

Abstract
Objective:
To identify inherited or de novo mutations associated with a suite of neurodevelopmental abnormalities in a 10-year-old patient displaying ataxia, motor and speech delay, and intellectual disability.
Methods:
We performed whole-exome sequencing of the proband and her parents. A pathogenic gene variant was identified as damaging based on sequence conservation, gene function, and association with disorders having similar phenotypic profiles. Functional characterization of the mutated protein was performed in vitro using a heterologous expression system.
Results:
A single de novo point mutation in the GRIK2 gene was identified as causative for the neurologic symptoms of the proband. The mutation is predicted to change a codon for alanine to that of a threonine at position 657 (A657T) in the GluK2 kainate receptor (KAR) subunit, a member of the ionotropic glutamate receptor gene family. Whole-cell voltage-clamp recordings revealed that KARs incorporating the GluK2(A657T) subunits show profoundly altered channel gating and are constitutively active in nominally glutamate-free extracellular media.
Conclusions:
In this study, we associate a de novo gain-of-function mutation in the GRIK2 gene with deficits in motor and higher order cognitive function. These results suggest that disruption of physiologic KAR function precludes appropriate development of the nervous system.
Neurodevelopmental disorders manifest as a diverse array of neurologic and neuropsychiatric symptoms that can be challenging to link to a discrete genetic basis. Identification of causative genetic variants for a growing number of these neurodevelopmental disorders can reduce complexity by uncovering common biological pathways affected in the disease state.1 The objective of this study was to elucidate pathologic genetic alterations underlying the neurologic symptoms of a 10-year-old child displaying congenital gait ataxia, developmental delay, and learning deficits.
Genetic testing detected a missense variant in the GRIK2 gene, which encodes the kainate receptor (KAR) subunit GluK2. KARs are channels activated by the neurotransmitter l-glutamate that play complex roles in neural development and CNS function.2 Loss-of-function mutations in GRIK2 have been previously associated with intellectual disability.3–5 In this study, we show that the GluK2 mutation identified in the proband yields GluK2-containing KARs with altered gating kinetics and constitutive activity. These findings associate a gain-of-function mutation in a GRIK gene with neurologic disease in humans.
METHODS
Participants.
The proband is a 10-year-old Caucasian female born without complications to healthy nonconsanguineous parents. Clinical and behavioral assessments were performed at the Center for Rare Childhood Disorders at the Translational Research Institute in Arizona, United States. MRI was performed to identify possible neuroanatomic abnormalities.
Whole-exome sequencing.
Genomic DNA was extracted from blood taken from the proband and both parents. Whole-exome sequencing was performed at the Translational Genomics Research Institute in Arizona when the patient was aged 6 years. Exomic libraries were prepared with the Illumina TruSeq Exome Enrichment Kit v2. Sequencing was performed by 101 bp paired-end sequencing on a HiSeq2000 instrument (Illumina Inc., San Diego, CA). Filtered reads were aligned to the human genome (Hg19/GRC37) using the Burrows-Wheeler transform. Reads were sorted, and PCR duplicates were removed using Picard, and base-quality recalibration and indel realignment were performed using the Genome Analysis Toolkit. Variants were called jointly with UnifiedGenotyper and annotated with dbNSFP and SnpEff for protein-coding events. Prediction scores were loaded from dbNSFP and used for filtering. An annotated variant file containing variants in 3 family members was filtered to include novel, private, or rare variants according to the Exome Aggregation Consortium (ExAC) database and the Genome Aggregation Database (gnomAD). Variants predicted to be damaging by the Combined Annotation Dependent Depletion (CADD) tool or to disrupt protein structure or function by Polymorphism Phenotyping v2 (PolyPhen2) algorithms were considered as candidate genes responsible for the child's phenotype.
Cell culture and electrophysiology.
Rat GluK2a(A657T) and Neto2 complementary DNAs (cDNAs) were provided by Michisuke Yuzaki (Keio University) and Susumu Tomita (Yale University), respectively. HEK293T/17 cells were maintained and transfected as described previously.6 After transfection, the cell media were supplemented with 1-naphthyl acetyl spermine trihydrochloride (NASPM, 20 μM) to prevent cell death by excitotoxicity. Plasmid DNAs were cotransfected at ratios of 1:4 for enhanced green fluorescent protein (eGFP) and GluK2(A657T); 1:3:0.5 for GluK2(A657T), Neto2, and eGFP, and 1:3:0.5 for GluK2(A657T), GluK5, and eGFP cDNAs, respectively.
Whole-cell voltage-clamp recordings were performed 1–2 days after transfection as described previously.6 The external solution contained the following: 150 mM NaCl, 2.8 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM Hepes, pH 7.3. The internal solution was composed of the following: 110 mM CsF, 30 mM CsCl, 10 mM Cs-Hepes, 5 mM EGTA, 4 mM NaCl, and 0.5 mM CaCl2, pH 7.3. Cells were held at −70 mV while in voltage clamp, and glutamate (10 mM) was fast-applied with a piezo-bimorph system. This recording configuration elicited currents with 10%–90% rise times of 1–3 milliseconds (ms). Desensitization rates were analyzed with Clampfit 10 (Molecular Devices, Sunnyvale, CA) by fitting the current decay during a 1-second application using single or biexponential parameters.
Statistical analysis.
Data are represented as mean ± SEM. Unpaired Student t test group analysis was performed using Prism 6.0 (GraphPad Software, La Jolla, CA) on receptor biophysical parameters with statistical significance denoted as follows: *p < 0.05, **p < 0.01, and ***p < 0.001.
Standard protocol approvals, registrations, and patient consents.
Written consent for the acquisition and disclosure of data generated by the study was obtained from the guardians, as verbal assent from the proband was not possible. The participating family enrolled into a research protocol sponsored by the Translational Genomics Research Institute, which was approved by Western Institutional Review Board (WIRB Protocol No. 20120789).
RESULTS
Identification of a de novo single-point mutation in the GRIK2 gene in a patient with neurodevelopmental delays and intellectual disability.
Neurologic evaluations of a 10-year-old female yielded a general diagnosis of severe neurodevelopmental delays and intellectual disability (figure 1A). She had congenital gait ataxia accompanied by impairments in motor and speech development (figure 1B, video at Neurology.org/ng). At 1 month of age, her parents reported generalized hypotonia and later noted significant delays in her fine and gross motor skill development. The patient underwent physical therapy beginning at 12 months of age. She was able to sit up on her own at 16 months of age and started walking short distances at 5.5 years, albeit with an ataxic gait and poor balance. Her muscle tone and bulk are currently within the normal range, and her fine motor coordination skills have improved slowly. She started using single words (up to 4 words) by 2–3 years of age, currently understands a limited set of words, and can identify colors and animals with 80% accuracy. She also communicates nonverbally using gestures. Neurologic evaluations revealed that her speech impairments were a result of intellectual disability rather than dysarthria. The proband also exhibits sialorrhea and sensory alterations such as aversion to loud sounds and light touch. Approximately 3 times a year, as reported by the parents, she experiences episodes of constant tongue protrusion that last for 2 weeks.
Figure 1. Summary of the clinical picture of the proband.
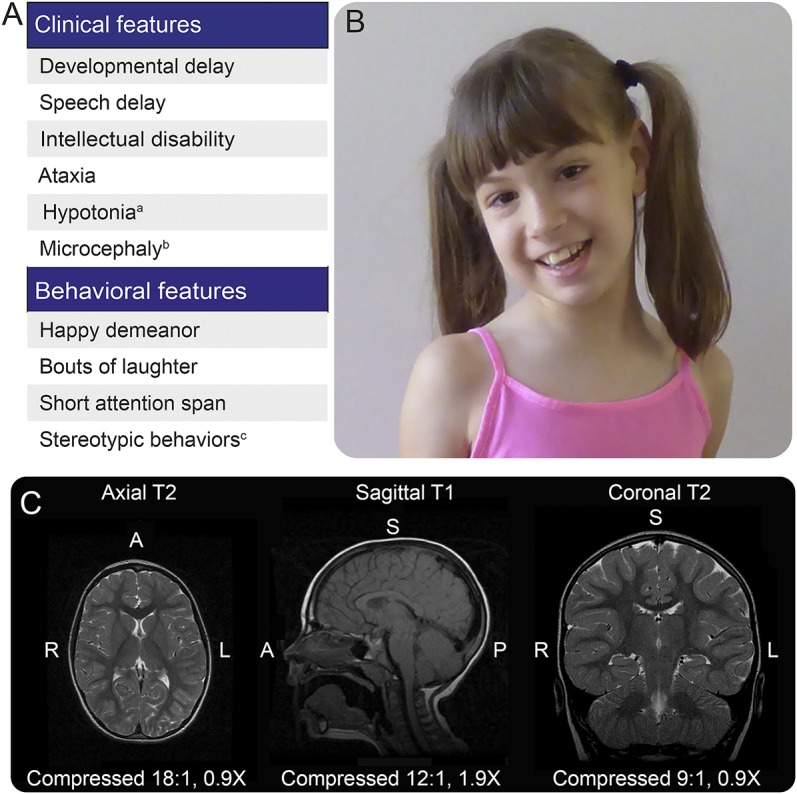
(A) Clinical and behavioral characteristics of the proband: aonly during the perinatal period; bat 22 months, her head circumference was 43 cm (<second percentile), and at 7 years, it was 47 ½ cm (<second percentile); ctongue thrusting. (B) Facial appearance of the proband. (C) Selected MRIs of the proband (sagittal T1, axial T2, and coronal T2) demonstrating normal anatomy of the cerebrum, cerebellum, and brainstem.
Behavioral assessments revealed that the patient had a short attention span, stereotyped behaviors, and a happy demeanor, which, together with the early-age hypotonia and ataxia, bore some similarity to children with Angelman syndrome. In testing her muscle tone, she did not display rigidity, spasticity, or dystonia, although she displayed adventitious (choreoathetoid) movements while walking (video). No seizures have been documented in the patient. She has not regressed in her development and does not display features suggestive of neurodegeneration. In accordance, MRI showed normal gross anatomy of the brain, including the cerebellum (figure 1C). Her head circumference was 43 and 47 ½ cm at 22 months and 7 years of age, respectively, which in both cases was below the second percentile. Neither parent exhibited microcephaly (father: 59 cm; mother: 58.4 cm).
Genetic testing ruled out several neurodevelopmental and neuromuscular diseases including Angelman syndrome, Prader-Willi syndrome, fragile X syndrome, Rett syndrome, spinal muscular atrophy, and myotonic dystrophy. A chromosomal microarray (Lineagen, Salt Lake City, UT) test performed at age 7 years revealed an interstitial deletion in chromosome 6q22.33: 129,669,542–129,734,572 (hg build 19), which includes the LAMA2 gene associated with merosin-deficient congenital muscular dystrophy (table). This deletion in chromosome 6q22.33 was subsequently identified in the paternal genome during analysis of exome sequencing data.
Table.
Potential causative missense or nonsense variation

Whole-exome sequencing was performed, and highly deleterious or damaging gene variations were identified by CADD and PolyPhen2 analysis and cross-referenced with the ExAC database and gnomAD to determine the prevalence of these mutations. Three heterozygous inherited or de novo mutations were classified as detrimental by 1 or both of the algorithms used (table). All other variants were seen at high frequencies in public databases, had poor coverage, or were predicted to be benign. Four inherited variants in 2 genes, NEB and OR1S2, showed compound heterozygosity. The NEB variants were excluded because loss-of-function mutations in this gene lead to congenital nemaline myopathies,7 characterized by hypotonia and hypoventilation, symptoms not present in the proband. The OR1S2 (olfactory receptor 1 subfamily S2) variant was excluded because no pathogenic mutations have been identified for this gene and there is no evidence of olfactory deficits in the proband. In addition, while the CADD scores for the 2 OR1S2 compound heterozygous mutations were quite high, both parents were asymptomatic and heterozygous carriers were reported in the ExAC database and gnomAD. GRIK2 was the only gene that contained a heterozygous de novo mutation (table).
The elimination of other variants and the fact that ionotropic glutamate receptor (iGluR) genes have been linked to neurodevelopmental disorders makes the GRIK2 mutations the most probable to be causative for the child's symptoms. Two heterozygous variants in GRIK2 were identified. The first is a paternally inherited c.2601G>A variant that alters methionine 867 to an isoleucine (M867I) within a highly conserved region in the cytoplasmic C-terminal region. This variant has been associated with autism spectrum disorders.8 We eliminated it as a candidate gene, however, because it was found in 52 homozygotes in the gnomAD, it was present in the asymptomatic father of the proband, and it has minimal effect on receptor function.9 The second de novo GRIK2 variant, c.1969G>A, is predicted to change alanine 657, a highly conserved amino acid within the pore-forming M3 transmembrane domain, to a threonine (A657T; figure 2, A and B). This variant had a CADD score of 32, placing it among the top 0.1% most deleterious mutations in the human genome, and it was not found in the ExAC database or gnomAD (table).
Figure 2. Description of the GRIK2 variant.

(A) Location of the single point mutation in the GRIK2 gene responsible for the alanine to threonine amino acid substitution at position 657 within the highly conserved region of the GluK2 protein. (B) Cartoon depiction of a kainate receptor and crystal structure of an α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) showing the location of the (A657T) mutation within the M3 domain of ionotropic glutamate receptors. NTD and LBD stand for N-terminal domain and ligand-binding domain, respectively. Crystal structure for GluA2 AMPAR was obtained from the Protein Data Bank code 3H5V.
The A657T mutation in other iGluRs greatly affects channel kinetics and function.10–12 Rodents expressing delta receptors bearing the A657T mutation, also known as Lurcher mutant mice, show ataxia and cerebellar neurodegeneration,13 symptoms which are phenocopied in a human patient with the equivalent mutation in the GRID2 gene.14 Together, these data support the identification of the GRIK2 A657T variant as causative for some or all of the deficits in the proband.
The GluK2(A657T) mutation alters the biophysical properties of KARs.
KARs are glutamate-gated ion channels that play diverse roles in neuronal development, synaptic plasticity, and modulation of network excitability.2 It is unknown how the GluK2(A657T) mutation alters one or more of these functions to produce the developmental and behavioral deficits in the proband. Preliminary biophysical studies suggested that GluK2(A657T)-containing channels exhibited constitutive activation in the absence of agonist when analyzed in heterologous expression systems.10 Neuronal KARs are heteromeric combinations of distinct subunits together with auxiliary proteins that differ in stoichiometry in distinct neuronal populations. Thus, a critical first step toward relating the patient's symptoms to dysregulated receptor function is to more fully describe the biophysical properties of KARs that incorporate GluK2(A657T) subunits. We therefore performed whole-cell voltage-clamp recordings of HEK293T/17-expressing GluK2(A657T)-containing channels in various stoichiometries.
Homomeric receptors containing either the wild-type GluK2 or the GluK2(A657T) subunit responded to 10 mM glutamate applied for 1 second with robust inward currents (mean peak amplitudes: GluK2, 7.1 ± 1.1 nA, n = 8; GluK2(A657T), 1.7 ± 0.4 nA, n = 12; p < 0.001; figure 3A). During the glutamate application, GluK2(A657T) receptors desensitized less than the wild-type GluK2 (percent desensitization: GluK2, 99.8% ± 0.1%, n = 8; GluK2(A657T), 86.8% ± 2.1%, n = 12; p < 0.001; figure 3A), and the entry into the desensitized state was significantly slower than wild-type receptor currents (mean weighted tau for desensitization: GluK2, 4.4 ± 0.3 ms, n = 8; GluK2(A657T), 30.1 ± 7.9 ms, n = 11; p < 0.01; figure 3A).
Figure 3. Biophysical properties of kainate receptors bearing the GluK2(A657T) subunit.

Representative traces, peak amplitudes, tau of desensitization, and percent desensitization of inward currents evoked by a 1-second 10 mM glutamate (glu) application in (A) homomeric kainate receptors (KARs) bearing the GluK2 (A657T) subunit. (B) Similar traces and data for GluK2(A657T)/Neto2 KARs. (C) Representative traces, peak amplitudes, tau of desensitization, and difference current evoked by a 1-second 10 mM glutamate (glu) application in heteromeric KARs composed of the GluK2(A657T) and GluK5 subunits.
The auxiliary protein neuropilin and tolloid-like protein-2 (Neto2) alters KAR function and is a likely component of native KAR complexes.15 Glutamate-evoked currents from GluK2(A657T)/Neto2 KARs also had smaller amplitude currents and desensitized ∼10-fold more slowly than GluK2/Neto2 currents (amplitudes: GluK2/Neto2, 7.1 ± 1.0 nA, n = 7; GluK2(A657T)/Neto2, 1.7 ± 0.5 nA, n = 8; p < 0.001; tau desensitization: GluK2/Neto2, 14.5 ± 2.1 ms, n = 7; GluK2(A657T)/Neto2, 192.8 ± 24.9 ms, n = 8; p < 0.001; figure 3B). GluK2(A657T) reduced the percent desensitization of Neto2-containing receptors (GluK2/Neto2, 99.9 ± 0.02%, n = 7; GluK2(A657T)/Neto2, 75.0 ± 3.5%, n = 8; p < 0.001; figure 3B).
GluK2 and GluK5 subunits in part comprise postsynaptic KARs at the dentate granule cell to CA3 pyramidal cell synapse.16,17 We therefore compared the biophysical properties of heteromeric GluK2/GluK5 KARs-containing GluK2 wild-type or A657T subunits. Glutamate-evoked currents with unusual kinetic features from heteromeric GluK2(A657T)/GluK5 KARs (figure 3C). When glutamate was applied, the peak current from GluK2(A657T)/GluK5 receptors desensitized to a steady-state current that was more positive than the baseline holding current. The constitutive current amplitude, as measured by the difference between the holding current amplitude and the amplitude of the steady-state current, was 96.7 ± 43.3 pA (n = 8; figure 3C); by contrast, wild-type GluK2/GluK5 receptors did not exhibit a positive steady-state current and instead desensitized profoundly, by 99.4%, to steady-state current amplitudes similar to the baseline holding current. The atypical behavior of GluK2(A657T)/GluK5 KARs has been observed previously18 and results from partial activation of a subset of component subunits in nominally glutamate-free artificial cerebrospinal fluid (ACSF)-bathing solution. The peak amplitude of currents from wild-type and mutant GluK2/GluK5 receptors was similar (mean peak amplitude: GluK2/GluK5, 0.6 ± 0.2 nA, n = 8; GluK2(A657T)/GluK5, 0.6 ± 0.3 nA, n = 8, p = 0.88; figure 3C), but GluK2(A657T)/GluK5 receptors desensitized with a time course slower than that of GluK2/GluK5 receptors (mean weighted tau: GluK2/GluK5, 2.7 ± 0.3, n = 8; GluK2(A657T)/GluK5, 14.8 ± 5.6, n = 7, p < 0.05; figure 3C).
The unusual currents observed from GluK2(A657T)/GluK5 receptors strongly suggested that KARs incorporating the mutant subunit are constitutively activated or are acutely sensitive to trace levels of glutamate in nominally free ACSF.12 This hypothesis was consistent with our observation that mean holding currents of recordings from GluK2(A657T)-containing receptors were greater than those of their wild-type counterparts (Ihold: GluK2, 108 ± 20 pA vs GluK2(A657T), 458 ± 66, n = 8 and 12; p < 0.001; GluK2/Neto2, 36 ± 10 pA vs GluK2(A657T)/Neto2, 432 ± 126 pA, n = 7 and 8; p < 0.05; GluK2/GluK5, 117 ± 25 pA vs GluK2(A657T)/GluK5, 196 ± 57 pA, n = 8 and 8; p = 0.2; figure 4A).
Figure 4. Kainate receptors bearing the GluK2 subunit are constitutively active.

(A) Amplitude of holding current at −70 mV for HEK293T/17 cells expressing homomeric GluK2(A567T) kainate receptors (KARs) (left panel), GluK2(A657T)/Neto2 (middle panel), or heteromeric GluK2(A657T)/GluK5 KARs (right panel). (B) Representative traces demonstrating the change in holding current after a 1-second application of 20 μM NASPM in cells expressing homomeric GluK2(A657T) KARs (left panel, n = 4), GluK2(A657T)/Neto2 KARs (middle panel, n = 4), or heteromeric GluK2(A657T)/GluK5 KARs (right panel, n = 4). NASPM = 1-naphthyl acetyl spermine trihydrochloride.
As more direct means of detecting constitutive activation, we measured the effects of the open-channel blocker, NASPM, on the holding current amplitude. For homomeric GluK2(A657T) KARs, NASPM application displaced the holding current amplitude by 105 ± 44 pA from baseline, which corresponded to a 35% decrease in holding current amplitude and was 7.3 ± 1.8% of the peak glutamate-evoked currents in the same recordings (figure 4B). Thus, the constitutive current arises from activation of a substantial fraction of the receptors. This was even more striking for GluK2(A657T)/Neto2 receptors, in which NASPM reduced the holding current by 266 ± 64 pA (a 62% decrease from the holding current amplitude). The constitutive current was 10.2 ± 0.4% of the peak glutamate currents in these cells. Finally, NASPM also reduced the holding currents of heteromeric GluK2(A657T)/GluK5 KARs by 11%, with a positive average displacement of 8.1 ± 3.3 pA. The NASPM response represented a 1.7 ± 0.7% of the glutamate-evoked peak amplitude. In sum, these results demonstrate that incorporation of the GluK2(A657T) subunit into KARs produces constitutively active channels with significantly altered gating kinetics.
DISCUSSION
The genetic underpinnings of neurodevelopmental disorders and intellectual disability are varied.1 Genes coding for KAR subunits have been associated with intellectual disability,3–5,19–21 but these were all predicted to be loss-of-function mutations. By contrast, here we identified a de novo variant in the GRIK2 gene predicted to result in KARs with a gain of function; we propose that the aberrant KAR activity results in the congenital gait ataxia, developmental delays, and intellectual disability displayed by the child. No other de novo genetic variation plausibly accounted for the spectrum of neurodevelopmental deficits.
The single nucleotide mutation in GRIK2 is predicted to alter the naturally encoded A657 to a threonine. A657 is contained within the third transmembrane (M3) domain, a key determinant of receptor function15 and a highly conserved motif in the primary protein sequence of all members of the iGluR gene family. An analogous A657T “Lurcher” mutation in the GluD2 subunit of iGluRs in the delta family causes congenital gait ataxia in the eponymous Lurcher mouse model and in humans.14,22 In our study, we extended earlier biophysical analyses of GluK2(A657T)-containing receptors10,11 to show that the gating kinetics of KARs incorporating either Neto2 or GluK5 were altered by the incorporation of the mutated subunit. In addition, we showed that the constitutive activation of iGluRs bearing the GluK2(A657T) mutation is also affected by the stoichiometry of KAR complexes, as demonstrated by the varying efficacy of NASPM in reducing the basal constitutive current. Constitutive activation of GluK2(A657T)-containing KARs might be due to increased sensitivity to glutamate rather than by opening of the channel in the absence of ligand, as has been shown for α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors.12 Regardless of the precise mechanisms, our findings suggest that the GluK2(A657T) mutation identified in the proband is a putative gain-of-function mutation and that the effects of this mutation on KAR function in vivo depend on the precise subunit composition of the receptor complex.
The specific makeup of iGluR complexes in the CNS is tightly regulated; as a consequence, the stoichiometry, subunit composition, and association with different auxiliary proteins can vary between different brain regions and neuronal populations. GluK2-containing KARs are found throughout the nervous system including regions associated with cognition and motor coordination, such as hippocampal and cortical principal neurons, striatal projection neurons, and cerebellar granule cells.23,24 Thus, we expect that incorporation of the GluK2(A657T) subunit will cause aberrant KAR signaling to varying degrees in the CNS. The broad distribution of GluK2 contrasts with the neuroanatomic specificity identified for the GluD2 Lurcher subunit; in that, the constitutive activation induced by the A657T variant caused abnormal development and increased neuronal death only in the cerebellum25 because GluD2 expression is high and nearly exclusive to cerebellar Purkinje neurons. Because the proband in our study has mild microcephaly with grossly normal brain anatomy, we postulate that the GluK2(A657T) mutation causes subtle alterations in neuronal development and synaptic connectivity rather than excessive cell death by excitotoxicity. Genetic deletion of the mouse Grik2 gene delays the functional maturation of the hippocampal mossy fiber to CA3 pyramidal cell synapse,26,27 and pharmacologic activation of KARs modulates excitatory and inhibitory transmission in the developing brain.28–30 GluK2 knockout mice also show decreased associative learning of fear-inducing stimuli31 and an autistic-like phenotype characterized by deficits in social behaviors and reduced behavioral flexibility.32 Excitatory-inhibitory imbalances can result in improper network connectivity, profound deficits in synaptic plasticity, and cognitive impairments.33 Thus, the neurodevelopmental and learning disabilities identified in the proband might be explained by subtle alterations in neuronal development induced by faulty KAR function.
We report here that a gain-of-function mutation in a GRIK gene likely underlies developmental and learning disabilities. Future work should characterize the effects of this mutation on KAR function in vivo, as well as its effect on neuronal development and learning. Together, these studies might shed some insight into mechanisms underlying the human pathology.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge the help and cooperation of the many families involved with the Center for Rare Childhood Disorders, and in particular, they acknowledge the generous assistance of the family of the child reported in this study. They also acknowledge the contributions of all members (current and past) of the C4RCD Research Group. The C4RCD Research Group includes the clinical team and laboratory research team involved in patient enrollment, sample processing, exome sequencing, data processing, preparation of variant annotation files, data analysis, validation of data, and return of research data to families. Candidate genes are identified and discussed at data analysis meetings of the entire group. The following members of the group have contributed significantly to this work: Newell Belnap, Ana Claasen, Jason J. Corneveaux (deceased), Amanda Courtright, David W. Craig, Matt de Both, Brooke Hjelm, Matthew J. Huentelman, Ahmet Kurdoglu, Vinodh Narayanan, Keri M. Ramsey, Sampathkumar Rangasamy, Ryan Richholt, Isabelle Schrauwen, Ashley L. Siniard, and Szabolcs Szelinger.
GLOSSARY
- ACSF
artificial cerebrospinal fluid
- cDNA
complementary DNA
- eGFP
enhanced green fluorescent protein
- iGluR
ionotropic glutamate receptor
- NASPM
1-naphthyl acetyl spermine trihydrochloride
- PolyPhen2
Polymorphism Phenotyping v2
- WIRB
Western Institutional Review Board
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Yomayra F. Guzmán performed the in vitro experiments that characterized kainate receptors containing the GluK2(A657T) variant, analyzed the data, performed the statistical analysis for all in vitro experiments, and wrote and edited the manuscript. Jacob R. Stolz performed in vitro experiments characterizing kainate receptors containing the GluK2(A657T) variant and edited the manuscript. Keri Ramsey, David W. Craig, Mathew J. Huentelman, and C4RCD Research Group identified the GRIK2 gene variant, coordinated clinical research efforts, and edited the manuscript. Vinodh Narayanan performed clinical evaluation of the proband, identified the GRIK2 variant, and edited the manuscript. Geoffrey T. Swanson designed in vitro experiments and wrote and edited the manuscript.
STUDY FUNDING
This work was supported by a grant from the National Institute of Neurological Disease and Stroke to G.T.S. (R01 NS080598) and Y.F.G. (R01 NS080598-S1), donations to the Center for Rare Childhood Disorders, including private anonymous donations, and support from the State of Arizona to Translational Genomics Research Institute. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
DISCLOSURE
Dr. Guzmán has received research support from NIH/National Institute of Neurological Disease and Stroke. Ms. Ramsey has received research support from TGen Foundation. Mr. Stolz reports no disclosures. Dr. Craig has received research support from the Science Foundation Arizona, the Arizona Alzheimer's Research Center, NIH/NHLBI, and NIHNCI. Dr. Huentelman has received research support from NIH/National Institute of Neurological Disease and Stroke. Dr. Narayanan has served on the editorial board of the Journal of Pediatric Neurology and has received research support from Novartis. Dr. Swanson has received research support from Pfizer and NIH. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet 2016;17:9–18. [DOI] [PubMed] [Google Scholar]
- 2.Contractor A, Mulle C, Swanson GT. Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci 2011;34:154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cordoba M, Rodriguez S, Gonzalez Moron D, Medina N, Kauffman MA. Expanding the spectrum of Grik2 mutations: intellectual disability, behavioural disorder, epilepsy and dystonia. Clin Genet 2015;87:293–295. [DOI] [PubMed] [Google Scholar]
- 4.Motazacker MM, Rost BR, Hucho T, et al. A defect in the ionotropic glutamate receptor 6 gene (GRIK2) is associated with autosomal recessive mental retardation. Am J Hum Genet 2007;81:792–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonaglia MC, Ciccone R, Gimelli G, et al. Detailed phenotype-genotype study in five patients with chromosome 6q16 deletion: narrowing the critical region for Prader-Willi-like phenotype. Eur J Hum Genet 2008;16:1443–1449. [DOI] [PubMed] [Google Scholar]
- 6.Griffith TN, Swanson GT. Identification of critical functional determinants of kainate receptor modulation by auxiliary protein Neto2. J Physiol 2015;593:4815–4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lehtokari VL, Pelin K, Sandbacka M, et al. Identification of 45 novel mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Hum Mutat 2006;27:946–956. [DOI] [PubMed] [Google Scholar]
- 8.Jamain S, Betancur C, Quach H, et al. Linkage and association of the glutamate receptor 6 gene with autism. Mol Psychiatry 2002;7:302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han Y, Wang C, Park JS, Niu L. Channel-opening kinetic mechanism for human wild-type GluK2 and the M867I mutant kainate receptor. Biochemistry 2010;49:9207–9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kohda K, Wang Y, Yuzaki M. Mutation of a glutamate receptor motif reveals its role in gating and delta2 receptor channel properties. Nat Neurosci 2000;3:315–322. [DOI] [PubMed] [Google Scholar]
- 11.Taverna F, Xiong ZG, Brandes L, Roder JC, Salter MW, MacDonald JF. The Lurcher mutation of an alpha-amino-3-hydroxy-5-methyl- 4-isoxazolepropionic acid receptor subunit enhances potency of glutamate and converts an antagonist to an agonist. J Biol Chem 2000;275:8475–8479. [DOI] [PubMed] [Google Scholar]
- 12.Klein RM, Howe JR. Effects of the lurcher mutation on GluR1 desensitization and activation kinetics. J Neurosci 2004;24:4941–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vogel MW, Caston J, Yuzaki M, Mariani J. The Lurcher mouse: fresh insights from an old mutant. Brain Res 2007;1140:4–18. [DOI] [PubMed] [Google Scholar]
- 14.Coutelier M, Burglen L, Mundwiller E, et al. GRID2 mutations span from congenital to mild adult-onset cerebellar ataxia. Neurology 2015;84:1751–1759. [DOI] [PubMed] [Google Scholar]
- 15.Traynelis SF, Wollmuth LP, McBain CJ, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 2010;62:405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Copits BA, Swanson GT. Dancing partners at the synapse: auxiliary subunits that shape kainate receptor function. Nat Rev Neurosci 2012;13:675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Contractor A, Sailer AW, Darstein M, Maron C, Xu J, Swanson GT, Heinemann SF. Loss of kainate receptor-mediated heterosynaptic facilitation of mossy- fiber synapses in KA2-/- mice. J Neurosci 2003;23:422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swanson GT, Green T, Sakai R, et al. Differential activation of individual subunits in heteromeric kainate receptors. Neuron 2002;34:589–598. [DOI] [PubMed] [Google Scholar]
- 19.Haldeman-Englert CR, Chapman KA, Kruger H, et al. A de novo 8.8-Mb deletion of 21q21.1-q21.3 in an autistic male with a complex rearrangement involving chromosomes 6, 10, and 21. Am J Med Genet A 2010;152A:196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pickard BS, Malloy MP, Christoforou A, et al. Cytogenetic and genetic evidence supports a role for the kainate-type glutamate receptor gene, GRIK4, in schizophrenia and bipolar disorder. Mol Psychiatry 2006;11:847–857. [DOI] [PubMed] [Google Scholar]
- 21.Takenouchi T, Hashida N, Torii C, Kosaki R, Takahashi T, Kosaki K. 1p34.3 deletion involving GRIK3: further clinical implication of GRIK family glutamate receptors in the pathogenesis of developmental delay. Am J Med Genet A 2014;164A:456–460. [DOI] [PubMed] [Google Scholar]
- 22.Zuo J, De Jager PL, Takahashi KA, Jiang W, Linden DJ, Heintz N. Neurodegeneration in Lurcher mice caused by mutation in delta2 glutamate receptor gene. Nature 1997;388:769–773. [DOI] [PubMed] [Google Scholar]
- 23.Wisden W, Seeburg PH. A complex mosaic of high-affinity kainate receptors in rat brain. J Neurosci 1993;13:3582–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bahn S, Volk B, Wisden W. Kainate receptor gene expression in the developing rat brain. J Neurosci 1994;14:5525–5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishiyama J, Matsuda K, Kakegawa W, et al. Reevaluation of neurodegeneration in lurcher mice: constitutive ion fluxes cause cell death with, not by, autophagy. J Neurosci 2010;30:2177–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lanore F, Labrousse VF, Szabo Z, Normand E, Blanchet C, Mulle C. Deficits in morphofunctional maturation of hippocampal mossy fiber synapses in a mouse model of intellectual disability. J Neurosci 2012;32:17882–17893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marchal C, Mulle C. Postnatal maturation of mossy fibre excitatory transmission in mouse CA3 pyramidal cells: a potential role for kainate receptors. J Physiol 2004;561:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kidd FL, Coumis U, Collingridge GL, Crabtree JW, Isaac JT. A presynaptic kainate receptor is involved in regulating the dynamic properties of thalamocortical synapses during development. Neuron 2002;34:635–646. [DOI] [PubMed] [Google Scholar]
- 29.Lauri SE, Segerstråle M, Vesikansa A, et al. Endogenous activation of kainate receptors regulates glutamate release and network activity in the developing hippocampus. J Neurosci 2005;25:4473–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lauri SE, Vesikansa A, Segerstråle M, Collingridge GL, Isaac JT, Taira T. Functional maturation of CA1 synapses involves activity-dependent loss of tonic kainate receptor-mediated inhibition of glutamate release. Neuron 2006;50:415–429. [DOI] [PubMed] [Google Scholar]
- 31.Ko S, Zhao MG, Toyoda H, Qiu CS, Zhuo M. Altered behavioral responses to noxious stimuli and fear in glutamate receptor 5 (GluR5)- or GluR6-deficient mice. J Neurosci 2005;25:977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Micheau J, Vimeney A, Normand E, Mulle C, Riedel G. Impaired hippocampus-dependent spatial flexibility and sociability represent autism-like phenotypes in GluK2 mice. Hippocampus 2014;24:1059–1069. [DOI] [PubMed] [Google Scholar]
- 33.Nelson SB, Valakh V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 2015;87:684–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.