromal factors involved in prostate carcinoma metastasis to bone. Cancer. 2003;97(3 Suppl):739–47. doi: 10.1002/cncr.11181. [DOI] [PubMed] [Google Scholar]
- 103.Zhang XH, et al. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. 2013;154(5):1060–73. doi: 10.1016/j.cell.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Taichman RS, et al. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62(6):1832–7. [PubMed] [Google Scholar]
- 105.Singh S, et al. CXCL12-CXCR4 interactions modulate prostate cancer cell migration, metalloproteinase expression and invasion. Lab Invest. 2004;84(12):1666–76. doi: 10.1038/labinvest.3700181. [DOI] [PubMed] [Google Scholar]
- 106.Park SI, Soki FN, McCauley LK. Roles of bone marrow cells in skeletal metastases: no longer bystanders. Cancer Microenviron. 2011;4(3):237–46. doi: 10.1007/s12307-011-0081-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nishimori, H., et al. Prostate cancer cells and bone stromal cells mutually interact with each other through bone morphogenetic protein-mediated signals. J Biol Chem. 2012;287(24):20037-46. [DOI] [PMC free article] [PubMed]
- 108.Joseph J, et al. Disseminated prostate cancer cells can instruct hematopoietic stem and progenitor cells to regulate bone phenotype. Mol Cancer Res. 2012;10(3):282–92. doi: 10.1158/1541-7786.MCR-11-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41(16):2502–12. doi: 10.1016/j.ejca.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 110.Thomas X, et al. Interdependence between cytokines and cell adhesion molecules to induce interleukin-6 production by stromal cells in myeloma. Leuk Lymphoma. 1998;32(1–2):107–19. doi: 10.3109/10428199809059251. [DOI] [PubMed] [Google Scholar]
- 111.Michigami T, et al. Cell-cell contact between marrow stromal cells and myeloma cells via VCAM-1 and alpha(4)beta(1)-integrin enhances production of osteoclast-stimulating activity. Blood. 2000;96(5):1953–60. [PubMed] [Google Scholar]
- 112.Ara T, et al. Interleukin-6 in the bone marrow microenvironment promotes the growth and survival of neuroblastoma cells. Cancer Res. 2009;69(1):329–37. doi: 10.1158/0008-5472.CAN-08-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Brocke-Heidrich K, et al. Interleukin-6-dependent gene expression profiles in multiple myeloma INA-6 cells reveal a Bcl-2 family-independent survival pathway closely associated with Stat3 activation. Blood. 2004;103(1):242–51. doi: 10.1182/blood-2003-04-1048. [DOI] [PubMed] [Google Scholar]
- 114.Klopp AH, et al. Concise review: dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells. 2011;29(1):11–9. doi: 10.1002/stem.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Farmer P, et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med. 2009;15(1):68–74. doi: 10.1038/nm.1908. [DOI] [PubMed] [Google Scholar]
- 116.Finak G, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14(5):518–27. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 117.Skolekova S, et al. Cisplatin-induced mesenchymal stromal cells-mediated mechanism contributing to decreased antitumor effect in breast cancer cells. Cell Commun Signal. 2016;14(1):4. doi: 10.1186/s12964-016-0127-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.