

ABSTRACT
mTOR has important roles in regulation of both innate and adaptive immunity, but whether and how mTOR modulates humoral immune responses have yet to be fully understood. To address this issue, we examined the effects of rapamycin, a specific inhibitor of mTOR, on B cell and CD4 T cell responses during acute infection with lymphocytic choriomeningitis virus. Rapamycin treatment resulted in suppression of virus-specific B cell responses by inhibiting proliferation of germinal center (GC) B cells. In contrast, the number of memory CD4 T cells was increased in rapamycin-treated mice. However, the drug treatment caused a striking bias of CD4 T cell differentiation into Th1 cells and substantially impaired formation of follicular helper T (Tfh) cells, which are essential for humoral immunity. Further experiments in which mTOR signaling was modulated by RNA interference (RNAi) revealed that B cells were the primary target cells of rapamycin for the impaired humoral immunity and that reduced Tfh formation in rapamycin-treated mice was due to lower GC B cell responses that are essential for Tfh generation. Additionally, we found that rapamycin had minimal effects on B cell responses activated by lipopolysaccharide (LPS), which stimulates B cells in an antigen-independent manner, suggesting that rapamycin specifically inhibits B cell responses induced by B cell receptor stimulation with antigen. Together, these findings demonstrate that mTOR signals play an essential role in antigen-specific humoral immune responses by differentially regulating B cell and CD4 T cell responses during acute viral infection and that rapamycin treatment alters the interplay of immune cell subsets involved in antiviral humoral immunity.
IMPORTANCE mTOR is a serine/threonine kinase involved in a variety of cellular activities. Although its specific inhibitor, rapamycin, is currently used as an immunosuppressive drug in transplant patients, it has been reported that rapamycin can also stimulate pathogen-specific cellular immunity in certain circumstances. However, whether and how mTOR regulates humoral immunity are not well understood. Here we found that rapamycin treatment predominantly inhibited GC B cell responses during viral infection and that this led to biased helper CD4 T cell differentiation as well as impaired antibody responses. These findings suggest that inhibition of B cell responses by rapamycin may play an important role in regulation of allograft-specific antibody responses to prevent organ rejection in transplant recipients. Our results also show that consideration of antibody responses is required in cases where rapamycin is used to stimulate vaccine-induced immunity.
KEYWORDS: B cell responses, CD4 T cells, Th1/Tfh response, humoral immunity, immunization, mTOR, rapamycin, viral immunity, viral infection
INTRODUCTION
As an evolutionarily conserved serine/threonine kinase, mTOR regulates several key cellular processes, including cell growth, proliferation, and metabolism, in response to various environmental cues (1–3). Rapamycin, a drug that specifically inhibits mTOR function, is currently used in transplant patients as an immunosuppressant to prevent immune responses against allografts (4, 5). One potential mechanism underlying this treatment is that rapamycin inhibits the proliferation of alloreactive T cells (6), but recent findings suggest that rapamycin has a variety of effects on the immune system and that there may be more complex mechanisms resulting in immunosuppression (7). A better understanding of the exact target cells and mechanisms of rapamycin-mediated immune suppression will facilitate the development of more effective and rational regimens to inhibit alloreactive immune responses with minimal suppression of pathogen-specific immunity.
Despite generally being considered an immunosuppressive drug, rapamycin also exhibits immunostimulatory effects in certain circumstances. Several reports have shown that rapamycin enhances inflammatory cytokine secretion and antigen presentation in macrophages and myeloid dendritic cells (8, 9). Moreover, inhibiting mTOR in CD8 T cells can promote memory T cell differentiation in response to infection and vaccination (10, 11). Thus, modulating mTOR activity has now become an attractive strategy to enhance dendritic cell- or CD8 T cell-mediated immunity during vaccination. However, considering that most successful vaccines confer protective immunity by inducing antibody responses (12), it is critical to understand whether and how mTOR regulates the generation of antigen-specific antibodies. Recent studies examined the role of mTOR in B cells and found that mTOR signals are required for production of high-affinity antibodies (13–15). These studies focused on B cell responses (13–15), but the interplay between CD4 T cells and B cells remains unclear in cases where mTOR signals are inhibited in both CD4 T cells and B cells in vivo, such as by rapamycin treatment. Thus, examining the relationship between CD4 T cell and B cell responses is important for a comprehensive understanding of the effects of rapamycin on humoral immunity, since antibody responses are coordinated by both CD4 T cells and B cells.
Following acute viral infections, antigen-specific naive CD4 T cells predominantly differentiate into Th1 and follicular helper T (Tfh) cells (16–19). Th1 cells play a critical role in mediating cellular immunity, while Tfh cells are the principal T cells that provide necessary help to B cells for the initiation and maintenance of germinal center (GC) reaction. Although the mTOR pathway has been shown to regulate differentiation of naive CD4 T cells into effector CD4 T cells (20–23), how in vivo rapamycin treatment influences effector and memory CD4 T cell differentiation has yet to be fully understood. Similar to that in CD4 T cells, the function of mTOR in B cell responses also remains to be determined.
In the present study, we attempted to examine how rapamycin influences B cell and CD4 T cell responses in vivo by using a mouse model of acute infection with lymphocytic choriomeningitis virus (LCMV). Our results showed that rapamycin treatment inhibited the generation of long-term antibody responses by reducing germinal center B cell formation. We also found that Tfh responses were significantly inhibited in rapamycin-treated mice, although the drug treatment enhanced overall memory CD4 T cell development. To further dissect the effect of rapamycin, we investigated the role of mTOR intrinsically in CD4 T cells and B cells in this study. Our results show that mTOR promotes antiviral humoral immunity by differentially regulating CD4 helper T cell and B cell responses.
RESULTS
Rapamycin inhibits B cell responses during viral infection and vaccination.
To understand the role of mTOR in humoral immunity during acute viral infections, rapamycin was administered to mice infected with LCMV strain Armstrong, which causes a systemic acute infection, with virus being cleared within 8 days after infection. Serum IgM and IgG antibodies specific for LCMV were examined at days 8, 15, and 60 postinfection (p.i.). We found comparable serum IgM titers between treated and untreated mice at day 8 postinfection (Fig. 1A, left panel). Although rapamycin-treated mice had slightly higher levels of virus-specific IgM titers on day 15 after infection, IgM responses in both groups were transient and were below the detection limit on day 60 after infection (Fig. 1A, left panel). In sharp contrast, rapamycin treatment led to reduced LCMV-specific IgG titers (Fig. 1A, right panel). The significant reduction in LCMV-specific IgG in rapamycin-treated mice was already seen at an early stage of infection (day 8) (Fig. 1A, right panel). Although IgG titers were increased at day 15 postinfection compared to those on day 8 for rapamycin-treated mice, they were much lower than those of control animals (Fig. 1A, right panel), suggesting that rapamycin inhibits or delays B cell activation/proliferation during the early stage of B cell responses after viral infection. Importantly, this reduction was maintained at the memory stage, and LCMV-specific IgG titers in rapamycin-treated mice were 10-fold lower than those in vehicle controls at day 60 postinfection (Fig. 1A, right panel). The lower IgG titers during the memory stage for the rapamycin treatment group suggest that the drug may negatively regulate GC reaction, because the generation of long-term LCMV-specific IgG responses is strictly GC B cell dependent (24). In agreement with this notion, rapamycin-treated mice exhibited a much lower frequency of PNAhigh FAShigh GC B cells at both days 8 and 15 postinfection (Fig. 1B, left panel), and the total number of PNAhigh FAShigh B cells in rapamycin-treated mice was about 10-fold lower than that in vehicle-treated mice at both time points (Fig. 1B, right panel). Likewise, the drug treatment blocked the formation of normal GC clusters (Fig. 1C). In keeping with these data, GC-derived LCMV-specific antibody-secreting cells (ASCs) in the bone marrow and memory B cells in the spleen were found to be greatly decreased in rapamycin-treated mice compared to vehicle-treated mice at day 60 postinfection (Fig. 1D and E, respectively). We also measured affinity maturation of antibody by using an enzyme-linked immunosorbent assay (ELISA) combined with a urea wash after the antibody had bound to the antigen on the plate (Fig. 1F and G). We found that rapamycin modestly impaired affinity maturation of LCMV-specific IgG (Fig. 1F and G).
FIG 1.

Rapamycin (Rapa) inhibits B cell responses during acute viral infection. B6 mice were infected with LCMV Armstrong, and rapamycin was administered from days −1 to 30 postinfection. (A) Analysis of LCMV-specific serum IgM (left) and IgG (right) levels at the indicated time points postinfection. (B) (Left) Frequencies of PNAhigh FAShigh B220+ germinal center (GC) B cells are shown in flow cytometry plots. (Right) Numbers of GC B cells in the spleen at the indicated time points. (C) GC histology in spleen sections from LCMV-infected mice treated with either vehicle or rapamycin from days −1 to 9 and sacrificed on day 10 postinfection. Bars, 10 μm. (D) Numbers of LCMV-specific antibody-secreting cells (ASCs) in bone marrow on day 60 postinfection (rapamycin treatment was performed on days −1 to 30 postinfection), calculated by ELISPOT assay. Individual representative wells for vehicle- and rapamycin-treated mice are shown above the bar graph. (E) LCMV-specific memory B cell numbers, analyzed on day 60 postinfection (rapamycin treatment was performed on days −1 to 30 postinfection). (F) LCMV NP-specific IgG in serum on day 60 after LCMV infection was detected by ELISA for mice treated or not with rapamycin. For affinity measurements, NP-captured IgG was washed with PBS or 8 M urea before detection. (G) To determine relative affinity, the ratio of the OD with urea to the OD without urea was calculated at a 1:200 serum dilution. Data show means and standard errors of the means (SEM) (n = 4 mice for each time point) and are representative of two independent experiments for panels A to E. For panels F and G, data from two independent experiments were combined (n = 8 mice for vehicle group and n = 5 mice for rapamycin group).
We next tested whether the inhibitory effects of rapamycin on B cell responses could be generalized to other immunization systems. To examine this, mice were immunized with NP-KLH in the presence or absence of rapamycin, and nucleoprotein (NP)-specific IgG titers were measured at days 8, 15, and 30 postimmunization. Significantly lower IgG titers were observed in rapamycin-treated mice than in vehicle-treated mice at day 8 postimmunization (Fig. 2A). Similar to the results for LCMV infection, rapamycin-treated mice showed increased IgG titers at days 15 and 30 postimmunization, but the titers never reached the levels seen for vehicle-treated mice (Fig. 2A). We also examined the effect of rapamycin during vaccination with a nonreplicating adenovirus serotype 5 vector (Ad5). Consistently, rapamycin treatment resulted in reduced Ad5-specific IgG titers throughout the observation period, and the drug-treated mice showed about 5-fold lower IgG titers than those of the vehicle controls at day 30 postvaccination (Fig. 2B). Collectively, these data indicate that rapamycin treatment significantly inhibits IgG B cell responses after viral infection or vaccinations.
FIG 2.
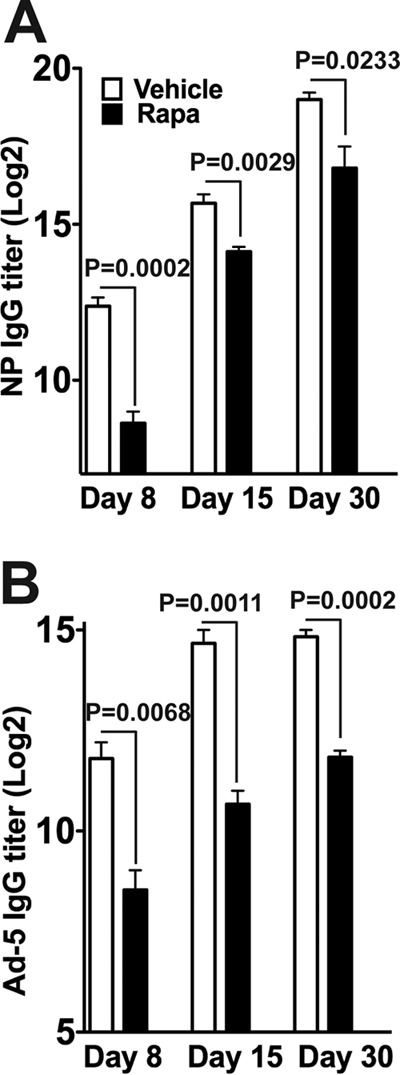
Rapamycin inhibits IgG responses in protein and Ad5 immunization models. B6 mice were immunized with either NP-KLH (precipitated in alum) (A) or Ad5 (B). Rapamycin was injected intraperitoneally daily for 30 days. Serum was harvested on the indicated days, and the antigen-specific IgG titer was quantified by ELISA. Graphs show means and SEM (n = 5 mice/time point for each group). Data representative of two independent experiments are presented.
Rapamycin inhibits early but not late B cell responses.
Next, in order to examine which stage of the B cell response is inhibited by rapamycin, we treated LCMV-infected mice with rapamycin during the early phase (days −1 to 14 p.i.), the late phase (days 16 to 30 p.i.), or the memory phase (days 90 to 120 p.i.) of B cell responses. In contrast to the severe inhibition of LCMV-specific IgG responses with rapamycin administered from days −1 to 14, rapamycin treatment at the late phase or memory phase of B cell responses had minimal to no effect on antibody titers as well as the formation of memory B cells/long-lived plasma cells (Fig. 3A to D). These data suggest that mTOR is dispensable during the late and memory phases of B cell responses but is crucial during the early phase, when B cells are actively proliferating with antigen stimulation and form GC.
FIG 3.

Rapamycin inhibits early but not late B cell responses. B6 mice were infected with LCMV Armstrong, and rapamycin was injected at the indicated times. (A) LCMV-specific IgG titers in serum for the indicated rapamycin treatment periods. The titer was measured at day 31 postinfection (rapamycin treatment days −1 to 14 or 16 to 30) or day 121 postinfection (rapamycin treatment days 90 to 120). (B) Numbers of LCMV-specific antibody-secreting cells (ASCs), analyzed at day 31 postinfection. (C) Numbers of memory B cells (MBCs), analyzed at day 31 postinfection. (D) Numbers of LCMV-specific ASCs, analyzed at day 121 postinfection. Graphs show means and SEM (n = 5 mice/group). Data representative of two independent experiments are presented.
Effect of rapamycin on expression of survival/apoptosis-related molecules in GC B cells.
The reduction in GC B cell number by rapamycin treatment may be attributed to lower survival of these cells than of cells exposed to vehicle treatment. To test this, we analyzed the expression of the survival/apoptosis-associated molecules Mcl-1, Bcl-xl, Bim, and active pan-caspase on GC B cells in spleens from rapamycin- or vehicle-treated mice on day 10 after infection. We observed comparable expression levels of Mcl-1, Bcl-xl, and Bim on GC B cells from both groups and lower levels of active pan-caspase for the rapamycin-treated group than for the vehicle-treated group (Fig. 4A to D). Since active pan-caspase is a marker of cells undergoing apoptosis, our data indicate that apoptosis of GC B cells is not the primary reason for the impaired B cell responses induced by rapamycin treatment during acute viral infection.
FIG 4.

Effect of rapamycin on expression of survival/apoptosis-related molecules in germinal center (GC) B cells. LCMV Armstrong-infected B6 mice were treated daily with rapamycin from day −1 postinfection. Flow cytometry analysis of the survival/apoptosis-related molecules Bim (A), Bcl-xl (B), Mcl-1 (C), and activated pan-caspase (D) on PNAhigh FAShigh B220+ germinal center B cells from vehicle- or rapamycin-treated mice was performed on day 10 postinfection. Data representative of two independent experiments are presented (n = 4 mice/group).
Rapamycin inhibits the proliferation of germinal center B cells.
One of the key features of GC B cells is their ability to proliferate rapidly and extensively during the early phase of the B cell response. To examine whether rapamycin inhibits GC B cell proliferation, LCMV-infected mice that had been treated with rapamycin or vehicle were given bromodeoxyuridine (BrdU) for 3 h before sacrifice on days 9 and 11 postinfection. The amount of BrdU incorporated into PNAhigh Fashigh B cells was 2- to 3-fold lower in rapamycin-treated mice at both time points (Fig. 5A), indicating that the proliferation of GC B cells was inhibited by rapamycin. Moreover, we found that compared to control mice, rapamycin-treated mice had a smaller fraction of CD86low CXCR4high dark-zone GC B cells (Fig. 5B), which possess intrinsically higher proliferation potential than their light-zone counterparts (25). We also analyzed the expression of the transcription factor B cell lymphoma 6 (Bcl-6; increases the proliferation of PNAhigh Fashigh B cells), and we found that rapamycin-treated mice had fewer Bcl-6+ PNAhigh Fashigh B cells than vehicle-treated mice (Fig. 5C). Finally, rapamycin treatment also impaired ongoing GC B cell proliferation when administered from days 9 to 14 postinfection (Fig. 5D). Together, these data show that rapamycin inhibits the proliferation of GC B cells, and this seems to be a primary cause of the impaired formation of memory B cells and long-lived plasma cells.
FIG 5.

Rapamycin inhibits germinal center B cell proliferation. B6 mice were infected with LCMV Armstrong. (A) Rapamycin was administered daily from day −1 of infection, and BrdU was given intraperitoneally 3 h prior to sacrifice. The percentages of BrdU+ cells in PNAhigh FAShigh germinal center (GC) B cells at the indicated time points are shown in the flow plots. (B) Flow cytometry analysis of dark-zone GC B cells (CXCR4high CD86low; gated on GC B cells) at day 8 postinfection (rapamycin treatment was performed on days −1 to 7). The bar graph shows percentages of dark-zone B cells among total GC B cells. (C) Frequencies of Bcl-6+ cells within the PNAhigh FAShigh B220+ GC B cell population on day 8 postinfection (rapamycin treatment was performed on days −1 to 7) as analyzed by flow cytometry and summarized in a bar graph. (D) The frequency of PNAhigh FAShigh GC B cells is shown for LCMV-infected mice treated with rapamycin or vehicle from days 9 to 14 postinfection (left; gated on B cells). BrdU staining is shown on the right for these PNAhigh FAShigh GC B cells (3-h pulse of BrdU) at 15 days postinfection. Graphs show means and SEM (n = 4 or 5 mice/group). Data representative of three independent experiments are presented.
Rapamycin enhances the formation of virus-specific CD4 T cell memory.
The results described above clearly establish that rapamycin treatment inhibits B cell responses. Because CD4 T cell help is required for optimal B cell responses, we next examined if rapamycin-treated mice would develop normal CD4 T cell responses. LCMV-specific CD4 T cell responses were monitored using the I-Ab GP66–77 tetramer. Interestingly, the administration of rapamycin from day −1 to day 30 postinfection substantially enhanced the number of virus-specific CD4 T cells at day 60 postinfection (Fig. 6A). We observed similar numbers of antigen-specific CD4 T cells at the peak of the responses (days 8 to 15 p.i.) between both groups, but rapamycin treatment increased the survival of antigen-specific CD4 T cells during the contraction phase (Fig. 6A). This reduced contraction is consistent with previously published data showing that rapamycin enhanced memory CD8 T cell formation (10). To examine the functionality of these memory CD4 T cells induced in the presence of rapamycin, we stimulated spleen cells with the LCMV GP61–77 peptide. Mice treated with rapamycin had around 3-fold higher frequencies of IFN-γ+ TNF-α+ and IFN-γ+ IL-2+ CD4 T cells than mice treated with the vehicle control (Fig. 6B). Thus, rapamycin was shown here to enhance the formation of functional antigen-specific memory CD4 T cells.
FIG 6.
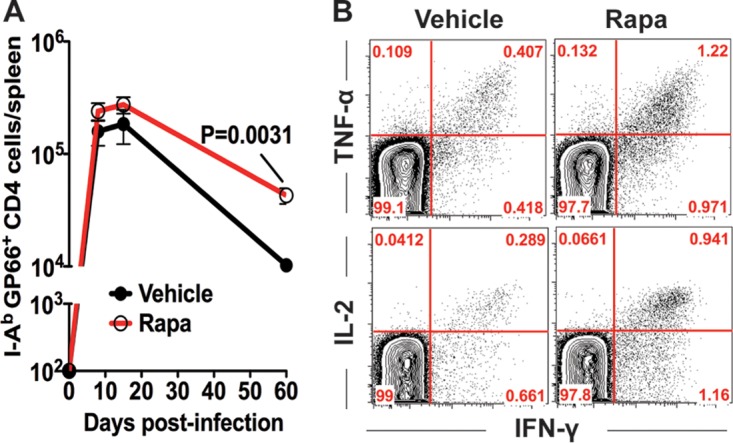
Rapamycin enhances virus-specific CD4 T cell responses. B6 mice were infected with LCMV Armstrong, and rapamycin was administered from days −1 to 30 postinfection. (A) Numbers of I-Ab GP66–77 tetramer-positive LCMV-specific CD4 T cells in the spleen (n = 4 mice per group). (B) Cytokine production by LCMV-specific CD4 T cells following stimulation with the GP61 peptide at 35 days postinfection (gated on CD4 T cells). Data representative of three independent experiments are presented.
Rapamycin treatment alters the balance between Th1 and Tfh cell differentiation.
The observation that rapamycin markedly impaired antigen-specific B cell responses despite a larger number of memory CD4 T cells prompted us to investigate if the drug treatment would modulate the differentiation program of antigen-specific CD4 T cells. Acute LCMV infection is known to induce two distinct CD4 T cell subsets: Th1 and Tfh cells (17). Therefore, a critical question to address was whether the balance of the differentiation of antigen-specific Th1 and Tfh subsets would be altered in rapamycin-treated LCMV-infected mice in which GC B cell responses were compromised. To extensively analyze Tfh responses, we took advantage of the LCMV I-Ab GP66–77-specific T cell receptor (TCR)-transgenic Smarta CD4 T cell system. Smarta chimeric mice were made by adoptively transferring naive Smarta CD4 T cells into B6 mice, and these chimeric mice were subsequently infected with LCMV. Consistent with the results seen with endogenous virus-specific memory CD4 T cells, rapamycin increased the number of memory Smarta CD4 T cells (data not shown). Using this system, we first analyzed CXCR5 expression, which is one of the key features of Tfh cells, directing the migration of primed Tfh cells to B cell follicles (26). We found that rapamycin strongly inhibited CXCR5 expression on Smarta cells at day 8 postinfection (Fig. 7A), suggesting that Tfh cell generation was impaired in rapamycin-treated mice. To further characterize Tfh differentiation in the presence of rapamycin, the expression of several other canonical markers of Tfh cells was examined. ICOS and PD-1 are well-known cell surface markers of Tfh cells, and we observed markedly decreased numbers of ICOShigh CXCR5high and PD-1high CXCR5high antigen-specific CD4 T cells in rapamycin-treated mice (Fig. 7B and C). Such decreased numbers of antigen-specific Tfh cells were also confirmed by lower expression of Bcl-6 (Fig. 7D), the transcriptional factor governing Tfh lineage differentiation (27). Together, these data indicate that rapamycin treatment impairs formation of the Tfh cell population.
FIG 7.

Rapamycin treatment modulates helper CD4 T cell differentiation. LCMV-specific transgenic CD4 T cells (Smarta cells; Thy1.1+) were adoptively transferred to B6 mice (Thy1.2+), followed by infection with LCMV Armstrong. These mice were treated with rapamycin from day −1 to day 7. Smarta cells in the spleen were analyzed at day 8 postinfection. (A) CXCR5 expression on Smarta cells. MFI, mean fluorescence intensity. (B) Flow cytometry analysis of ICOShigh CXCR5+ and PD-1high CXCR5+ Tfh cells (gated on Smarta cells). (C) Numbers of Tfh cells (ICOShigh CXCR5+), summarized from the data in panel B. (D) Percentages of Bcl-6high CXCR5+ Tfh cells among total Smarta cells. (E) Production of IFN-γ by Smarta cells after stimulation with the LCMV GP61 peptide. (F) Frequencies of Th1 cells (CXCR5− granzyme B+) in Smarta cells. For all experiments, n = 4 to 6 mice/group. Graphs show means and SEM. Data are representative of 3 independent experiments.
Given that only Th1 and Tfh subsets are induced in response to LCMV infection, the negative role of rapamycin in Tfh differentiation suggested that it may instead promote Th1 differentiation. To test this hypothesis, we compared Th1 responses between rapamycin-treated and untreated mice. We first measured the frequencies of gamma interferon (IFN-γ)-producing cells in antigen-specific CD4 T cells after ex vivo stimulation with peptide. In the presence of rapamycin, around 75% of antigen-specific CD4 T cells were found to secrete IFN-γ, whereas only about 50% of their counterparts from vehicle-treated mice secreted IFN-γ (Fig. 7E). Next, the expression of another Th1 marker, granzyme B (GZB), was also examined on antigen-specific CD4 T cells obtained from LCMV-infected mice in the presence or absence of rapamycin treatment. Likewise, the percentage of GZB+ CXCR5low cells was substantially increased by rapamycin (Fig. 7F). Together, these data indicate that rapamycin skews virus-specific effector CD4 T cell differentiation from Tfh to Th1 subsets.
CD4 T cell-intrinsic inhibition of mTOR signals impairs Th1 formation.
Our data clearly show that inhibiting mTOR with rapamycin reduces both Tfh and B cell responses. Since Tfh and GC B cells reciprocally regulate each other (28), it is important to understand whether rapamycin intrinsically inhibits B cell or CD4 T cell responses. To examine this, we inhibited mTOR signaling exclusively in antigen-specific CD4 T cells by using a retroviral gene knockdown system (Fig. 8A). Retrovirus-transduced or nontransduced Smarta cells were adoptively transferred into B6 mice, followed by LCMV infection (Fig. 8A). This system allowed us to directly compare Th1 and Tfh differentiation patterns of transduced (GFP+) and nontransduced (GFP−) Smarta cells within the same animal (Fig. 8B). In contrast to the effect of rapamycin administration, we found that mTOR knockdown in antigen-specific CD4 T cells resulted in inhibition of Th1 differentiation, as demonstrated by lower frequencies of the CXCR5low GZBhigh population (Fig. 8C). Furthermore, mTOR knockdown also inhibited the expression of the Th1 lineage-associated transcription factor T-bet (Fig. 8D). Conversely, mTOR knockdown increased the frequencies of Tfh cells, characterized as CXCR5high PD-1high cells (Fig. 8E). Notably, all these results were faithfully recapitulated by the knockdown of raptor (a necessary component of mTORC1), with a decreased CXCR5low GZBhigh Th1 subset as well as reduced T-bet and increased CXCR5+ PD-1high Tfh populations (Fig. 8F to H).
FIG 8.

Inhibiting mTOR signals intrinsically in antigen-specific CD4 T cells impairs Th1 cell formation. (A) Experimental design for mTOR or raptor knockdown in LCMV-specific transgenic CD4 T cells (Smarta cells). Retrovirus-transduced Smarta cells were transferred into B6 mice, followed by LCMV Armstrong infection. The differentiation of Smarta cells into Th1 and Tfh cells in the spleen was analyzed at day 10 postinfection. (B) Retrovirus-transduced (GFP+) and nontransduced (GFP−) Smarta cells were compared in the same animal. (C to E) In mTOR knockdown experiments, the frequency of CXCR5− granzyme B+ Th1 cells (C), the MFI of T-bet (D), and the frequency of PD-1high CXCR5+ Tfh cells (E) were analyzed. (F to H) In raptor knockdown experiments, similar analyses were done. For these experiments, n = 4 to 6 animals per group. Data representative of 3 independent experiments are presented. (I) Experimental design for the experiments performed for panels J to L. Retrovirus-transduced Smarta cells were cultured for 3 days. GFP+ Smarta cells were sorted and then transferred to B6 mice, followed by LCMV Armstrong infection. Total numbers of GFP+ Smarta cells (J), Th1 cells (K), and Tfh cells (L) were calculated. Data representative of two independent experiments are presented (n = 4 mice/group).
Since we transferred Smarta transgenic CD4 T cells immediately after retrovirus transduction (before expressing green fluorescent protein [GFP]) in the above-described experiments, how many retrovirus-transduced Smarta cells were adoptively transferred into the recipient mice was unknown. This prevented us from examining whether the number of GFP+ Tfh cells was increased by mTOR or raptor knockdown compared to the control level in this experimental setting. To address this issue, we sorted GFP+ retrovirus-transduced Smarta cells (raptor-specific short hairpin RNA [raptor-shRNA] or vector control) and then transferred the same number of these cells into recipient mice, followed by LCMV infection. The total number of raptor-shRNA-transduced Smarta cells was much lower than that of vector-transduced Smarta cells on day 8 after infection (Fig. 8J). In particular, the reduction in cell numbers was much more severe for Th1 cells (Fig. 8K and L). Taken together, consistent with previous observations (21, 23), the data show that selective attenuation of the mTORC1 signaling pathway in antigen-specific CD4 T cells inhibits the generation of Th1 cells after acute viral infection. In addition, our results also suggest that the increased Th1 cell formation observed in rapamycin-treated mice was not due to reduced mTOR activity in antigen-specific CD4 T cells.
mTOR acts intrinsically in B cells to regulate germinal center responses.
The above-described RNA interference (RNAi) experiments indicate that Tfh cells are not the primary target cells of rapamycin for the impaired humoral immunity observed in drug-treated mice after viral infection or immunization. Therefore, we next asked if rapamycin has any intrinsic effects on B cell responses. To address this issue, we generated rapamycin-insensitive B cells by knocking down FKBP12, which is an essential binding partner for rapamycin to inhibit mTORC1 signals (2). We transduced bone marrow (BM) hematopoietic stem cells (HSCs) with an FKBP12 shRNA retrovirus and subsequently transferred the transduced HSCs into lethally irradiated recipient mice (Fig. 9A). After reconstitution, these bone marrow chimeric mice allowed us to analyze the intrinsic effect of rapamycin on B cells (Fig. 9A). In this experimental setting, we observed that approximately 10% to 30% of total donor B cells were transduced, as they were GFP positive (Fig. 9B). In addition, we observed normal matured B cell populations (Fig. 9C). To validate the effect of FKBP12 knockdown on B cells in response to rapamycin, we compared mTOR activities by measuring phosphorylated S6 in FKBP12 shRNA-transduced or non-shRNA-transduced B cells upon anti-B cell receptor (anti-BCR) stimulation in the absence or presence of rapamycin. FKBP12 shRNA-transduced GFP+ B cells showed increased S6 phosphorylation in the presence of rapamycin, while the drug strikingly inhibited the phosphorylation of S6 in nontransduced B cells upon anti-BCR stimulation (Fig. 9D), indicating the reliability of our system. These BM chimeric mice were then infected with LCMV in the presence of rapamycin, and GC B cell differentiation levels between transduced (GFP+) and nontransduced (GFP−) donor B cells (Ly5.1+ B220+) were compared within the same individual animals. In mice with HSCs transduced with control retrovirus, both nontransduced (GFP−) and transduced (GFP+) cells were unable to efficiently differentiate into GC B cells (around 2% of total donor B cells) (Fig. 9E). However, in BM chimeric mice with HSCs harboring FKBP12 shRNA, rapamycin-insensitive B cells (Ly5.1+ B220+ GFP+) gave rise to a significantly higher frequency of GC B cells than that seen for rapamycin-sensitive, nontransduced donor B cells (Ly5.1+ B220+ GFP−) (Fig. 9E). In addition, higher LCMV-specific IgG titers were seen in BM chimeric mice harboring the FKBP12 knockdown than in control mice (Fig. 9F), suggesting that rapamycin-insensitive B cells triggered a higher functional B cell response in the presence of rapamycin. Furthermore, Tfh responses were improved in rapamycin-treated LCMV-infected BM chimeric mice with HSCs transduced with the FKBP12 shRNA retrovirus (Fig. 9G). This indicates that GC B cell formation is important for Tfh responses and also suggests that the decreased Tfh responses seen in rapamycin-treated normal B6 mice (Fig. 7) are likely due to inhibition of GC B cell formation. Thus, these data demonstrate that B cells are the primary target cells of rapamycin for the impairment of antibody responses and that B cell-autonomous mTOR signaling plays a critical role in promoting GC B cell responses.
FIG 9.

mTOR acts intrinsically in B cells to regulate germinal center (GC) responses. (A) Experimental design. Bone marrow hematopoietic stem cells (HSCs; Ly5.1+) were transduced with retroviruses encoding scramble control or FKBP12 shRNA, and then these cells were transferred into irradiated B6 (Ly5.2+) mice. After reconstitution, mice were infected with LCMV and treated with rapamycin for 11 days. Mice were sacrificed at day 12 postinfection, and splenocytes were analyzed. (B to D) Validation of retrovirus-transduced B cells in chimeric mice before infection. (B) Retrovirus transduction (GFP expression) of B cells is shown in the flow plot before LCMV infection. After reconstitution, transduced donor B cells in peripheral blood mononuclear cells (PBMC) were demonstrated as GFP+ CD19+ Ly5.1+ cells. (C) B cell maturity was shown in PBMC before LCMV infection. The maturation of transduced donor B cells (GFP+) was compared to that of nontransduced B cells in terms of surface IgM and IgD expression by flow cytometry. (D) Flow cytometry analysis of phosphorylated S6 (pS6) of FKBP12 shRNA-transduced B cells from BM chimeric mice. Total B cells were stimulated with anti-mouse IgM μ chain for 1 h in the presence or absence of rapamycin, followed by flow cytometry analysis of pS6. (E) Frequencies of germinal center B cells in transduced (GFP+; green squares) and nontransduced (GFP−; open circles) donor B cells (Ly5.1+ B220+) from the spleen. Each line represents a comparison of germinal center B cell frequencies between transduced and nontransduced B cells in the same animal. (F) LCMV-specific IgG titers in sera from mice with control or FKBP12 shRNA retrovirus-transduced HSCs. (G) Frequencies of Tfh cells (ICOShigh CXCR5high) among CD44high CD4 T cells in the control and FKBP12 shRNA groups (n = 5 mice/group). Data show means and SEM. Data are representative of two independent experiments.
mTOR activity is dispensable for LPS-induced B cell responses.
In contrast to BCR-dependent antibody responses to viral infection or protein immunization, mitogens, such as lipopolysaccharide (LPS), can induce fast B cell proliferation and plasma cell differentiation independently of BCR engagement (29). We wondered whether rapamycin would also block B cell responses under such conditions. To examine this question, LPS was administered intravenously to mice, which were treated with vehicle or rapamycin from days −3 to 2, and B cell responses were analyzed at days 1 and 3 post-LPS injection. We found that LPS injection upregulated S6 phosphorylation on day 1 and that rapamycin treatment efficiently blocked its phosphorylation (Fig. 10A). However, the activation of polyclonal B cells was not inhibited by rapamycin, and we observed a comparable upregulation of both CD86 and major histocompatibility complex class II (MHC II) molecules on day 1 (Fig. 10B). More intriguingly, BrdU incorporation experiments showed that the proliferation of these activated B cells was also not influenced by rapamycin on both days 1 and 3 (Fig. 10C). Thus, we detected comparable numbers of BrdU+ B cells between vehicle- and rapamycin-treated mice (Fig. 10C and D). Lastly, we analyzed the differentiation of activated B cells (CD86high MHC IIhigh) into plasma cells (IgD− CD138+). We found that rapamycin-treated mice had plasma cells in numbers similar to those in vehicle-treated mice (Fig. 10E). Therefore, these data indicate that in contrast to viral infection or protein immunization, rapamycin has minimal effects on mitogen-induced B cell activation, proliferation, and plasma cell differentiation.
FIG 10.

mTOR activity is dispensable for LPS-induced B cell responses. B6 mice were given LPS intravenously and were treated daily with rapamycin from day −3 to day 2 of LPS injection. BrdU was administered 3 h before mouse sacrifice. On either day 1 or day 3, mice were sacrificed and their splenocytes analyzed. (A) LPS induced mTOR activation, as indicated by phospho-S6 (pS6) staining on day 1 post-LPS administration. (B) B cell activation on day 1, identified by CD86 and MHC II staining. (C) Proliferation of B cells as revealed by flow cytometry analysis of BrdU incorporation at the indicated time points. (D)Total BrdU+ B cell numbers at day 3. (E) Flow cytometry analysis of the frequency of antibody-secreting cells (ASCs; IgD− CD138high) in the vehicle or rapamycin treatment group (gated on CD4− CD8− cells). (F) Total numbers of ASCs. The presented data are representative of 2 independent experiments.
DISCUSSION
In this study, we analyzed the effects of rapamycin on B cell responses as well as Th1 versus Tfh differentiation during acute viral infection and immunization. Our data revealed that rapamycin treatment acted intrinsically in B cells to inhibit GC B cell responses and that this led to the subsequent impairment of B cell responses. Despite such reduced B cell responses, rapamycin was able to enhance memory CD4 T cell formation. However, the drug treatment resulted in strikingly biased CD4 T cell differentiation toward Th1 cells and decreased Tfh cell formation. This skewed CD4 T cell differentiation was likely due to impaired GC B cell responses in the presence of rapamycin treatment. Indeed, when we targeted mTOR or raptor intrinsically in antigen-specific CD4 T cells by RNAi without disturbing mTOR signals in B cells, Th1 cell formation was inhibited. Thus, these findings demonstrate that mTOR signals differentially regulate CD4 T cell and B cell responses and that rapamycin predominantly blocked germinal center B cell reaction.
Consistent with a previous study, we found that inhibition of mTORC1 activity in antigen-specific CD4 T cells by RNAi had a minimal impact on the generation of Tfh cells (23). In contrast, recent studies using conditional knockout mice showed that mTORC1 signaling was essential for induction of Tfh responses (30, 31). These contradictory results may be explained by the different approaches used to silence mTOR activity. RNAi usually inhibits expression of 70 to 95% of target transcripts, but 5 to 30% of them can remain in the cells, whereas conditional knockout genetically deletes target genes. Thus, these two silencing methods result in different degrees of inhibition of mTOR signals, and very low levels of mTOR activity might be sufficient to generate Tfh cells after viral infection.
Another important issue that remains unresolved is that of Th1 responses in LCMV-infected rapamycin-treated mice. As shown in Fig. 8, inhibiting mTOR signaling intrinsically in CD4 T cells by RNAi inhibited Th1 responses. However, we observed that LCMV-infected rapamycin-treated mice generated normal Th1 responses. Since mTOR is expressed ubiquitously in many cells, the normal Th1 responses may have been mediated by other types of cells, such as innate immune cells, stimulated by rapamycin treatment (32). In addition, since rapamycin treatment during acute LCMV infection causes a delay of viral clearance for 2 days, this may also affect CD4 T cell differentiation. Thus, such environmental factors changed by rapamycin might override the intrinsic effect of the drug on antigen-specific CD4 T cells.
Our findings may shed some light on the effect of rapamycin in transplantation patients. Although it has long been postulated that the immunosuppressive activity of rapamycin in transplant patients is mediated by its antiproliferative effect on T cells (4), a growing body of evidence suggests that rapamycin may employ multiple mechanisms to regulate immune responses (7). More paradoxically, rapamycin has been found to enhance memory CD8 T cell differentiation (10, 11). In this study, our data demonstrated that rapamycin treatment also promoted memory CD4 T cell differentiation. Together, these facts raise the possibility that additional mechanisms may exist for rapamycin-mediated inhibition of allograft rejection. We also show here that rapamycin efficiently inhibited in vivo B cell responses in both viral infection and protein immunization models. Given the important role of B cell responses in mediating both acute and chronic allograft rejection (33), these data suggest that rapamycin may exert its effect in preventing organ rejection at least partly through the inhibition of alloantigen-specific B cell responses. Consistent with this notion, it was reported that germinal center B cells were involved in a mouse model of graft-versus-host disease and that everolimus (a rapamycin analog) was able to suppress the disease (34). Thus, our present study provides novel insight into the potential immunosuppressive effects of rapamycin in transplant patients and warrants more detailed investigations regarding rapamycin's effects on B cell-mediated allorejection.
The data presented here demonstrate that the impaired GC B cell response induced by rapamycin treatment primarily accounts for the inhibition of Tfh differentiation as well as antibody responses. Consistent with previous papers (13–15), we identified that rapamycin-mediated GC B cell impairment was caused by inhibition of GC B cell proliferation. Importantly, dark-zone centroblasts that have a higher proliferative potential were more strikingly inhibited by rapamycin treatment. Interestingly, inhibition of antigen-specific IgG responses by rapamycin was observed as early as day 8 postinfection or postimmunization, when short-lived extrafollicular plasma cells provide a majority of serum IgG and GC B cells have a minimal contribution to antibody responses. These data suggest that proliferation of activated B cells during the early phase of infection (days 0 to 8) may be inhibited or delayed by rapamycin, similar to what was seen for GC B cell proliferation. In contrast to such early-stage B cell responses, our results show that rapamycin has minimal to no effect during the differentiation of GC B cells into long-lived plasma cells as well as on the maintenance of already-differentiated long-lived plasma cells. Together, these lines of evidence suggest that GC B cells demand higher mTOR activity to sustain their rapid and extensive growth and proliferation, both of which are heavily dependent on mTOR-mediated glycolytic metabolism to provide substrates necessary for protein, lipid, and DNA biosynthesis (35, 36). In addition, Rajewsky and colleagues showed that tonic BCR-mediated phosphatidylinositol 3-kinase (PI3K)–Akt activation, one of the signaling events upstream of mTOR, is important for the survival of resting mature B cells (37), and our data extend the role of the PI3K-Akt-mTOR axis as the major regulator of GC B cell formation when BCR is engaged with a cognate antigen and T cell help. Interestingly, we did not observe a measurable impairment of B cell proliferation by rapamycin treatment in response to in vivo LPS stimulation. We speculate that mTOR signaling is required for BCR-mediated B cell proliferation while being dispensable for BCR-independent, mitogen-mediated proliferation, which may depend more strictly on the Toll-like receptor-induced canonical NF-κB pathway.
Overall, our data suggest that B cell responses are a concern for the use of rapamycin to enhance vaccine-induced immunity, although there are clear positive effects on the formation of memory CD8 T cells as well as memory CD4 T cells. To overcome this issue, it is important to study the molecular mechanisms for how mTOR signaling pathways regulate BCR-dependent B cell responses in vivo. Furthermore, we examined the role of mTOR in primary B cell responses in this study, but it is unclear whether mTOR activity is required for secondary B cell responses, in which memory B cells are activated upon recognition of cognate antigens. Many existing vaccine regimens utilize prime-boost strategies to maximize protective immunity. Thus, for improvements of vaccine-induced immunity by modulation of mTOR activity, it is essential to examine if rapamycin inhibits secondary B cell responses similarly to primary responses or if mTOR signals have a minimal contribution to secondary B cell responses.
In summary, we characterized the complex role of mTOR activity in regulating Tfh versus Th1 differentiation as well as B cell responses during viral infection and immunization. These findings provide valuable insights into the precise modulation of mTOR signals in certain immune cell types to improve vaccination effectiveness or to alleviate pathogenic immune responses. Furthermore, our study suggests that inhibiting B cell responses by use of rapamycin is a potential immunosuppressive mechanism and that rapamycin may prevent the generation of allograft-reactive B cell responses in transplant recipients.
MATERIALS AND METHODS
Mice, adoptive transfer, viral infection, and rapamycin treatment.
Eight- to 12-week-old C57BL/6J (B6) (Thy1.2+ or Ly5.1+) mice were purchased from Jackson Laboratory. Thy1.1+ or Ly5.1+ Smarta TCR (specific to LCMV I-Ab GP66–77)-transgenic mice were fully backcrossed to C57BL/6 mice in our animal facility. For Smarta cell transfer, 2 × 104 naive Smarta CD4 T cells were transferred intravenously into naive B6 mice. Mice were infected intraperitoneally with 2 × 105 PFU of LCMV Armstrong. Rapamycin was administered intraperitoneally daily (75 μg/kg of body weight) (10). Control mice were given vehicle without rapamycin. All mice were used in accordance with approved animal protocols at Emory University.
NP-KLH and Ad5 immunization and in vivo LPS stimulation.
For NP-KLH immunization, 50 μg of NP-KLH (Biosearch) was adsorbed by use of alum (Pierce) and subsequently injected into B6 mice. Ad5 immunization was performed by intramuscularly immunizing mice with 1 × 108 viral particles of replication-incompetent Ad5 in both hind leg muscles. For LPS experiments, 15 μg of LPS (Enzo Life Sciences) was given intravenously to mice.
Retroviral transduction in Smarta cells.
The pMKO.1-GFP plasmid (Addgene plasmid 10676, provided by W. Hahn), with insertions of short hairpin RNA (shRNA) sequences targeting mTOR and raptor, was reported previously (10). The recombinant retrovirus package was achieved by cotransfection of the pMKO.1 plasmid and pCl-Eco (Imgenex) into 293T cells by use of TranIT-293 reagent (Mirus). Forty-eight hours after transfection, the supernatant containing packaged virus was harvested. Smarta cells were activated in vivo by injection of 200 μg of GP61–77 peptide into transgenic mice. Eighteen hours later, activated Smarta cells were isolated from spleens and purified by use of a CD4 T cell negative-selection column (Miltenyi). Activated Smarta cells were mixed with freshly harvested retrovirus supernatant containing 8 μg/ml Polybrene (Sigma) and 10 ng/ml of interleukin-2 (IL-2) (R&D), and then the cells were spin transduced at 37°C for 90 min. A total of 2 × 104 transduced Smarta cells were transferred to naive mice, followed by LCMV infection. In some experiments, empty vector- or shRNA-transduced CD45.1+ GFP+ Smarta cells were sorted after 72 h of culture in the presence of 10 ng/ml IL-2, and 2 × 104 cells were transferred to wild-type recipient mice (CD45.2), followed by LCMV infection.
Retrovirus transduction of bone marrow hematopoietic stem cells.
Bone marrow HSCs were transduced with retrovirus by use of previously described methods (38), with minor modifications. Briefly, bone marrow cells were isolated from Ly5.1+ 5-fluorouracil-treated mice, and then the cells were cultured overnight in the presence of IL-3, IL-6, and stem cell factor (R&D). To generate retroviruses expressing scramble shRNA or FKBP12 RNA, we used a modified pMKO.1 GFP vector in which the promoter of the GFP marker was changed from the simian virus 40 (SV40) promoter to the PGK promoter. The retroviruses were made as described above and were concentrated using Retro-Concentin (System Biosciences) to improve transduction efficiency. Bone marrow cells stimulated with cytokines were spin transduced at 37°C for 90 min with concentrated retrovirus containing 4 μg/ml of Polybrene and the three cytokines (IL-3, IL-6, and stem cell factor). These transduced bone marrow cells were cultured overnight once again in the presence of the three cytokines, followed by a second round of spin infection. Retrovirus-transduced bone marrow cells were adoptively transferred into lethally irradiated (2 doses of 550 cGy, 3 h apart) Ly-5.2+ B6 mice.
Flow cytometry.
Flow cytometry data were acquired by use of a Canto II flow cytometer (BD Biosciences) and analyzed with Flow Jo (Tree Star). An MHC II tetramer was provided by the NIH tetramer core facility. Antibodies used for staining were obtained from BD Biosciences, Biolegends, eBiosciences, and Vector Lab. Tfh staining was performed as described previously (19). Bcl-6 staining was performed with Foxp3 staining buffer from eBiosciences. MHC II tetramer staining was done by incubating the tetramer with cells at 37°C for 3 h, followed by surface staining with other markers. For in vivo BrdU incorporation, mice were given BrdU (3 mg BrdU in 0.5 ml phosphate-buffered saline [PBS] for a 3-h pulse experiment) intraperitoneally. BrdU in B cells was stained by use of a BrdU flow kit according to the manufacturer's instructions (BD Biosciences).
ELISA, ELISPOT assay, and memory B cell assay.
For ELISA experiments, plates were coated with NP-OVA, Ad5 particles, or LCMV-infected cell lysates, and antigen-specific antibodies were detected with horseradish peroxidase (HRP)-conjugated anti-mouse IgG or IgM secondary antibodies. To determine relative affinity, a recombinant LCMV nucleoprotein was used as described previously (39). Briefly, after antibody binding, the ELISA plates were incubated with 8 M urea, and the ratio of optical density (OD) with urea to the OD without urea was calculated at a 1:200 serum dilution to examine the reduction in NP-specific antibody titer. An enzyme-linked immunosorbent spot (ELISPOT) assay to detect antibody-secreting cells as well as memory B cells was performed as previously described (24).
Immunofluorescence assay.
Immunofluorescence staining of germinal center B cells was performed as described previously (40). Briefly, frozen tissue sections were cold fixed in acetone for 3 min at 4°C, blocked with 10% normal goat serum, and stained with biotin-peanut agglutinin (PNA), Alexa 555 dye-labeled anti-IgD, and Alexa 647 dye-labeled anti-CD4, followed by Alexa 488 dye-labeled avidin. After each step, cells were washed at least three times with 0.1% Tween 20 in PBS. Coverslips were mounted on slides by use of an antifade kit (Molecular Probes) and then examined using a Zees LSM 510 confocal fluorescence microscope. Images were processed by using Adobe Photoshop 7.0.
Statistical analysis.
Statistical analysis was conducted with Prism 5.0 (GraphPad). A two-tailed unpaired Student t test and the 95% confidence interval were used to calculate P values.
ACKNOWLEDGMENTS
We thank the NIH tetramer core facility (Emory University, Atlanta, GA) for providing the MHC II tetramer I-Ab GP66–77 for LCMV. We also thank W. Hahn for providing the pMKO.1 GFP vector.
This work was supported by the National Institutes of Health (NIH) (grant AI030048 to R.A.) and by the National Basic Research Program of China (973 Program; grant 2013CB531500 to L.Y.).
REFERENCES
- 1.Albert V, Hall MN. 2015. mTOR signaling in cellular and organismal energetics. Curr Opin Cell Biol 33:55–66. doi: 10.1016/j.ceb.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Laplante M, Sabatini DM. 2012. mTOR signaling in growth control and disease. Cell 149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dazert E, Hall MN. 2011. mTOR signaling in disease. Curr Opin Cell Biol 23:744–755. doi: 10.1016/j.ceb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 4.Abraham RT, Wiederrecht GJ. 1996. Immunopharmacology of rapamycin. Annu Rev Immunol 14:483–510. doi: 10.1146/annurev.immunol.14.1.483. [DOI] [PubMed] [Google Scholar]
- 5.Saunders RN, Metcalfe MS, Nicholson ML. 2001. Rapamycin in transplantation: a review of the evidence. Kidney Int 59:3–16. doi: 10.1046/j.1523-1755.2001.00460.x. [DOI] [PubMed] [Google Scholar]
- 6.Halloran PF. 2004. Immunosuppressive drugs for kidney transplantation. N Engl J Med 351:2715–2729. doi: 10.1056/NEJMra033540. [DOI] [PubMed] [Google Scholar]
- 7.Powell JD, Pollizzi KN, Heikamp EB, Horton MR. 2012. Regulation of immune responses by mTOR. Annu Rev Immunol 30:39–68. doi: 10.1146/annurev-immunol-020711-075024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohtani M, Nagai S, Kondo S, Mizuno S, Nakamura K, Tanabe M, Takeuchi T, Matsuda S, Koyasu S. 2008. Mammalian target of rapamycin and glycogen synthase kinase 3 differentially regulate lipopolysaccharide-induced interleukin-12 production in dendritic cells. Blood 112:635–643. doi: 10.1182/blood-2008-02-137430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weichhart T, Costantino G, Poglitsch M, Rosner M, Zeyda M, Stuhlmeier KM, Kolbe T, Stulnig TM, Horl WH, Hengstschlager M, Muller M, Saemann MD. 2008. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity 29:565–577. doi: 10.1016/j.immuni.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 10.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. 2009. mTOR regulates memory CD8 T-cell differentiation. Nature 460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. 2009. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plotkin SA. 2008. Vaccines: correlates of vaccine-induced immunity. Clin Infect Dis 47:401–409. doi: 10.1086/589862. [DOI] [PubMed] [Google Scholar]
- 13.Keating R, Hertz T, Wehenkel M, Harris TL, Edwards BA, McClaren JL, Brown SA, Surman S, Wilson ZS, Bradley P, Hurwitz J, Chi H, Doherty PC, Thomas PG, McGargill MA. 2013. The kinase mTOR modulates the antibody response to provide cross-protective immunity to lethal infection with influenza virus. Nat Immunol 14:1266–1276. doi: 10.1038/ni.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang S, Pruitt M, Tran D, Du Bois W, Zhang K, Patel R, Hoover S, Simpson RM, Simmons J, Gary J, Snapper CM, Casellas R, Mock BA. 2013. B cell-specific deficiencies in mTOR limit humoral immune responses. J Immunol 191:1692–1703. doi: 10.4049/jimmunol.1201767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones DD, Gaudette BT, Wilmore JR, Chernova I, Bortnick A, Weiss BM, Allman D. 2016. mTOR has distinct functions in generating versus sustaining humoral immunity. J Clin Invest 126:4250–4261. doi: 10.1172/JCI86504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hale JS, Youngblood B, Latner DR, Mohammed AU, Ye L, Akondy RS, Wu T, Iyer SS, Ahmed R. 2013. Distinct memory CD4(+) T cells with commitment to T follicular helper- and T helper 1-cell lineages are generated after acute viral infection. Immunity 38:805–817. doi: 10.1016/j.immuni.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA, Rutishauser R, Cui W, Kleinstein SH, Craft J, Kaech SM. 2011. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity 35:633–646. doi: 10.1016/j.immuni.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pepper M, Pagan AJ, Igyarto BZ, Taylor JJ, Jenkins MK. 2011. Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity 35:583–595. doi: 10.1016/j.immuni.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu L, Cao Y, Xie Z, Huang Q, Bai Q, Yang X, He R, Hao Y, Wang H, Zhao T, Fan Z, Qin A, Ye J, Zhou X, Ye L, Wu Y. 2015. The transcription factor TCF-1 initiates the differentiation of TFH cells during acute viral infection. Nat Immunol 16:991–999. doi: 10.1038/ni.3229. [DOI] [PubMed] [Google Scholar]
- 20.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. 2009. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, Powell JD. 2011. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol 12:295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, Magnuson MA, Boothby M. 2010. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity 32:743–753. doi: 10.1016/j.immuni.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ray JP, Staron MM, Shyer JA, Ho PC, Marshall HD, Gray SM, Laidlaw BJ, Araki K, Ahmed R, Kaech SM, Craft J. 2015. The interleukin-2-mTORc1 kinase axis defines the signaling, differentiation, and metabolism of T helper 1 and follicular B helper T cells. Immunity 43:690–702. doi: 10.1016/j.immuni.2015.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crotty S, Kersh EN, Cannons J, Schwartzberg PL, Ahmed R. 2003. SAP is required for generating long-term humoral immunity. Nature 421:282–287. doi: 10.1038/nature01318. [DOI] [PubMed] [Google Scholar]
- 25.Victora GD, Nussenzweig MC. 2012. Germinal centers. Annu Rev Immunol 30:429–457. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 26.Haynes NM, Allen CD, Lesley R, Ansel KM, Killeen N, Cyster JG. 2007. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol 179:5099–5108. doi: 10.4049/jimmunol.179.8.5099. [DOI] [PubMed] [Google Scholar]
- 27.Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, Crotty S. 2009. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325:1006–1010. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, Sallusto F. 2013. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity 38:596–605. doi: 10.1016/j.immuni.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 29.Martin F, Oliver AM, Kearney JF. 2001. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity 14:617–629. doi: 10.1016/S1074-7613(01)00129-7. [DOI] [PubMed] [Google Scholar]
- 30.Yang J, Lin X, Pan Y, Wang J, Chen P, Huang H, Xue HH, Gao J, Zhong XP. 2016. Critical roles of mTOR complex 1 and 2 for T follicular helper cell differentiation and germinal center responses. eLife 5:e17936. doi: 10.7554/eLife.17936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown SA, Cloer C, Kishton RJ, Gao X, Youngblood B, Do M, Li MO, Locasale JW, Rathmell JC, Chi H. 2016. mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity 45:540–554. doi: 10.1016/j.immuni.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weichhart T, Hengstschlager M, Linke M. 2015. Regulation of innate immune cell function by mTOR. Nat Rev Immunol 15:599–614. doi: 10.1038/nri3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kittleson MM, Kobashigawa JA. 2012. Antibody-mediated rejection. Curr Opin Organ Transplant 17:551–557. doi: 10.1097/MOT.0b013e3283577fef. [DOI] [PubMed] [Google Scholar]
- 34.Shao L, Lie AK, Zhang Y, Wong CH, Kwong YL. 2015. Aberrant germinal center formation, follicular T-helper cells, and germinal center B-cells were involved in chronic graft-versus-host disease. Ann Hematol 94:1493–1504. doi: 10.1007/s00277-015-2394-z. [DOI] [PubMed] [Google Scholar]
- 35.Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, Clish CB, Murphy LO, Manning BD. 2010. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, Wang Y, Jing Y, Yang H, Chen R, Chang L, Zhang Y, Goto J, Onda H, Chen T, Wang MR, Lu Y, You H, Kwiatkowski D, Zhang H. 2011. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci U S A 108:4129–4134. doi: 10.1073/pnas.1014769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, Kutok JL, Kearney JF, Otipoby KL, Rajewsky K. 2009. PI3 kinase signals BCR-dependent mature B cell survival. Cell 139:573–586. doi: 10.1016/j.cell.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jordan MS. 2006. Genetic reconstitution of bone marrow for the study of signal transduction ex vivo. Methods Mol Biol 332:331–342. [DOI] [PubMed] [Google Scholar]
- 39.Rasheed MA, Latner DR, Aubert RD, Gourley T, Spolski R, Davis CW, Langley WA, Ha SJ, Ye L, Sarkar S, Kalia V, Konieczny BT, Leonard WJ, Ahmed R. 2013. Interleukin-21 is a critical cytokine for the generation of virus-specific long-lived plasma cells. J Virol 87:7737–7746. doi: 10.1128/JVI.00063-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ye L, Zeng R, Bai Y, Roopenian DC, Zhu X. 2011. Efficient mucosal vaccination mediated by the neonatal Fc receptor. Nat Biotechnol 29:158–163. doi: 10.1038/nbt.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]